
Abstract
From their observation of necrotic areas around bacterial foci in Buruli ulcers (BUs), Connor and co-workers were the first, back in 1965, to suggest that M. ulcerans may produce a diffusible cytotoxin [1]. This hypothesis was later confirmed by injecting mycobacterial culture filtrates into the skin of guinea pigs, showing that this causes focal necrosis resembling that of naturally occurring human infections [1–3]. In 1999, George et al. succeeded in isolating a cytotoxic factor from M. ulcerans lipid extracts, and deciphered its chemical nature [4]. The M. ulcerans toxin was named mycolactone, based on its mycobacterial origin and macrolactone structure: a 12-membered lactone ring, to which a C5-O-linked polyunsaturated acyl side chain and a C-linked upper side chain comprising C12–C20 are appended (Fig. 1). Follow-up studies showed that M. ulcerans-derived mycolactone was in reality a mixture of two stereoisomers called A and B [6, 7] (Fig. 1). Since the initial discovery of mycolactone A/B, eight additional mycolactone congeners have been identified that are either produced by M. ulcerans strains of different geographical origins or genetically related fish and frog pathogens, which were initially given different species designations such as M. pseudoshottsii and M. liflandii. Comparative genome analysis later revealed that all mycolactone-producing mycobacteria evolved from a common M. marinum-like progenitor, and are therefore ecovars of a single M. ulcerans species [8]. While the macrolide core structure and upper side chain are conserved, mycolactone populations from different M. ulcerans sub-lineages vary in the length, number and localization of hydroxyl groups and number of double bonds of the lower side chain. These modifications of the lower polyunsaturated acyl side chain cause pronounced changes in cytotoxicity [9–11]. The origin and chemistry of natural mycolactones, structure-activity relationships and the various approaches that were developed to generate synthetic mycolactones have been reviewed recently [12, 13]. These aspects will therefore not be further discussed here. Instead, this chapter provides an update on our understanding of mycolactone (A/B) biology, and discusses its proposed mechanisms of action in relation with BU pathogenesis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


From their observation of necrotic areas around bacterial foci in Buruli ulcers (BUs), Connor and co-workers were the first, back in 1965, to suggest that M. ulcerans may produce a diffusible cytotoxin [1]. This hypothesis was later confirmed by injecting mycobacterial culture filtrates into the skin of guinea pigs, showing that this causes focal necrosis resembling that of naturally occurring human infections [1,2,3]. In 1999, George et al. succeeded in isolating a cytotoxic factor from M. ulcerans lipid extracts, and deciphered its chemical nature [4]. The M. ulcerans toxin was named mycolactone, based on its mycobacterial origin and macrolactone structure: a 12-membered lactone ring, to which a C5-O-linked polyunsaturated acyl side chain and a C-linked upper side chain comprising C12–C20 are appended (Fig. 1). Follow-up studies showed that M. ulcerans-derived mycolactone was in reality a mixture of two stereoisomers called A and B [6, 7] (Fig. 1). Since the initial discovery of mycolactone A/B, eight additional mycolactone congeners have been identified that are either produced by M. ulcerans strains of different geographical origins or genetically related fish and frog pathogens, which were initially given different species designations such as M. pseudoshottsii and M. liflandii. Comparative genome analysis later revealed that all mycolactone-producing mycobacteria evolved from a common M. marinum-like progenitor, and are therefore ecovars of a single M. ulcerans species [8]. While the macrolide core structure and upper side chain are conserved, mycolactone populations from different M. ulcerans sub-lineages vary in the length, number and localization of hydroxyl groups and number of double bonds of the lower side chain. These modifications of the lower polyunsaturated acyl side chain cause pronounced changes in cytotoxicity [9,10,11]. The origin and chemistry of natural mycolactones, structure-activity relationships and the various approaches that were developed to generate synthetic mycolactones have been reviewed recently [12, 13]. These aspects will therefore not be further discussed here. Instead, this chapter provides an update on our understanding of mycolactone (A/B) biology, and discusses its proposed mechanisms of action in relation with BU pathogenesis.

Structure of mycolactone A/B. The red line indicates the region where stereoisomers differ (from [5])
1 Mycolactone and BU Disease
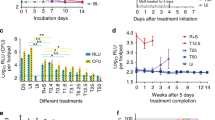
1.1 Pharmacodistribution
Mycolactone’s diffusion in infected hosts was initially believed to coincide with its cytocidal action. However, in mice experimentally infected with M. ulcerans in footpads, the distinctive mass spectrometric signature of the toxin was detected in peripheral blood cells, spleen, liver and kidneys [14], showing that bacterially-produced mycolactone distributes far beyond infected tissues. The presence of mycolactone was assessed by extracting total lipids from homogenized organs with organic solvents, and analyzing the resulting acetone-soluble fractions by liquid chromatography tandem-mass spectrometry (LC-MS/MS). The body-wide distribution of mycolactone was further supported by animal studies using a radiolabeled form of the toxin, generated by feeding M. ulcerans cultures with [1–14C] propionic acid and [1,2–14C] acetic acid. In mice injected with 14C-mycolactone via the subcutaneous, intravenous or intraperitoneal routes, radioactivity was indeed detected in peripheral blood, spleen, liver and kidneys after 24 h [14]. Using the LC-MS/MS approach, structurally-intact mycolactone was detected in human skin biopsies harvested from the center of pre-ulcerative BU lesions or the edge of viable skin around ulcers [15], in ulcer exudates and in serum samples from newly diagnosed patients and patients undergoing antibiotic treatment [16]. While toxin concentrations within lesions declined upon M. ulcerans killing by antibiotic treatment, mycolactone’s presence was still detected several weeks after completion of therapy [15, 16], indicating a slow elimination rate. By spiking cell pellets or serum samples with purified toxin, we noted the poor yield of mycolactone extraction with organic solvents (around 10%). Assuming that 90% of mycolactone is lost during the process, serum concentrations of mycolactone would thus be estimated to fall within the 0–20 nM range at the start of antibiotic therapy [16]. These numbers should nevertheless be considered with caution, as they are based on a limited number of measurements. How mycolactone traffics from bacteria infecting the skin to peripheral blood and distant organs, and whether it accumulates preferentially in certain cell populations or tissues during the course of BU disease remains largely unknown.
At the cellular level, mycolactone is currently believed to diffuse passively through the plasma membrane. In support of this model, fluorescently labeled mycolactones were shown to penetrate fibroblasts in a non-saturable and non-competitive manner within minutes [17] and to accumulate in the cytoplasmic compartment [9, 18]. Two studies using molecular dynamics simulations on the one hand, and Langmuir monolayers as membrane models on the other hand, recently suggested that the passage of mycolactone across cellular membranes may nevertheless alter their dynamic properties, and cause mechanical and physical perturbations [19, 20].
