
Abstract
Background
Dysregulated lipid oxidation occurs in several pathological processes characterized by cell proliferation and migration. Nonetheless, the molecular mechanism of lipid oxidation is not well appreciated in liver fibrosis, which is accompanied by enhanced fibroblast proliferation and migration.
Methods
We investigated the causes and consequences of lipid oxidation in liver fibrosis using cultured cells, animal models, and clinical samples.
Results
Increased ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP1) expression caused increased lipid oxidation, resulting in the proliferation and migration of hepatic stellate cells (HSCs) that lead to liver fibrosis, whereas fibroblast-specific ENPP1 knockout reversing these results. Elevated ENPP1 and N6-methyladenosine (m6A) levels were associated with high expression of Wilms tumor 1 associated protein (WTAP). Mechanistically, WTAP-mediated m6A methylation of the 3'UTR of ENPP1 mRNA and induces its translation dependent of YTH domain family proteins 1 (YTHDF1). Additionally, ENPP1 could interact with hypoxia inducible lipid droplet associated (HILPDA) directly; overexpression of ENPP1 further recruits HILPDA-mediated lipid oxidation, thereby promotes HSCs proliferation and migration, while inhibition of ENPP1 expression produced the opposite effect. Clinically, increased expression of WTAP, YTHDF1, ENPP1, and HILPDA, and increased m6A mRNA content, enhanced lipid oxidation, and increased collagen deposition in human liver fibrosis tissues.
Conclusions
We describe a novel mechanism in which WTAP catalyzes m6A methylation of ENPP1 in a YTHDF1-dependent manner to enhance lipid oxidation, promoting HSCs proliferation and migration and liver fibrosis.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liver fibrosis [1], an umbrella term for a range of liver diseases associated with varying degrees of injury and fibrosis [2], remains an unresolved problem in liver diseases [3]. Liver fibrosis is characterized by the proliferation and migration of hepatic stellate cells (HSCs) and subsequent collagen deposition [4]. HSCs proliferation and migration are affected by lipid metabolic reprogramming, in which lipid oxidation are vital processes [5, 6]. Indeed, excessive lipid oxidation may impair cellular functions, including cell proliferation and migration [7,8,9]. Nevertheless, lipid oxidation, distinctive characteristics of liver fibrosis, remain poorly understood at the molecular level.
Excessive lipid oxidation, promoting the activation of pathological mechanisms leading to steatosis and fibrosis [10, 11]. Notably, ectonucleotide pyrophosphatase/ phosphodiesterase (ENPP1) is an energy metabolism factor that plays a crucial role in lipid metabolism [12]. ENPP1 is expressed in a range of human diseases, including osteoarthritis [13], diabetes [14], cancer [15], and cardiovascular disease [16]. Specifically, the ENPP1 gene has been closely associated with obesity in recent years [17, 18]. However, whether ENPP1 regulates lipid oxidation, thereby contributing to HSCs proliferation and migration, has yet to be determined.
Dysregulation of ENPP1 expression may be linked to epitranscriptomic mechanisms. N6-methyladenosine (m6A) is the most abundant internal mark on mammalian messenger RNA [19, 20]. The m6A modification is mainly mediated by m6A regulators: writers, erasers, and readers [21, 22]. The core writers that catalyze m6A modification are Wilms’ tumor 1-associating protein (WTAP) and methyltransferase-like 3/14 (METTL3/14) [23]. Readers include YT521-B homology (YTH) domain-containing family proteins (YTHDF1/2/3) and YTH domain-containing proteins (YTHDC1/2) [24]. Interestingly, the WTAP-mediated m6A modification specifically cooperates with the m6A reader YTHDF1, and this interaction is essential for mRNA stability and transcription [25, 26]. Nonetheless, whether ENPP1 expression, as regulated by WTAP-mediated m6A modification, contributes to liver fibrosis remains unclear.
In this study, we demonstrated that WTAP boosts lipid oxidation as well as promotes HSCs proliferation and migration by increasing the ENPP1 mRNA methylation rate via the YTHDF1-dependent m6A modification. These findings reveal that aberrant m6A methylation of ENPP1 mRNA may be a novel and critical regulatory factor in liver fibrosis, thus providing insight into the underlying molecular mechanism, which can be leveraged for the prevention and treatment of liver fibrosis.
