
Abstract
Adaptive immune receptor repertoires (AIRRs) are rich with information that can be mined for insights into the workings of the immune system. Gene usage, CDR3 properties, clonal lineage structure, and sequence diversity are all capable of revealing the dynamic immune response to perturbation by disease, vaccination, or other interventions. Here we focus on a conceptual introduction to the many aspects of repertoire analysis and orient the reader toward the uses and advantages of each. Along the way, we note some of the many software tools that have been developed for these investigations and link the ideas discussed to chapters on methods provided elsewhere in this volume.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others
Key words
1 Introduction
Once an adaptive immune receptor repertoire (AIRR) experiment has been carried out and the data has been appropriately preprocessed and annotated (see chapter “AIRR Community Guide to TR and IG Gene Annotation”), the next step is to plan a course of analysis to answer the questions posed by the experiment. As AIRRs are complex datasets that can contain thousands or even millions of sequences, it is important to have a working familiarity with the type of information each analysis can provide, as well as the limitations of an analysis. Here we provide an introduction to a variety of widely used techniques and discuss their applicability. In other chapters in this volume, we provide detailed experimental protocols and instructions to perform such analyses for the purpose of addressing specific biological questions. For a definition of terms used throughout this chapter, please see the AIRR Community glossary of terms, available at https://zenodo.org/record/5095381.
2 Materials
A breathtaking array of computational tools are available for repertoire analysis. These range from bespoke command line tools written in various programming languages that require facility in a Linux terminal to software with fully developed graphical interfaces and no requirement for programming skills of any kind. Thus, a key factor in choosing which programs to use will be the skill level and comfort of the user. Moreover, most tools have a narrow scope of the types of analysis they can perform, so matching the implementation to the desired goal is also a critical consideration. In addition, thought must be given to the computational resources necessary for repertoire analysis, including both storage and processing.
A comprehensive listing of the available software is out of the scope of this conceptual introduction, but the interested reader is directed to some recent reviews [1,2,3,4]. Here we focus on a small selection of commonly used tools, especially those which comply with AIRR Community guidelines for reproducibility and interoperability (https://docs.airr-community.org/en/stable/swtools/airr_swtools_standard.html). These are highlighted in Table 1, and several are discussed in more detail below and in other chapters in this volume, where we demonstrate their application to common analytical tasks.
3 Methods
In this section we introduce some of the most frequently used methods to analyze AIRRs and suggest computational tools that can perform such analysis. Some of the methods are applicable to both IG and TR , and some are specific. In addition, the selection of the method and the interpretation of the results can depend on the specific biological state; for instance, some samples might be expanded from solid tumors, others from antigen-specific cells isolated from peripheral blood or from whole blood from healthy and diseased patients. The theoretical framework presented here can be used to interpret the results of the practical methods detailed in the AIRR Community chapters “Bulk gDNA Sequencing of Antibody Heavy Chain Gene Rearrangements for Detection and Analysis of B-Cell Clone Distribution,” “Bulk Sequencing From mRNA With UMI for Evaluation of B-Cell Isotype and Clonal Evolution and Single-Cell Analysis,” and “Tracking of Antigen-Specific T Cells: Integrating Paired-Chain AIRR-Seq and Transcriptome Sequencing,” all in this volume.
3.1 Gene Usage
The V gene is the most diverse gene of the TR and IG loci. This is driven especially by variation in the first and second complementarity-determining regions (CDR1 and CDR2) of the genes, which contribute to the specificity and affinity of the immune receptor. Differences in the distribution of V genes used in the rearranged repertoire can indicate an antigen-specific response or unusual clonal expansions and can be evaluated with the function compareVGeneDistributions of the sumrep R package [20] (https://github.com/matsengrp/sumrep). The D and J gene strongly contributes to the CDR3 and can be compared using compareDGeneDistributions and compareJGeneDistributions. Skewing of the V-J usage can be revealed by plotting the V-J combination as a heatmap (Fig. 1a). The distribution of V-J and V-D-J usage can be compared between two repertoires using the functions compareVJDistributions and compareVDJDistributions in sumrep.

Data visualizations. Examples of different data visualizations to gain insights into the AIRR. The plot title describes the basic analysis. Smp = sample. For further details, please refer to the main text
3.2 Properties of the CDR3
The CDR3 is the most variable part of the rearranged IG /TR , and is a key contributor to the overall specificity of the receptor [26]. Therefore, analyzing the properties of this region is of great interest.
Due to the randomness in addition and deletion of nucleotides during the rearrangement of the receptor, CDR3 lengths will be distributed around a mean value (Fig. 1b). Any changes to this distribution signifies an expansion of cells with a particular immune receptor.
Different receptors specific for the same epitope can be expected to share motifs [27, 28]. Such motifs can be a few identical amino acids or amino acids with similar physical properties. Apart from properties like size, charge, and polarity, the properties of amino acids can be described by different factors derived through dimensionality reduction of a larger number of properties. Atchley [29] factors comprise five numerical descriptions, and Kidera [30] factors comprise ten numerical descriptions.
The R package sumrep [20] provides functions to compare the CDR3 properties of two repertoires, such as the CDR3 length and a number of amino acid physicochemical properties [31, 32].
3.3 Clonal Lineages
A clone or a clonal lineage comprises a group of T or B cells descended from the same original naive ancestor. As such, all cells in a clone contain the same set of rearrangements. An important part of AIRR-seq analysis is computationally reconstructing these relationships from the sequences obtained. For TRs, the exercise is relatively straightforward, as only PCR and sequencing error need to be accounted for. With IGs, however, somatic hypermutation can significantly obscure the ancestry of a particular sequence [33], and so more complex strategies are required (see Subheading 3.9.2).
When analyzing bulk AIRR-seq data, in which native pairing between heavy and light, alpha and beta, and gamma and delta chains is lost, clones are sometimes defined based on a single chain. This may be sufficient for IGH and TRB rearrangements, which are more diverse and contain most of the information needed to group sequences into clonal lineages [34]. However, care should still be taken in interpreting such data. Many different definitions of clonally related sequences have been offered in the literature (e.g., see the work by Kotouza and co-workers [35]), and methods to infer clones from AIRR-seq data are under active investigation [6, 12, 14, 18, 36].