1.2 Mycolactone Contribution to BU Disease Manifestations
1.2.1 Pathogenesis and Histopathology of BU Lesions
BU is a chronic necrotizing skin disease affecting primarily subcutaneous adipose tissue. Lesions usually start as single, painless, subcutaneous nodule or papule. The dermis and epidermis overlying the lesion eventually degenerates and sloughs off, leading to the development of ulcers with undermined edges and a necrotic slough at the base. Contiguous coagulation necrosis of the deep dermis and subcutaneous fat tissue with destruction of blood vessels and interstitial edema, epidermal hyperplasia and the presence of fat cell ghosts and extracellular clusters of acid fast bacilli (AFB) constitute hallmarks of BU disease. In advanced lesions the necrotic process may extend through deep fascia and expose deeper structures like muscle or bone. In early lesions, necrosis of dermal collagen is leading to a fibrillary appearance, but gradually all cellular and structural details are disappearing and an amorphous coagulum is developing in the center of the lesion [21]. Clusters of extracellular acid-fast bacilli are primarily found in the deep layers of the necrotic adipose tissue [22], but tissue destruction extends far beyond (Fig. 2), which is ascribed to the diffusion of mycolactone. In the center of active lesions, inflammatory cells are rarely found (Fig. 2) and cellular responses are defective at both the local and the systemic levels. There is multiple evidence, summarized below, that mycolactone contributes directly to these disease manifestations.

Histopathological features of a tissue punch biopsy from an untreated human BU plaque lesion. Histological sections were stained with Haematoxylin-Eosin (HE) or Ziehl-Neelsen (ZN). (a) The subcutaneous tissue presents with the typical signs of BU histopathology: massive necrosis, fat cell ghosts and edema. The epidermis presents with epidermal hyperplasia. (b) A band of extracellular ZN positive AFB is present focally in the deep layer of the necrotic subcutis (adapted from [23])
The most commonly used experimental infection models for BU is the mouse model, in which inoculation of M. ulcerans into the footpads, ears or the tail causes lesions that develop over several weeks. Evaluation of antibiotic treatment modalities in this model has been shown to be predictive for treatment efficacy in humans. However the mouse model has some limitations for the investigation of the pathogenesis of BU in humans, since the composition of the tissue at the injection sites differs markedly from human skin. Therefore the pig has been recently evaluated as a model to study the early pathogenesis of BU [24]. Pig and human skin share many similarities like thickness, general structure of the epidermis, dermis and the subcutaneous tissue [25], making porcine skin a preferred model for burn and wound healing studies [26]. Also guinea pigs have been used to study early host-pathogen interaction and in particular the effect of mycolactone [27]. However both in pigs and guinea pigs no chronic infections are establishing [28].
The key role of mycolactone in the pathogenesis of M. ulcerans infection has been first demonstrated by injection of this molecule into the skin of experimental animals. In guinea pigs injection reproduced cell death and lack of acute inflammatory response observed after challenge with viable bacteria [27]. In pigs a necrotic lesion is developing that is surrounded by an infiltration belt after mycolactone injection (Fig. 3b), resembling closely the histopathological picture found in early human lesions (Fig. 3a). The key role of mycolactone in BU pathogenesis has been further demonstrated by infection of experimental animals with mycolactone negative M. ulcerans mutants. While wild-type bacteria cause an extracellular infection, the mycolactone negative mutants produce an intracellular inflammatory infection [30, 31] similar to that of M. marinum, the progenitor of M. ulcerans [8].

Formation of a necrotic lesion core surrounded by an infiltration belt. Histological sections were stained with Ziehl-Neelsen (ZN)/Methyleneblue (A1) or Methyleneblue alone (B1), the “In situ cell death detection kit” (A2) or by immunohistochemistry for the macrophage marker CD68 (B2). (a) Early human BU lesion presenting with a necrotic core containing extracellular clusters of AFB (location indicated by red stars) surrounded by a belt of infiltrating leukocytes demonstrated by the blue staining of nuclei (A1). Strong TUNEL staining inside the necrotic core and in some of the infiltrating cells of the infiltration belt (B1). (b) Lesions in the skin of a pig (Sus scrofa) induced by a single subcutaneous injection of 50 μg of synthetic mycolactone A/B. The toxin caused formation of a lesion with a necrotic core (B1 and B2) surrounded by a dense belt of infiltrating cells consisting primarily of CD68 positive macrophages (B2) (adapted from [29])
M. ulcerans is multiplying in the sanctuary of the central necrotic core of the lesion, which is usually free of living infiltrating leukocytes, although large numbers of macrophages and lymphocytes are collecting already in early lesions at the margin of the necrotic center of the lesions [29]. These infiltrates form a belt, which separates the highly confined necrotic core from the still intact tissue surrounding the lesion (Fig. 3a). Infiltration of leukocytes may be reduced by sub-lethal concentrations of mycolactone affecting chemokine production by skin dendritic cells (DCs) [32] (see Sect. 1.2.3), while cells entering the necrotic core may undergo apoptosis caused by mycolactone’s diffusion from bacterial foci in the center of the lesions (see below). Some of the leukocytes in the infiltration belt are TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining positive [29], which is regarded as a sign for double-strand DNA breaks generated during apoptosis. The necrotic core of BU lesions shows a very strong, but diffuse TUNEL staining (Fig. 3a). At the periphery of BU lesions, interaction of phagocytes with AFBs and intracellular M. ulcerans can be observed and in particular in resolving BU lesions, granulomatous inflammation can be found. During antibiotic treatment of BU, intra-lesional concentrations of mycolactone decline [15]. This allows leukocytes to reach the extracellular mycobacteria located in the necrotic core of the lesion, leading to their phagocytosis and destruction [33]. In turn, chronic leukocyte infiltration cumulates in the development of ectopic lymphoid structures [34]. Gathering of DCs and development of defined granuloma structures indicate that antigen recognition and processing is leading to active M. ulcerans specific immune responses. Three main types of mixed infiltrates have been identified in antibiotic treated lesions, highly organized epithelioid granulomas, diffuse infiltrates and dense lymphocyte clusters in proximity to vessels. AFB already start to be internalized by phagocytes after 2 weeks of treatment, when M. ulcerans can still be cultured from tissue homogenates. Antibiotic treatment may act rapidly on the bacterial production of mycolactone, whereas complete killing of bacteria may take longer.