Materials and methods
Detailed descriptions of the materials and methods are presented in the Supplemental Methods and Tables.
Results
ENPP1 upregulation accompanied by excessive lipid oxidation in vivo and in vitro
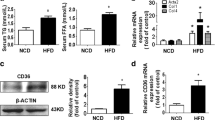
To explore the pathogenesis of liver fibrosis, we first established a carbon tetrachloride (CCl4)-induced mouse model of liver fibrosis as previously described [1] (Fig. S1A). As expected, the CCl4 mice showed marked increases in body weight, liver weight and liver index values compared with the vehicle mice. In addition, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were significantly elevated in the CCl4 mice (Fig. S1B, D). Additionally, we observed minor shifts in the levels of most lipid species and striking differences in the levels of serum hepatic triglyceride (TG) and total cholesterol (TC), which are neutral lipids, indicating abnormal lipid deposition (Fig. S1C, E). In addition, the CCl4 mice developed marked hepatic steatosis, collagen deposition, fibrosis, and impaired liver function, as evidenced by liver morphology, hematoxylin and eosin (H&E) staining, Masson’s trichrome staining and Sirius red staining (Fig. S1F). Additionally, oil red O staining indicated abnormal lipid accumulation (Fig. S1F). These results confirmed that the liver fibrosis models had been successfully established by CCl4 mice and that these mice showed abnormal lipid accumulation.
Next, we aimed to understand the changes of lipid metabolism in liver fibrosis, especially because LDs are dynamic organelles that are hubs of cellular lipid and energy metabolism [27]. First, we unexpectedly found abnormal ATP metabolism in the liver of the CCl4 mice but not in the mouse hearts, lungs or kidneys (Fig. S1G). Nucleotide pyrophosphatases/phosphodiesterases (NPPs) hydrolyze pyrophosphates (for example, in ATP) and phosphodiesters (for example, in oligonucleotides), respectively, to produce nucleoside 5’-monophosphate [28, 29], which are closely related to ENPP family genes [30, 31]. To detect abnormal phenomena, we comprehensively measured the expression levels of seven ENPP family genes in mice liver fibrosis tissues. The results showed that ENPP1 expression in the liver fibrosis group was significantly increased compared to that in the control group, but the changes in ENPP4 and ENPP6 expression were low, and in ENPP2, ENPP3 and ENPP7, they were negligible (Fig. 1A–B and Fig. S1H). Therefore, ENPP1 may play a role in lipid accumulation in liver fibrosis.

ENPP1 Upregulation Accompanied by Excessive Lipid Oxidation in vivo and in vitro. A The expressions of ENPP1-7 at the mRNA level were examined by RT-qPCR (n = 6). B The expression of ENPP1-7 at the protein level was verified by Western blotting analysis (n = 6). C The expression of Collagen l, PPARγ, PPARα, PPARβ and α-SMA at the protein level was detected by Western blotting analysis (n = 6). D The expression of Collagen l, PPARγ, PPARα, PPARβ and α-SMA at the mRNA level was detected by RT-qPCR (n = 6). E TEM images of LDs in the CCl4-induced mice model, and the number of LDs in 15.2 μm2 liver tissue was calculated (n = 6). F Fluorescence microscopy images of liver tissue showing the BODIPY 493/503 and ENPP1 co-stained with α-SMA (n = 6), Original scale bars, 50 μm. Colocalization was quantified using ImageJ with JACoP plugin and Manders’s correlation coefficient (MCC). G The protein level of Collagen l, PPARα, PPARβ, α-SMA, and ENPP1 were detected in HSC and AML-12 cells by western blot (n = 6). H The mRNA level of Collagen l, PPARα, PPARβ, α-SMA, and ENPP1 were detected in HSC and AML-12 cells by RT-qPCR (n = 6). I Fluorescence microscopy images of liver tissue showing the PPARβ co-stained with ENPP1 (n = 6), Original scale bars, 50 μm. Colocalization was quantified using ImageJ with JACoP plugin and Manders’s correlation coefficient (MCC). J MTT and K CCK-8 assays were applied to evaluate the proliferation abilities of HSC and AML-12 (n = 6). L Representative images of Edu staining of HSC and AML-12, and the percentage of Edu-positive cells were calculated (n = 6). M Cell populations at different phases were detected by cell cycle analysis in HSC and AML-12 cells (n = 6). All data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Vehicle group and Control group. ns no significance, two-tailed Student’s t-test
Second, transmission electron microscopy (TEM) analysis found that the number of LDs was increased in liver fibrosis mice compared with control (Fig. 1E and Fig. S1J). As indicated by tissue staining, highly abundant lipids and abnormal LD accumulation were identified in liver fibrosis samples (Fig. S1F). Interestingly, the level of lipid oxidation marker peroxisome proliferators activated receptor (PPARα and PPARβ) were significantly increased, while PPAR γ was slightly decreased in liver fibrosis mice compared to control mice (Fig. 1C–D, and Fig. S1I). These data indicate abnormal lipid deposition and excessive lipid oxidation in liver fibrosis.
