
Abstract
The Ensembl Genome Browser provides a wealth of freely available genomic data that can be accessed for many purposes by genetics, genomics, and molecular biology researchers. Herein we present two protocols for exploring different aspects of these data: a phenotype and its associated variants and genes, and a promoter and the epigenetic marks and protein-binding activity associated with it. These workflows illustrate a subset of the data types available through the Ensembl Browser, and can be considered a springboard for further exploration.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
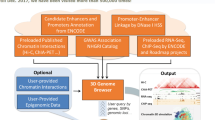
Genome browsers are resources that integrate data at the genomic level, thereby allowing visualization of related genomic information in one space. These data can include genes, noncoding elements that regulate gene expression , genetic variation and the results of comparative genomics analyses, among other forms of annotation (Fig. 1) [1,2,3,4]. Commonly used genome browsers include Ensembl, the UCSC Genome Browser [5] and IGV [6].

Ensembl features. Ensembl integrates together gene annotation, genetic variation, gene regulation data, and comparative genomics onto a single genomic platform. Gene annotation is carried out in house, annotating the full intron–exon structure of coding and noncoding transcripts. Short variants, such as SNPs and indels, are pulled into Ensembl from external databases, alongside structural variants and copy-number variants . ChIP-seq and DNase-seq data is used for in-house prediction of regions of open chromatin and regulatory elements such as promoters, enhancers and CTCF binding sites on the genome and their activity in different cell types. Whole genome alignments and gene tree analysis is carried out in house to compare species in Ensembl. These data are presented alongside each other on the genome in the Ensembl browser, and can also be accessed for bulk export through BioMart , programmatically through APIs and as flat-files on the Ensembl FTP site
The Ensembl project was initially launched in 1999 with the aim of developing methodologies for automatic annotation of (human) genomic sequence with genes and their constituent transcripts [7]. Since that time, the project has broadened substantially in scope; the Ensembl Genome Browser [8], which came online in 2000, now includes reference genomic sequence and annotation for nearly 100 chordate organisms. Ensembl is rapidly incorporating new data, including whole clades of new species’ genomes and reference sequence for multiple strains of existing species, such as mouse . In addition, existing annotation is regularly augmented by the inclusion of new data sets. Ensembl’s sister site, Ensembl Genomes, provides access to nonvertebrate genomes through dedicated portals for Bacteria, Fungi , Plants, Metazoa, and Protists [9, 10].
Ensembl data, annotations, and analyses are updated every 2−3 months, alongside software updates to both the public-facing website and the underlying databases. Prior releases are frozen as archive sites, and from Dec 2013 (Ensembl version 74) will remain accessible via our web interface for at least 5 years following their initial release. A dedicated site is also maintained for the GRCh37 reference human genome assembly , which is annotated with new data on a limited basis (Fig. 2) [11]; partial data from ongoing genome annotation can be accessed via the preview Pre! site.

The Ensembl homepage. The Ensembl homepage provides access to a search function, which can retrieve information associated with, for example, genes, transcripts, proteins, variants , phenotypes, and ontology terms. In addition, links are available to Ensembl’s sister site, Ensembl Genomes, as well as to the most-searched genomes and a complete list of annotated genomes. Fully annotated genomes are available on the main Ensembl site, while genomes whose annotation is in process can be browsed on the Ensembl Pre! site. Ensembl maintains web interfaces of archived versions for 5 years. These can be accessed from a link in the lower right-hand corner. Documentation and help pages can be accessed from the homepage, as well as in-house and external tools integrated into the Ensembl web interface. A dedicated page describing data-download strategies is also available and presents links to the point-and-click tool BioMart , which permits bulk download of Ensembl datasets with no requirement for programming expertise, as well as APIs and FTP site
Data from Ensembl can be accessed at multiple scales. In this chapter, we describe data access through the browser web pages and via BioMart [12], a web-based tool that allows customized retrieval of data from the Ensembl databases. However, data can also be accessed programmatically via our Perl and REST APIs [13, 14] Files containing genome-wide data are available for all species represented in Ensembl via an FTP site [15]; data from all releases of Ensembl can be retrieved from the FTP site, or from our databases via the Perl APIs, in perpetuity.
Beyond providing access to data related to publicly available genome annotation , Ensembl integrates a number of tools designed to process or analyze your own data. The ID History Converter converts Ensembl IDs from a previous release into their current equivalents, while the Assembly Converter maps genomic coordinates from one version of a genome assembly to another. The Variant Effect Predictor predicts the functional consequences of a set of known and/or novel variants [16]. Sequence alignment using BLAST and BLAT against Ensembl genes, genomes and proteins is also available [17, 18], along with a suite of tools developed as part of the 1000 Genomes Project [19] that can be accessed on the dedicated GRCh37 browser site [11].
In this chapter we describe two workflows showcasing a subset of the data available in the Ensembl browser and indicating possible routes to access them. First, we demonstrate a phenotype -centric search highlighting variation data associated with genes and transcripts. Secondly, we present a gene-centric search illustrating gene and transcript models, and the exploration of regulatory features in the region of a gene. In each case we also indicate strategies for data export via BioMart . Those interested in our annotation methods, in programmatic access to Ensembl data, or in exploring other forms of data and annotation are encouraged to refer to our publications [20].
