
Abstract
Increasing evidence implicates astrocytic dysfunction in Alzheimer’s disease (AD), a neurodegenerative disorder characterised by progressive cognitive loss. The accumulation of amyloid-β (Aβ) plaques is a histopathological hallmark of AD and associated with increased astrocyte reactivity. In APP/PS1 mice modelling established AD (9 months), we now show an altered astrocytic morphology and enhanced activity of astrocytic hemichannels, mainly composed by connexin 43 (Cx43). Hemichannel activity in hippocampal astrocytes is also increased in two models of early AD: (1) mice with intracerebroventricular (icv) administration of Aβ1-42, and (2) hippocampal slices superfused with Aβ1-42 peptides. In hippocampal gliosomes of APP/PS1 mice, Cx43 levels were increased, whereas mice administered icv with Aβ1-42 only displayed increased Cx43 phosphorylation levels. This suggests that hemichannel activity might be differentially modulated throughout AD progression. Additionally, we tested if adenosine A2A receptor (A2AR) blockade reversed alterations of astrocytic hemichannel activity and found that the pharmacological blockade or genetic silencing (global and astrocytic) of A2AR prevented Aβ-induced hemichannel dysregulation in hippocampal slices, although A2AR genetic silencing increased the activity of astroglial hemichannels in control conditions. In primary cultures of astrocytes, A2AR-related protective effect was shown to occur through a protein kinase C (PKC) pathway. Our results indicate that the dysfunction of hemichannel activity in hippocampal astrocytes is an early event in AD, which is modulated by A2AR.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterised by a progressive decline of cognitive functions, namely learning and memory, which is linked with an abnormal accumulation of amyloid-β peptides (Aβ). Although the presence of Aβ plaques is a major histopathological hallmark of established AD, it is considered that the extracellular accumulation of soluble Aβ oligomers is a causative agent of neurodegeneration, mainly of synaptic deterioration that is usually associated with the first signs of memory impairments preceding the formation of Aβ deposits that are characteristic of early AD (reviewed in [1]). The hippocampus is particularly affected in early stages of AD, undergoing structural and functional changes typified by alterations of synaptic plasticity that are considered the neurophysiological basis of learning and memory encoding (reviewed in [2]). Astrocytes are glial cells with multiple processes that establish contact with 60–70% of hippocampal synapses and regulate synaptic plasticity, mainly through their capacity to release gliotransmitters, such as ATP and glutamate, and to uptake glutamate and GABA from the synaptic cleft [3,4,5]. Accordingly, increasing evidence supports a role of astrocytes in AD onset and progression, as heralded by several morphological and molecular changes of astrocytes in AD mouse models (e.g. APP/PS1) and in human patients [6, 7] as well as by the link between Aβ plaques formation and changes in astrocytes morphology and activity [8, 9].
Astrocytic responses depend on the functions of connexins (Cx), which are proteins forming gap junction channels and hemichannels that allow inter-cellular fluxes of ions and small molecules, such as IP3, ATP, glutamate, and energy metabolites (reviewed in [10, 11]). Astrocytic hemichannels are mainly composed of hexamers of Cx43 and Cx30, which accumulate in astrocyte processes allowing the release of gliotransmitter and ion fluxes that can regulate hippocampal synaptic transmission and plasticity, and consequently memory [12, 13]. Astrocytic Cx43 levels are upregulated in animal models of AD, namely in transgenic APP/PS1 and 5xFAD mice [14,15,16]. Moreover, there are evidences of dysfunctional astrocytic hemichannels in animal models of AD that do not exhibit alterations on gap junction astrocytic communication [17,18,19]. Furthermore, in adult APP/PS1 mice with 8–18 months of age, it was reported that activated microglial cells did not contribute to the elevated connexin immunoreactivity that was concentrated in astroglial processes infiltrating the amyloid plaques [14], implying that the AD-related alteration of connexins hemichannels is intrinsic to astrocytes.
Our group previously showed that cultured astrocytes exposed to Aβ peptides display an enhancement of Cx43 hemichannels and that adenosine A2A receptors (A2AR), which are closely associated with Cx43, regulate the levels and activity of hemichannels [20]. A2AR modulate key astrocytic functions that are affected by Aβ exposure, such as glutamate uptake [21], ATP release and hemichannel activity [20], and Ca2+ signalling [22], and we recently showed that the genetic deletion of astrocytic A2AR in the hippocampus of adult mice impairs synaptic plasticity and cause memory deficits [23]. Moreover, several studies support the idea that A2AR are a promising target to manage AD, since the pharmacological or genetic blockade of A2AR confers neuroprotection [24,25,26,27,28]. Likewise, as occurs for Cx43, astrocytic A2AR are increased in the hippocampus of mice exposed intra-cerebro-ventricularly to Aβ [21], mice expressing human amyloid precursor protein [29], 3xTg-AD mice [28] and APP/PS1 mice [26] as well as in AD patients [29]. However, it remains to be clarified if the activity of Cx43 hemichannels is modified in early AD and whether A2AR regulate the activity of hemichannels in hippocampal astrocytes. Thus, in the present study, we resorted to in vivo and ex vivo models of early AD to investigate alterations in astrocytic hemichannel activity in hippocampal slices and tested if A2AR modulate Cx43 hemichannel activity under physiological and AD-like conditions.
Materials and methods
Animals and surgeries
Animals were kept with food and water available ad libitum under a controlled environment (23 ± 2 °C, 50 ± 10% relative humidity and 12 h light/dark cycle). Mice were handled following the “3R” principles, and all experiments were approved by the Ethical Committee of the Center for Neuroscience and Cell Biology of the University of Coimbra (ORBEA_300_2021/24092021) and certified by Direção Geral de Alimentação e Veterinária (DGAV; Portuguese National Authority for Animal Health and Well-Being, 0421/000/000/2021).
A line of double transgenic mice, APPswe/PS1dE9 (APP/PS1), with B6C3F1/J genetic background (Jackson Laboratory, Bar Harbor, Maine, strain B6;C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax RRID:MMRRC_034832-JAX) and wild type (WT) littermates were used. We chose to use male mice, to avoid estrous cycle influences, with 9 months old, as the deficits of reference memory and of synaptic plasticity are already evident in both males and females at this age [25, 30]. We also used C57Bl/6 male mice obtained from Charles River Laboratories Barcelona, Spain (MGI catalogue number (Cat#) 5811150, Research Resource Identifier, RRID:MGI:5811150), global A2AR knockout (GbA2ARKO) and forebrain neuron conditional knockout mice (FbA2ARKO) with C57Bl/6 genetic background that were generated by Jiang-Fan Chen (Boston University School of Medicine, MA). The selective deletion of A2AR in astrocytes was accomplished in A2AR floxed (A2Aflox/flox) mice through the Cre-lox method. Briefly, an AAV5-GFAP-GFP-CRE viral construct (1 µL from 4.8 × 1012 particles/mL, obtained from Vector Core, University of North Carolina, USA) was bilaterally administrated into the CA1 region of the dorsal hippocampus (GFAP-CRE-A2AR mice), whereas control (GFAP-CTR) mice received a similar construct without Cre recombinase, AAV5-GFAP-eGFP (Vector Core, University of North Carolina, USA), as previously described [23].
Some C57Bl/6 mice (5 mice) were subjected to an intra-cerebro-ventricular (icv) administration of the synthetic peptide Aβ1-42 (Bachem), carried out as previously described [24, 31]. Aβ1-42 was dissolved in sterile water to obtain a solution mostly composed of soluble monomers and low molecular weight oligomers (2.25 mg/mL) and 4 μL of Aβ1-42 (2 nmol) or water (vehicle) were administered icv, which translates into 5–30 pmol levels of Aβ1-42 within the hippocampus [24]. This Aβ1-42-icv model recapitulates two main features of early AD, namely the impairment of reference memory and of synaptic function, as we previous reported [24, 31].
Morris water maze
The Morris water maze test was performed as previously described [32] in order to evaluate hippocampal-dependent spatial learning and memory. The test was done using a 105 cm diameter circular pool filled with water opacified with non-toxic white paint. A platform was submerged 1 cm underneath water surface. The acquisition training phase consisted of four swimming trials of 60 s per day with a 20 min inter-trial interval. In each trial, mice were placed in the pool from a different drop location and given 60 s to find the location of the hidden platform. To complete the trial, the animals must remain 10 s on the platform. This stage was completed when the average of escape latency from the four drop points reached 20 s in WT mice. It was then followed by a retention/probe trial in which the platform was removed and mice were placed on the pool from a random drop location and allowed to swim freely for 60 s. The number of crossings of the hidden platform location was recorded with Any-Maze version 4.99 tracking software (Stoelting, Wood Dale, USA, RRID:SCR_014289). The strategies used by mice to find the platform location in the retention trial were analysed offline and classified as hippocampus-dependent (allocentric) or hippocampus-independent (egocentric), as previously described [33].
Pharmacological treatments in hippocampal slices
Mice were sacrificed by decapitation after deep anaesthesia with halothane. The brain was removed and the hippocampus dissected in ice-cold, oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid solution (aCSF, composition: 124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, 1 mM MgSO4, and 2 mM CaCl2; pH 7.4). Transversal hippocampal slices (400 μm-thick) were obtained with a McIlwain tissue chopper, and then allowed to recover functional and energetically in gassed ACSF for 90 min at 32 °C.
Control hippocampal slices (CTR) were only superfused with aCSF. For Aβ1-42 challenge, hippocampal slices were superfused with Aβ1-42 (50 nM in aCSF) for 60 min, a concentration and time of peptide exposure that we previously showed to affect synaptic plasticity (see [34]), before assaying hemichannel activity. Aβ1-42 solution contained mainly soluble monomers and oligomers, as previously reported by us [21, 24]. When testing the effect of A2AR, slices were incubated for 30 min with the A2AR selective antagonist, SCH58261, [(2-(2-furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine, 50 nM in aCSF, Tocris Bioscience] prior to the challenge with Aβ1-42, and continuously superfused with SCH58261 during the exposure to Aβ1-42. In experiments with APP/PS1 mice, hippocampal slices were superfused with Gap19 (250 μM in aCSF; Tocris Bioscience) for 90 min before assaying hemichannel activity.
Ethidium bromide uptake assay and immunolabelling of astrocytes
The activity of hemichannels was carried out with an ethidium bromide (EtBr) assay as previously described [35, 36]. Briefly, after the pharmacological treatments, hippocampal slices were incubated with EtBr (20 μM; Sigma-Aldrich) in aCSF solution in an oxygenated chamber for 5 min at room temperature (RT) and rinsed with gassed aCSF for 15 min to stop EtBr uptake. Hippocampal slices were then fixed by immersion overnight at 4 °C in a 4% paraformaldehyde (PFA) solution in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 1.9 mM KH2PO4, pH 7.4) and then rinsed with a glycine solution (0.5 M) for 5 min to block unreacted aldehydes that produce background signals. This step was followed by several washes with PBS. Slices were protected from light throughout the entire procedure and were further processed for GFAP immunolabelling, as previously described [23]. Briefly, hippocampal slices were permeabilized for 30 min with 0.1% Triton X-100 in PBS and then with blocking solution (10% horse serum, 0.1% Triton X-100 in PBS) for 2 h. Slices were then incubated for 2 days at 4 °C with the primary antibody, rabbit anti-GFAP (1:1000, Millipore Cat# AB5804, RRID:AB_2109645) diluted in blocking solution. After washing, slices were incubated first with a blocking solution for 30 min and then with a donkey anti-rabbit 488 (1:1000, Thermo Fisher Scientific Cat# A-21206, RRID:AB_2535792) secondary antibody for 4 h at RT. In the case of hippocampal slices obtained from GFAP-CRE-A2AR mice, we also used as a primary antibody goat anti-GFP (1:500, Abcam Cat# ab6673, RRID:AB_305643) and anti-goat 488 (1:1000, Molecular Probes Cat# A-11055, RRID:AB_2534102) and an anti-rabbit 647 (1:1000, Molecular Probes Cat# A-21245, RRID:AB_141775) as secondary antibodies. Hippocampal slices were then washed three times with PBS during 10 min, incubated with Hoechst33258 (1:2000, Sigma Aldrich) for 30 min at RT to stain the nuclei, washed again three times during 10 min with PBS and mounted onto Ibidi μ-slide 8 well treated using Fluoromount Aqueous Mounting Medium (Sigma-Aldrich). Hippocampal slices were visualised with a LSM 710 confocal inverted microscope (Zeiss, RRID:SCR_018063) with a 40× objective (Plan-Apochromat 40×/1.4 Oil DIC M27 objective) and Z-stacks were acquired using Black Zen software (RRID:SCR_018163).