The distribution of clone sizes in an AIRR can be informative of underlying biology. One visualization is to plot ranks from high to low on the x-axis and associated frequency on the y-axis (Fig. 1c) to reveal clonal expansion. A closer look at the top x (Fig. 1d) helps likewise to identify clonal expansion. When plotting the log of rank and the frequency (Fig. 1e), the slope reveals the distribution of clones, such that the steeper the slope, the less evenly distributed the repertoire. The function estimateAbundance in the R package Alakazam [6] can estimate clonal abundance with confidence intervals obtained by bootstrapping.
An alternative visualization makes use of division of clonal frequencies into different groups (“binning”) and sums the frequencies in each bin (Fig. 1f). The binning is essentially arbitrary, but binning the clone frequencies into the bins [0.0, 0.001], [0.001, 0.01], [0.01, 0.1], and [0.1, 1] are widely used. Binning by rank is an alternative where the bins [1, 10], [11, 100], [101, 1000], and [1001, inf] are common.
3.4 Diversity
The concept of diversity unites two properties of a repertoire, namely, the number of distinct clones and their distribution. As such, diversity describes the composition and state of a repertoire. For instance, a repertoire derived from a completely naive cell population is much more diverse both in terms of distinct clones and their distribution compared to the repertoire of antigen-specific memory cells.
There are numerous sampling factors that are important to consider when measuring diversity. Perhaps the most important is whether a sample is derived from gDNA or mRNA [37]. As discussed in Subheading 3.1 in chapter “AIRR Community Guide to TR and IG Gene Annotation,” in the case of gDNA, each sampled cell contributes one or two templates, while the number of templates in mRNA data will be skewed by cell subset-specific transcript abundance. In the case of the former, diversity measures will be influenced substantially less by the underlying subset distribution than the latter. For both, one can measure diversity weighted by copy number or by clone number. For DNA data, using copy number-weighted diversity measures can give a sense of how similar sequences are in the underlying repertoire while using an unweighted measure will indicate how similar clones are. With RNA, using copy number-weighted measures will give a general measure of how similar large clones are, and unweighted measures will give a measure of how similar all clones are.
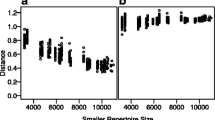
Another consideration when analyzing diversity is the depth of sequencing, that is, the proportion of clones that were sequenced compared to how many were actually in the sample. Assessing appropriate sequencing depth is no trivial task, but very important as undersampling can lead to false conclusions. Rarefaction curves [38] can help to evaluate if a repertoire is near full sampling depth. In this visualization, the number of distinct clones are plotted for a given subsample size (Fig. 1g). If the numbers of distinct clones plateau, the repertoire is near full sampling depth. Conversely, the absence of a plateau is an indication that the sampling depth of repertoire is shallow.
Another use of rarefaction is an estimation of the total number of clones from the sample. To achieve this, libraries from the sample of interest must be run in replicates, where more replicates give a more accurate estimate of total clones [39].
There are a large number of diversity metrics. These different metrics are all united in Hill numbers which are calculated over a range of diversities to generate a smooth curve (Fig. 1h) [40,41,42]. The function calcDiversity in the R package Alakazam estimates the Hill numbers for a repertoire. The same function also makes calculation of particular diversity indices straightforward. The function compareHillNumbers in the R package sumrep compares one or more Hill numbers of two repertoires. Newer approaches toward diversity metrics specific for AIRR make use of Hill numbers combined with a functional similarity matrix [43].
3.5 Similarity of AIRR Sequences
The similarity of AIRR sequences directly influences antigen recognition breadth: the more dissimilar the receptors are, the larger is the antigen space covered. One major approach to interrogate and measure AIRR sequence similarity is network analysis (Fig. 1i) [44,45,46,47,48,49,50]. Networks allow investigation of sequence similarity and thereby add a complementary layer of information to repertoire diversity analysis. Sequence networks are built by defining each nucleotide or amino acid sequence as a node. Two nodes are connected with an edge if a certain similarity condition is satisfied, which is typically defined as a string distance (e.g., Levenshtein/edit distance). A commonly used distance for both IG and TR is one amino acid difference [44]. For B cells, networks representing amino acid distances of up to 12 amino acids have been reported [47]. Building a sequence similarity network is computationally expensive. This challenge has been approached by at least two methods that allow the construction of large-scale networks from millions of AIRR sequences [47, 51].
Although networks of a few thousand nodes may be visualized using software suites such as igraph, Cytoscape, and Gephi [52, 53], and the visual interpretation of networks becomes indiscernible with a size of >102 nodes. Furthermore, the visualization of networks does not provide quantitative information regarding the network similarity architecture. To address this problem, graph properties and network analysis have recently been employed to quantify the architecture of large-scale AIRR networks [47]. Architecture analytics may be subdivided into properties that capture the repertoire at the global level (generally one coefficient per network), and those that describe the repertoire at the local level (one coefficient per sequence per repertoire). These network measures may be used to identify enrichment of network clusters (Fig. 1i), potentially originating from an ongoing immune response [46, 47].
To increase precision in isolating immune-associated AIRR sequences and clusters therefore, network analysis may be coupled with AIRR generation probabilities [45]. More generally, it has been observed that sequences that tend to show increased sharing across individuals (discussed in the see Subheading 3.7), are also more connected within a repertoire [45, 47, 48] and confer robustness on its architecture with respect to network properties [47].
Recently, sequence similarity and diversity analysis have been combined, providing further insights into AIRR architecture [43].
3.6 Similarity among Repertoires
Similarity indices measure the similarity of two populations by not only considering the number of shared clones but also taking clone count or frequency into account (Fig. 1j). Similarity is sometimes calculated as dissimilarity (for historical reasons), but the index is always in the range of [0, 1]. It is therefore important to indicate the meaning of 0 and 1 to avoid confusion. One of the most popular indices is called Morisita-Horn, implemented in the function vegdist in the R package vegan [54]. Numerically, the observed overlaps are usually small, but considering the potential repertoire being sampled, the upfront chance of an overlap is very small. Alternatively, the CDR3s shared between samples can be plotted as a true/false heatmap (Fig. 1k). This is particularly useful when tracking clones over time or assessing the specificity of transplant infiltrating cells [55, 56].
Similarities on other parameters such as different amino acid properties as well as pairwise CDR3 distance and GC content can be compared between repertoires by the function compareRepertoires in the R package sumrep.
Other proposed similarity measures make use of feature counting [57], while another B-cell-specific similarity metric focuses on identical CDR3 length together with identical V and J genes considered within and between repertoires [58].