The cellular mechanism(s) of mycolactone-induced ulceration of the skin were investigated by exposing fibroblasts, epithelial cells and endothelial cells to purified mycolactone in vitro and monitoring dose-response effects on cell viability. Prolonged exposure (>48 h) to nanomolar concentrations (>10 nM) of mycolactone A/B induced the detachment and death of all skin cells studied. This included mouse (L929) and human (HDF) fibroblast cell lines, human epithelial (HeLa) and keratinocyte (HaCaT) cell lines [17, 18, 27, 35,36,37], and human dermal microvascular endothelial cells [38]. Primary human keratinocytes were relatively more susceptible to mycolactone-mediated cytotoxicity, with cell mortality being detected after only 24 h of treatment [39, 40]. Notably, cell detachment was preceded by alterations of the actin cytoskeleton [17, 18, 35]. In HeLa cells exposed to mycolactone, production of spike-like protrusions was activated after 30 min, while actin polymerization abnormally localized in the perinuclear area after 4 h, coinciding with a near-complete incapacity of the cells to adhere to plastic wells [18]. Longer treatments (16–24 h) induced defects in epithelial cell capacity to establish cell-cell contacts and close wounds in vitro [18, 41]. In the mouse ear model, intradermal injection of mycolactone caused a dramatic loss of E-cadherin–adhesive contacts in the stratum spinosum, coupled to thinning of the external stratum granulosum and lucidum, resulting in a significant decrease in epidermis width after 54 h [18]. In accordance with the coagulative necrosis in BU lesions, exposing human dermal microvascular endothelial cells to mycolactone depleted thrombomodulin from the cell surface [38]. Mycolactone also decreased the production of collagen biosynthesis enzymes by L929 fibroblasts [35]. Together, these studies suggest that mycolactone provokes BU formation by concomitantly reducing the viability and the healing potential of skin cells, altering the strength of cell-cell and cell-extracellular matrix adhesion, and affecting coagulation control.
1.2.2 Local Analgesia
Early reports favored the hypothesis that BU-associated analgesia was primarily due to nerve destruction. Indeed, the histopathological examination of BU biopsies showed local axonal damages, with loss of myelin in 24% of the patients [42]. Consistently, mouse footpad infection with M. ulcerans induced nerve fiber degeneration in advanced ulcers [43], and injection of purified mycolactone in mouse footpads triggered neurological damages associated with hyposensitivity [44]. The possibility that mycolactone causes analgesia by directly killing sensory neurons and Schwann cells, the peripheral nervous system (PNS) cells generating and transmitting pain signals, was investigated in vitro. Anand and co-workers observed significant neurite degeneration in rat and human primary dorsal root ganglion (DRG) sensory neurons after 24 h of exposure to a 100 nM concentration of toxin [45]. Consistently, we found that exposing mouse DRG to 25 nM mycolactone for 16 h downregulated the production of multiple proteins mediating interactions between neurons and the extracellular matrix [46]. Longer treatments (>48 h) induced massive mortality of primary DRGs in both studies [45, 47], although a third study reported a minimal loss of viability following exposure to mycolactone doses of up to 70 μM [48]. Schwann cells, the principal glia of the PNS, are important in many aspects of peripheral nerve biology including the conduction of nervous impulses along axons, nerve regeneration upon injury and production of the nerve extracellular matrix. Prolonged (>48 h) exposure to mycolactone also caused significant mortality in the mouse Schwann cell line SW10 [49], and primary rat Schwann cells (IC50 of 1 nM) [47]. Of note, microglia showed comparable susceptibility to mycolactone-induced toxicity, with decreased viability manifesting after 48 h of treatment with mycolactone concentrations varying between 3 and 10 nM [47]. Whether mycolactone diffuses into peripheral nerves and reaches the central nervous system (CNS) in vivo is unknown. However, these cellular studies fully support the view that BU-associated analgesia may be due, at least partially, to cytopathic effects at the lesion level.
Notably, Marsollier, Brodin and co-workers showed that infection with M. ulcerans can induce local hypoesthesia at early stages of disease, in the absence of apparent nerve destruction [50]. Here, pain was generated by applying a focused radiant heat source onto infected footpads, and pain perception was monitored by an adaptation of the tail-flick procedure, in mice that had been previously anesthetized. Injection of 5 μg mycolactone in mouse footpads produced the same effects after 2 h and during 2 days, with normal nociceptive abilities being recovered after 8 days. Parallel to this, we showed that systemic delivery of mycolactone (2 μg) limits the development of distantly generated inflammatory pain [10]. Mycolactone was administered intraperitoneally 1 h before formalin injection into the hind paws, and stereotypical licking and biting behaviors were monitored after 10 min, the time that it takes for inflammatory processes to develop. In addition to demonstrating the intrinsic analgesic properties of mycolactone, these animal studies indicated that bacterially-derived mycolactone reduces pain perception in infected hosts by other mechanisms than cytotoxicity. Section 2 reviews the molecular targets that have been proposed to explain these observations.
1.2.3 Local and Systemic Immunomodulation
Transient defects in the systemic production of IFN-γ have been repeatedly reported in BU patients with progressive ulcers [51,52,53,54,55]. The relative lack of IFN-γ in ex vivo-stimulated blood samples from BU patients reversed at late stages of disease, which may explain why it was not observed in two studies [56, 57]. Of note, IFN-γ production defects resolved after surgical excision of the lesions, indicating their association with M. ulcerans bacteria [54]. These defects were observed with antigen-specific [51, 52] and non-specific [53,54,55] activation stimuli (such as the T-cell mitogen PHA), suggesting that M. ulcerans infection induces T cell anergy. In line with this hypothesis, multi-analyte profiling of PHA-stimulated whole blood culture supernatants revealed that most T cell-derived cytokines were suppressed during BU progression [55]. Moreover, patients with BU displayed a distinctive immunosuppressive signature in their serum, marked by a downregulation of multiple mediators of inflammation [55, 58]. In all, these studies revealed that M. ulcerans impairs the normal development of inflammatory and cellular immune responses to infection.