Third, aggravated liver fibrosis is related to collagen deposition and α-SMA overexpression [32]. In this study, the collagen I and α-SMA levels were significantly increased in the liver fibrosis mice ((Fig. 1C–D, and Fig. S1I). These findings suggest that the elevation in ENPP1 level was accompanied by liver fibrosis and impaired liver function.
Crucially, to identify whether a similar specific effect on fibroblasts and lipid oxidation was evident at the cellular level, we performed immunofluorescence co-staining assays, and the results showed that the LD was positively correlated with the expression levels of ENPP1 and α-SMA, which are marker proteins of HSCs activation, indicatng the important role played by HSC in regulating lipid oxidation in liver fibrosis (Fig. 1F and Fig. S1K). Nonetheless, we did not find increased co-localization between ENPP1 and hepatocyte marker Alb by co-staining assays in the liver fibrosis tissue (Data not display). Hence, these results indicated that ENPP1 upregulation accompanied by excessive lipid oxidation in liver fibrosis.
To further investigate whether the change of ENPP1 in vitro is similar to that in the liver fibrosis mice, we explored the role of ENPP1 in mediating lipid oxidation in HSCs and hepatocytes (AML-12) (Fig. S1L). First, we sought to determine whether ENPP1 is involved in liver fibrosis. We found that the levels of ENPP1, collagen l and α-SMA were substantially increased in TGF-β1-treated HSCs. TGF-β1 is the most potent fibrogenic cytokine and a key driver of HSCs activation and liver fibrosis [33]; nonetheless, the level of ENPP1, collagen l and α-SMA was only slightly changed in AML-12 cells (Fig. 1G–H and Fig. S1M). These results revealed that ENPP1 upregulation may play a critical role in TGF-β1-induced HSCs activation, but not hepatocyte. In addition, the level of PPARα and PPARβ was increased, while that of PPARα and PPARβ was only slightly changed in AML-12 cells. Moreover, PPARβ were colocalized with ENPP1, as determined by immunofluorescence staining, indicating that ENPP1 was closely related to lipid oxidation in TGF-β1-treated HSCs but not in AML-12 cells (Fig. 1G–I, and Fig. S1M–N).
Next, MTT, cell counting kit-8 (CCK-8) and EdU assays results suggested that induction of TGF-β1 markedly increased the proliferation of HSCs, and flow cytometry revealed that HSCs proliferation, but not that of AML-12 cells, was promoted via regulation of the G0/G1-S transition (Fig. 1J–M and Fig. S1O). Furthermore, wound healing and Transwell migration assays indicated that TGF-β1-treated HSCs migrated much more quickly than control cells, but only slight changes in AML-12 cell migration rates were observed (Fig. S1P–Q). To eliminate the influence of the culture medium, we found no changes in the proliferation and migration properties of HSCs and AML-12 cells without TGF-β1 and under the same culture conditions (Fig. S1R). Increased proliferation was observed only with HSCs after adding TGF-β1 (Fig. S1S). These results suggest that increased ENPP1-mediated lipid oxidation may underlie the induction of liver fibrosis, which is caused by HSCs proliferation and migration. Therefore, we hypothesized that ENPP1 promotes lipid oxidation might be key to TGF-β1-induced HSCs and liver fibrosis.