3.7 Public Clones
Though not clones in a true biological sense, the existence of identical TRs and identical or closely similar IGs in multiple individuals due to convergent rearrangement has been noted on several occasions [59,60,61]. Such rearrangements are termed public clones and can yield insights into common selection patterns, which in turn can elucidate how the immune system responds to disease and if there are commonalities between individuals. The ability to identify public clones in an AIRR depends on the sequencing depth and the number of individuals tested [62, 63]. In addition, the meaning of a public immune receptor must be assessed in the context of the likelihood for it to be generated [8, 13]. Receptors with shorter CDR3s are more likely to be generated by chance and can overlap even between individuals with no exposures in common [60, 64, 65] and do not necessarily indicate a convergent response in multiple individuals to similar antigens. Sequences that share the same (preferably longer) CDR3 amino acid sequence but have different nucleotide sequences are more convincing as candidate public clones, as differences in the nucleotide sequences may indicate independent generation with convergent selection [66].
Functionally identical IG can be identified by allowing some degree of difference in the CDR3 . There is no well-defined cutoff to ensure the capture of a majority of receptors with identical specificities without including IGs of unrelated specificity into a particular collection of public IGs. A commonly used cutoff is 10–20% amino acid difference in the CDR3 [67,68,69,70]. Although a less restrictive cutoff might detect more divergent public clones [71], care must be taken to avoid identification of spurious public immune receptors [72]. Cross-contamination and index hopping on the sequencer further complicate the identification of public clones [73], and suitable definitions and analysis parameters may be helpful.
3.8 Detection and Monitoring of Cross-Sample Contamination Events
Despite strict quality assurance and control measures, PCR-based sample cross-contamination can occur at any time. Environmental contamination events are expected to arise from the presence of remaining DNA amplicons, which can be re-amplified and incorporated into new, unrelated libraries [74]. PCR contaminations can lead to major losses of reagents, time, and samples, and rapid detection and isolation are critical to the health of an AIRR-seq research laboratory. There are several experimental precautions that can reduce contamination, including separate work areas and different sample barcodes, as illustrated in the AIRR Community chapter “Quality Control: Chain Pairing Precision and Monitoring of Cross-Sample Contamination.”
3.9 B-Cell-Specific Aspects
3.9.1 IG SHM Analysis
SHM is the process driving the affinity maturation of IGs during the adaptive immune response [75]. Mutations are introduced at a rate of ~10−3 mutations per base pair per division. These mutations are not randomly distributed along the IG but accumulate more in hotspots and CDRs, whereas coldspots and framework regions are disfavored for mutation. Furthermore, substitution profiles may be germline gene-directed [76,77,78,79], possibly as a consequence of specific features of the encoded protein sequence. Understanding SHM biases is key to develop better tools to reconstruct lineages, quantify selection pressure, and generate realistic simulated sequence data [9, 79, 80].
To better understand the distribution of targets for SHM , it is, for instance, possible to use the R package sumrep that provides two functions getHotspotCountDistribution and getColdspotCountDistribution to the distribution of the hot- and coldspot motifs in the repertoire. In addition, sumrep interfaces with the R package SHazaM [6], which calculates a mutability model for the likelihood for the center base in a 5-mer to be mutated (the function getMutabilityModel). The associated function getSubstitutionModel provides the relative probabilities that the center base in a 5-mer is mutated into each of the other three nucleotides. SHazaM also provides methods for quantification of selection pressure and whether it has contributed to the nature of the specific IG repertoire during antigenic stimulation [81].
3.9.2 Identification of B-Cell Clones
As noted above, B-cell clones can be inferred from AIRR-seq data by analyzing their CDR3s and/or mutation patterns (Fig. 1l). Repertoires usually consist of hundreds or thousands of clonal lineages. Due to the presence of SHM , members of a B-cell clone cannot be identified solely based on identical CDR3s. There are many methods available to group IGs into clonal lineages (Table 1), but all generally attempt to computationally group sequences which likely share a common progenitor. However, different approaches can drastically change the interpretation of the underlying IG immune repertoire.
Some approaches begin by grouping sequences by their CDR3 independent of their V, D, or J gene usage [22]. Other software first groups sequences by gene (generally just V and J due to the difficulty in D gene annotation) and CDR3 length after which sequences similar in the CDR3 are grouped into clonal lineages [12, 19, 82, 83]. SCOPer does a similar grouping, but then evaluates the similarity by analyzing shared SHM in the V and J genes [84]. Finally, some pipelines use common mutations in the body of the V gene to group sequences from the same clonal lineage [36, 85]. It is also possible to combine these approaches, but this section focuses on each independently.
Each approach has potential benefits and flaws. Initially grouping sequences by CDR3 , either by identity or hierarchical clustering, can result in inflated copy number and sequence counts for common CDR3s (in particular those of short length that incorporate few non-templated bases) which may have arisen independently and utilize different genes. However, this method can be beneficial as some gene calls may be incorrect (in particular when annotation of sequences has not been made using a personalized repertoire as defined above), and similar CDR3 amino-acid sequences, especially those with long lengths, can indicate that sequences are related.
Grouping sequences by both gene annotation and CDR3 length prior to inferring clonal lineages can be beneficial for a number of reasons. Because V gene annotation is generally robust to sequencing error, sequences with similar CDR3s but different V gene assignments are unlikely to derive from the same rearrangement. Binning by gene annotation can therefore prevent erroneous clonal groupings. It also eases the computational burden, as CDR3 identity only needs calculation among smaller sets of sequences. Similar advantages apply to binning by CDR3 length as well, since distance metrics can be calculated more efficiently without the need for alignment. While insertions and deletions can occur as part of SHM , they are relatively rare [86, 87] and can be neglected in many cases.
Once sequences have been binned, hierarchical clustering is a common technique for identifying clonally related sequences [82]. This requires a choice of linkage (e.g., single, average) to define the distance between groups of sequences and a threshold for cutting the hierarchy into discrete groups. A convenient way to set the threshold is to analyze the distribution of distances between nearest neighbors. This distribution is typically bimodal, with the first mode representing sequences in the same clonal lineage, while the second mode represents sequences that do not have any relatives in the data. If the distribution for a particular sample is not bimodal, a set of external sequences from a different subject can be used to establish the threshold [82]. While the threshold for separating the two modes can sometimes be established by visual inspection of the distribution, there are algorithmic methods to determine it more consistently [18].