Downregulation of cytokines and chemokines in patient sera was still observed 1 month after the end of antibiotic therapy [55, 58]. Since mycolactone was detected in lesions and serum of patients several weeks after they had completed antibiotic treatment [15, 16], the concept emerged that mycolactone may be the bacterial factor causing immunological defects in infected individuals. This hypothesis was supported by mouse studies showing that infection with wild-type M. ulcerans, but not with a mycolactone-deficient mutant, causes systemic defects in IL-2 production [14]. Pioneering work conducted by Foxwell and co-workers showed that mycolactone prevents the LPS-induced release of TNF and IL-10 by human monocytes, and production of IL-2 by activated T lymphocytes, under conditions not altering cell viability [59]. Subsequent studies by our group and others examined in detail the cell-type specific effects of mycolactone on immune functions, with the following conclusions. Comparably to skin cells and cells of the nervous system, human primary macrophages and DCs were susceptible to prolonged (>48 h) treatment with mycolactone (>10 nM) [10, 32]. In contrast, the viability of human primary T cells and polymorphonuclear neutrophils treated under the same conditions was minimally affected [10], showing that certain cell types are more resistant than others against mycolactone toxicity. Notably, non-cytotoxic treatment with mycolactone prevented the phenotypic and functional maturation of DCs, limiting their ability to activate T cells and produce inflammatory chemokines in response to TLR stimulation [32]. It decreased DC expression of MHC class I and II, leading to impaired direct and indirect antigen presentation [60]. In monocytes and macrophages, mycolactone blocked the activation-induced production of cytokines and chemokines post-transcriptionally and irrespective of the activation stimulus [61, 62]. Mycolactone also downregulated the basal expression of TCR and homing receptor L-selectin (CD62L) by resting T cells, leading to altered responsiveness to TCR stimulation and impaired capacity to reach peripheral lymph nodes in vivo [10, 63, 64]. It also limited the T cell capacity to produce cytokines in response to activation stimuli that bypass the TCR, at the post-transcriptional level. In mice, the systemic administration of mycolactone conferred protection against chronic skin inflammation and rheumatoid arthritis, demonstrating the anti-inflammatory potency of mycolactone in vivo [10]. Mycolactone also displayed potent anti-inflammatory activity on the nervous system, with nanomolar concentrations of mycolactone preventing the activation-induced production of pro-inflammatory mediators by activated DRGs, Schwann cells and microglia in conditions that did not affect cell viability [47]. In intrathecally-injected rats, mycolactone decreased significantly the production of inflammatory cytokines in the spinal cord without inducing detectable cytotoxicity [47]. The overall conclusion of these studies was that mycolactone represents a potent natural immunosuppressor operating by a novel molecular mechanism. They supported the view that M. ulcerans produces mycolactone to evade recognition and control by the host immune system.
2 Molecular Targets and Mechanisms of Action
In recent years, three distinct molecular targets have been proposed to explain the diverse biological effects of mycolactone. Proposed mechanisms of action are discussed below, starting from the most recently discovered, the Sec61 translocon, as we feel it is important to re-examine previous findings in the light of these new data.
2.1 Sec61 Blockade
Simmonds and co-workers made a breakthrough in 2014 by discovering that mycolactone blocks the production of TNF and other model secretory proteins by preventing protein translocation into the endoplasmic reticulum (ER), leading to their subsequent degradation in the cytosol by the ubiquitin:proteasome system [62]. Using cell-free systems, High and co-workers then showed that mycolactone causes selective defects in the cotranslational insertion of Sec61 secretory substrates [65]. Sec61 is a heterotrimeric complex which in eukaryotes mediates the transport of almost all secretory proteins (secreted proteins and most ER-, Golgi-, endosome- and lysosome-resident proteins, as well as proteins containing glycosylphosphatidylinositol-anchors) and integral transmembrane proteins (TMPs) into the ER [66]. We found that mycolactone operates by targeting the pore-forming subunit of the Sec61 translocon, and identified a single amino acid mutation conferring resistance to mycolactone, localizing its interaction site near the lumenal plug of Sec61 [67]. Expression of this Sec61 mutant in mycolactone-treated T cells rescued their homing potential and effector functions, providing definitive genetic evidence that Sec61 is the host receptor mediating the immunomodulatory effects of mycolactone [67]. Compared to previously known Sec61 inhibitors (cotransin and derivatives, Apratoxin A), mycolactone was more effective at blocking the production of cytokines and homing receptors by immune cells [46, 67]. Mycolactone is therefore the first Sec61 inhibitor that is produced by a human pathogen, and the most potent inhibitor to date.
TMPs can be divided into Type I, II or III, according to the presence of a signal peptide (SP) and the orientation of the protein’s N-terminus at the ER membrane [68]. Using in vitro assays of single-pass TMP translocation, High and co-workers observed that secretory proteins, Type I and Type II TMPs were generally susceptible to mycolactone-mediated Sec61 blockade, while in contrast, mycolactone had no effect on Type III TMP integration [65, 69]. This led the authors to hypothesize that protein resistance to mycolactone essentially depends on how the protein initially engages the translocon. Our profiling of mycolactone’s signature in the proteome of CD4+ T lymphocytes, DCs and DRG neurons broadly supported this mechanistic model for the effects of mycolactone on different classes of single-spanning TMPs, and extended its validity to multi-pass TMPs [46, 60, 67]. Importantly, these global analyses also highlighted proteomic alterations beyond Sec61 substrates, resulting from secondary effects of protein translocation blockade. For instance, by blocking the production of both IFN-γ and IFN-γ receptor in activated lymphocytes, mycolactone affected the autocrine activation of multiple IFN-responsive genes [67]. In macrophages, it resulted in altered IFN-γ-induced production of nitric oxide synthase (iNOS) [67], which is essential for the control of M. ulcerans infection. In addition to highlighting the critical importance of Sec61 activity for immune cell function, migration and communication, these data thus provided a molecular explanation for the immunological defects of BU patients.