2 Materials
Computer, Internet connection.
An Internet browser: recent versions of Firefox, Chrome, Safari, and Internet Explorer are supported.
3 Methods
These workflows were written using Ensembl release 88 (March 2017). There may be updates to the data or interfaces if you are using a more recent release.
3.1 WF1: Phenotype-Based Searches and Identification of Associated Genetic Variation
The Ensembl browser can be searched using a variety of terms, including genomic regions, genes, variants , or phenotypes; the following workflow describes a phenotype-based search that highlights data and annotations collated in the Phenotype, Variant, Gene, and Transcript tabs.
Non-melanoma skin cancer—principally basal cell and squamous cell carcinomas—is a relatively common pathology associated with variants in several genes [21].
-
1.
Getting started: To explore the phenotype in more detail, type “non-melanoma skin cancer” into the search box on the Ensembl home page, www.ensembl.org , and click the “Go” button. The search autocomplete may retrieve direct links to suggested results; this will allow you to proceed immediately to step 2.
A list of search results will be generated, with “Non-melanoma skin cancer (Human Phenotype)” appearing first. Options on the left-hand side of the page permit restriction by species and/or other categories: click on the different filters individually to apply them to the search results.
-
2.
Studying loci associated with a phenotype: Click the “Non-melanoma skin cancer (Human Phenotype)” link to open the Phenotype tab.
The loci associated with non-melanoma skin cancer are presented in tabular form; their external identifiers, genomic coordinates and associated genes, and the publications in which they were initially described are all listed. Links are provided to further information about the annotation source and relevant publications (in this case, the GWAS catalog [22] and PubMed [23]; Fig. 3 ).
-
3.
Studying a variant: One of the variants associated with non-melanoma skin cancer, rs1805007, falls within the MC1R gene. Click the “rs1805007” link to load the Variant tab.
The Variant tab collates data relating specifically to the variant of interest (A full list of the databases from which Ensembl imports variation data can be found in the documentation [24].).
An overview of the data is found at the top of the Variant tab (Fig. 4A), while a table indicating the phenotypes associated with the variant can be found lower down the page.
The most severe consequence linked to rs1805007 is “missense_variant”, indicating that the alternative allele at this locus lead to an amino acid substitution. All consequences of the rs1805007 variant can be explored by clicking on the “See all predicted consequences” link. Ensembl uses Sequence Ontology terms to describe variant consequences [25].
Below the consequence, you can see that the reference allele of rs1805007 at the genomic position 16:89919709 is C, and one alternative allele, T, has been observed. Minor allele frequency (MAF) has been calculated for the alternative allele, which was observed in 1000 Genomes Project participants: it was identified in 2% of participants in that study [2, 26].
Navigating to the Variant tab from the Phenotype tab automatically loads a table containing the phenotype data relating to this variant, as mentioned above. Tanning ability, sensitivity to sun, and fair hair and skin color have all been associated with the variant, as has basal cell carcinoma, a form of non-melanoma skin cancer. Collectively, these phenotypes are consistent with the observed linkage between fair complexions and sensitivity to sun exposure.
-
4.
The menu on the left presents additional options. Click “Population genetics” to view allele frequencies in global populations.
On this page, data from the 1000 Genomes [26], HapMap [27], and NHLBI Exome Sequencing [28] Projects and the Exome Aggregation Consortium (ExAC) [29] are displayed. The data from the 1000 Genomes Project are shown at the top (Fig. 4B); the pie-charts represent allele frequencies for different superpopulations. Allele frequencies for subpopulations within each superpopulation can be viewed by clicking the “Sub-populations” link beneath the corresponding superpopulation. Allele and genotype frequencies among 1000 Genomes Project participants can also be found in tabular form immediately below the graphical views.
The frequency of the T variant allele in 1000 Genomes Project participants is highest among European subgroups, and individuals homozygous for the variant also occur only in these subgroups. This is expected given the phenotypes associated with the variant (Fig. 4B).
-
5.
To explore genes and transcripts with which the variant is associated, click “Genes and regulation” in the menu on the left.
As we saw previously, the variant lies within the MC1R gene; the summary table here indicates that it overlaps two independent transcripts of this gene as a missense variant and is a downstream gene variant of a third transcript. Other genes and transcripts affected by the variant, as well as the associated consequences, are also shown (Fig. 4C).
In a second table, called “ Gene expression correlations,” you can find a list of genes whose expression has been found by the GTEx Project to be affected by the variant of interest [30].
Finally, any regulatory features or motifs in which the variant falls will be listed in two separate tables at the bottom of the page. There are no regulatory features or motifs that overlap the variant rs1805007.
-
6.
Studying a gene and its transcripts: Click “ENSG00000258839” in the Genes and regulation table to go directly to the Gene tab, which collates gene-related information, for MC1R.