Analysis of EtBr fluorescence signal in astrocytes involved the processing of the images obtained by confocal microscopy through deconvolution using Huygens software (RRID:SCR_014237) to reduce out-of-focus information, thus increasing spatial resolution through a classic maximum likelihood estimation (CMLE) algorithm. Then, a normalisation operation was conducted in the GFAP channel in order to improve visualisation of GFAP+ cells with Imaris software (RRID:SCR_007370), which was used in the subsequent processing stages and analysis. The next step was a nuclei segmentation through creation of surfaces in the nuclei channel. Astrocyte nuclei were manually selected according to the following criteria: (i) cells positive for GFAP which comprehend a single nucleus enwrapped by a GFAP immuno-labelled structure, (ii) nucleus not truncated and (iii) nucleus exhibiting EtBr fluorescence above background. An additional criterion was considered when analysing hippocampal slices from GFAP-CRE-A2AR mice: only GFAP-positive cells with GFP immuno-labelling were evaluated. EtBr uptake was quantified as a ratio between EtBr fluorescence intensity (in arbitrary units, au) and nucleus volume (µm3) for each astrocyte (i.e. GFAP+ cell); we then calculated, for each experimental condition, the mean of EtBr uptake normalised (ratio) to the corresponding control condition. It should be mentioned that all GFAP+ cells take up EtBr and that we checked for differences in the volume of astrocyte nuclei amongst the different experimental conditions analysed and no statistically significant differences were found (data not shown).
Aβ immunohistochemistry and thioflavin-S staining
APP/PS1 and WT mice were transcardially perfused with ice-cold PBS followed by 4% PFA in PBS. Brains were removed, post-fixed for 24 h in PFA, dehydrated in 30% sucrose solution for 72 h at 4 °C and cryopreserved at −80 °C. Coronal brain sections (30 μm) were obtained using a cryostat (CryoStar NX50, ThermoScientific, RRIDD:SCR_022732) for Aβ immunolabelling and thioflavin-S staining following a previously described protocol with minor modifications [37]. First, antigen retrieval was performed through the incubation of air-dried sections in citrate buffer (10 mM sodium citrate tribasic dehydrate, 0.05% Tween 20, pH 6.0) at 95 °C for 15 min. Then, sections were rinsed three times with PBS for 5 min, permeabilized with a 0.2% Tween 20 solution in PBS for 15 min and non-specific binding blocked by incubation of 1% bovine serum albumin (BSA) 0.05% Tween 20 in PBS for 30 min. Sections were incubated overnight in an airtight humidity chamber with the mouse anti-β-amyloid (βA) primary antibody (1:250, Covance Cat# SIG-39320, RRID:AB_662798) in blocking solution. Sections were washed again three times in PBS for 5 min prior to incubation for 2 h with the secondary anti-mouse Alexa594 antibody (1:1000, Molecular Probes Cat# A-21203, RRID:AB_141633). Following three rinses with PBS for 5 min, sections were stained with 1% thioflavin-S solution for 10 min, differentiated in 70% ethanol, rinsed thrice with water, mounted with DAKO mounting medium and cover-slipped. Finally, hippocampal sections were visualised by fluorescence microscopy (Zeiss, Axio Imager Z2 microscope, RRID:SCR_018856) and the pictures were captured using the AxioVision Imaging System (RRID:SCR_002677 version 4.8).
Immunohistochemistry in brain slices
Free floating coronal brain sections (30 μm-thick) from APP/PS1 and WT mice were obtained as described above. For GFAP immunolabelling, brain sections were rinsed with PBS, incubated with permeabilization solution (0.1% Triton-X100 in PBS) for 15 min and then with blocking solution (10% horse serum + 0.1% Triton-X100 solution in PBS) for 2 h. Afterwards, sections were incubated with the rabbit anti-GFAP (1:1000, Millipore Cat# AB5804, RRID:AB_2109645) primary antibody overnight at 4 °C. Sections were then washed with PBS, further incubated with anti-rabbit Alexa594 (1:1000, Thermo Fisher Scientific Cat# A-21207, RRID:AB_141637) for 2 h at RT and washed again with PBS. Nuclei were stained with DAPI (1:5000, Invitrogen) for 10 min at RT. Following rinsing with PBS, sections were mounted onto gelatin-coated slides using ProLong™ Antifade mounting medium (Cell Signaling Technology). Z-stack images were captured with intervals of 0.5 μm with a LSM 710 confocal inverted microscope (Zeiss, RRID:SCR_018063) with a 63 × objective (Plan-Apochromat 63x/1.40 Oil DIC M27) for the tridimensional reconstruction of the astrocytic structure.
3D reconstruction of astrocytes
The morphology of astrocytes was studied as previously described [23, 38, 39] by tridimensional reconstruction of astrocytic structures using an open access tool, Simple Neurite Tracer (SNT, RRID:SCR_016566) plugin available in Fiji-ImageJ software. The tridimensional reconstruction of astrocytic processes within the stratum radiatum of the CA1 subregion of the dorsal hippocampus was carried out using Z-stack images as reported above. Astrocytes were selected for the tridimensional reconstruction of arbors of astrocytic processes according with the following criteria: (i) a single nucleus enwrapped by a GFAP-immunolabelled structure, (ii) the main structure of the astrocyte did not present truncated processes and (iii) reconstruction was carried out in the first five astrocytes fulfilling the previously mentioned criteria in each animal of both genotypes. The morphometric analysis of astrocytic arbor complexity was performed by quantifying the number of processes, their total length and the number of intersections with concentric spheres starting at the centre of the soma with intervals of 4 μm (Sholl analysis).
Preparation of gliosomes
Gliosomes were obtained from hippocampal tissue through a discontinuous Percoll gradient as previously described [40, 41]. Briefly, hippocampal tissue was homogenised in ice-cold isolation sucrose solution (0.25 M sucrose, 10 mM HEPES, pH 7.4 at 4 °C) using a glass-Teflon tissue grinder. Nuclei and debris were removed by centrifugation (1000g, 5 min at 4 °C) and the supernatant was carefully placed on top of the discontinuous gradient composed by 23, 10, 6 and 2% v/v of Percoll in a sucrose solution (0.32 M sucrose, 1 mM EDTA, pH 7.4 at 4 °C), which was stratified by centrifugation (31,000g for 5 min at 4 °C), turning off the centrifuge brake for the last 2000g to avoid a sudden stop. Gliosomes were collected between the 2% and 6% v/v Percoll layers, whereas synaptosomes (purified synapses) were collected in the interface between the 23% and 10% of Percoll layers. Each fraction was washed with isotonic physiological solution 140 mM NaCl, 5 mM KCl, 5 mM NaHCO3, 1.2 mM NaH2PO4, 1 mM MgCl2, 10 mM glucose and 10 mM HEPES, pH 7.4 at 4 °C) and further centrifuged (30,000g for 20 min at 4 °C). Pellets were washed again in isotonic physiological solution and centrifuged (22,000g for 20 min at 4 °C), the supernatant was discarded and the pellets solubilised in RIPA lysis buffer supplemented with 1 mM dithiothreitol (Sigma Aldrich), 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma Aldrich), 0.001% of a protease inhibitor cocktail (CLAP; Sigma Aldrich) and phosphatases inhibitor cocktail phosphoSTOP (Roche). The comparative analysis of gliosomes and synaptosomes by Western blot allowed confirming the enrichment of our gliosomal preparation in proteins found in perisynaptic astrocytic processes, such as Cx43 and glutamate transporters (GLT-1 and GLAST), as can be observed in Figure S1, the gliosomes preparation of adult wild-type mice had an enrichment not only in GFAP levels, but also of Cx43, GLT-1 and GLAST, as compared with synaptosomal preparations that were enriched in synaptophysin, a widely used synaptic marker.
Western blot
Western blot was performed as previously described [20]. The amount of protein samples used to quantify Cx43 and phosho-Cx43 at Ser368 was 3 and 30 µg, respectively; the amount of protein samples used to quantify other proteins was as follows: 3 µg GFAP, 20 µg for GLT-1 and GLAST and 10 µg for synaptophysin. Membranes were probed with rabbit antibodies against Cx43 (1:8000, Sigma-Aldrich Cat# C6219, RRID:AB_476857), anti-phospho-Cx43-Ser368 (1:1000, Cell Signaling Technology Cat# 3511, RRID:AB_2110169), GLT-1 (1:1000 ThermoFisher Scientific PA5-17099 RRID:AB_10978571) GLAST (1:1000 Abcam Ab416 RRID:AB_304334), GFAP (1:20,000, Millipore Ab5804 RRID:AB_2109645) or with mouse anti-synaptophysin antibody (1:20,000 Sigma S5768, RRID:AB_477523). These primary antibodies were diluted in Tris-buffered saline (137 mM NaCl, 20 mM Tris, pH 7.6) containing 0.1% Tween 20 (TBS-T) and 5% non-fat dry milk or 3% BSA overnight at 4 °C. After washing with TBS-T, membranes were incubated with IgG secondary antibodies (anti-rabbit, 1:10,000, RRID: AB_228338; anti-mouse 1:10,000, RRID:AB_228302, both from ThermoFisher Scientific) for 2 h at RT. Membranes were then washed, revealed using enhanced chemiluminescence substrate (ECL; GE Healthcare) and visualised with an imaging system (Chemidoc, RRID:SCR_019037). Then, membranes were re-probed for anti-α-tubulin antibody (1:20,000, Sigma-Aldrich Cat# T6074, RRID:AB_477582) for protein load control. Densitometric analysis of protein bands was performed using Image Lab Software (BioRad, RRID: SCR_014210). The relative densities of Cx43 and phospho-Cx43 were normalised to α-tubulin and expressed as percentage of the respective control condition.
Astrocytic cultures
Primary cultures of astrocytes were prepared following a previously used protocol [20]. Briefly, mixed glial cultures were obtained from the cerebral cortex of 2–5 days Wistar rats, grown in astrocyte medium [DMEM, supplemented with 10% foetal bovine serum, penicillin (100 U/mL), streptomycin (100 μg/ mL), HEPES (6 g/L), and sodium bicarbonate (0.84 g/L)] and maintained at 37 °C in a humified 5% CO2 incubator for 10–15 days until reaching confluency. Microglia cells were removed by mechanical shaking of the mixed cultures in an orbital shaker for 4 h at 200 rpm and astrocytes detached by a mild trypsinization procedure, re-seeded at a density of 5 × 104 cells/coverslip, and maintained in culture for 2–3 days prior to EtBr uptake.