Mycolactone was recently shown to promote Bim-dependent cell apoptosis through the mTORC2-Akt-FoxO3 axis [36]. Using L929 fibroblasts as a model, Bieri et al. found that mycolactone inhibits the mechanistic Target of Rapamycin (mTOR) signaling pathway, leading to inactivation of the serine/threonine kinase Akt, subsequent activation of the transcription factor FoxO3, and up-regulation of pro-apoptotic Bim. The importance of Bim-dependent apoptosis in the pathogenesis was demonstrated by M. ulcerans infection of Bim knock-out mice, which did not develop necrotic BU lesions and was able to contain the mycobacterial multiplication [36]. How mycolactone interferes with mTORC2 signaling nevertheless remained elusive. Subsequent studies by our group showed that expression of mycolactone-resistant mutants of Sec61 in HEK293 cells protected them against the cytopathic effects of the toxin [67], establishing the critical role of Sec61 blockade in mycolactone-induced cell death. Recently, Simmonds and co-workers reported that inhibition of Sec61-dependent translocation by mycolactone rapidly activates an integrated stress response (ISR) in RAW264.7 and HeLa cells without activating ER stress sensors, driving cytotoxicity via the ATF4/CHOP/Bcl-2/Bim route [70]. Our studies in DCs led to comparable conclusions with regard to mycolactone stimulating ATF4/CHOP signaling, however we detected ER stress-specific activation signals within hours of treatment [46]. Mycolactone-driven ER stress nevertheless differed from conventional unfolded protein response (UPR) by the downregulation of GRP78/BIP, a master regulator of the UPR that is normally induced by canonical ER stress. Whether mycolactone-driven ATF4 induction results primarily from ISR or UPR may thus depend on the cell type. Of note, there is convincing evidence that chronic UPR causes Akt inhibition through complex interactions with mTORC1/C2 signaling [71], providing a possible explanation for how Sec61 blockade activates the mTORC2-Akt-FoxO3 axis, leading to apoptosis by a parallel route downstream of the UPR [36]. Importantly, both studies support the view that mycolactone-mediated cytotoxicity is a late consequence of Sec61 blockade, resulting from the induction of chronic stress triggering apoptosis via the ATF4/CHOP/Bcl-2/Bim signaling pathway.
In summary, by blocking Sec61, mycolactone downregulates the production of a large array of secretory and membrane receptors. One major effect of this molecular blockade is that the host is unable to mount an effective immune response to the underlying mycobacterial infection. Prolonged cell exposure to mycolactone is however cytotoxic, since it triggers stress responses inducing apoptosis.
2.2 AT2R Stimulation
In 2014, Marsollier, Brodin and co-workers identified type 2 angiotensin II receptors (AT2R) as additional targets of mycolactone, mediating its analgesic properties [50]. In primary hippocampal neurons, mycolactone (>7 nM) provoked cell hyperpolarization within 20 min, through activation of KCN4 (TRAAK) potassium channels. The authors showed that mycolactone operates by activating AT2R, leading to phospholipase A2-mediated arachidonic acid (AA) liberation, and generation of prostaglandin E2 (PGE2) from AA by cyclooxygenase-1, which activates KCN4. Mycolactone also caused the hyperpolarization of primary DRGs, at concentrations superior to 1 μM [48]. The importance of the AT2R signaling cascade in mycolactone-driven hypoesthesia was demonstrated by use of an AT2R selective blocker in mice infected with M. ulcerans, or injected with mycolactone, and AT2R-KO animals as controls [50]. AT2R silencing in HeLa cells had no effect on mycolactone-driven defects in IFN-γ receptor expression nor MCP-1 chemokine production, indicating that AT2R is not involved in mycolactone-mediated immunomodulation [67]. Based on our current understanding of mycolactone-mediated Sec61 blockade, the biogenesis of the multi-spanning membrane protein AT2R may resist its inhibitory effects on protein translocation [69]. Together with the recent evidences of Sec61-dependent anti-inflammatory activity of mycolactone on the immune and nervous systems, and toxicity in neurons (see Sect. 2.1), these data suggest that mycolactone-mediated activation of AT2R and inhibition of Sec61 are two independent mechanisms contributing to BU-associated analgesia.
2.3 N-WASP Activation
In 2013, we reported that mycolactone activates the Wiskott-Aldrich syndrome protein (WASP) family of actin-nucleating factors, leading to uncontrolled assembly of actin [18]. WASP and N-WASP belong to a family of scaffold proteins mediating the dynamic remodeling of the actin cytoskeleton, via interaction of their C-terminal verprolin-cofilin-acidic (VCA) domain with the ARP2/3 actin-nucleating complex. While WASP expression is restricted to hematopoietic cells, N-WASP is widely expressed. In basal conditions, WASP and N-WASP are auto-inhibited by intramolecular interactions sequestering the VCA domain from ARP2/3. Binding of activated GTPases or phosphoinositide lipids to N-terminal target sequences triggers conformational changes resulting in release of the VCA, thereby enabling binding and activation of ARP2/3. Using in vitro assays of actin polymerization, we showed that mycolactone mimics endogenous GTPase CDC42 in disruption of WASP/N-WASP auto-inhibition. The possible contribution of WASP/N-WASP activation to mycolactone immunosuppressive effects was ruled out by silencing or inhibiting the proteins in cellular assays of cytokine production [18]. In contrast, N-WASP inhibition by wiskostatin in HeLa epithelial cells partly relieved mycolactone-induced alterations in actin polymerization and cell adhesion. A Sec61 blockade is unlikely to explain such early, wiskostatin-sensitive effects of mycolactone. We propose that a fraction of mycolactone may bind to cytosolic WASP/N-WASP following its diffusion through the plasma membrane, leading to the uncontrolled assembly of actin and defective cell-matrix adhesion. In support of this model, a limited, yet significant co-localization of mycolactone with active WASP was observed after 1 h of treatment in HeLa cells [18]. Mycolactone injection in mouse epidermis induced structural changes in the multilayered organization of this tissue that were suppressed by wiskostatin, suggesting that mycolactone-driven activation of N-WASP and subsequent defects in cell adhesion occur in vivo. While this mechanism may not be central to BU pathogenesis, we speculate that it may synergize with Sec61 blockade to impair skin integrity.
3 Conclusions
Recent years have witnessed tremendous progress in our understanding of the molecular mechanisms underpinning mycolactone biology (summarized in Fig. 4), and therefore BU pathogenesis. Notably, studies on mycolactone have revealed a novel mechanism of immunomodulation based on protein translocation blockade, which may be exploited therapeutically to limit inflammation and pain. Meanwhile, attempts have been made to develop a live vaccine for BU based on immunization with mycolactone-negative M. ulcerans strains [8]. However, vaccination only transiently delayed the onset of pathology induced by a highly virulent M. ulcerans strain [72]. While mycolactone is an obvious target antigen for a BU subunit vaccine, initial attempts by several groups to develop an immunogenic formulation of the toxoid failed [73]. Recent studies nevertheless indicated that toxin neutralizing antibodies can be elicited by protein-carrier conjugates of synthetic mycolactone [37], providing new prospects for the development of a BU vaccine.