Navigating to the Gene tab from the Variant tab loads the Variant table, which lists all variants in the Ensembl database that fall within the gene itself or in the region 5 kb upstream or downstream of the gene. The top of the page presents a short overview of MC1R, including a description of the gene, its genomic location and synonyms, and an option to show a table of all its transcripts. This information can also be found at the top of all subsequent views within the Gene tab. As in other tabs in the Ensembl browser, the menu to the left of the Gene tab presents links to a variety of additional data and annotations (Fig. 5A).
-
7.
Click “Summary” in the left-hand menu.
General information about the gene, including a description, synonyms and the genomic location, can be found in this view. A graphical model of the gene’s transcripts is shown at the bottom (Fig. 5A).
-
8.
For the complete set of phenotypes associated with MC1R, click the “Phenotypes” link in the left-hand menu.
The three tables list phenotypes associated with the gene, with variants in the gene, and with other species’ orthologues of the gene, as predicted by the Ensembl comparative genomics pipeline [3]. Several phenotypes have been linked to rs1805007, and the MC1R gene also plays a role in coat and skin pigmentation in other organisms, suggesting a conserved function.
-
9.
Click on the “GO: Biological process” link in the left-hand menu.
The GO, or Gene Ontology , terms related to biological processes which have been associated with the transcripts of the MC1R gene are displayed in the table (Fig. 5B) [31, 32]. Each row of the table contains the GO accession number, a description of the GO term , and the evidence codes, annotation source and stable IDs of transcripts associated with that GO term. Hover over the evidence codes to see their definitions.
MC1R-encoded proteins are involved in signal transduction and the melanin biosynthesis pathway , and are located in the plasma membrane, consistent with a role in pigmentation.
Two further links in the menu at left provide GO term associations regarding the Molecular Function and Cellular Component corresponding to transcripts of the MC1R gene (Fig. 5A).
-
10.
Click on the “External references” link in the left-hand menu.
Links to records in external databases such as EntrezGene [33], HGNC [34], and MIM Gene and MIM Morbid [35] can be found on this page.
-
11.
Studying a transcript: Click the “Show transcript table” button in the “Transcripts” section at the top of the page.
A tabular view of the individual transcripts comprising the gene model can be seen (for more information on the Ensembl gene annotation strategy, see ref. 1 ). This table displays information about transcript length and biotype, as well as links to the entries in the CCDS [36], UniProt [37], and RefSeq [33] databases that correspond to particular transcripts.
The level of support for a transcript prediction, and its biological relevance, can be inferred from the matching evidence records and associated flags.
-
12.
Click the “ENST00000555147.1” link in the Transcript table; ENST00000555147.1 is the Ensembl stable ID for the MC1R-001 transcript.
The MC1R-001 transcript’s biotype is listed as “protein-coding,” and the transcript is colored golden in the graphical view. This indicates that it has been independently annotated with identical coordinates by both the Ensembl automated gene annotation and the HAVANA manual gene annotation methods [1] (Fig. 5A).
We are now located in the Transcript tab, which is visible in the blue navigation bar at the top of the page, next to the Gene tab. From the left-hand menu of the Transcript tab you can access complete, spliced or translated transcript sequences (“Exons,” “cDNA,” and “Protein,” respectively), as well as graphical and tabular representations of annotated protein domains (“Protein summary” and “Domains & features,” respectively). “General identifiers” provides links to related records in external repositories (Fig. 6A).
You can now click on “Hide Transcript table” in the Gene section at the top of the page to remove the Transcript table from the page view.
-
13.
Click on the “Supporting evidence” link in the left-hand menu.
This page displays the records used in the annotation in graphical form; all records are hyperlinked to the original data in RefSeq, UniProt, and ENA [38] (Fig. 6B).
-
14.
Click the “Variant table” link in the left-hand menu.

The Ensembl phenotype tab. The Ensembl phenotype tab allows you to explore the phenotype ontology associated with a phenotype and any loci (variants , QTLs, or genes) linked to the phenotype. Loci associated with the phenotype shown in a table on the Associated loci page. The buttons above the table allow filtering. Links take you to the database and/or paper where the link between locus and phenotype was made

The Ensembl variation tab. The Ensembl variation tab provides a wealth of information about a particular variant, such as a SNP or indel. (A) A variant summary shown on all pages in the variant tab, including alleles , MAF , and evidence status. The menu at the left-hand side provides links to all the pages providing information on the variant. (B) Pie charts from the Population Genetics page, showing the allele frequencies for the variant in the 1000 Genomes populations. (C) The Genes and Regulation table, listing all genes affected by the variant with details of sequence ontology consequences, position in the gene and protein, and SIFT and Polyphen scores for amino acid changes (where relevant)

The Ensembl gene tab. The Ensembl gene tab provides a number of views to look at different aspects of a gene. (A) The gene summary page includes a graphical depiction of the transcripts of the gene, shownFig. 5 (continued) against the genome. The central contig indicates the genome. Positive stranded genes, such as MC1R are depicted above the contig. Strand is also indicated by an arrow alongside the transcript name indicating the direction of transcription, and by introns, which are shown pointing upward on positive stranded genes and downward on negative stranded genes. Some transcripts have been removed from this image for size. On all pages in the gene tab, a menu on the left-hand side lists all the pages available for looking at a gene. (B) Three pages are available for looking at the GO terms associated with a gene, conforming to the three categories of terms, Biological process, Molecular function, and Cellular component. These are listed for each gene, including which transcript they are associated with and how they were annotated

The Ensembl transcript tab. The transcript tab contains all views for looking at a transcript and its associated protein, where relevant. (A) The left-hand menu on the transcript tab lists all the pages for looking at transcripts and proteins, and differs subtly from the gene tab menu. It has three different sequence views, allowing you to view the exon and intron sequences in a table, an alignment of the cDNA, CDS and peptide sequences, and the protein sequence only. As you open different features, such as genes, transcripts, and variants, tabs appear in the top bar, allowing easy navigation between the different features you’ve been looking at. (B) The Supporting evidence page shows which cDNA and protein evidence was used to annotate the transcript model
This table displays the set of variants associated with the MC1R-001 transcript (Fig. 7A).