ENPP1 promotes HSCs proliferation and migration
We further investigated the mechanism underlying ENPP1-mediated lipid oxidation and the promotion of HSCs proliferation and migration (Fig. 2A). First, we infected HSCs with ENPP1 short interfering RNA (siRNA) to knockdown ENPP1 expression (Table S1), and the silencing efficiency was demonstrated at the mRNA and protein levels (Fig. 2B–C and Fig. S2A). We observed a significant decrease in the mRNA and protein levels of collagen l and α-SMA in the si-ENPP1 group compared with the si-NC group (Fig. 2B–C and Fig. S2A), which suggested that ENPP1 knockdown significantly reduced HSCs activation.

ENPP1 Promotes HSCs Proliferation and Migration. A The schema diagram for Fig. 2. B The mRNA level of Collagen l, PPARα, PPARβ, α-SMA, and ENPP1 were detected in ENPP1 knockdown HSC cells by RT-qPCR (n = 6). C The protein level of Collagen l, PPARα, PPARβ, α-SMA, and ENPP1 were detected in ENPP1 knockdown HSC cells by western blot (n = 6). D The MDA and ROS levels in ENPP1 knockdown HSC is shown (n = 6). E Representative images of Edu staining of ENPP1 knockdown HSC cells, and the percentage of Edu-positive cells were calculated (n = 6). F Cell populations at different phases were detected by cell cycle analysis in ENPP1 knockdown HSC cells (n = 6). G Wound healing assays were conducted to compare the migration capabilities of ENPP1 knockdown HSC cells (n = 6), original scale bars, 200 μm. H Trans-well assays of ENPP1 knockdown HSC cells were applied to measure their migration abilities (n = 6), original scale bars, 100 μm. I The mRNA level of Collagen l, PPARα, PPARβ, α-SMA, and ENPP1 were detected in ENPP1 overexpression HSC cells by RT-qPCR (n = 6). J The protein level of Collagen l, PPARα, PPARβ, α-SMA, and ENPP1 were detected in ENPP1 overexpression HSC cells by western blot (n = 6). K The MDA and ROS levels in ENPP1 overexpression HSC is shown (n = 6). L MTT and M CCK-8 assays were applied to evaluate the proliferation abilities of ENPP1 overexpression HSC cells (n = 6). N Representative images of Edu staining of ENPP1 overexpression HSC cells, and the percentage of Edu-positive cells were calculated (n = 6). O Cell populations at different phases were detected by cell cycle analysis in ENPP1 overexpression HSC cells (n = 6). P Wound healing assays were conducted to compare the migration capabilities of ENPP1 overexpression HSC cells (n = 6), original scale bars, 200 μm. Q Trans-well assays of ENPP1 overexpression HSC cells were applied to measure their migration abilities (n = 6), original scale bars, 100 μm. All data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. si-NC group and OE-NC group. ns no significance, two-tailed Student’s t-test
Second, knocking down ENPP1 significantly inhibited PPARα and PPARβ expression (Fig. 2B–C and Fig. S2A), and markedly suppressed MDA and ROS in HSCs (Fig. 2D). Moreover, the MTT, CCK-8, EdU and flow cytometry assays showed that knocking down ENPP1 expression decreased the proliferation rate of HSCs (Fig. 2E–F and Fig. S2B-D). Furthermore, wound healing and trans-well assays showed that inhibition of ENPP1 expression decreased the migration rate of HSCs (Fig. 2G–H and Fig. S2E-F). Meanwhile, we found using another potent siRNA that similarly significant knockdown of ENPP1 decreased the proliferation and migration capacity of HSCs (Fig. S2K-M). In summary, the loss of ENPP1 expression led to suppressed lipid oxidation, reducing the HSCs proliferation and migration rates.
To further demonstrate the role played by ENPP1 overexpression in lipid oxidation and HSCs activation, we used a plasmid to construct an ENPP1 overexpression model (Fig. 2A and Table S1). Intriguingly, ENPP1 overexpression was demonstrated, ENPP1 expression was elevated in HSCs treated with overexpressed ENPP1 (Fig. 2I–J and Fig. S2G). Next, we determined that the expression levels of collagen l and α-SMA were significantly increased after ENPP1 was overexpressed (Fig. 2I–J and Fig. S2G), further supporting our initial observation suggesting that ENPP1 overexpression aggravates HSCs activation.