The last common approach is to group sequences into clones by common mutations in the body of the V gene. This can be done by constructing clonal lineages directly or by inspecting the k-mers of each sequence [36, 88]. Unlike methods that first separate sequences by gene call and junction length, this method takes advantage of infrequent mutations to group sequences into clones. This can be beneficial for a number of reasons in certain circumstances. First, this method does not rely on proper gene calling or sequence alignment, which can be difficult in samples containing highly mutated populations or more generally due to sequencing error. Additionally, it is not sensitive to junction length, allowing sequences that have accumulated insertions and deletions to be grouped into clones [89, 90]. This method necessitates one to define the minimum number of mutations required to group two sequences into the same clone. A fixed value can be used, or the value can be dynamically determined based on the distribution of distances between each pair of sequences.
3.9.3 IG Affinity Maturation
The reconstruction and analysis of IG clonal lineages trees is a powerful method to understand the immune response, affinity maturation, and the generation of broadly neutralizing antibodies (bNAb) [91,92,93]. Within a B-cell clonal lineage, B cells descended from a shared common ancestor evolve through SHM and antigen-driven selection. While standard algorithms for inferring phylogenetic trees using maximum parsimony and maximum likelihood [94] are often employed, these approaches can be improved [80]. In particular, the unique biology of B cells can present problems for standard phylogenetic approaches and has led to the development of B-cell-specific phylogenetic tools. One cause of the problems is that SHM is enzymatically driven and biased by hotspot and coldspot motifs. This violates the assumption of independent evolution among sites that many likelihood-based phylogenetics methods rely on. To address this challenge, more context-aware phylogenetic methods, such as IgPhyML [9, 10], have been developed. While context-aware models of SHM clearly improve estimates of phylogenetic model parameters used to detect antigen-driven selection [10], it is less clear how much they improve estimates of tree topology and branch lengths [95]. Another problem is that while standard phylogenetic models consider clonal lineages individually, IG repertoires often contain hundreds of independent clones. The use of repertoire-wide models, which allow some parameters to be shared among these multiple clonal lineages, can improve model precision significantly [10]. One important application of B-cell phylogenetics is estimating the series of mutations leading from a clone’s unmutated germline ancestor to a sequence of interest, such as a known bnAb sequence. While standard phylogenetic methods can reconstruct intermediate sequences, they are less appropriate for reconstructing the germline ancestral sequence because they do not take into account the biology of V(D)J rearrangement. This has led to the development of tools such as Clonalyst and linearham [96, 97] that improve the reconstruction of these sequences by combining phylogenetic models with models of V(D)J rearrangement. Another feature of B-cell clonal lineages is that reconstructed intermediate sequences are often identical to observed IG sequences. Some tools, such as IgTree [98] and Alakazam [6], use this fact to simplify the visualization of these lineage trees by collapsing observed and sampled intermediate nodes. Finally, lineage trees containing B cells from multiple tissues, isotypes, and timepoints have the potential to be used to make inferences about how B-cell migration, isotype switching, and evolution over time occur. Multiple analyses have used lineage trees for this purpose [33, 40, 99, 100], and generalized tools for making these inferences from B-cell repertoires, such as Dowser and PopTree, are an area of active development [7].
3.10 T-Cell-Specific Aspects
There is growing evidence that TR repertoire perturbations can serve as a biomarker of immune response toward some solid tumors [101,102,103] and pathogens such as Epstein-Barr virus (EBV), cytomegalovirus (CMV), Ebola, and SARS-CoV-2 [104,105,106,107,108]. Challenges with studying T-cell repertoires include the dependence of T-cell interactions on the major histocompatibility complex (MHC) [109], changes in TRBV usage based on MHC and significant differences in TRBV usage, and clonality in CD4+ and CD8+ repertoires [110,111,112].
Antigen-specific TCRs can be isolated either by sorting of MHC-tetramer-positive cells or activated cells after stimulation with overlapping peptide pools. Staining with tetramers requires knowledge of the correct epitope in the right MHC context, and T cells with high affinity tend to be recovered with the highest efficiency. Therefore, tetramer staining sometimes fails to identify some of the relevant TCRs [113]. Stimulation with overlapping peptide pools, on the other hand, can lead to isolation of non-peptide-specific T cells due to bystander activation [114]. The TR of the antigen-enriched cells can be compared to samples from different timepoints to track the frequency of clones of interest [104, 106].
4 Conclusion
In this chapter, we have provided a brief overview of diverse, widely used techniques to uncover biological information in AIRR-seq data. These techniques can be applied to all of the AIRR-seq data created using the methodologies described in this book. They further form the basis for selecting the optimal experimental protocol to address the biological question and choosing the computational methods used in the analysis.
References
Zhang Y, Yang X, Zhang Y, Zhang Y, Wang M, Ou JX et al (2020) Tools for fundamental analysis functions of TCR repertoires: a systematic comparison. Brief Bioinform 21:1706–1716. https://doi.org/10.1093/bib/bbz092
López-Santibáñez-Jácome L, Avendaño-Vázquez SE, Flores-Jasso CF (2019) The pipeline repertoire for Ig-seq analysis. Front Immunol 10:899. https://doi.org/10.3389/fimmu.2019.00899