-
15.
Filter the table to view missense variants between amino acid coordinates 150–160.
-
(a)
Filter the table for missense variants by clicking “Consequence” in the Filter section, then “Turn All Off” and “Missense variant.”
-
(b)
Filter the table to view variants at a specific amino acid coordinate within the translated sequence of the transcript by clicking on “Filter Other Columns,” then “AA Coord.” Use the sliders to restrict the area for which variants are shown to 150–160.
You can filter this table in numerous ways, including by consequence, source, and genomic or amino-acid coordinates (Fig. 7B). For missense variants, there are also options to filter by predicted pathogenicity score, as determined by SIFT [39] and/or PolyPhen [40] (PolyPhen calculations are available only for human variants). SIFT and PolyPhen pathogenicity predictions have been calculated for rs1805007 and the amino acid substitution is considered deleterious (An additional variant, rs149922657, has been observed at the same position of MC1R, but has not been associated with any phenotype.).
-
(a)
-
16.
Click “Haplotypes ” in the left-hand menu.
This page allows you to view linked variants that tend to be coinherited. As a default, the amino acid identities and coordinates of each haplotype are shown, along with their frequencies in different 1000 Genomes Project populations [26]; however, clicking “switch to CDS view” at the top of the table will show nucleotide sequences instead (Fig. 8A). The fifth haplotype listed in the protein-haplotype table represents our variant of interest. The frequency of this haplotype is, as already seen, higher in the European subgroup. Lower in the table can be found the 151R>H haplotype corresponding to rs149922657, the other variant observed at position 151; this variant was recovered in only two 1000 Genomes Project participants.
Clicking on any haplotype will load a table indicating its frequencies in different 1000 Genomes populations in more detail (Fig. 8B), as well as a sequence view highlighting the nucleotide and amino-acid positions altered, if applicable (Fig. 8C).
-
17.
Exporting Ensembl variation data: Data can be exported from Ensembl at multiple scales. A link to the BioMart tool, which permits the download of customized datasets at intermediate scale, can be found in the navigation bar at the top of all Ensembl pages (Fig. 2) [12]. In the BioMart interface, select the Dataset “Ensembl Variation” (this will also include the release number, which is 88 at the time of writing), then “Human Short Variants (SNPs and indels excluding flagged variants).” To download all variants of ≤50 bp lying within MC1R, as well as 5 kb upstream and downstream of the gene, filter by “Gene-associated Variant Filters,” selecting “Gene stable IDs” and inputting “ENSG00000258839,” the stable ID for the MC1R gene. You can choose attributes of interest under “Variant” or “Flanking sequences”—for example, the variant name, source, consequence, start and end coordinates, and pathogenicity predictions—which will be listed next to each variant in the output table. Click the “Results” button to view and download the results table (Fig. 9).

Table of short variants found within a transcript. The variant table lists all the variants found within a transcript. A similar page can be found in the gene tab listing all the variants in a gene. The table lists the variants, which are links to the variant tab, with their positions, alleles , SO consequences, and predicted protein effects. Buttons above the table allow you to filter to table to only show variants of interest. (A) The unfiltered table for MC1R-001. (B) The same table, filtered to only show missense variants between residues 150 and 160. The applied filters are shown above the table and can be easily removed

Representation of protein haplotypes found in 1000 Genomes individuals. For each of the individuals in the 1000 Genomes population, the complete protein and CDS sequences were calculated. Sets of cosegregating variants were defined as protein and transcript haplotypes, their frequencies determined and listed in the Transcript haplotype page. (A) The table lists all the haplotypes found by the amino acid change. Click on the haplotype for more details (shown in panels B and C). By default, the page shows the protein haplotypes, but can be switched to show the CDS haplotypes. (B) The frequency of the selected haplotype across 1000 Genomes subpopulations. (C) An alignment of the reference and haplotype protein and CDS sequences

The BioMart interface. BioMart allows easy export of tables of gene, variant , or regulatory feature data. A video tutorial for BioMart is available at https://www.youtube.com/watch?v=QvGT2G0-hYA&ab_channel=EnsemblHelpdesk
3.2 WF2: Gene-Based Searches and Identification of Regulatory Features in a Genomic Region
The following workflow describes a gene-based search and indicates some of the data and annotations collated in the Gene tab and Regulation tab.