Proposed molecular targets and mechanisms of action for mycolactone-mediated ulcerative, immunosuppressive and analgesic properties
References
Connor DH, Lunn HF (1965) Mycobacterium ulcerans infection (with comments on pathogenesis). Int J Lepr 33(Suppl):698–709
Krieg RE, Hockmeyer WT, Connor DH (1974) Toxin of Mycobacterium ulcerans. Production and effects in guinea pig skin. Arch Dermatol 110:783–788
Read JK, Heggie CM, Meyers WM, Connor DH (1974) Cytotoxic activity of Mycobacterium ulcerans. Infect Immun 9:1114–1122
George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, Lee R, Small PL (1999) Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283:854–857
Kishi Y (2011) Chemistry of mycolactones, the causative toxins of Buruli ulcer. Proc Natl Acad Sci U S A 108:6703–6708
Benowitz AB, Fidanze S, Small PLC, Kishi Y (2001) Stereochemistry of the core structure of the mycolactones. J Am Chem Soc 123:5128–5129
Fidanze S, Song FB, Szlosek-Pinaud M, Small PLC, Kishi Y (2001) Complete structure of the mycolactones. J Am Chem Soc 123:10117–10118
Doig KD, Holt KE, Fyfe JA, Lavender CJ, Eddyani M, Portaels F, Yeboah-Manu D, Pluschke G, Seemann T, Stinear TP (2012) On the origin of Mycobacterium ulcerans, the causative agent of Buruli ulcer. BMC Genomics 13:258
Chany AC, Casarotto V, Schmitt M, Tarnus C, Guenin-Mace L, Demangel C, Mirguet O, Eustache J, Blanchard N (2011) A diverted total synthesis of mycolactone analogues: an insight into Buruli ulcer toxins. Chemistry 17:14413–14419
Guenin-Mace L, Baron L, Chany AC, Tresse C, Saint-Auret S, Jonsson F, Le Chevalier F, Bruhns P, Bismuth G, Hidalgo-Lucas S, Bisson JF, Blanchard N, Demangel C (2015) Shaping mycolactone for therapeutic use against inflammatory disorders. Sci Transl Med 7:289ra285
Scherr N, Gersbach P, Dangy JP, Bomio C, Li J, Altmann KH, Pluschke G (2013) Structure-activity relationship studies on the macrolide exotoxin mycolactone of Mycobacterium ulcerans. PLoS Negl Trop Dis 7:e2143
Gehringer M, Altmann KH (2017) The chemistry and biology of mycolactones. Beilstein J Org Chem 13:1596–1660
Saint-Auret S, Chany AC, Casarotto V, Tresse C, Parmentier L, Abdelkafi H, Blanchard N (2017) Total syntheses of Mycolactone A/B and its analogues for the exploration of the biology of Buruli ulcer. Chimia (Aarau) 71:836–840
Hong H, Coutanceau E, Leclerc M, Caleechurn L, Leadlay PF, Demangel C (2008) Mycolactone diffuses from Mycobacterium ulcerans-infected tissues and targets mononuclear cells in peripheral blood and lymphoid organs. PLoS Negl Trop Dis 2:e325
Sarfo FS, Phillips RO, Zhang J, Abass MK, Abotsi J, Amoako YA, Adu-Sarkodie Y, Robinson C, Wansbrough-Jones MH (2014) Kinetics of mycolactone in human subcutaneous tissue during antibiotic therapy for Mycobacterium ulcerans disease. BMC Infect Dis 14:202
Sarfo FS, Le Chevalier F, Aka N, Phillips RO, Amoako Y, Boneca IG, Lenormand P, Dosso M, Wansbrough-Jones M, Veyron-Churlet R, Guenin-Mace L, Demangel C (2011) Mycolactone diffuses into the peripheral blood of buruli ulcer patients - implications for diagnosis and disease monitoring. PLoS Negl Trop Dis 5:e1237
Snyder DS, Small PL (2003) Uptake and cellular actions of mycolactone, a virulence determinant for Mycobacterium ulcerans. Microb Pathog 34:91–101
Guenin-Mace L, Veyron-Churlet R, Thoulouze MI, Romet-Lemonne G, Hong H, Leadlay PF, Danckaert A, Ruf MT, Mostowy S, Zurzolo C, Bousso P, Chretien F, Carlier MF, Demangel C (2013) Mycolactone activation of Wiskott-Aldrich syndrome proteins underpins Buruli ulcer formation. J Clin Invest 123(4):1501–1512
Lopez CA, Unkefer CJ, Swanson BI, Swanson JMJ, Gnanakaran S (2018) Membrane perturbing properties of toxin mycolactone from Mycobacterium ulcerans. PLoS Comput Biol 14:e1005972
Nitenberg M, Benarouche A, Maniti O, Marion E, Marsollier L, Gean J, Dufourc EJ, Cavalier JF, Canaan S, Girard-Egrot AP (2018) The potent effect of mycolactone on lipid membranes. PLoS Pathog 14:e1006814
Hayman J (1993) Out of Africa: observations on the histopathology of Mycobacterium ulcerans infection. J Clin Pathol 46:5–9
Ruf MT, Bolz M, Vogel M, Bayi PF, Bratschi MW, Sopho GE, Yeboah-Manu D, Boock AU, Junghanss T, Pluschke G (2016) Spatial distribution of Mycobacterium ulcerans in Buruli ulcer lesions: implications for laboratory diagnosis. Plos Negl Trop Dis 10:e0004767
Ruf MT, Sopoh GE, Brun LV, Dossou AD, Barogui YT, Johnson RC, Pluschke G (2011) Histopathological changes and clinical responses of Buruli ulcer plaque lesions during chemotherapy: a role for surgical removal of necrotic tissue? PLoS Negl Trop Dis 5:e1334
Bolz M, Ruggli N, Borel N, Pluschke G, Ruf MT (2016) Local cellular immune responses and pathogenesis of Buruli ulcer lesions in the experimental Mycobacterium ulcerans pig infection model. PLoS Negl Trop Dis 10:e0004678
Liu Y, Chen JY, Shang HT, Liu CE, Wang Y, Niu R, Wu J, Weiv H (2010) Light microscopic, Electron microscopic, and Immunohistochemical comparison of Bama Minipig (Sus scrofa domestica) and human skin. Comp Med 60:142–148
Sullivan TP, Eaglstein WH, Davis SC, Mertz P (2001) The pig as a model for human wound healing. Wound Repair Regen 9:66–76
George KM, Pascopella L, Welty DM, Small PL (2000) A Mycobacterium ulcerans toxin, mycolactone, causes apoptosis in guinea pig ulcers and tissue culture cells. Infect Immun 68:877–883
Silva-Gomes R, Marcq E, Trigo G, Goncalves CM, Longatto-Filho A, Castro AG, Pedrosa J, Fraga AG (2015) Spontaneous healing of Mycobacterium ulcerans lesions in the guinea pig model. PLoS Negl Trop Dis 9:e0004265