Lees WD (2020) Tools for adaptive immune receptor repertoire sequencing. Curr Opin Syst Biol 24:86–92. https://doi.org/10.1016/j.coisb.2020.10.003
Smakaj E, Babrak L, Ohlin M, Shugay M, Briney B, Tosoni D et al (2020) Benchmarking immunoinformatic tools for the analysis of antibody repertoire sequences. Bioinformatics 36:1731–1739. https://doi.org/10.1093/bioinformatics/btz845
Martin ACR (2010) Protein sequence and structure analysis of antibody variable domains. In: Kontermann R, Dübel S (eds) Antibody engineering. Springer, Berlin, pp 33–51
Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH (2015) Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31:3356–3358. https://doi.org/10.1093/bioinformatics/btv359
Hoehn KB, Pybus OG, Kleinstein SH (2020) Phylogenetic analysis of migration, differentiation, and class switching in B cells. Immunology
Marcou Q, Mora T, Walczak AM (2018) High-throughput immune repertoire analysis with IGoR. Nat Commun 9:561. https://doi.org/10.1038/s41467-018-02832-w
Hoehn KB, Lunter G, Pybus OG (2017) A phylogenetic codon substitution model for antibody lineages. Genetics 206:417–427. https://doi.org/10.1534/genetics.116.196303
Hoehn KB, Vander Heiden JA, Zhou JQ, Lunter G, Pybus OG, Kleinstein SH (2019) Repertoire-wide phylogenetic models of B cell molecular evolution reveal evolutionary signatures of aging and vaccination. Proc Natl Acad Sci U S A 116:22664–22672. https://doi.org/10.1073/pnas.1906020116
ImmunoMind Team (2019) immunarch: an R Package for painless analysis of large-scale immune repertoire data
Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV et al (2015) MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 12:380–381. https://doi.org/10.1038/nmeth.3364
Sethna Z, Elhanati Y, Callan CG, Walczak AM, Mora T (2019) OLGA: fast computation of generation probabilities of B- and T-cell receptor amino acid sequences and motifs. Bioinformatics 35:2974–2981. https://doi.org/10.1093/bioinformatics/btz035
Ralph DK, Matsen FA (2016) Likelihood-based inference of B cell clonal families. PLoS Comput Biol 12:e1005086. https://doi.org/10.1371/journal.pcbi.1005086
Ralph DK, Matsen FA (2020) Using B cell receptor lineage structures to predict affinity. PLoS Comput Biol 16:e1008391. https://doi.org/10.1371/journal.pcbi.1008391
Gidoni M, Snir O, Peres A, Polak P, Lindeman I, Mikocziova I, IMI test presentation (2019) Mosaic deletion patterns of the human antibody heavy chain gene locus shown by Bayesian haplotyping. Nat Commun 10:628. https://doi.org/10.1038/s41467-019-08489-3
Sturm G, Szabo T, Fotakis G, Haider M, Rieder D, Trajanoski Z, IMI test presentation (2020) Scirpy: a Scanpy extension for analyzing single-cell T-cell receptor-sequencing data. Bioinformatics 36:4817–4818. https://doi.org/10.1093/bioinformatics/btaa611
Nouri N, Kleinstein SH (2018) A spectral clustering-based method for identifying clones from high-throughput B cell repertoire sequencing data. Bioinformatics 34:i341–i349. https://doi.org/10.1093/bioinformatics/bty235
Schramm CA, Sheng Z, Zhang Z, Mascola JR, Kwong PD, Shapiro L (2016) SONAR: a high-throughput pipeline for inferring antibody ontogenies from longitudinal sequencing of B cell transcripts. Front Immunol 7:372. https://doi.org/10.3389/fimmu.2016.00372
Olson BJ, Moghimi P, Schramm CA, Obraztsova A, Ralph D, Vander Heiden JA et al (2019) Sumrep: a summary statistic framework for immune receptor repertoire comparison and model validation. Front Immunol 10:2533. https://doi.org/10.3389/fimmu.2019.02533
Lees WD, Shepherd AJ (2015) Utilities for high-throughput analysis of B-cell clonal lineages. J Immunol Res 2015:1–9. https://doi.org/10.1155/2015/323506
Giraud M, Salson M, Duez M, Villenet C, Quief S, Caillault A et al (2014) Fast multiclonal clusterization of V(D)J recombinations from high-throughput sequencing. BMC Genomics 15:409. https://doi.org/10.1186/1471-2164-15-409
Duez M, Giraud M, Herbert R, Rocher T, Salson M, Thonier F (2016) Vidjil: a web platform for analysis of high-throughput repertoire sequencing. PLoS One 11:e0166126. https://doi.org/10.1371/journal.pone.0166126
Christley S, Scarborough W, Salinas E, Rounds WH, Toby IT, Fonner JM, IMI test presentation (2018) VDJServer: a cloud-based analysis portal and data commons for immune repertoire sequences and rearrangements. Front Immunol 9:976. https://doi.org/10.3389/fimmu.2018.00976
Rosenfeld AM, Meng W, Luning Prak ET, Hershberg U (2018) ImmuneDB, a novel tool for the analysis, storage, and dissemination of immune repertoire sequencing data. Front Immunol 9:2107. https://doi.org/10.3389/fimmu.2018.02107
Xu JL, Davis MM (2000) Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity 13:37–45. https://doi.org/10.1016/S1074-7613(00)00006-6
Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F et al (2017) Identifying specificity groups in the T cell receptor repertoire. Nature 547:94–98. https://doi.org/10.1038/nature22976
Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A et al (2017) Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547:89–93. https://doi.org/10.1038/nature22383
Atchley WR, Zhao J, Fernandes AD, Druke T (2005) Solving the protein sequence metric problem. Proc Natl Acad Sci U S A 102:6395–6400. https://doi.org/10.1073/pnas.0408677102
Kidera A, Konishi Y, Oka M, Ooi T, Scheraga HA (1985) Statistical analysis of the physical properties of the 20 naturally occurring amino acids. J Protein Chem 4:23–55. https://doi.org/10.1007/BF01025492
Haigh OL, Grant EJ, Nguyen THO, Kedzierska K, Field MA, Miles JJ (2021) Genetic bias, diversity indices, physiochemical properties and CDR3 motifs divide auto-reactive from Allo-reactive T-cell repertoires. Int J Mol Sci 22:1625. https://doi.org/10.3390/ijms22041625
Sankar K, Hoi KH, Hötzel I (2020) Dynamics of heavy chain junctional length biases in antibody repertoires. Commun Biol 3:207. https://doi.org/10.1038/s42003-020-0931-3
Wu X, Zhang Z, Schramm CA, Joyce MG, Kwon YD, Zhou T et al (2015) Maturation and diversity of the VRC01-antibody lineage over 15 years of chronic HIV-1 infection. Cell 161:470–485. https://doi.org/10.1016/j.cell.2015.03.004