The POU5F1 gene, formerly known as OCT4, encodes one of the so-called “Yamanaka factors” implicated in cellular de-differentiation and induction of pluripotency [41, 42]. We can search Ensembl to view the POU5F1 gene model and associated annotation, including predicted regulatory features.
-
1.
Getting started: Type “POU5F1” into the search box on the homepage, www.ensembl.org, or in the upper right corner of any browser page, and click “Go.” This will generate a search-results page with “POU5F1 (Human Gene)” as the top hit. Click the title link to navigate directly to the POU5F1 Gene tab.
The gene “Summary” containing a graphical representation of the gene model loads by default following navigation from the search results.
-
2.
Downloading gene sequences: The sequence of the gene and flanking regions can be downloaded from the Gene tab in two ways.
-
(a)
To download the sequence in FASTA format for processing in an external tool, simply click the “Export data” button below the left-hand menu.
This will open a pop-up window that presents customization options.
-
(b)
To view POU5F1 sequence in the browser, click “Sequence” in the left-hand menu.
This opens a display in FASTA format ; buttons to download and to BLAST the sequence are shown on this page, and download customization options are similarly available (Fig. 10).
-
(a)
-
3.
Exploring regulatory features: Select “Summary” in the left-hand menu. Scroll down to the graphical view of the gene model and locate the Regulatory Build track.
The Regulatory Build depicts regulatory features that have been annotated based on epigenome-scale data imported from sources such as ENCODE [43], Roadmap Epigenomics [44] and Blueprint [45]. These motifs are color-coded according to the predicted function of the element (Fig. 11A).
-
4.
Click on the red promoter overlapping the 5′ end of the longest transcript of POU5F1, POU5F1-004, to open a pop-up box with the stable ID (“ENSR00000195510”), type (“Promoter”), and genomic coordinates of the core element and flanking sequences. Click the stable ID to open the Regulation tab.
Note: POU5F1 is transcribed from the reverse strand, and thus the 5’ sequences containing the promoter are located to the right of the gene.
The Regulation tab displays a graphical representation of the genomic region surrounding the element and a table of the 68 cell types with regulation data currently in Ensembl, organized by activity state. In addition to the Regulatory Build, several tracks are shown by default; these include CRISPR/Cas9 genome-editing sites predicted by the Wellcome Trust Sanger Institute (WTSI) [30], transcription start sites identified by FANTOM5 [46], miRNA binding sites imported from Tarbase [31], and enhancers identified by VISTA [29]. Tracks with no data in the immediate region of the feature are not shown (Fig. 11B) (The term “track” refers to a data type that can be plotted against the genome.).
Feature activity by cell type can be viewed in graphical form by clicking the “Select cells” button and, in the resulting pop-up, choosing “All on” or selecting individual cell types.
-
5.
To view the element’s activation state in individual cell types, click the “Details by cell type” button at the top of the Regulation tab or the link in the left-hand menu. Click the “Select cells” button and then choose “A549” (repressed), “Placenta” (poised), “Pancreas” (inactive), “GM12878” (active). Next, click “Select evidence,” then “All on,” to load the experimental data available for the cell types of interest.
You are now viewing data from cell types in which the element is active, inactive, poised, and repressed (Fig. 12). These activation states are determined on the basis of the histone modifications observed in the region, along with transcription factor and RNA polymerase II or III binding, as well as areas of DNase I hypersensitivity indicating open chromatin [4].
Additional tracks can be accessed by clicking the “Configure this page” button, at left, or the cogwheel at the top of the image. These include the evidence underlying the Regulatory Build, as well as comparative genomics analyses and variation data that may provide additional context for the annotated feature.
-
6.
Ensure that both “Peaks” and “Signal” buttons are selected.
This will display a summary of the aligned reads (signal) as well as the peaks for each assay. Annotated features are clickable; for example, clicking on a predicted promoter will indicate any transcription factors known to bind it, along with links to the JASPAR database [47], where further information on motifs is presented. For other elements, the position of the apex is indicated with black arrowheads (Fig. 12).
-
7.
To view regulatory features across a larger genomic region, navigate to the Location tab, available to the left of the Gene tab in the navigation bar.
The Location tab displays three images: a global view of the chromosome of interest, an intermediate-scale view providing an overview of the region flanking the relevant genomic locus (in this case that of POU5F1), and a final view that presents gene-annotation, comparative genomics and variation tracks by default, along with the Regulatory Build.
It is possible to configure the page to view the activity of local regulatory features by cell type, along with the evidence underlying these determinations. As in the Regulation tab, tracks depicting other Ensembl annotations can be added to provide context to the elements shown.