Ruf MT, Steffen C, Bolz M, Schmid P, Pluschke G (2017) Infiltrating leukocytes surround early Buruli ulcer lesions, but are unable to reach the mycolactone producing mycobacteria. Virulence 8:1918–1926
Coutanceau E, Marsollier L, Brosch R, Perret E, Goossens P, Tanguy M, Cole ST, Small PL, Demangel C (2005) Modulation of the host immune response by a transient intracellular stage of Mycobacterium ulcerans: the contribution of endogenous mycolactone toxin. Cell Microbiol 7:1187–1196
Adusumilli S, Mve-Obiang A, Sparer T, Meyers W, Hayman J, Small PLC (2005) Mycobacterium ulcerans toxic macrolide, mycolactone modulates the host immune response and cellular location of M-ulcerans in vitro and in vivo. Cell Microbiol 7:1295–1304
Coutanceau E, Decalf J, Martino A, Babon A, Winter N, Cole ST, Albert ML, Demangel C (2007) Selective suppression of dendritic cell functions by Mycobacterium ulcerans toxin mycolactone. J Exp Med 204:1395–1403
Schutte D, Pluschke G (2009) Immunosuppression and treatment-associated inflammatory response in patients with Mycobacterium ulcerans infection (Buruli ulcer). Expert Opin Biol Ther 9:187–200
Schutte D, Um-Boock A, Mensah-Quainoo E, Itin P, Schmid P, Pluschke G (2007) Development of highly organized lymphoid structures in Buruli ulcer lesions after treatment with rifampicin and streptomycin. PLoS Negl Trop Dis 1:e2
Gama JB, Ohlmeier S, Martins TG, Fraga AG, Sampaio-Marques B, Carvalho MA, Proenca F, Silva MT, Pedrosa J, Ludovico P (2014) Proteomic analysis of the action of the Mycobacterium ulcerans toxin mycolactone: targeting host cells cytoskeleton and collagen. PLoS Negl Trop Dis 8:e3066
Bieri R, Scherr N, Ruf MT, Dangy JP, Gersbach P, Gehringer M, Altmann KH, Pluschke G (2017) The macrolide toxin mycolactone promotes bim-dependent apoptosis in Buruli ulcer through inhibition of mTOR. ACS Chem Biol 12:1297–1307
Dangy JP, Scherr N, Gersbach P, Hug MN, Bieri R, Bomio C, Li J, Huber S, Altmann KH, Pluschke G (2016) Antibody-mediated neutralization of the exotoxin Mycolactone, the main virulence factor produced by Mycobacterium ulcerans. PLoS Negl Trop Dis 10:e0004808
Ogbechi J, Ruf MT, Hall BS, Bodman-Smith K, Vogel M, Wu HL, Stainer A, Esmon CT, Ahnstrom J, Pluschke G, Simmonds RE (2015) Mycolactone-dependent depletion of endothelial cell thrombomodulin is strongly associated with fibrin deposition in Buruli ulcer lesions. PLoS Pathog 11:e1005011
Bozzo C, Tiberio R, Graziola F, Pertusi G, Valente G, Colombo E, Small PL, Leigheb G (2010) A Mycobacterium ulcerans toxin, mycolactone, induces apoptosis in primary human keratinocytes and in HaCaT cells. Microbes Infect 12:1258–1263
Gronberg A, Zettergren L, Bergh K, Stahle M, Heilborn J, Angeby K, Small PL, Akuffo H, Britton S (2010) Antioxidants protect keratinocytes against M. ulcerans mycolactone cytotoxicity. PLoS One 5:e13839
Graziola F, Colombo E, Tiberio R, Leigheb G, Bozzo C (2017) Mycobacterium ulcerans mycolactone interferes with adhesion, migration and proliferation of primary human keratinocytes and HaCaT cell line. Arch Dermatol Res 309:179–189
Zavattaro E, Boccafoschi F, Borgogna C, Conca A, Johnson RC, Sopoh GE, Dossou AD, Colombo E, Clemente C, Leigheb G, Valente G (2012) Apoptosis in Buruli ulcer: a clinicopathological study of 45 cases. Histopathology 61:224–236
Goto M, Nakanaga K, Aung T, Hamada T, Yamada N, Nomoto M, Kitajima S, Ishii N, Yonezawa S, Saito H (2006) Nerve damage in Mycobacterium ulcerans-infected mice - probable cause of painlessness in Buruli ulcer. Am J Pathol 168:805–811
En J, Goto M, Nakanaga K, Higashi M, Ishii N, Saito H, Yonezawa S, Hamada H, Small PL (2008) Mycolactone is responsible for the painlessness of Mycobacterium ulcerans infection (buruli ulcer) in a murine study. Infect Immun 76:2002–2007
Anand U, Sinisi M, Fox M, MacQuillan A, Quick T, Korchev Y, Bountra C, McCarthy T, Anand P (2016) Mycolactone-mediated neurite degeneration and functional effects in cultured human and rat DRG neurons: mechanisms underlying hypoalgesia in Buruli ulcer. Mol Pain 12:1744806916654144
Morel JD, Paatero AO, Wei J, Yewdell JW, Guenin-Macé L, Van Haver D, Impens F, Pietrosemoli N, Paavilainen VO, Demangel C (2018) Proteomics reveals scope of mycolactone-mediated Sec61 blockade and distinctive stress signature. Mol Cell Proteomics 17(9):1750–1765. https://doi.org/10.1074/mcp.RA118.000824
Isaac C, Mauborgne A, Grimaldi A, Ade K, Pohl M, Limatola C, Boucher Y, Demangel C, Guenin-Mace L (2017) Mycolactone displays anti-inflammatory effects on the nervous system. PLoS Negl Trop Dis 11:e0006058
Song OR, Kim HB, Jouny S, Ricard I, Vandeputte A, Deboosere N, Marion E, Queval CJ, Lesport P, Bourinet E, Henrion D, Oh SB, Lebon G, Sandoz G, Yeramian E, Marsollier L, Brodin P (2017) A bacterial toxin with analgesic properties: hyperpolarization of DRG neurons by Mycolactone. Toxins (Basel) 9:E227
En J, Kitamoto S, Kawashima A, Yonezawa S, Kishi Y, Ishii N, Goto M (2017) Mycolactone cytotoxicity in Schwann cells could explain nerve damage in Buruli ulcer. PLoS Negl Trop Dis 11:e0005834
Marion E, Song OR, Christophe T, Babonneau J, Fenistein D, Eyer J, Letournel F, Henrion D, Clere N, Paille V, Guerineau NC, Saint Andre JP, Gersbach P, Altmann KH, Stinear TP, Comoglio Y, Sandoz G, Preisser L, Delneste Y, Yeramian E, Marsollier L, Brodin P (2014) Mycobacterial toxin induces analgesia in buruli ulcer by targeting the angiotensin pathways. Cell 157:1565–1576