Based on our previous experience [42], we manipulated different transducing systems to explore the mechanisms involved in the regulation of astrocytic hemichannels, namely using the cAMP analogue and activator of protein kinase A (PKA) 8-Br-cAMP (8-bromoadenosine-3',5'-cyclic monophosphate, 5 µM; Tocris), the protein kinase C (PKC) inhibitor GF109203X (2-[1-(3-dimethylaminopropyl)indol-3-yl]-3-(indol-3-yl)maleimide, 5 µM; Tocris), and the PKC activator PMA (phorbol 12-myristate 13-acetate, Ascent Scientific, 10 ng/mL). The concentrations of the compounds used were supramaximal, but selective for their targets (see [42]). All drugs were applied to cultured astrocytes 30 min prior to challenge with Aβ1-42 (1 μM, 24 h), a concentration higher than that used in slices but that we previously defined to be required to trigger astrocyte reactivity and dysfunction in cells obtained from newborn rodents [20,21,22].
Following the exposure to these pharmacological agents, the activity of hemichannels was assessed through EtBr uptake as previously described [20]. Briefly, astrocytes were exposed for 10 min to EtBr (5 μM) in HBSS solution (137 mM NaCl, 5.4 mM KCl, 0.34 mM Na2HPO4, 0.44 mM KH2PO4, 2.7 mM glucose, 1.2 mM CaCl2, pH 7.4), washed, fixed with 4% PFA solution and the nuclei stained with Hoechst 33,258. Following three rinses, coverslips were mounted with Fluoromount™ aqueous mounting medium. Images were taken from five random fields under epifluorescence microscopy (Zeiss, Axio Imager Z2 microscope, RRID:SCR_018856 with AxioVision Imaging System, RRID:SCR_002677 version 4.8) and analysed by FIJI-ImageJ software (RRID:SCR_002285).
Statistical analysis
Data are presented as mean ± SEM of the indicated number (n) of animals (in the case of behaviour and Western blot analysis) or hippocampal slices from different animals (for EtBr uptake quantification). In the case of data from tri-dimensional reconstructions of astrocytic processes, n refers to the number of cells reconstructed. Comparisons between experimental groups were performed with two-way ANOVA followed by Tukey's or Sidak's multiple comparisons test. Comparison between two experimental conditions was performed using either a paired or unpaired Student's t test. To identify differences between strategies used in the Morris water maze test, a table of contingency was built and a Fisher’s exact test of independence was performed. Differences in astrocytic morphometric analysis between WT and APP/PS1 mice (Sholl analysis) were assessed by multiple Student's t test using the Holm–Sidak method for correction of multiple comparisons. Statistical significance was set for p values < 0.05. All statistical tests were carried out using Graphpad Prism software (version 8.0.1, RRID:SCR_002798).
Results
APP/PS1 mice exhibiting memory deficits present morphological alterations of hippocampal astrocytes
Learning and memory assessment using the Morris water maze test showed that WT mice displayed a normal learning pattern as they improved their performance throughout the acquisition stage reaching the escape latency criterium (20 s) on day 3 (day 1: 38.43 ± 4.07 s vs. day 3: 10.73 ± 1.57 s, n = 10); in contrast, APP/PS1 mice did not reach the escape latency criterium (day 1: 46.72 ± 2.12 s vs. day 3: 38.94 ± 2.20 s, n = 9). Two-way ANOVA identified the genotype as a source of variation (F1,17 = 56.63, p < 0.0001), and post hoc Sidak’s multiple comparisons test showed a statistically significant difference between WT and APP/PS1 mice on day 2 (20.15 ± 3.30 s vs. 41.90 ± 4.55 s, p < 0.0001) and on day 3 (10.73 ± 1.57 s vs. 38.94 ± 2.20 s, p < 0.0001, Fig. 1A). In the probe trial, 24 h following the acquisition stage, APP/PS1 mice crossed the platform location significantly fewer times than WT mice (WT: 2.70 ± 0.34, n = 10, vs. APP/PS1: 0.56 ± 0.18, n = 9, p < 0.0001, t17 = 5.482, Fig. 1B). Thus, APP/PS1 mice displayed impaired hippocampal-dependent memory. When analysing the search patterns, we observed that APP/PS1 preferentially resorted to non-hippocampal-dependent strategies (only 33% of APP/PS1 mice used strategies dependent on the hippocampus) whereas 90% of WT mice used hippocampal-dependent strategies to find the location of the hidden platform (p < 0.0001, Fig. 1C).

APP/PS1 mice displayed deficits in hippocampal-dependent memory, an accumulation of Aβ and of Aβ plaques and an increased astrocytic arbor complexity in the hippocampus. A In the Morris water maze test, APP/PS1 mice displayed a higher latency to find the hidden platform location in the acquisition learning curve than WT mice. ****p < 0.0001, two-way ANOVA followed by a post hoc Sidak’s multiple comparisons test. B APP/PS1 mice showed an impairment in spatial memory since the number of crossings of the platform location was significantly lower than WT mice in probe trial of the Morris water maze test. ****p < 0.0001, unpaired Student’s t test. C Analysis of the search strategy patterns unveiled that WT mice preferentially used strategies implicating the hippocampus, whereas APP/PS1 mice mostly used strategies not dependent on the hippocampus to discover the hidden platform location in the probe trial of the Morris water maze. ****p < 0.0001, Fisher’s exact test of independence. Data are presented as mean ± SEM of n = 9–10 mice. D Representative images of thioflavin-S staining (left column, green) and Aβ immunolabelling (right column, red) disclosed the presence of amyloid-β plaques in the hippocampus of APP/PS1 but not of WT mice. Scale bar: 100 μm. E Representative images of GFAP immunolabelling (red) using a magnification of 63 × in slices from WT and APP/PS1 mice showing the morphology of hippocampal astrocytes. Nuclei were stained with DAPI (blue). Scale bar: 20 μm. Representative fillings of tri-dimensionally reconstructed astrocytes are shown for each group: APP/PS1 mice exhibited an increase in morphological complexity by evaluating F total length, G number of processes, and H Sholl analysis as compared with astrocytes from WT mice. Data are mean ± SEM of 15 astrocytes (from three different mice). ****p < 0.0001, unpaired Student's t test and *p < 0.05, **p < 0.01 in Sholl analysis through multiple t tests
Next, we checked for the presence of Aβ accumulation and deposition in the hippocampus of APP/PS1 mice. Figure 1D illustrates the accumulation of Aβ in the hippocampus of APP/PS1 mice, and thioflavin-S staining further supports the existence of an accumulation of these peptides, suggesting the presence of amyloid plaques. By contrast, in the hippocampus of WT mice no thioflavin-S staining was observed. Additionally, we investigated whether APP/PS1 mice displayed alterations in astrocytic arbor complexity through tri-dimensional reconstructions of astrocytes in the CA1 hippocampal region. Astrocytes of APP/PS1 mice displayed a significant increase in the total length of processes (APP/PS1: 1651.15 ± 43.18 µm vs WT: 1171.43 ± 54.27 µm, n = 15 astrocytes from three mice, p < 0.0001, t28 = 6.917, Fig. 1F) and also in the number of processes (APP/PS1: 98.07 ± 3.30 vs WT: 61.47 ± 2.30, n = 15 astrocytes from three mice, p < 0.0001, t28 = 9.098, Fig. 1G) relatively to hippocampal astrocytes from WT mice. This suggests an increment in morphologic complexity of hippocampal astrocytes in APP/PS1 compared to WT mice. These results were supported by Sholl analysis which unveiled a significant augmentation of the astrocytic arbor complexity of APP/PS1 compared to WT mice at 8 µm and at 16–24 µm from the central soma (8 µm: p = 0.0312, 16 µm: p = 0.0023, 20 µm: p = 0.0035 and 24 µm: p = 0.0033, Fig. 1H).
Astrocytic Cx43 hemichannel activity is increased in the hippocampus of APP/PS1 mice
The activity of astrocytic hemichannels was assessed in hippocampal slices using the ethidium bromide (EtBr) uptake assay. Data showed a significant effect of the genotype in EtBr uptake by hippocampal astrocytes (p = 0.0179, F1,8 = 8.809) and further analysis with a post hoc multiple comparisons test unveiled a significant increase of EtBr uptake in hippocampal astrocytes of APP/PS1 compared with WT mice (1.00 ± 0.036 for WT vs 1.69 ± 0.087 for APP/PS1, n = 5, p = 0.0004, Fig. 2B). To investigate the contribution of Cx43 to the altered hemichannel activity in APP/PS1 mice, Cx43 hemichannels were selectively blocked with Gap19 [43]. Two-way ANOVA showed a significant effect of Gap19 (p = 0.0014, F1,7 = 26.13) and an interaction between Gap19 and genotype (p = 0.0015, F1,7 = 25.35). Sidak’s multiple comparisons tests disclosed a significant effect of Gap19 on EtBr uptake by hippocampal astrocytes of APP/PS1 mice (1.685 ± 0.087 for CTR vs 1.066 ± 0.163 for Gap19, n = 5, p = 0.0003). Thus, Cx43 hemichannels were major contributors to the EtBr uptake and their blockade rescued the dysfunctional hemichannel activity, restoring astrocytic EtBr uptake to levels similar to WT (Fig. 2B).

APP/PS1 mice showed an increase of Cx43 hemichannel activity in hippocampal astrocytes and an increase of Cx43 levels. A Representative images of EtBr fluorescence signal (red) in astrocytes in hippocampal slices of WT and of APP/PS1 mice. Astrocytes were immuno-labelled with anti-GFAP (green) and nuclei were stained with Hoechst 33,258 (blue). Scale bars: 20 μm. B EtBr uptake was significantly higher in hippocampal astrocytes of APP/PS1 mice than in WT mice. The selective blockade of Cx43 hemichannels significantly reduced EtBr uptake in astrocytes of APP/PS1 mice, indicating the involvement of these channels in EtBr uptake. Hippocampal slices from APP/PS1 mice were treated with the Cx43 hemichannel inhibitor, Gap19 (250 µM, 90 min), prior to EtBr uptake assay. ***p < 0.001, two-way ANOVA followed by post hoc Sidak’s multiple comparisons test. The activity of hemichannels was assessed through the fluorescence intensity of EtBr signal, per astrocyte (GFAP+cells) nucleus volume (µm3) from WT or APP/PS1 mice and expressed as a ratio of control condition. Data are mean ± SEM of five independent experiments. C Levels of total Cx43, but not of phospho-Cx43 at Ser 368 (D), were increased in gliosomes (membranes from astrocytic processes) obtained from the hippocampus of APP/PS1 relatively to WT mice. The ratio between immunoreactivities for total Cx43 or for phospho-Cx43 at Ser 368 to α-tubulin levels were expressed as a percentage of values obtained in WT mice. Data are mean ± SEM of 3–4 independent experiments. *p < 0.05, unpaired Student’s t test. Representative immunoblots for Cx43, phospho-Cx43 and α-tubulin are shown below the average bar graphs
Given the relevant role of Cx43 hemichannels in the altered EtBr uptake in APP/PS1 mice, we further evaluated putative alterations of Cx43 levels in gliosomes (membranes of astrocytic processes) obtained from the hippocampus of APP/PS1 compared to WT mice. Data showed a substantial enhancement of Cx43 levels in gliosomes from APP/PS1 mice when compared with WT mice (100.00 ± 4.68% for WT vs. 251.33 ± 55.02% for APP/PS1 mice, n = 4, p = 0.0337, t6 = 2.741, Fig. 2D). Since the activity of hemichannels is modulated by phosphorylation, we also evaluated alterations of Cx43 phosphorylation at Ser368 and no alterations were observed between gliosomes of WT and APP/PS1 mice (100.00 ± 43.85% for WT vs. 87.95 ± 32.30% for APP/PS1, n = 4, p = 0.8323, t6 = 0.2212, Fig. 2E). The ratio between pCx43 and total Cx43 was 1.00 ± 0.12 for WT mice and of 0.64 ± 0.12 for APP/PS1 mice, supporting an increase in total Cx43 levels.