-
8.
Click on the blue “Configure this page” button to add regulatory data tracks for the same cell types: A549, placenta, pancreas, and GM12878.
This opens a menu listing the many possible tracks available to display on the genome. Categories of tracks are listed on the left. Tracks can be turned on and off by clicking on the box alongside them. To see the activity of regulatory features in different cell types, turn them on within the “Regulatory features” section. In the “Histones & polymerases” and “Open chromatin & TFBS” sections, you will find that tracks are displayed as a matrix, with cell types along the top and evidence to the side (Fig. 13).
-
9.
Exporting regulatory features with BioMart: A list of regulatory features, by type, in a genomic region can also be exported via BioMart . Navigate to BioMart, then select “Ensembl Regulation” > “Human Regulatory Features” (it may be necessary to refresh the window by clicking “New” if you have performed a previous query). For features within 5 kb up- and downstream of POU5F1, filter for Chromosome 6, Base pair start: 31159337, Base pair end: 31185731. As defaults, “Chromosome Name,” “Start (bp),” “End (bp),” and “Feature Type” are selected as Attributes. Add “Regulatory Stable ID” and generate your results.
Nine features are returned for this genomic region, including the promoter we explored, ENSR00000195510.

Exporting gene sequence from Ensembl. All sequence views in Ensembl allow download in either plain FASTA or annotated rich text format (RTF)

The Ensembl regulatory build and regulatory features. (A) The regulatory build is shown as a track on the gene image. Clicking on a feature in the track opens a pop-up menu, with a link to the regulatory feature tab. Some transcripts have been removed from this image for size. (B) The summary page of the regulatory feature tab contains a table listing activity in different cell types. The graphic shows the feature in context, along with genes, CRISPR-Cas9 sites and FANTOM5 annotation

Evidence for and activity of regulatory features in different cell types. The Details by cell type page in the regulatory feature tab can be manipulated to show the activity of the feature in cell types of interest using the buttons at the top. For each cell type, the feature is shown colour-coded to indicate its activity, with the evidence shown below. The evidence is ChIP-seq and DNase-seq data, and is shown as peaks of significant activity and as signal giving the number of reads. The top of the peak is indicated in the peak bar by pairs of black arrows. Black blocks in the regulatory features indicate the position of transcription factor binding motifs, which are listed in a pop-up when clicked on

Adding regulation tracks to a region view. The Region in detail view displays a genomic region and can be customized to show tracks of interest using the Configure this page button. This opens a detailed menu listing all the available tracks, using categories on the left, including regulatory features and evidence. Regulatory evidence can be added using a matrix selector, listing the cell type along the top and type of evidence down the left
4 Discussion
Here, we describe methods to navigate variation and regulation data in the Ensembl browser, focusing on human, although the principle of navigation is relevant to queries in all species.
The typical entry point to a query in the browser is the search function. The Ensembl search is versatile and can retrieve information linked to a variety of inputs—including, but not limited to, genomic locations; gene, transcript, protein and regulatory feature IDs; GO terms ; variant IDs; and phenotypes. Unless otherwise specified in the query, search results for human will be returned first; filters displayed on the left-hand side of the results page permit the restriction of results by category (e.g., gene, variant) and by organism.
Selecting a search result will open a tab that collates information on the entity: in the two workflows presented above, we present strategies for accessing the Phenotype, Variant, Gene, Transcript, Location, and Regulation tabs following phenotype- and gene-based searches. As you move from tab to tab in a single query, previously accessed tabs will remain open in the blue navigation bar at the top of the page to facilitate seamless data-retrieval in a minimal number of steps; you can reenter a previous tab simply by clicking on the tab header in the navigation bar (Fig. 6A).
By default, tabs will open with a summary of the information available for each entity (e.g., a transcript or variant), although herein we indicate a few cases where other data are loaded: for example, the Gene Variant table is presented immediately upon navigation from the Variant to the Gene tab. Should a view not be as expected, links to all data and annotations available in a tab can be found in the menu on the left; for Location, Gene and Transcript tabs, these links adhere to a similar framework but present annotations at different scales.
Tabs can be customized by clicking on the blue “Configure this page” button below the left-hand menu, or the cogwheel icons that appear in the upper borders of graphical displays (Fig. 13). Customization allows you to add or remove data tracks that may be useful to interpretation or analysis; for example, to view the evidence underlying an activation-state prediction for a regulatory feature in a cell type of interest (in the Location or Regulation tabs). Other examples of customization include, in the Location tab, the addition of tracks containing the data, imported from external repositories, that were used to annotate transcripts in a genomic region (ENA, UniProt, and RefSeq tracks accessible in Location tab) [11]. Public datasets can also be added from the Track Hub Registry [48], and you can import your own data, in multiple formats [49], for examination in the context of the browser.