Zhou JQ, Kleinstein SH (2019) Cutting edge: Ig H chains are sufficient to determine most B cell clonal relationships. J Immunol 203:1687–1692. https://doi.org/10.4049/jimmunol.1900666
Kotouza MT, Gemenetzi K, Galigalidou C, Vlachonikola E, Pechlivanis N, Agathangelidis A et al (2020) TRIP - T cell receptor/immunoglobulin profiler. BMC Bioinformatics 21:422. https://doi.org/10.1186/s12859-020-03669-1
Lindenbaum O, Nouri N, Kluger Y, Kleinstein SH (2021) Alignment free identification of clones in B cell receptor repertoires. Nucleic Acids Res 49:e21–e21. https://doi.org/10.1093/nar/gkaa1160
Bashford-Rogers RJM, Palser AL, Idris SF, Carter L, Epstein M, Callard RE et al (2014) Capturing needles in haystacks: a comparison of B-cell receptor sequencing methods. BMC Immunol 15:29. https://doi.org/10.1186/s12865-014-0029-0
Gotelli NJ, Colwell RK (2001) Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol Lett 4:379–391. https://doi.org/10.1046/j.1461-0248.2001.00230.x
Greiff V, Menzel U, Haessler U, Cook SC, Friedensohn S, Khan TA et al (2014) Quantitative assessment of the robustness of next-generation sequencing of antibody variable gene repertoires from immunized mice. BMC Immunol 15:40. https://doi.org/10.1186/s12865-014-0040-5
Stern JNH, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ et al (2014) B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med 6:248ra107. https://doi.org/10.1126/scitranslmed.3008879
Greiff V, Bhat P, Cook SC, Menzel U, Kang W, Reddy ST (2015) A bioinformatic framework for immune repertoire diversity profiling enables detection of immunological status. Genome Med 7:49. https://doi.org/10.1186/s13073-015-0169-8
Hill MO (1973) Diversity and evenness: a unifying notation and its consequences. Ecology 54:427–432. https://doi.org/10.2307/1934352
Arora R, Burke HM, Arnaout R (2018) Immunological diversity with similarity. Immunology
Miho E, Yermanos A, Weber CR, Berger CT, Reddy ST, Greiff V (2018) Computational strategies for dissecting the high-dimensional complexity of adaptive immune repertoires. Front Immunol 9:224. https://doi.org/10.3389/fimmu.2018.00224
Pogorelyy MV, Minervina AA, Shugay M, Chudakov DM, Lebedev YB, Mora T et al (2019) Detecting T cell receptors involved in immune responses from single repertoire snapshots. PLoS Biol 17:e3000314. https://doi.org/10.1371/journal.pbio.3000314
Ben-Hamo R, Efroni S (2011) The whole-organism heavy chain B cell repertoire from zebrafish self-organizes into distinct network features. BMC Syst Biol 5:27. https://doi.org/10.1186/1752-0509-5-27
Miho E, Roškar R, Greiff V, Reddy ST (2019) Large-scale network analysis reveals the sequence space architecture of antibody repertoires. Nat Commun 10:1321. https://doi.org/10.1038/s41467-019-09278-8
Madi A, Poran A, Shifrut E, Reich-Zeliger S, Greenstein E, Zaretsky I et al (2017) T cell receptor repertoires of mice and humans are clustered in similarity networks around conserved public CDR3 sequences. eLife 6:e22057. https://doi.org/10.7554/eLife.22057
Madi A, Shifrut E, Reich-Zeliger S, Gal H, Best K, Ndifon W et al (2014) T-cell receptor repertoires share a restricted set of public and abundant CDR3 sequences that are associated with self-related immunity. Genome Res 24:1603–1612. https://doi.org/10.1101/gr.170753.113
Bashford-Rogers RJM, Palser AL, Huntly BJ, Rance R, Vassiliou GS, Follows GA et al (2013) Network properties derived from deep sequencing of human B-cell receptor repertoires delineate B-cell populations. Genome Res 23:1874–1884. https://doi.org/10.1101/gr.154815.113
Valkiers S, Van Houcke M, Laukens K, Meysman P (2021) clusTCR: a python interface for rapid clustering of large sets of CDR3 sequences. Bioinformatics
Csardi G, Nepusz T (2006) The igraph software package for complex network research. InterJournal Complex Systems 1695
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. https://doi.org/10.1101/gr.1239303
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D et al (2019) Vegan: community ecology package
Stervbo U, Nienen M, Hecht J, Viebahn R, Amann K, Westhoff TH et al (2020) Differential diagnosis of interstitial allograft rejection and BKV nephropathy by T-cell receptor sequencing. Transplantation 104:e107–e108. https://doi.org/10.1097/TP.0000000000003054
Nienen M, Stervbo U, Mölder F, Kaliszczyk S, Kuchenbecker L, Gayova L et al (2019) The role of pre-existing cross-reactive central memory CD4 T-cells in vaccination with previously unseen influenza strains. Front Immunol 10:593. https://doi.org/10.3389/fimmu.2019.00593
Bolen CR, Rubelt F, Vander Heiden JA, Davis MM (2017) The repertoire dissimilarity index as a method to compare lymphocyte receptor repertoires. BMC Bioinformatics 18:155. https://doi.org/10.1186/s12859-017-1556-5
Greiff V, Menzel U, Miho E, Weber C, Riedel R, Cook S et al (2017) Systems analysis reveals high genetic and antigen-driven predetermination of antibody repertoires throughout B cell development. Cell Rep 19:1467–1478. https://doi.org/10.1016/j.celrep.2017.04.054
Bradley P, Thomas PG (2019) Using T cell receptor repertoires to understand the principles of adaptive immune recognition. Annu Rev Immunol 37:547–570. https://doi.org/10.1146/annurev-immunol-042718-041757
Soto C, Bombardi RG, Branchizio A, Kose N, Matta P, Sevy AM et al (2019) High frequency of shared clonotypes in human B cell receptor repertoires. Nature 566:398–402. https://doi.org/10.1038/s41586-019-0934-8
Greiff V, Weber CR, Palme J, Bodenhofer U, Miho E, Menzel U et al (2017) Learning the high-dimensional Immunogenomic features that predict public and private antibody repertoires. J Immunol 199:2985–2997. https://doi.org/10.4049/jimmunol.1700594
Elhanati Y, Sethna Z, Callan CG, Mora T, Walczak AM (2018) Predicting the spectrum of TCR repertoire sharing with a data-driven model of recombination. Immunol Rev 284:167–179. https://doi.org/10.1111/imr.12665
Greiff V, Miho E, Menzel U, Reddy ST (2015) Bioinformatic and statistical analysis of adaptive immune repertoires. Trends Immunol 36:738–749. https://doi.org/10.1016/j.it.2015.09.006