Hemichannel activity is affected in hippocampal astrocytes at early AD stages
The aforementioned data showed that adult APP/PS1 mice displayed hippocampal-dependent memory deficits at 9 months of age, exhibiting also Aβ plaques in parallel with altered astrocytic morphology, heighten hemichannel activity and increased Cx43 levels in the hippocampus. However, several evidences suggest that soluble Aβ oligomer rather than Aβ deposits primarily contribute to neuronal dysfunction, synaptic loss and early memory deficits (reviewed in [1, 44]) as well as with astrocytic dysfunction in early AD stages [34, 35]. Therefore, we next evaluated hemichannel activity in conditions mimicking early AD, using hippocampal slices directly superfused with Aβ1-42 (50 nM, 60 min) and hippocampal slices collected from adult mice intracerebroventricularly injected with Aβ1-42 (icv-Aβ1-42), which we previously showed to cause hippocampal synaptic plasticity and memory impairment after 15 days of peptide administration without evidence of Aβ deposits [24, 34]. In hippocampal slices exposed to Aβ1-42, the EtBr uptake by astrocytes was significantly higher than that of control slices (2.03 ± 0.185, n = 5, for Aβ1-42 vs. 1.00 ± 0.075, n = 4, for CTR, p = 0.0475, t3 = 3.250, Fig. 3A). Similarly, the icv Aβ1-42 administration also resulted in an enhancement of EtBr uptake by hippocampal astrocytes compared to vehicle-injected control mice (icv-VEH: 1.00 ± 0.075, n = 4 vs. icv-Aβ1–42: 1.42 ± 0.127, n = 5, p = 0.0323, t7 = 2.664, Fig. 3B).

Hemichannel activity was also enhanced in astrocytes of models mimicking early AD. The activity of hemichannels was augmented in astrocytes of hippocampal slices obtained from A WT mice and incubated with Aβ1-42 (50 nM, 60 min) and B from WT mice administrated icv with Aβ1-42 (icv-Aβ1-42), compared to control (or icv-vehicle administration) WT mice, being the EtBr uptake assay performed after 15 days of Aβ1-42- or vehicle-icv administration. The activity of hemichannels was evaluated by the mean fluorescence intensity of EtBr signal per astrocyte nucleus volume and expressed as a ratio of control for each experimental condition. Data are mean ± SEM of 4–5 independent experiments. *p < 0.05, paired for (A) or unpaired for (B) Student's t test
icv-Aβ1-42 peptides administration affects Cx43 phosphorylation at Ser368
Since in APP/PS1 mice, the enhancement of the activity of astrocytic hemichannels was linked with an increase of Cx43 levels, we next analysed if Aβ1-42-icv administration affected Cx43 levels in hippocampal gliosomes. Curiously, although no alterations were found in Cx43 levels (WT: 100.00 ± 2.41% vs. Aβ: 93.29 ± 15.45%, n = 5, p = 0.6836, t8 = 0.4227, Fig. 4A), a significant increase in the levels of Cx43 phosphorylated at Ser368 residue was observed in gliosomes from the hippocampus of Aβ1-42-icv injected compared to vehicle-injected control mice (WT: 100.00 ± 6.30% vs. Aβ: 171.28 ± 16.27%, n = 4, p = 0.0065, t6 = 4.085, Fig. 4B). Accordingly, the ratio phospho-Cx43(Ser368)/total Cx43 was 0.98 ± 0.05 for icv-VEH mice and 1.56 ± 0.05 for icv-Aβ1-42 mice.

Aβ1-42-icv administration increased Cx43 phosphorylation at Ser368 in hippocampal gliosomes of mice mimicking early AD stages. Although no alterations were observed in A Cx43 levels, an enhancement in B phospho-Cx43 levels (at Ser368) was observed in gliosomes obtained from the hippocampus of mice icv administrated with Aβ1-42 in comparison with icv-vehicle (VEH)-injected control mice. Data are ratios between Cx43 or phospho-Cx43 and α-tubulin (loading control protein) immunoreactivities expressed as percentage of the control condition (icv-VEH). Data are mean ± SEM of 4–5 independent experiments. **p < 0.01, unpaired Student’s t test. Representative immunoblots for Cx43, phospho-Cx43 and α-tubulin are shown
A2AR modulate the activity of hemichannel activity in hippocampal astrocytes
Next, we investigated whether astrocytic A2AR were involved in the modulation of the activity of hemichannels in astrocytes of hippocampal slices. For this purpose, we used a mouse model with astrocytic A2AR genetically silenced in the hippocampus, as described above (“Animals and surgeries" section). Data showed that silencing astrocytic A2AR increased EtBr uptake by astrocytes when compared with the control mice (1.25 ± 0.044 for GFAP-CRE-A2AR vs. 1.00 ± 0.031 for GFAP-CTR, n = 4, p = 0.0035, t6 = 4.641, Fig. 5A). Additionally, we investigated if silencing astrocytic A2AR affected the total and phosphorylated levels of Cx43. Data showed a significant increase in the levels of Cx43 (GFAP-CRE-A2AR: 147.97 ± 16.06% vs. GFAP-CTR: 100.00 ± 1.78% n = 5, p = 0.0179, t8 = 2.968, Fig. 5B) and of phospho-Cx43 at Ser368 (GFAP-CRE-A2AR: 140.78 ± 15.70% vs. GFAP-CTR: 100.00 ± 1.49% n = 7, p = 0.0238, t12 = 2.586, Fig. 5C), being the ratio phosphoCx43/Cx43 similar for both groups (GFAP-CTR: 1.00 ± 0.09 vs. GFAP-CRE-A2AR: 0.098 ± 0.12). These data confirm that A2AR control the activity of hemichannels composed by Cx43 in hippocampal astrocytes, increasing Cx43 levels and Cx43 phosphorylation.

Astrocytic A2AR genetic silencing increased the activity of hemichannels and the levels of Cx43 and of phospho-Cx43 at Ser368 in hippocampal astrocytes. A Deletion of A2AR selectively in astrocytes led to the enhancement of EtBr uptake in hippocampal astrocytes. Hemichannel activity was evaluated by the mean fluorescence intensity of EtBr signal per astrocyte nucleus volume expressed as a ratio of control condition (GFAP-CTR) in hippocampal slices from GFAP-CTR and GFAP-CRE-A2AR mice. The levels of B Cx43 and of C phospho-Cx43 at Ser368 were increased in gliosomes obtained from the hippocampus of GFAP-CRE-A2AR compared to GFAP-CTR mice. Immunoreactivity ratio of Cx43 and of phospho-Cx43 to α-tubulin were expressed as percentage of GFAP-CTR. Representative immunoblots for Cx43, phospho-Cx43 and α-tubulin are shown. Data are mean ± SEM of 4–7 independent experiments. *p < 0.05, **p < 0.01 unpaired Student’s t test
A2AR regulate alterations in astrocytic hemichannel activity triggered by an acute challenge with Aβ1-42
To further explore the role of A2AR in the dysregulation of astrocytic hemichannel activity triggered by Aβ1-42 peptides, we resorted to different approaches. First, a pharmacological strategy was employed where hippocampal slices were treated ex vivo with A2AR selective antagonist, SCH58261, prior to Aβ1-42 challenge. Two-way ANOVA revealed an interaction between Aβ1-42 and SCH58261 (F1,14 = 5.241, p = 0.0381), and post hoc analysis with Tukey’s multiple comparisons test revealed that A2AR selective blockade prevented the increase in EtBr uptake triggered by Aβ1-42 (Aβ1-42: 2.03 ± 0.185, n = 5, vs. SCH58261 + Aβ1-42: 1.24 ± 0.158, n = 5, p = 0.0066, Fig. 6A, of noting that the Aβ1-42/SAL data are the same presented in Fig. 3A). The administration of SCH58261 per se did not significantly affect EtBr uptake, as compared with control conditions (SCH: 0.891 ± 0.112 vs CTR SAL: 1.000 ± 0.075; p = 0.9604, Fig. 6A). Moreover, we further resorted to A2AR genetic silencing. In GbA2ARKO mice, two-way ANOVA showed an interaction between genotype and Aβ1-42 (p = 0.0035, F1.4 = 38.24) and Sidak’s multiple comparisons tests unveiled that the amount of EtBr taken by astrocytes in hippocampal slices from GbA2ARKO mice was significantly higher than in WT mice (KO: 1.34 ± 0.087 vs. WT: 1.00 ± 0.088, n = 3, p = 0.0493). Furthermore, Aβ1-42 superfusion of hippocampal slices enhanced EtBr uptake in WT mice (Aβ1-42: 1.42 ± 0.109 vs. CTR: 1.00 ± 0.088, n = 3, p = 0.0023), but did not affect EtBr uptake by astrocytes in hippocampal slices from GbA2ARKO mice (CTR: 1.34 ± 0.087 vs. Aβ1-42: 1.32 ± 0.051, n = 3, p = 0.9069, Fig. 6B). Interestingly, despite no significant differences were detected in Cx43 levels (WT: 108.65 ± 10.92% vs. KO: 106.26 ± 9.88%, n = 6, p = 0.8746, t10 = 0.1619, supplementary data Figure S2A), a downregulation in Cx43 phosphorylation at Ser368 was observed in gliosomes obtained from the hippocampus of GbA2ARKO mice (WT: 100.00 ± 9.74 vs. KO: 67.24 ± 3.57, n = 4, p = 0.0196, t6 = 3.158, supplementary data Figure S2B). When A2AR genetic deletion is selectively carried out in astrocytes, two-way ANOVA showed an interaction between Aβ1-42 and the effective viral construct administrated (p = 0.0183, F1,6 = 10.34) and Sidak’s multiple comparisons test showed a lack of effect of Aβ1-42 superfusion in EtBr uptake by astrocytes in hippocampal slices of GFAP-CRE-A2AR mice (CTR: 1.25 ± 0.040 vs. Aβ1-42: 1.28 ± 0.084, n = 4, p = 0.9320), whereas in GFAP-CTR mice Aβ1-42 significantly increased the amount of EtBr taken up by astrocytes (Aβ1-42: 1.38 ± 0.058, n = 4 vs. CTR: 1.00 ± 0.031, n = 4, p = 0.0054, Fig. 6C, noting that CTR data of GFAP-CRE-A2AR and of GFAP-CTR mice are the same data present in Fig. 5A). In contrast, in FbA2ARKO mice, two-way ANOVA did not reveal an interaction between genotype and Aβ1-42 treatment (p = 0.8714, F1,6 = 0.0286); notwithstanding, it is pertinent to emphasise that a significant effect was observed in the genotype (p = 0.0276, F1,6 = 8.364) and Aβ1-42 treatment (p = 0.0020, F1,6 = 27.24). Additionally, Sidak’s multiple comparisons test revealed a significant enhancement in EtBr uptake by hippocampal astrocytes triggered by Aβ1-42 in both WT mice (CTR: 1.00 ± 0.031 vs. Aβ1-42: 1.38 ± 0.058, n = 4, p = 0.0177) and FbA2ARKO mice (CTR: 1.39 ± 0.120 vs. Aβ1-42: 1.74 ± 0.157, n = 4, p = 0.0234). Curiously, a significant increase in EtBr uptake was also observed in hippocampal astrocytes of FbA2ARKO mice when compared with WT mice (KO: 1.39 ± 0.120 vs. WT: 1.00 ± 0.031, n = 4, p = 0.0443, Fig. 6D), but the Aβ1-42 superfusion similarly increased (p > 0.05) EtBr uptake in WT (0.38 ± 0.03) and FbA2ARKO (0.35 ± 0.04) mice (Fig. 6D). Surprisingly, the analysis of gliosomal preparations from the hippocampus of FbA2ARKO mice revealed neither alterations of Cx43 levels (WT: 100.00 ± 2.47% vs. KO: 102.35 ± 7.58%, n = 5, p = 0.7760, t8 = 0.2943, supplementary data Figure S2C) nor of Cx43 phosphorylation at Ser368 (WT: 100.00 ± 8.53% vs. KO: 81.81 ± 16.02%, n = 6, p = 0.3397, t10 = 1.003, supplementary data Figure S2D). Taken together, these data identified the importance of astrocytic A2AR in the modulation of hemichannel activity of hippocampal astrocytes in pathological conditions mimicking early AD stages.