Data can be exported directly from the browser by clicking the blue “Export data” buttons found below the left-hand menu in most tabs, or the “Download sequence” buttons above FASTA sequences in the Gene and Transcript tabs. In addition, the BioMart tool described in the workflows presented herein can be used to retrieve custom datasets from our Gene, Variation and Regulation databases, and data can be accessed programmatically from our Perl APIs and REST service. Data from all Ensembl releases can also be downloaded en masse from our FTP site.
Species’ sequence data and annotations may be updated several times a year. You should therefore be attentive, when querying Ensembl data, to the current browser version, as annotations are subject to change. Data can, however, still be retrieved directly from archived versions of the browser, as well as via BioMart , while the browser web interface remains online. Following the decommissioning of any browser version, the data remain accessible from our FTP site and APIs, as mentioned above.
A dedicated email helpdesk is available to field any inquiries about Ensembl and we typically reply to messages within two days of receipt. We also hold training workshops upon invitation by research institutes; from 2013 to 2016 we participated in an average of 86 workshops, and trained 2150 students, per year. Our training materials are accessible online [50], along with a number of courses that are available on the Train Online Platform of the European Bioinformatics Institute (EMBL-EBI) [51], and we have published a blogpost outlining the process of hosting your own workshop [52]. Short help videos can be found both on our YouTube channel [53] and, for those who cannot access YouTube, on Youku [54]. We invite the community to contact us via helpdesk@ensembl.org for more information about workshops, with questions regarding the browser, and to suggest features and resources which would assist their work.
Funding/Acknowledgments
Ensembl receives majority funding from the Wellcome Trust (grant number WT108749/Z/15/Z) with additional funding for specific project components from the National Human Genome Research Institute (U41HG007234 and U41HG007823), the Biotechnology and Biological Sciences Research Council (BB/L024225/1, BB/M011615/1, and BB/M020398/1), Open Targets, the Wellcome Trust (WT104947/Z/14/Z, WT200990/Z/16/Z, and WT201535/Z/16/Z) and the European Molecular Biology Laboratory. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement n° 634143 (MedBioinformatics). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement n° 733161 (MultipleMS).
References
Aken BL, Ayling S, Barrell D et al (2016) The Ensembl gene annotation system. Database (Oxford) 2016. https://doi.org/10.1093/database/baw093
Chen Y, Cunningham F, Rios D et al (2010) Ensembl variation resources. BMC Genomics 11:293. https://doi.org/10.1186/1471-2164-11-293
Herrero J, Muffato M, Beal K et al (2016) Ensembl comparative genomics resources. Database (Oxford) 2016. https://doi.org/10.1093/database/baw053
Zerbino DR, Johnson N, Juetteman T et al (2016) Ensembl regulation resources. Database (Oxford) 2016. https://doi.org/10.1093/database/bav119
Kent WJ, Sugnet CW, Furey TS et al (2002) The human genome browser at UCSC. Genome Res 12(6):996–1006. https://doi.org/10.1101/gr.229102. Article published online before print in May 2002
Robinson JT, Thorvaldsdottir H, Winckler W et al (2011) Integrative genomics viewer. Nat Biotechnol 29(1):24–26. https://doi.org/10.1038/nbt.1754
Hubbard T, Barker D, Birney E et al (2002) The Ensembl genome database project. Nucleic Acids Res 30(1):38–41
The Ensembl Browser. http://www.ensembl.org
Kersey PJ, Allen JE, Armean I et al (2016) Ensembl Genomes 2016: more genomes, more complexity. Nucleic Acids Res 44(D1):D574–D580. https://doi.org/10.1093/nar/gkv1209
The Ensembl Genomes Browser. http://www.ensemblgenomes.org
Aken BL, Achuthan P, Akanni W et al (2017) Ensembl 2017. Nucleic Acids Res 45(D1):D635–D642. https://doi.org/10.1093/nar/gkw1104
Kinsella RJ, Kahari A, Haider S et al (2011) Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011:bar030. https://doi.org/10.1093/database/bar030
Ruffier M, Kahari A, Komorowska M et al (2017) Ensembl core software resources: storage and programmatic access for DNA sequence and genome annotation. Database (Oxford) 2017(1). https://doi.org/10.1093/database/bax020
Yates A, Beal K, Keenan S et al (2015) The Ensembl REST API: Ensembl data for any language. Bioinformatics 31(1):143–145. https://doi.org/10.1093/bioinformatics/btu613
The Ensembl FTP site. ftp://ftp.ensembl.org
McLaren W, Gil L, Hunt SE et al (2016) The Ensembl variant effect predictor. Genome Biol 17(1):122. https://doi.org/10.1186/s13059-016-0974-4
Kent WJ (2002) BLAT–the BLAST-like alignment tool. Genome Res 12(4):656–664. https://doi.org/10.1101/gr.229202. Article published online before March 2002
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Clarke L, Zheng-Bradley X, Smith R et al (2012) The 1000 genomes project: data management and community access. Nat Methods 9(5):459–462. https://doi.org/10.1038/nmeth.1974
Ensembl Publications. http://www.ensembl.org/info/about/publications.html