Briney B, Inderbitzin A, Joyce C, Burton DR (2019) Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566:393–397. https://doi.org/10.1038/s41586-019-0879-y
Soto C, Bombardi RG, Kozhevnikov M, Sinkovits RS, Chen EC, Branchizio A et al (2020) High frequency of shared clonotypes in human T cell receptor repertoires. Cell Rep 32:107882. https://doi.org/10.1016/j.celrep.2020.107882
Venturi V, Quigley MF, Greenaway HY, Ng PC, Ende ZS, McIntosh T et al (2011) A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J Immunol 186:4285–4294. https://doi.org/10.4049/jimmunol.1003898
Nielsen SCA, Yang F, Hoh RA, Jackson KJL, Roeltgen K, Lee J-Y et al (2020) B cell clonal expansion and convergent antibody responses to SARS-CoV-2. Res Sq
Nielsen SCA, Yang F, Jackson KJL, Hoh RA, Röltgen K, Jean GH (2020) Human B cell clonal expansion and convergent antibody responses to SARS-CoV-2. Cell Host Microbe 28:516–525.e5. https://doi.org/10.1016/j.chom.2020.09.002
Kim SI, Noh J, Kim S, Choi Y, Yoo DK, Lee Y et al (2021) Stereotypic neutralizing V H antibodies against SARS-CoV-2 spike protein receptor binding domain in patients with COVID-19 and healthy individuals. Sci Transl Med 13:eabd6990. https://doi.org/10.1126/scitranslmed.abd6990
Galson JD, Schaetzle S, Bashford-Rogers RJM, Raybould MIJ, Kovaltsuk A, Kilpatrick GJ et al (2020) Deep sequencing of B cell receptor repertoires from COVID-19 patients reveals strong convergent immune signatures. Front Immunol 11:605170. https://doi.org/10.3389/fimmu.2020.605170
Ohlin M (2014) A new look at a poorly immunogenic neutralization epitope on cytomegalovirus glycoprotein B. Is there cause for antigen redesign? Mol Immunol 60:95–102. https://doi.org/10.1016/j.molimm.2014.03.015
Japp AS, Meng W, Rosenfeld AM, Perry DJ, Thirawatananond P, Bacher RL et al (2021) TCR+/BCR+ dual-expressing cells and their associated public BCR clonotype are not enriched in type 1 diabetes. Cell 184:827–839.e14. https://doi.org/10.1016/j.cell.2020.11.035
Costello M, Fleharty M, Abreu J, Farjoun Y, Ferriera S, Holmes L et al (2018) Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genomics 19:332. https://doi.org/10.1186/s12864-018-4703-0
Seitz V, Schaper S, Dröge A, Lenze D, Hummel M, Hennig S (2015) A new method to prevent carry-over contaminations in two-step PCR NGS library preparations. Nucleic Acids Res 43(20):e135. https://doi.org/10.1093/nar/gkv694
Methot SP, Di Noia JM (2017) Molecular mechanisms of somatic Hypermutation and class switch recombination. Adv Immunol 133:37–87
Sheng Z, Schramm CA, Kong R, Comparative Sequencing Program NISC, Mullikin JC, Mascola JR et al (2017) Gene-specific substitution profiles describe the types and frequencies of amino acid changes during antibody somatic Hypermutation. Front Immunol 8:537. https://doi.org/10.3389/fimmu.2017.00537
Schramm CA, Douek DC (2018) Beyond hot spots: biases in antibody somatic hypermutation and implications for vaccine design. Front Immunol 9:1876. https://doi.org/10.3389/fimmu.2018.01876
Kirik U, Persson H, Levander F, Greiff L, Ohlin M (2017) Antibody heavy chain variable domains of different germline gene origins diversify through different paths. Front Immunol 8:1433. https://doi.org/10.3389/fimmu.2017.01433
Zhou JQ, Kleinstein SH (2020) Position-dependent differential targeting of somatic Hypermutation. J Immunol 205:3468–3479. https://doi.org/10.4049/jimmunol.2000496
Yermanos A, Greiff V, Krautler NJ, Menzel U, Dounas A et al (2017) Comparison of methods for phylogenetic B-cell lineage inference using time-resolved antibody repertoire simulations (AbSim). Bioinformatics 33:3938–3946. https://doi.org/10.1093/bioinformatics/btx533
Yaari G, Uduman M, Kleinstein SH (2012) Quantifying selection in high-throughput immunoglobulin sequencing data sets. Nucleic Acids Res 40:e134–e134. https://doi.org/10.1093/nar/gks457
Gupta NT, Adams KD, Briggs AW, Timberlake SC, Vigneault F, Kleinstein SH (2017) Hierarchical clustering can identify B cell clones with high confidence in Ig repertoire sequencing data. J Immunol 198:2489–2499. https://doi.org/10.4049/jimmunol.1601850
Aouinti S, Malouche D, Giudicelli V, Kossida S, Lefranc M-P (2015) IMGT/HighV-QUEST statistical significance of IMGT Clonotype (AA) diversity per gene for standardized comparisons of next generation sequencing immunoprofiles of immunoglobulins and T cell receptors. PLoS One 10:e0142353. https://doi.org/10.1371/journal.pone.0142353
Nouri N, Kleinstein SH (2020) Somatic hypermutation analysis for improved identification of B cell clonal families from next-generation sequencing data. PLoS Comput Biol 16:e1007977. https://doi.org/10.1371/journal.pcbi.1007977
Briney B, Le K, Zhu J, Burton DR (2016) Clonify: unseeded antibody lineage assignment from next-generation sequencing data. Sci Rep 6:23901. https://doi.org/10.1038/srep23901
Briney BS, Willis JR, Crowe JE (2012) Location and length distribution of somatic hypermutation-associated DNA insertions and deletions reveals regions of antibody structural plasticity. Genes Immun 13:523–529. https://doi.org/10.1038/gene.2012.28
Briney BS, Willis JR, Crowe JE (2012) Human peripheral blood antibodies with long HCDR3s are established primarily at original recombination using a limited subset of germline genes. PLoS One 7:e36750. https://doi.org/10.1371/journal.pone.0036750
Yaari G, Benichou JIC, Vander Heiden JA, Kleinstein SH, Louzoun Y (2015) The mutation patterns in B-cell immunoglobulin receptors reflect the influence of selection acting at multiple time-scales. Philos Trans R Soc B Biol Sci 370:20140242. https://doi.org/10.1098/rstb.2014.0242
Wilson PC, de Bouteiller O, Liu Y-J, Potter K, Banchereau J, Capra JD et al (1998) Somatic Hypermutation introduces insertions and deletions into immunoglobulin V genes. J Exp Med 187:59–70. https://doi.org/10.1084/jem.187.1.59
Ohlin M, Borrebaeck CAK (1998) Insertions and deletions in hypervariable loops of antibody heavy chains contribute to molecular diversity. Mol Immunol 35:233–238. https://doi.org/10.1016/S0161-5890(98)00030-3
Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB, Rothstein TL, Weigert MG (1987) The role of clonal selection and somatic mutation in autoimmunity. Nature 328:805–811. https://doi.org/10.1038/328805a0
Haynes BF, Kelsoe G, Harrison SC, Kepler TB (2012) B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol 30:423–433. https://doi.org/10.1038/nbt.2197
Liao H-X, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD et al (2013) Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496:469–476. https://doi.org/10.1038/nature12053
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376. https://doi.org/10.1007/BF01734359
Davidsen K, Matsen FA (2018) Benchmarking tree and ancestral sequence inference for B cell receptor sequences. Front Immunol 9:2451. https://doi.org/10.3389/fimmu.2018.02451
Kepler TB (2013) Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors. F1000Res 2:103. https://doi.org/10.12688/f1000research.2-103.v1
Dhar A, Ralph DK, Minin VN, Matsen FA (2020) A Bayesian phylogenetic hidden Markov model for B cell receptor sequence analysis. PLoS Comput Biol 16:e1008030. https://doi.org/10.1371/journal.pcbi.1008030
Barak M, Zuckerman NS, Edelman H, Unger R, Mehr R (2008) IgTree: creating immunoglobulin variable region gene lineage trees. J Immunol Methods 338:67–74. https://doi.org/10.1016/j.jim.2008.06.006
Horns F, Vollmers C, Croote D, Mackey SF, Swan GE, Dekker CL et al (2016) Lineage tracing of human B cells reveals the in vivo landscape of human antibody class switching. eLife 5:e16578. https://doi.org/10.7554/eLife.16578
Vieira MC, Zinder D, Cobey S (2018) Selection and neutral mutations drive pervasive mutability losses in long-lived anti-HIV B-cell lineages. Mol Biol Evol 35:1135–1146. https://doi.org/10.1093/molbev/msy024
Cui J-H, Lin K-R, Yuan S-H, Jin Y-B, Chen X-P, Su X-K et al (2018) TCR repertoire as a novel indicator for immune monitoring and prognosis assessment of patients with cervical cancer. Front Immunol 9:2729. https://doi.org/10.3389/fimmu.2018.02729
Vollmer T, Schlickeiser S, Amini L, Schulenberg S, Wendering DJ, Banday V et al (2021) The intratumoral CXCR3 chemokine system is predictive of chemotherapy response in human bladder cancer. Sci Transl Med 13:eabb3735. https://doi.org/10.1126/scitranslmed.abb3735
Li N, Yuan J, Tian W, Meng L, Liu Y (2020) T-cell receptor repertoire analysis for the diagnosis and treatment of solid tumor: a methodology and clinical applications. Cancer Commun (Lond) 40:473–483. https://doi.org/10.1002/cac2.12074
Dziubianau M, Hecht J, Kuchenbecker L, Sattler A, Stervbo U, Rödelsperger C et al (2013) TCR repertoire analysis by next generation sequencing allows complex differential diagnosis of T cell-related pathology: NGS allows complex differential diagnosis. Am J Transplant 13:2842–2854. https://doi.org/10.1111/ajt.12431
Wolf K, Hether T, Gilchuk P, Kumar A, Rajeh A, Schiebout C et al (2018) Identifying and tracking low-frequency virus-specific TCR Clonotypes using high-throughput sequencing. Cell Rep 25:2369–2378.e4. https://doi.org/10.1016/j.celrep.2018.11.009
Pogorelyy MV, Minervina AA, Touzel MP, Sycheva AL, Komech EA, Kovalenko EI et al (2018) Precise tracking of vaccine-responding T cell clones reveals convergent and personalized response in identical twins. Proc Natl Acad Sci U S A 115:12704–12709. https://doi.org/10.1073/pnas.1809642115
Schober K, Buchholz VR, Busch DH (2018) TCR repertoire evolution during maintenance of CMV-specific T-cell populations. Immunol Rev 283:113–128. https://doi.org/10.1111/imr.12654
Gittelman RM, Lavezzo E, Snyder TM, Zahid HJ, Elyanow R, Dalai S, IMI test presentation (2020) Diagnosis and tracking of SARS-CoV-2 infection By T-cell receptor sequencing. Infectious diseases (except HIV/AIDS)
Klein L, Kyewski B, Allen PM, Hogquist KA (2014) Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol 14:377–391. https://doi.org/10.1038/nri3667
Logunova NN, Kriukova VV, Shelyakin PV, Egorov ES, Pereverzeva A, Bozhanova NG, IMI test presentation (2020) MHC-II alleles shape the CDR3 repertoires of conventional and regulatory naïve CD4+ T cells. Proc Natl Acad Sci U S A 117:13659–13669. https://doi.org/10.1073/pnas.2003170117
Lu J, Van Laethem F, Bhattacharya A, Craveiro M, Saba I, Chu J, IMI test presentation (2019) Molecular constraints on CDR3 for thymic selection of MHC-restricted TCRs from a random pre-selection repertoire. Nat Commun 10:1019. https://doi.org/10.1038/s41467-019-08906-7
Migalska M, Sebastian A, Radwan J (2019) Major histocompatibility complex class I diversity limits the repertoire of T cell receptors. Proc Natl Acad Sci U S A 116:5021–5026. https://doi.org/10.1073/pnas.1807864116
Rius C, Attaf M, Tungatt K, Bianchi V, Legut M, Bovay A, IMI test presentation (2018) Peptide-MHC class I tetramers can fail to detect relevant functional T cell clonotypes and underestimate antigen-reactive T cell populations. J Immunol 200:2263–2279. https://doi.org/10.4049/jimmunol.1700242
Martin MD, Jensen IJ, Ishizuka AS, Lefebvre M, Shan Q, Xue H-H, IMI test presentation (2019) Bystander responses impact accurate detection of murine and human antigen-specific CD8 T cells. J Clin Invest 129:3894–3908. https://doi.org/10.1172/JCI124443
Acknowledgments
The authors would like to thank Mats Ohlin for the constructive criticism of the manuscript. US was supported by grants from Mercator Stiftung, Germany; German Research Foundation, Germany (DFG, grant 397650460); BMBF e:KID, Germany (01ZX1612A); and BMBF NoChro, Germany (FKZ 13GW0338B).
Author information
Authors and Affiliations
Consortia
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2022 The Author(s)
About this protocol

Cite this protocol
Marquez, S. et al. (2022). Adaptive Immune Receptor Repertoire (AIRR) Community Guide to Repertoire Analysis. In: Langerak, A.W. (eds) Immunogenetics. Methods in Molecular Biology, vol 2453. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2115-8_17
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2115-8_17
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2114-1
Online ISBN: 978-1-0716-2115-8
eBook Packages: Springer Protocols