A2AR regulated alterations in hemichannel activity caused by an acute challenge with Aβ1-42. A The enhancement in astrocytic hemichannel activity driven by the acute exposure of hippocampal slices to Aβ1-42 was prevented by A2AR selective blockade. Hippocampal slices were pre-incubated with SCH58261 (SCH, 50 nM) for 30 min prior to challenge with Aβ1-42 (50 nM, 60 min), before the EtBr uptake assay. B In GbA2ARKO and C GFAP-CRE-A2AR mice, Aβ1-42 failed to increase EtBr uptake in hippocampal astrocytes, whereas D in FbA2ARKO mice, Aβ1-42 augmented astrocytic EtBr uptake. The activity of hemichannels was assessed through the fluorescence intensity of EtBr signal, per astrocyte (GFAP+cells) nucleus volume, expressed as the mean of each animal (n) and normalised to the control condition. Data are mean ± SEM of 3–5 independent experiments, corresponding to different mice *p < 0.05, **p < 0.01, two-way ANOVA post hoc Sidak’s multiple comparisons test. Please note that the control values in panel A is the same as in Fig. 3A and in panel C is the same as Fig. 5A
The dysfunctional hemichannel activity triggered by Aβ seems to involve a PKC-mediated signalling pathway
Although A2AR are pleiotropic receptors, they are mainly recognised to engage the PKA-mediated transducing system (reviewed in [46]). However, since we previously reported that the Aβ1-42-induced increase of hemichannel activity is paralleled by increased Cx43 phosphorylation in residue Ser368, which is phosphorylated by PKC [47, 48], and this effect was mimicked by A2AR activation and prevented by selective A2AR blockade [20], we next characterised the transducing system involved in the control of hemichannel activity in cultured hippocampal astrocytes. We observed that PKA activation with the cAMP analogue 8-Br-cAMP (5 µM) did not affect EtBr uptake (101.16 ± 9.01% vs. CTR: 100%, t3 = 0.1284, p = 0.9060), whereas the activation of PKC with a phorbol ester analogue (PMA, phorbol 12-myristate 13-acetate, 10 ng/mL) increased hemichannel activity (128.10 ± 4.52% vs. CTR: 100%, t3 = 6.222, p = 0.0084, supplementary data Figure S3A). Moreover, PMA significantly enhanced Cx43 phosphorylation at Ser368 (135.1 ± 10.82% vs. CTR: 99.00 ± 6.74%, t6 = 2.829, p = 0.0300, supplementary data Figure S3B). These data suggest that the enhancement of hemichannel activity triggered by Aβ1-42 might involve the recruitment of a PKC-mediated pathway. In support of this hypothesis, PKC inhibition with GF109203X (5 µM) prevented the enhancement of hemichannel activity caused by Aβ1-42 (Fig. 7B). Thus, two-way ANOVA unveiled a significant effect of GF109203X (p = 0.0002, F1,11 = 30.14) and of Aβ1-42 (p = 0.0031, F1,11 = 14.16) in EtBr taken up by cultured astrocytes. Further analysis with Tukey multiple comparisons test showed that GF109203X significantly reduced EtBr uptake in astrocytes exposed to Aβ1-42 (87.51 ± 5.36% vs. 138.05 ± 9.14% for Aβ1-42, p = 0.0016) when compared to astrocytes exposed only to Aβ1-42. Interestingly, GF109203X appeared to decrease EtBr uptake by hippocampal astrocytes, but this effect did not reach statistical significance (71.13 ± 11.39%, p = 0.0805). Moreover, PKC activation with PMA mimicked the effect of Aβ1-42 on EtBr uptake in cultures astrocytes (Fig. 7A). Three-way ANOVA disclosed a significant effect of PMA (F1,31 = 83.02, p < 0.0001) and Tukey multiple comparisons tests revealed a significant increase of EtBr uptake relatively to non-treated control (100%), in astrocytes challenged with either Aβ1-42 alone (142.56 ± 3.94%, p = 0.0279), PMA alone (172.58 ± 14.77%, p = 0.0008) and PMA + Aβ1-42 (165.29 ± 7.42%, p = 0.0009). Noteworthy, no additivity was observed between the effects of PMA and of Aβ1-42 (p = 0.9998). To further validate the involvement of a PKC-mediated pathway, PKC was activated followed by A2AR blockade prior to Aβ1-42 challenge (Fig. 7B). Tukey multiple comparisons tests disclosed that astrocytes exposed to the PKC activator PMA and to the selective A2AR antagonist SCH58261 displayed a significant increased EtBr uptake in the absence (170.64 ± 10.46%, p = 0.0001) and in the presence of Aβ1-42 (181.23 ± 21.07%, p < 0.0001) when compared with control astrocytes. Moreover, as can be seen in Fig. 7B, A2AR blockade per se did not affect EtBr uptake (95.18 ± 3.29%, p > 0.999) by cultured astrocytes, but prevented alterations triggered by Aβ1-42 (99.95 ± 2.22%, p = 0.0409), similarly to that previously observed by us [20]. Noteworthy, when astrocytes were exposed to PMA, SCH58261 and Aβ1-42, the amount of EtBr taken up by astrocytes was significantly higher than in astrocytes exposed to SCH58261 and Aβ1-42 (p < 0.0001), but as expected not different (p > 0.05) to that observed in astrocytes exposed to PMA and SCH58261 (Fig. 7B). Altogether, the gathered results strongly support the involvement of a signalling pathway mediated by PKC in our experimental conditions, which can be recruited downstream A2AR activation.

The increased hemichannel activity in cultured astrocytes exposed to Aβ1-42 was regulated by PKC. A PKC inhibition with GF109203X (GF) prevented the effect of Aβ1-42 on EtBr uptake in astrocytes. Cultured astrocytes were exposed to GF (5 µM) for 30 min prior to challenge with Aβ1-42 (1 µM, 24 h). Data are mean ± SEM of 3–4 independent experiments. *p < 0.05, two-way ANOVA post hoc Tukey’s multiple comparisons test. B On the other hand, PKC activation with PMA (phorbol 12-myristate 13-acetate, 10 ng/mL) mimicked the effect of Aβ1-42 on EtBr uptake in astrocytes. In the presence of PMA, the selective A2AR antagonist SCH58261 (SCH) did not blunt the Aβ1-42-induced increase in EtBr uptake by astrocytes. The selective A2AR antagonist SCH58261 (SCH) was no longer able to blunt the increase in EtBr uptake in astrocytes pre-treated with PMA. Cultured astrocytes were exposed to PMA (10 ng/mL, 30 min), then SCH was added (50 nM, 30 min) and finally astrocytes were exposed to Aβ1-42 (1 µM, 24 h) in the presence of the previously added compounds, as depicted in the timeline shown. The activity of hemichannels was assessed through the mean fluorescence intensity of retained EtBr in the nucleus upon the subtraction of background values. Data are presented as percentage values relative to non-treated control cells (100%) and are mean ± SEM of 3–5 independent experiments. *p < 0.05, ***p < 0.001, three-way ANOVA post hoc Tukey’s multiple comparisons test
Discussion
The present study re-enforces the relevance of astrocytic alterations at the onset of memory deficits and in experimental models of established AD, by showing that the activity of astrocytic hemichannels composed by Cx43 is modified in animal models both of early stages as well as of established AD. Furthermore, the observed different alterations of total Cx43 levels and of phosphorylated Cx43 at Ser368 in different models mimicking different AD stages from in vitro to in vivo suggest that the enhancement in astroglial hemichannel activity might be due to different mechanisms throughout the evolution of AD.
The analysis of astrocyte morphology in the hippocampus of APP/PS1 mice when memory dysfunction became evident, clearly showed global alterations of astrocyte morphology, typified by an increase in arbor complexity and an enhancement in the number and total length of astrocytic processes. This finding is aligned with previous observations that an increase in astrocyte reactivity is linked to the accumulation of Aβ associated with AD, as assessed by the analysis of GFAP immunoreactivity in human samples and animal models of AD [6, 27, 49, 50], namely in APP/PS1 mice [7, 14, 51, 52]. Given the active role of astrocytes in synaptic communication and memory processes [38, 53, 54], these alterations of astrocytes emerge as potential contributors to the deficits in hippocampal-dependent learning and memory observed in the performance of APP/PS1 mice in the Morris water maze test.
Our present study also revealed that the increased reactivity of astrocytes in the AD brain was associated with an increase of astroglial hemichannel activity. In the present study, although we did not use scrambled peptides, we pinpointed the specific contribution of Cx43 to the measured astroglial hemichannel activity by resorting to Gap19, which was previously shown to selectively target Cx43 hemichannels without affecting gap junctions [43]. Accordingly, we observed increased total levels of Cx43 in APP/PS1 mice that display scarce amyloid plaques. This is in agreement with previous studies also reporting an increase of Cx43 protein and mRNA levels in the brain of both patients [49, 55] and animal models [14, 18, 56] with established AD. This increased density of total Cx43 was originally associated with sites of Aβ deposits [14, 49], but was later recognised to be widespread in the afflicted brain regions of AD [18]. It should be mentioned that although we observed an increase in astrocytic process length, number and complexity in APP/PS1 mice, the increase in total Cx43 levels does not seem to be a direct consequence of the increased number of astrocytic processes, because we quantified the levels of total and phosphorylated (at Ser 368) Cx43 in a similar amount of astrocytic processes membranes (gliosomes) of wild-type and APP/PS1 mice.