Zhang M, Song F, Liang L et al (2013) Genome-wide association studies identify several new loci associated with pigmentation traits and skin cancer risk in European Americans. Hum Mol Genet 22(14):2948–2959. https://doi.org/10.1093/hmg/ddt142
The GWAS catalog. https://www.ebi.ac.uk/gwas/
Europe PMC. https://europepmc.org/
Sources of Ensembl variation data. http://www.ensembl.org/info/genome/variation/sources_documentation.html
Eilbeck K, Lewis SE, Mungall CJ et al (2005) The sequence ontology: a tool for the unification of genome annotations. Genome Biol 6(5):R44. https://doi.org/10.1186/gb-2005-6-5-r44
Genomes Project Consortium, Auton A, Brooks LD et al (2015) A global reference for human genetic variation. Nature 526(7571):68–74. https://doi.org/10.1038/nature15393
Goldstein DB, Cavalleri GL (2005) Genomics: understanding human diversity. Nature 437(7063):1241–1242. https://doi.org/10.1038/4371241a
Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP). http://evs.gs.washington.edu/EVS/
Visel A, Minovitsky S, Dubchak I et al (2007) VISTA enhancer browser–a database of tissue-specific human enhancers. Nucleic Acids Res 35(Database issue):D88–D92. https://doi.org/10.1093/nar/gkl822
Hodgkins A, Farne A, Perera S et al (2015) WGE: a CRISPR database for genome engineering. Bioinformatics 31(18):3078–3080. https://doi.org/10.1093/bioinformatics/btv308
Vlachos IS, Paraskevopoulou MD, Karagkouni D et al (2015) DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res 43(Database issue):D153–D159. https://doi.org/10.1093/nar/gku1215
Gene Ontology Consortium (2015) Gene ontology consortium: going forward. Nucleic Acids Res 43(Database issue):D1049–D1056. https://doi.org/10.1093/nar/gku1179
O’Leary NA, Wright MW, Brister JR et al (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44(D1):D733–D745. https://doi.org/10.1093/nar/gkv1189
HGNC database of human gene names. http://www.genenames.org/
Online Mendelian Inheritance in Man. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). https://www.omim.org/
Pruitt KD, Harrow J, Harte RA et al (2009) The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 19(7):1316–1323. https://doi.org/10.1101/gr.080531.108
The UniProt Consortium (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res 45(D1):D158–D169. https://doi.org/10.1093/nar/gkw1099
Toribio AL, Alako B, Amid C et al (2017) European nucleotide archive in 2016. Nucleic Acids Res 45(D1):D32–D36. https://doi.org/10.1093/nar/gkw1106
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814
Adzhubei IA, Schmidt S, Peshkin L et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. https://doi.org/10.1038/nmeth0410-248
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. https://doi.org/10.1016/j.cell.2006.07.024
Okita K, Ichisaka T, Yamanaka S (2007) Generation of germline-competent induced pluripotent stem cells. Nature 448(7151):313–317. https://doi.org/10.1038/nature05934
ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74. https://doi.org/10.1038/nature11247
Roadmap epigenomics Consortium, Kundaje A, Meuleman W et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518(7539):317–330. https://doi.org/10.1038/nature14248
Fernandez JM, de la Torre V, Richardson D et al (2016) The BLUEPRINT data analysis portal. Cell Syst 3(5):491–495.e495. https://doi.org/10.1016/j.cels.2016.10.021
Fantom Consortium, Forrest AR, Kawaji H et al (2014) A promoter-level mammalian expression atlas. Nature 507(7493):462–470. https://doi.org/10.1038/nature13182
Bryne JC, Valen E, Tang MH et al (2008) JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res 36(Database issue):D102–D106. https://doi.org/10.1093/nar/gkm955
The Track Hub Registry. https://trackhubregistry.org
Data formats compatible with Ensembl. http://www.ensembl.org/info/website/upload/index.html - formats
The Ensembl Training Site. http://training.ensembl.org
EMBL-EBI’s Train Online Platform. https://www.ebi.ac.uk/training/online/
Hosting an Ensembl Workshop. http://www.ensembl.info/blog/2017/01/05/so-you-want-to-run-an-ensembl-workshop/
The Ensembl Helpdesk YouTube channel. https://www.youtube.com/user/EnsemblHelpdesk
The Ensembl Helpdesk Youku channel. http://i.youku.com/i/UMzM1NjkzMTI0?spm=a2h0j.8191423.subscription_wrap.DD~A
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons License and indicate if changes were made. The images or other third party material in this chapter are included in the chapter’s Creative Commons License, unless indicated otherwise in a credit line to the material. If material is not included in the chapter’s Creative Commons License and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2018 The Author(s)
About this protocol

Cite this protocol
Newman, V., Moore, B., Sparrow, H., Perry, E. (2018). The Ensembl Genome Browser: Strategies for Accessing Eukaryotic Genome Data. In: Kollmar, M. (eds) Eukaryotic Genomic Databases. Methods in Molecular Biology, vol 1757. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7737-6_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7737-6_6
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-7736-9
Online ISBN: 978-1-4939-7737-6
eBook Packages: Springer Protocols