Gooding TM, Johnson PD, Campbell DE, Hayman JA, Hartland EL, Kemp AS, Robins-Browne RM (2001) Immune response to infection with Mycobacterium ulcerans. Infect Immun 69:1704–1707
Prevot G, Bourreau E, Pascalis H, Pradinaud R, Tanghe A, Huygen K, Launois P (2004) Differential production of systemic and intralesional gamma interferon and interleukin-10 in nodular and ulcerative forms of Buruli disease. Infect Immun 72:958–965
Westenbrink BD, Stienstra Y, Huitema MG, Thompson WA, Klutse EO, Ampadu EO, Boezen HM, Limburg PC, van der Werf TS (2005) Cytokine responses to stimulation of whole blood from patients with buruli ulcer disease in Ghana. Clin Diagn Lab Immun 12:125–129
Yeboah-Manu D, Peduzzi E, Mensah-Quainoo E, Asante-Poku A, Ofori-Adjei D, Pluschke G, Daubenberger CA (2006) Systemic suppression of interferon-gamma responses in Buruli ulcer patients resolves after surgical excision of the lesions caused by the extracellular pathogen Mycobacterium ulcerans. J Leukoc Biol 79:1150–1156
Phillips R, Sarfo FS, Guenin-Mace L, Decalf J, Wansbrough-Jones M, Albert ML, Demangel C (2009) Immunosuppressive signature of cutaneous Mycobacterium ulcerans infection in the peripheral blood of patients with Buruli ulcer disease. J Infect Dis 200(11):1675–1684
Nausch N, Antwi-Berko D, Mubarik Y, Abass KM, Owusu W, Owusu-Dabo E, Debrah LB, Debrah AY, Jacobsen M, Phillips RO (2017) Analysis of Mycobacterium ulcerans-specific T-cell cytokines for diagnosis of Buruli ulcer disease and as potential indicator for disease progression. PLoS Negl Trop Dis 11:e0005415
Phillips R, Horsfield C, Kuijper S, Sarfo SF, Obeng-Baah J, Etuaful S, Nyamekye B, Awuah P, Nyarko KM, Osei-Sarpong F, Lucas S, Kolk AH, Wansbrough-Jones M (2006) Cytokine response to antigen stimulation of whole blood from patients with Mycobacterium ulcerans disease compared to that from patients with tuberculosis. Clin Vaccine Immunol 13:253–257
Phillips RO, Sarfo FS, Landier J, Oldenburg R, Frimpong M, Wansbrough-Jones M, Abass K, Thompson W, Forson M, Fontanet A, Niang F, Demangel C (2014) Combined inflammatory and metabolic defects reflected by reduced serum protein levels in patients with Buruli ulcer disease. PLoS Negl Trop Dis 8:e2786
Pahlevan AA, Wright DJ, Andrews C, George KM, Small PL, Foxwell BM (1999) The inhibitory action of Mycobacterium ulcerans soluble factor on monocyte/T cell cytokine production and NF-kappa B function. J Immunol 163:3928–3935
Grotzke JE, Kozik P, Morel JD, Impens F, Pietrosemoli N, Cresswell P, Amigorena S, Demangel C (2017) Sec61 blockade by mycolactone inhibits antigen cross-presentation independently of endosome-to-cytosol export. Proc Natl Acad Sci U S A 114:E5910–E5919
Simmonds RE, Lali FV, Smallie T, Small PL, Foxwell BM (2009) Mycolactone inhibits monocyte cytokine production by a posttranscriptional mechanism. J Immunol 182:2194–2202
Hall BS, Hill K, McKenna M, Ogbechi J, High S, Willis AE, Simmonds RE (2014) The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog 10:e1004061
Boulkroun S, Guenin-Mace L, Thoulouze MI, Monot M, Merckx A, Langsley G, Bismuth G, Di Bartolo V, Demangel C (2010) Mycolactone suppresses T cell responsiveness by altering both early signaling and posttranslational events. J Immunol 184:1436–1444
Guenin-Mace L, Carrette F, Asperti-Boursin F, Le Bon A, Caleechurn L, Di Bartolo V, Fontanet A, Bismuth G, Demangel C (2011) Mycolactone impairs T cell homing by suppressing microRNA control of L-selectin expression. Proc Natl Acad Sci U S A 108:12833–12838
McKenna M, Simmonds RE, High S (2016) Mechanistic insights into the inhibition of Sec61-dependent co- and post-translational translocation by mycolactone. J Cell Sci 129(7):1404–1415
Park E, Rapoport TA (2012) Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu Rev Biophys 41:21–40
Baron L, Paatero AO, Morel JD, Impens F, Guenin-Mace L, Saint-Auret S, Blanchard N, Dillmann R, Niang F, Pellegrini S, Taunton J, Paavilainen VO, Demangel C (2016) Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J Exp Med 213:2885–2896
Goder V, Spiess M (2001) Topogenesis of membrane proteins: determinants and dynamics. FEBS Lett 504:87–93
McKenna M, Simmonds RE, High S (2017) Mycolactone reveals the substrate-driven complexity of Sec61-dependent transmembrane protein biogenesis. J Cell Sci 130:1307–1320
Ogbechi J, Hall BS, Sbarrato T, Taunton J, Willis AE, Wek RC, Simmonds RE (2018) Inhibition of Sec61-dependent translocation by mycolactone uncouples the integrated stress response from ER stress, driving cytotoxicity via translational activation of ATF4. Cell Death Dis 9:397
Appenzeller-Herzog C, Hall MN (2012) Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol 22:274–282
Fraga AG, Martins TG, Torrado E, Huygen K, Portaels F, Silva MT, Castro AG, Pedrosa J (2012) Cellular immunity confers transient protection in experimental Buruli ulcer following BCG or mycolactone-negative Mycobacterium ulcerans vaccination. PLoS One 7:e33406
Einarsdottir T, Huygen K (2011) Buruli ulcer. Hum Vaccin 7:1198–1203
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this chapter

Cite this chapter
Guenin-Macé, L., Ruf, MT., Pluschke, G., Demangel, C. (2019). Mycolactone: More than Just a Cytotoxin. In: Pluschke, G., Röltgen, K. (eds) Buruli Ulcer. Springer, Cham. https://doi.org/10.1007/978-3-030-11114-4_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-11114-4_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11113-7
Online ISBN: 978-3-030-11114-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)