Interestingly, we now observed that the increased activity of hemichannels in hippocampal astrocytes is an early event of AD pathogenesis, which is maintained in advanced stages of the disease. In early AD modelled by Aβ-icv exposure, there was an increase in Cx43 phosphorylation at Ser368 without alterations of the total levels of Cx43, whereas in hippocampal gliosomes of 9-month-old APP/PS1 mice already displaying sparse Aβ plaques, there was an increase in Cx43 levels without alterations in Cx43 phosphorylation at Ser368. This is in agreement with the ability of soluble forms of Aβ to control Cx43 levels at cell membrane [57], bolstering gliotransmitters release from cultured astrocytes, namely ATP and glutamate [20, 56, 58], which might promote neuronal injury [58]. These findings further indicate that the onset of AD seems to be related with alterations in Cx43 phosphorylation, whereas advanced AD stages involve alterations in Cx43 levels, which is consistent with the progressive increase in Cx43 levels observed in APP/PS1 and 5xFAD mice [59] as well as in AD patients [55]. Although we have not directly explored the impact of increased Cx43-HC activity as a trigger of memory dysfunction, this increased of Cx43 might be a contributing factor for AD-related dysfunction of synaptic plasticity and memory since the genetic silencing of Cx43 affords a neuroprotection against Aβ-induced modifications [55] and improves memory deficits in APP/PS1 mice by increasing synaptic function without affecting amyloid plaque formation or the inflammatory response [18, 59].
We further explored the mechanism involved in the regulation of hemichannel activity by phosphorylation which remains largely unknown in astrocytes. The observed enhancement in Cx43 phosphorylation in Ser368 residue, which is mainly mediated by PKC [60, 61], in hippocampal astrocytes of mice icv administrated with Aβ peptides, is in line with alterations previously reported by us in cultured astrocytes exposed to Aβ peptides [20]. These data suggest an Aβ-induced increase in PKC activity, which was already reported in human astrocytes exposed to Aβ [62] as well as in cortical astrocytes of 5xFAD mice [47]. Thus, we postulate as a working hypothesis that the increased PKC activation and the subsequent enhancement of Cx43 phosphorylation at Ser368 might be responsible for the increased hemichannel activity under AD conditions in hippocampal astrocytes. Interestingly, a recent study showed that an inflammatory stimulus (Thy-1) caused astrocytic Cx43 phosphorylation at Ser373 through a PI3K/AKT signalling pathway, being this phosphorylation related with the increased opening of hemichannels and ATP release [63]. Some studies performed in endothelial and cardiac cells also showed a link between PKC-mediated phosphorylation of Cx43 at Ser368 residue leading to altered hemichannel activity [64] and of gap junctional intercellular communication [48, 64, 65]; however, little is known about the control of Cx43 phosphorylation at Ser368 in astrocytes [66].
A second major advance provided by the present study is the concept that adenosine A2A receptors (A2AR) modulate the impact of Aβ in the activity of astrocytic Cx43 hemichannels. Although, we had previously shown that A2AR control Cx43 hemichannels in cultured astrocytes exposed to Aβ peptides [20], we now extend our studies to a more complex system, namely mouse hippocampal slices modelling early or advanced stages of AD, in which A2AR were genetically silenced in astrocytes and/or in neurons. Our group and others showed that A2AR blockade ameliorates synaptic dysfunction and loss in addition to memory deficits in mice icv administered with Aβ1-42 [24, 31] and in APP/PS1 mice [25, 26, 67]. This A2AR-mediated control of AD-related dysfunction involves an overfunction of neuronal A2AR [25, 31, 67], in accordance with the predominant localization of A2AR in excitatory synapses in the limbic cortex [68, 69]. However, A2AR are also present in astrocytes and the selective manipulation of astrocytic A2AR has an impact on memory function [23, 29, 70]. Furthermore, A2AR modulate alterations triggered by Aβ in primary cultures of astrocytes, namely decreased glutamate uptake [21], altered Ca2+ dynamics [22], and increased hemichannel activity and subsequently ATP release [20], a danger signal in brain disease conditions [71]. We now extend these findings by showing that the Aβ-induced alterations in hemichannel activity in hippocampal slices were abrogated by the pharmacological blockade and genetic silencing of A2AR in astrocytes, whereas they were unaffected in hippocampal astrocytes of mice with a selective genetic silencing of A2AR in neurons (FbA2ARKO); this further re-enforces the conclusion that astrocytic A2AR are responsible for the regulation of Aβ-induced alterations in astrocytic hemichannel activity in the hippocampus. Interestingly, A2AR also have a relevant role in non-pathological conditions since global, neuronal and astrocytic A2AR genetic silencing significantly impacted the activity of astrocytic hemichannels in the hippocampus. Indeed, mice lacking astrocytic A2AR displayed an increased hemichannel activity, which is likely a consequence of an increase of total Cx43 and phospho-Cx43 levels (see also [72]). Thus, astrocytic A2AR emerge as key modulators of hemichannel activity, which is consistent with the reported physical association between A2AR and Cx43 in primary cultures of astrocytes [20]. This provides a molecular rationale to understand that the removal of A2AR from the complex with Cx43 can contribute to dysregulate hemichannel activity, which may underlie the observation that the selective silencing of astrocytic A2AR results in impairments of synaptic plasticity and deficits in hippocampal-dependent reference memory [23]. Interestingly, we also observed that the genetic silencing of neuronal A2AR (using FbA2ARKO mice) triggered an enhancement of the activity of hippocampal astrocytic hemichannels relatively to WT littermates, despite no alterations in Cx43 or phospho-Cx43 levels were detected, suggesting that alterations in neuronal function due to A2AR deletion can influence astrocytic activity. This idea is in agreement with a study reporting that, under physiological conditions, the activity of Cx43 hemichannels in astrocytes is promoted by neuronal activity, which, in turn, modulates neuronal network function via a purinergic pathway in the olfactory bulb [73]. Altogether our data indicate that astrocytic A2AR are crucial to modulate Cx43 hemichannel activity in hippocampal astrocytes under AD-like conditions. Furthermore, our data also provides a tentative rationale for a riddle regarding the impact of astrocytic A2AR on memory performance, whereby the genetic elimination of astrocytic A2AR is detrimental for memory performance in naïve animals [23, 70] but beneficial in AD mouse models [29]. This is in agreement with the observed increase of hemichannel activity upon deletion of astrocytic A2AR in naïve animals and with the lack of additional effects of Aβ on hemichannel activity in hippocampal slices of mice with a genetic deletion of astrocytic A2AR.
This opposite role of A2AR in naïve animals and in conditions of early AD is likely due to the upregulation of astrocytic A2AR [21]. Previous studies have shown that the upregulation of A2AR is coupled with an alteration of the transducing signalling system operated by A2AR [74]. In fact, whereas A2AR are canonically coupled to the activation of adenylate cyclase and generation of cAMP [46], these pleiotropic receptors seem to be mostly coupled to PKC when they are upregulated in disease conditions associated with increased glutamatergic signalling [74], such as in early AD (see e.g. [75,76,77]). And, in agreement with the impact of PKC on the activity of astrocytic hemichannels, we observed that A2AR blockade was not able to prevent Aβ-induced alterations of hemichannel activity when PKC was previously activated, which supports the contention that the recruitment of the PKC pathway is a downstream event to A2AR activation under conditions of Aβ exposure. This is in agreement with several previous reports indicating the involvement of PKC in the signalling of glial A2AR [74, 78, 79], as also occurs in neurons [80] and other cell types [81,82,83] under stressful conditions.
In conclusion, the present study reinforces the role of astrocytic dysfunction in AD pathogenesis. In transgenic APP/PS1 mice displaying diffuse Aβ plaques, deficits in hippocampal-dependent memory were accompanied by alterations in astrocytic morphology and by an enhancement of astrocytic Cx43 hemichannel activity in the hippocampus. Similarly, models of early AD also showed an increase in the activity of Cx43 hemichannels involving an increase of Cx43 phosphorylation at Ser368. This increased activity of astrocytic Cx43 hemichannel activity in the hippocampus was regulated by astrocytic A2AR, as their genetic silencing increased both channels activity and Cx43 and phospho-Cx43 levels. Additionally, astrocytic A2AR also modulate the impact of Aβ on Cx43 hemichannel activity through a PKC-dependent mechanism (Fig. 8). Overall, these findings re-enforce the contention that astrocytic A2AR modulate astrocyte-to-neuron communication, which is altered in AD-like conditions. This may contribute to the benefits afforded by A2AR antagonists in early AD.

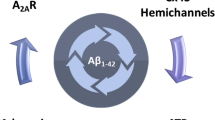
Hypothetical mechanistic interaction between Cx43 and A2AR in AD. In early and late stages of AD models, there is an enhancement in the activity of astroglial hemichannels. In early AD, the heighten activity of hemichannels seems to be due to an increase in Cx43 phosphorylation. Cx43 phosphorylation at Ser368 occurs through PKC, an event downstream of A2AR activation. On the other hand, in plaque-bearing APP/PS1 mice, the augment in astrocytic hemichannel activity is linked with an enhancement of Cx43 levels
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8(6):595–608. https://doi.org/10.15252/emmm.201606210
Martin SJ, Grimwood PD, Morris RG (2000) Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23:649–711. https://doi.org/10.1146/annurev.neuro.23.1.649
Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002) Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22(1):183–192. https://doi.org/10.1523/JNEUROSCI.22-01-00183.2002
Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG (2007) Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27(24):6473–6477. https://doi.org/10.1523/JNEUROSCI.1419-07.2007
Santello M, Toni N, Volterra A (2019) Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci 22(2):154–166. https://doi.org/10.1038/s41593-018-0325-8
Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB (2010) Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging 31(4):578–590. https://doi.org/10.1016/j.neurobiolaging.2008.05.015
Li K-Y, Gong P-F, Li J-T, Xu N-J, Qin S (2020) Morphological and molecular alterations of reactive astrocytes without proliferation in cerebral cortex of an APP/PS1 transgenic mouse model and Alzheimer’s patients. Glia 68(11):2361–2376. https://doi.org/10.1002/glia.23845
Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971(2):197–209. https://doi.org/10.1016/s0006-8993(03)02361-8
Verkhratsky A, Zorec R, Rodríguez JJ, Parpura V (2016) Astroglia dynamics in ageing and Alzheimer’s disease. Curr Opin Pharmacol 26:74–79. https://doi.org/10.1016/j.coph.2015.09.011
Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N (2010) Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11(2):87–99. https://doi.org/10.1038/nrn2757
Decrock E, De Bock M, Wang N, Bultynck G, Giaume C, Naus CC, Green CR, Leybaert L (2015) Connexin and pannexin signaling pathways, an architectural blueprint for CNS physiology and pathology? Cell Mol Life Sci 72(15):2823–2851. https://doi.org/10.1007/s00018-015-1962-7
Orellana JA, Retamal MA, Moraga-Amaro R, Stehberg J (2016) Role of astroglial hemichannels and pannexons in memory and neurodegenerative diseases. Front Integr Neurosci 10:26. https://doi.org/10.3389/fnint.2016.00026
Charvériat M, Naus CC, Leybaert L, Sáez JC, Giaume C (2017) Connexin-dependent neuroglial networking as a new therapeutic target. Front Cell Neurosci 11:174. https://doi.org/10.3389/fncel.2017.00174
Mei X, Ezan P, Giaume C, Koulakoff A (2010) Astroglial connexin immunoreactivity is specifically altered at β-amyloid plaques in β-amyloid precursor protein/presenilin1 mice. Neuroscience 171(1):92–105. https://doi.org/10.1016/j.neuroscience.2010.08.001
Angeli S, Kousiappa I, Stavrou M, Sargiannidou I, Georgiou E, Papacostas SS, Kleopa KA (2020) Altered expression of glial gap junction proteins Cx43, Cx30, and Cx47 in the 5XFAD model of Alzheimer’s disease. Front Neurosci 14:582934. https://doi.org/10.3389/fnins.2020.582934
Pechlivanidou M, Kousiappa I, Angeli S, Sargiannidou I, Koupparis AM, Papacostas SS, Kleopa KA (2022) Glial gap junction pathology in the spinal cord of the 5xFAD mouse model of early-onset Alzheimer’s disease. Int J Mol Sci 23(24):15597. https://doi.org/10.3390/ijms232415597
Cruz NF, Ball KK, Dienel GA (2010) Astrocytic gap junctional communication is reduced in amyloid-β-treated cultured astrocytes, but not in Alzheimer’s disease transgenic mice. ASN Neuro 2(4):e00041. https://doi.org/10.1042/AN20100017
Yi C, Mei X, Ezan P, Mato S, Matias I, Giaume C, Koulakoff A (2016) Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ 23(10):1691–1701. https://doi.org/10.1038/cdd.2016.63
Yi C, Ezan P, Fernández P, Schmitt J, Sáez JC, Giaume C, Koulakoff A (2017) Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 65(10):1607–1625. https://doi.org/10.1002/glia.23182
Madeira D, Dias L, Santos P, Cunha RA, Canas PM, Agostinho P (2021) Association between adenosine A2A receptors and connexin 43 regulates hemichannels activity and ATP release in astrocytes exposed to amyloid-β peptides. Mol Neurobiol 58(12):6232–6248. https://doi.org/10.1007/s12035-021-02538-z
Matos M, Augusto E, Machado NJ, Dos Santos-Rodrigues A, Cunha RA, Agostinho P (2012) Astrocytic adenosine A2A receptors control the amyloid-β peptide-induced decrease of glutamate uptake. J Alzheimers Dis 31(3):555–567. https://doi.org/10.3233/JAD-2012-120469
Dias L, Madeira D, Dias R, Tomé ÂR, Cunha RA, Agostinho P (2022) Aβ1-42 peptides blunt the adenosine A2A receptor-mediated control of the interplay between P2X7 and P2Y1 receptors mediated calcium responses in astrocytes. Cell Mol Life Sci 79(8):457. https://doi.org/10.1007/s00018-022-04492-y
Madeira D, Lopes CR, Simões AP, Canas PM, Cunha RA, Agostinho P (2023) Astrocytic A2A receptors silencing negatively impacts hippocampal synaptic plasticity and memory of adult mice. Glia 71(9):2137–2153. https://doi.org/10.1002/glia.24384
Canas PM, Porciúncula LO, Cunha GMA, Silva CG, Machado NJ, Oliveira JMA, Oliveira CR, Cunha RA (2009) Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci 29:14741–14751. https://doi.org/10.1523/JNEUROSCI.3728-09.2009
Viana da Silva S, Haberl MG, Zhang P, Bethge P, Lemos C, Gonçalves N, Gorlewicz A, Malezieux M, Gonçalves FQ, Grosjean N, Blanchet C, Frick A, Nägerl UV, Cunha RA, Mulle C (2016) Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat Commun 7:11915. https://doi.org/10.1038/ncomms11915
Faivre E, Coelho JE, Zornbach K, Malik E, Baqi Y, Schneider M, Cellai L, Carvalho K, Sebda S, Figeac M, Eddarkaoui S, Caillierez R, Chern Y, Heneka M, Sergeant N, Müller CE, Halle A, Buée L, Lopes LV, Blum D (2018) Beneficial effect of a selective adenosine A2A receptor antagonist in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Front Mol Neurosci 11:235. https://doi.org/10.3389/fnmol.2018.00235
Orr AG, Lo I, Schumacher H, Ho K, Gill M, Guo W, Kim DH, Knox A, Saito T, Saido TC, Simms J, Toddes C, Wang X, Yu G-Q, Mucke L (2018) Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol Dis 110:29–36. https://doi.org/10.1016/j.nbd.2017.10.014
Silva AC, Lemos C, Gonçalves FQ, Pliássova AV, Machado NJ, Silva HB, Canas PM, Cunha RA, Lopes JP, Agostinho P (2018) Blockade of adenosine A2A receptors recovers early deficits of memory and plasticity in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 117:72–81. https://doi.org/10.1016/j.nbd.2018.05.024
Orr AG, Hsiao EC, Wang MM, Ho K, Kim DH, Wang X, Guo W, Kang J, Yu G-Q, Adame A, Devidze N, Dubail DB, Masliah E, Conklin BR, Mucke L (2015) Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat Neurosci 18(3):423–434. https://doi.org/10.1038/nn.3930
Lopes CR, Silva JS, Santos J, Rodrigues MS, Madeira D, Oliveira A, Moreira-de-Sá A, Lourenço VS, Gonçalves FQ, Silva HB, Simões AP, Rolo AP, Canas PM, Tomé ÂR, Palmeira CM, Lopes JP, Cunha RA, Agostinho P, Ferreira SG (2023) Downregulation of sirtuin 1 does not account for the impaired long-term potentiation in the prefrontal cortex of female APPswe/PS1dE9 mice modelling Alzheimer’s disease. Int J Mol Sci 24(8):6968. https://doi.org/10.3390/ijms24086968
Gonçalves FQ, Lopes JP, Silva HB, Lemos C, Silva AC, Gonçalves N, Tomé ÂR, Ferreira SG, Canas PM, Rial D, Agostinho P, Cunha RA (2019) Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol Dis 132:104570. https://doi.org/10.1016/j.nbd.2019.104570
Moreira-de-Sá A, Gonçalves FQ, Lopes JP, Silva HB, Tomé ÂR, Cunha RA, Canas PM (2020) Adenosine A2A receptors format long-term depression and memory strategies in a mouse model of Angelman syndrome. Neurobiol Dis 146:105137. https://doi.org/10.1016/j.nbd.2020.105137
Garthe A, Kempermann G (2013) An old test for new neurons: refining the Morris water maze to study the functional relevance of adult hippocampal neurogenesis. Front Neurosci 7:63. https://doi.org/10.3389/fnins.2013.00063
Lopes CR, Amaral IM, Pereira MF, Lopes JP, Madeira D, Canas PM, Cunha RA, Agostinho P (2022) Impact of blunting astrocyte activity on hippocampal synaptic plasticity in a mouse model of early Alzheimer’s disease based on amyloid-β peptide exposure. J Neurochem 160(5):556–567. https://doi.org/10.1111/jnc.15575
Giaume C, Orellana JA, Abudara V, Sáez JC (2012) Connexin-based channels in astrocytes: how to study their properties. Methods Mol Biol 814:283–303. https://doi.org/10.1007/978-1-61779-452-0_19
Gajardo-Gómez R, Labra VC, Maturana CJ, Shoji KF, Santibañez CA, Sáez JC, Giaume C, Orellana JA (2017) Cannabinoids prevent the amyloid β-induced activation of astroglial hemichannels: a neuroprotective mechanism. Glia 65(1):122–137. https://doi.org/10.1002/glia.23080
Meadowcroft MD, Connor JR, Yang QX (2015) Cortical iron regulation and inflammatory response in Alzheimer’s disease and APPSWE/PS1ΔE9 mice: a histological perspective. Front Neurosci 9:255. https://doi.org/10.3389/fnins.2015.00255
Pereira MF, Amaral IM, Lopes C, Leitão C, Madeira D, Lopes JP, Gonçalves FQ, Canas PM, Cunha RA, Agostinho P (2021) L-α-aminoadipate causes astrocyte pathology with negative impact on mouse hippocampal synaptic plasticity and memory. FASEB J 35(8):e21726. https://doi.org/10.1096/fj.202100336R
Tavares G, Martins M, Correia JS, Sardinha VM, Guerra-Gomes S, das Neves SP, Marques F, Sousa N, Oliveira JF (2017) Employing an open-source tool to assess astrocyte tridimensional structure. Brain Struct Funct 222(4):1989–1999. https://doi.org/10.1007/s00429-016-1316-8
Dunkley PR, Jarvie PE, Robinson PJ (2008) A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc 3(11):1718–1728. https://doi.org/10.1038/nprot.2008.171
Matos M, Augusto E, Santos-Rodrigues AD, Schwarzschild MA, Chen J-F, Cunha RA, Agostinho P (2012) Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia 60(5):702–716. https://doi.org/10.1002/glia.22290
Cunha RA, Ribeiro JA (2000) Adenosine A2A receptor facilitation of synaptic transmission in the CA1 area of the rat hippocampus requires protein kinase C but not protein kinase A activation. Neurosci Lett 289(2):127–130. https://doi.org/10.1016/s0304-3940(00)01295-7-e
Abudara V, Bechberger J, Freitas-Andrade M, De Bock M, Wang N, Bultynck G, Naus CC, Leybaert L, Giaume C (2014) The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front Cell Neurosci 8:306. https://doi.org/10.3389/fncel.2014.00306
Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2(7):a006338. https://doi.org/10.1101/cshperspect.a006338
Zulfiqar S, Garg P, Nieweg K (2019) Contribution of astrocytes to metabolic dysfunction in the Alzheimer’s disease brain. Biol Chem 400(9):1113–1127. https://doi.org/10.1515/hsz-2019-014
Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM (2005) Adenosine and brain function. Int Rev Neurobiol 63:191–270. https://doi.org/10.1016/S0074-7742(05)63007-3
Muraleedharan A, Rotem-Dai N, Strominger I, Anto NP, Isakov N, Monsonego A, Livneh E (2021) Protein kinase C eta is activated in reactive astrocytes of an Alzheimer’s disease mouse model: evidence for its immunoregulatory function in primary astrocytes. Glia 69(3):697–714. https://doi.org/10.1002/glia.23921
Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF (2000) Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol 149(7):1503–1512. https://doi.org/10.1083/jcb.149.7.1503
Nagy JI, Li W, Hertzberg EL, Marotta CA (1996) Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res 717(1–2):173–178. https://doi.org/10.1016/0006-8993(95)01526-4
Vehmas AK, Kawas CH, Stewart WF, Troncoso JC (2003) Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging 24(2):321–331. https://doi.org/10.1016/S0197-4580(02)00090-8
Shrivastava AN, Kowalewski JM, Renner M, Bousset L, Koulakoff A, Melki R, Giaume C, Triller A (2013) β-amyloid and ATP-induced diffusional trapping of astrocyte and neuronal metabotropic glutamate type-5 receptors. Glia 61(10):1673–1686. https://doi.org/10.1002/glia.22548
Ceyzériat K, Haim LB, Denizot A, Pommier D, Matos M, Guillemaud O, Palomares M-A, Abjean L, Petit F, Gipchtein P, Gaillard M-C, Guillermier M, Bernier S, Gaudin M, Aurégan G, Joséphine C, Déchamps N, Veran J, Langlais V, Cambon K, Bemelmans AP, Baijer J, Bonvento G, Dhenain M, Deleuze J-F, Oliet SHR, Brouillet E, Hantraye P, Sauvage M-AC-d, Olaso R, Panatier A, Escartin C (2018) Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol Commun 6(1):104. https://doi.org/10.1186/s40478-018-0606-1
Lima A, Sardinha VM, Oliveira AF, Reis M, Mota C, Silva MA, Marques F, Cerqueira JJ, Pinto L, Sousa N, Oliveira JF (2014) Astrocyte pathology in the prefrontal cortex impairs the cognitive function of rats. Mol Psychiatry 19(7):834–841. https://doi.org/10.1038/mp.2013.182
Han H, Peng Y, Dong Z (2015) D-Serine rescues the deficits of hippocampal long-term potentiation and learning and memory induced by sodium fluoroacetate. Pharmacol Biochem Behav 133:51–56. https://doi.org/10.1016/j.pbb.2015.03.017
Kajiwara Y, Wang E, Wang M, Sin WC, Brennand KJ, Schadt E, Naus CC, Buxbaum J, Zhang B (2018) GJA1 (connexin43) is a key regulator of Alzheimer’s disease pathogenesis. Acta Neuropathol Commun 6(1):144. https://doi.org/10.1186/s40478-018-0642-x
Pham C, Hérault K, Oheim M, Maldera S, Vialou V, Cauli B, Li D (2021) Astrocytes respond to a neurotoxic Aβ fragment with state-dependent Ca2+ alteration and multiphasic transmitter release. Acta Neuropathol Commun 9(1):44. https://doi.org/10.1186/s40478-021-01146-1
Maulik M, Vasan L, Bose A, Dutta Chowdhury S, Sengupta N, Das Sarma J (2020) Amyloid-β regulates gap junction protein connexin 43 trafficking in cultured primary astrocytes. J Biol Chem 295(44):15097–15111. https://doi.org/10.1074/jbc.RA120.013705
Orellana JA, Shoji KF, Abudara V, Ezan P, Amigou E, Sáez PJ, Jiang JX, Naus CC, Sáez JC, Giaume C (2011) Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J Neurosci 31(13):4962–4977. https://doi.org/10.1523/JNEUROSCI.6417-10.2011
Ren R, Zhang L, Wang M (2018) Specific deletion connexin43 in astrocyte ameliorates cognitive dysfunction in APP/PS1 mice. Life Sci 208:175–191. https://doi.org/10.1016/j.lfs.2018.07.033
Bao X, Lee SC, Reuss L, Altenberg GA (2007) Change in permeant size selectivity by phosphorylation of connexin 43 gap-junctional hemichannels by PKC. Proc Natl Acad Sci USA 104(12):4919–4924. https://doi.org/10.1073/pnas.0603154104
Solan JL (1860) Lampe PD (2018) Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim Biophys Acta Biomembr 1:83–90. https://doi.org/10.1016/j.bbamem.2017.04.008
Hüll M, Müksch B, Akundi RS, Waschbisch A, Hoozemans JJM, Veerhuis R, Fiebich BL (2006) Amyloid β peptide (25–35) activates protein kinase C leading to cyclooxygenase-2 induction and prostaglandin E2 release in primary midbrain astrocytes. Neurochem Int 48(8):663–672. https://doi.org/10.1016/j.neuint.2005.08.013
Pérez-Núñez R, Chamorro A, González MF, Contreras P, Artigas R, Corvalán AH, van Zundert B, Reyes C, Moya PR, Avalos AM, Schneider P, Quest AFG, Leyton L (2023) Protein kinase B (AKT) upregulation and Thy-1-αvβ3 integrin-induced phosphorylation of Connexin43 by activated AKT in astrogliosis. J Neuroinflamm 20(1):5. https://doi.org/10.1186/s12974-022-02677-7
Bao X, Reuss L, Altenberg GA (2004) Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of Serine 368. J Biol Chem 279(19):20058–20066. https://doi.org/10.1074/jbc.M311137200
Liao CK, Cheng HH, Wang SD, Yeih DF, Wang SM (2013) PKCɛ mediates serine phosphorylation of connexin43 induced by lysophosphatidylcholine in neonatal rat cardiomyocytes. Toxicology 314(1):11–21. https://doi.org/10.1016/j.tox.2013.08.001
Nuriya M, Morita A, Shinotsuka T, Yamada T, Yasui M (2018) Norepinephrine induces rapid and long-lasting phosphorylation and redistribution of connexin 43 in cortical astrocytes. Biochem Biophys Res Commun 504(4):690–697. https://doi.org/10.1016/j.bbrc.2018.09.021
Temido-Ferreira M, Ferreira DG, Batalha VL, Marques-Morgado I, Coelho JE, Pereira P, Gomes R, Pinto A, Carvalho S, Canas PM, Cuvelier L, Buée-Scherrer V, Faivre E, Baqi Y, Müller CE, Pimentel J, Schiffmann SN, Buée L, Bader M, Outeiro TF, Blum D, Cunha RA, Marie H, Pousinha PA, Lopes LV (2020) Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol Psychiatry 25(8):1876–1900. https://doi.org/10.1038/s41380-018-0110-9
Rebola N, Canas PM, Oliveira CR, Cunha RA (2005) Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 132(4):893–903. https://doi.org/10.1016/j.neuroscience.2005.01.014
Kaster MP, Machado NJ, Silva HB, Nunes A, Ardais AP, Santana M, Baqi Y, Müller CE, Rodrigues AL, Porciúncula LO, Chen JF, Tomé ÂR, Agostinho P, Canas PM, Cunha RA (2015) Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc Natl Acad Sci USA 112(25):7833–7838. https://doi.org/10.1073/pnas.1423088112
Matos M, Shen HY, Augusto E, Wang Y, Wei CJ, Wang YT, Agostinho P, Boison D, Cunha RA, Chen JF (2015) Deletion of adenosine A2A receptors from astrocytes disrupts glutamate homeostasis leading to psychomotor and cognitive impairment: relevance to schizophrenia. Biol Psychiatry 78(11):763–774. https://doi.org/10.1016/j.biopsych.2015.02.026
Agostinho P, Madeira D, Dias L, Simões AP, Cunha RA, Canas PM (2020) Purinergic signaling orchestrating neuron-glia communication. Pharmacol Res 162:105253. https://doi.org/10.1016/j.phrs.2020.105253
Liu Y, Chu S, Hu Y, Yang S, Li X, Zheng Q, Ai Q, Ren S, Wang H, Gong L, Xu X, Chen NH (2021) Exogenous adenosine antagonizes excitatory amino acid toxicity in primary astrocytes. Cell Mol Neurobiol 41(4):687–704. https://doi.org/10.1007/s10571-020-00876-5
Roux L, Madar A, Lacroix MM, Yi C, Benchenane K, Giaume C (2015) Astroglial connexin 43 hemichannels modulate olfactory bulb slow oscillations. J Neurosci 35(46):15339–15352. https://doi.org/10.1523/JNEUROSCI.0861-15.2015
Dai SS, Zhou YG, Li W, An JH, Li P, Yang N, Chen XY, Xiong RP, Liu P, Zhao Y, Shen HY, Zhu PF, Chen JF (2010) Local glutamate level dictates adenosine A2A receptor regulation of neuroinflammation and traumatic brain injury. J Neurosci 30(16):5802–5810. https://doi.org/10.1523/JNEUROSCI.0268-10.2010
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55(5):697–711. https://doi.org/10.1016/j.neuron.2007.07.025
Ranasinghe KG, Kudo K, Hinkley L, Beagle A, Lerner H, Mizuiri D, Findlay A, Miller BL, Kramer JH, Gorno-Tempini ML, Rabinovici GD, Rankin KP, Garcia PA, Kirsch HE, Vossel K, Nagarajan SS (2022) Neuronal synchrony abnormalities associated with subclinical epileptiform activity in early-onset Alzheimer’s disease. Brain 145(2):744–753. https://doi.org/10.1093/brain/awab442
Targa DAH, Matosin N, Ooi L (2022) Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl Psychiatry 12(1):257. https://doi.org/10.1038/s41398-022-02024-7
Cristóvão-Ferreira S, Vaz SH, Ribeiro JA, Sebastião AM (2009) Adenosine A2A receptors enhance GABA transport into nerve terminals by restraining PKC inhibition of GAT-1. J Neurochem 109(2):336–347. https://doi.org/10.1111/j.1471-4159.2009.05963.x
Fu SY, Xiong RP, Peng Y, Zhang ZH, Chen X, Zhao Y, Ning YL, Yang N, Zhou YG, Li P (2019) PKC mediates LPS-induced IL-1β expression and participates in the pro-inflammatory effect of A2AR under high glutamate concentrations in mouse microglia. Neurochem Res 44(12):2755–2764. https://doi.org/10.1007/s11064-019-02895-1
Pinto-Duarte A, Coelho JE, Cunha RA, Ribeiro JA, Sebastião AM (2005) Adenosine A2A receptors control the extracellular levels of adenosine through modulation of nucleoside transporters activity in the rat hippocampus. J Neurochem 93(3):595–604. https://doi.org/10.1111/j.1471-4159.2005.03071.x
Gessi S, Bencivenni S, Battistello E, Vincenzi F, Colotta V, Catarzi D, Varano F, Merighi S, Borea PA, Varani K (2017) Inhibition of A2A adenosine receptor signaling in cancer cells proliferation by the novel antagonist TP455. Front Pharmacol 8:888. https://doi.org/10.3389/fphar.2017.00888
Singh BL, Chen L, Cai H, Shi H, Wang Y, Yu C, Chen X, Han X, Cai X (2019) Activation of adenosine A2A receptor accelerates and A2A receptor antagonist reduces intermittent hypoxia induced PC12 cell injury via PKC-KATP pathway. Brain Res Bull 150:118–126. https://doi.org/10.1016/j.brainresbull.2019.05.015
Sorrentino C, Hossain F, Rodriguez PC, Sierra RA, Pannuti A, Osborne BA, Minter LM, Miele L, Morello S (2019) Adenosine A2A receptor stimulation inhibits TCR-induced Notch1 activation in CD8+T-cells. Front Immunol 10:162. https://doi.org/10.3389/fimmu.2019.00162
Acknowledgements
The authors thank the funding agencies for research financial support as well as to research centres: CNC-UC and FMUC for the equipment and facilities, in particular the microscopy unit of CNC-UC.
Funding
Open access funding provided by FCT|FCCN (b-on). This work was supported by La Caixa Foundation (HP17/00523), Centro 2020 (CENTRO-01–0145 FEDER-000008: BrainHealth 2020 and CENTRO-01–0246-FEDER-000010) and FCT (PTDC/MED NEU/31274/2017 and UIDB/04539/2020. Thanks are also due to FCT for PhD grants of Daniela Madeira (SFRH/BD/139334/2018) and Cátia R. Lopes (2021.06954.BD).
Author information
Authors and Affiliations
Contributions
DM and CRL performed animal surgeries, behavioural tests and EtBr uptake assays, including brain slices preparation, under the supervision of PMC. JD did also EtBr uptake and immunohistochemistry and performed the data analysis of EtBr uptake. Astrocyte cell cultures, gliosomes preparation and Western blot assays were carried out by DM and CRL. Experimental planning, data interpretation, and manuscript writing were ensured by DM, RAC and PA.
Corresponding author
Ethics declarations
Conflict of interest
Rodrigo A. Cunha is a scientific advisor of the Institute for Scientific Information on Coffee (ISIC).
Ethics approval
This study was approved by the ORBEA_300_2021/24092021 and certified by Direção Geral de Alimentação e Veterinária (DGAV; Portuguese National Authority for Animal Health and Well Being, 0421/000/000/2021).
Consent for publication
This study does not have individual person’s data. All the authors read and consent to publish this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Madeira, D., Domingues, J., Lopes, C.R. et al. Modification of astrocytic Cx43 hemichannel activity in animal models of AD: modulation by adenosine A2A receptors. Cell. Mol. Life Sci. 80, 340 (2023). https://doi.org/10.1007/s00018-023-04983-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-04983-6