Furthermore, overexpression of ENPP1 markedly increased PPARα and PPARβ expression (Fig. 2I–J and Fig. S2G) and significantly promoted MDA and ROS levels (Fig. 2K). Notably, ENPP1 overexpression significantly accelerated HSCs proliferation and migration (Fig. 2L–Q and Fig. S2H-J). Collectively, these results indicated that ENPP1 overexpression enhanced the proliferation and migration of TGF-β1-treated HSCs, and indicated that ENPP1 play a crucial role in regulating lipid oxidation and HSCs proliferation and migration.
ENPP1 regulates lipid oxidation by targeting HILPDA
To more thoroughly investigate how ENPP1 affects lipid oxidation in HSCs, we performed transcriptome sequencing to identify transcriptome alterations of ENPP1 knockdown in HSCs treated with TGF-β1. GO classification term and KEGG pathway annotations were used to identify the molecular functions and biological pathways of genes that were differentially expressed genes in the control and ENPP1-knockdown HSCs groups (Fig. 3A–C). A volcano plot and hierarchical clustering indicated that 30 genes were upregulated and that 15 genes were downregulated after ENPP1 expression was abrogated (Fig. 3D-–E). Among these genes, the expression of the hypoxia-inducible lipid droplet-associated (HILPDA)-encoding gene was significantly changed, which has been previously observed to be associated with lipid metabolism [34]. Therefore, we further explored the relationship between ENPP1 and HILPDA.

ENPP1 Regulates Lipid Oxidation by targeting HILPDA. A GO classification of the source genes of the differentially expressed mRNA in the control and ENPP1 knockdown HSC groups. B GO assignments of the source genes of the differentially expressed mRNA in the control and ENPP1 knockdown HSC groups. C KEGG assignments of the source genes of the differentially expressed mRNA in the control and ENPP1 knockdown HSC groups. D Volcan plot of target and junction event records in the control and ENPP1 knockdown HSC groups. E Heat map of differential gene expression in the control and ENPP1 knockdown HSC groups. F The mRNA level of HILPDA were detected in liver tissues by RT-qPCR (n = 6). G The mRNA level of HILPDA were detected in HSC by RT-qPCR (n = 6). H The protein level of HILPDA were detected by western blot (n = 6). I The protein level of HILPDA were detected in HSC by western blot (n = 6). J HILPDA interacts with ENPP1 by the reciprocal Co-IP assays in HSC cells induced by TGF-β1. K HILPDA interacts with ENPP1 by the reciprocal Co-IP assays in ENPP1 knockdown HSC. L Pull-down bands were detected by immunoblotting with anti-Flag antibody. M–O The protein level of PPARα and PPARβ in HILPDA knockdown HSC were detected by western blot (n = 6). M HILPDA overexpression HSC (N), and HSC by FFA (O). P-S The MDA and ROS levels in HILPDA knockdown HSC is shown (n = 6). P HILPDA overexpression HSC (Q), and HSC by FFA (R), HILPDA knockdown HSC by FFA (S). T The protein level of PPARα and PPARβ in HILPDA overexpression HSC by FFA were detected by western blot (n = 6). All data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Vehicle group, Control group, si-NC group and OE-NC group. ns two-tailed Student’s t-test
Specifically, RT-qPCR and western blotting analyses revealed that the level of HILPDA was elevated in liver fibrosis mice and in TGF-β1-treated HSCs (Fig. 3F–H and Fig. S3A). Furthermore, knocking down ENPP1 led to a notable decrease in the HILPDA level, whereas ENPP1 overexpression typically led to an increase in the level of HILPDA (Fig. 3I and Fig. S3B). These results implied that ENPP1 may play a regulatory role in HILPDA expression. To confirm that ENPP1 and HILPDA interact, we performed coimmunoprecipitation (Co-IP) assays and verified that ENPP1 binds to HILPDA in activated HSCs stimulated with TGF-β1 and that the amount of HILPDA protein that binds to ENPP1 was increased after ENPP1 was overexpressed (Fig. 3J–K and Fig. S3C-D). In addition, we showed that GST-tagged ENPP1 pulled down HILPDA in vitro (Fig. 3L and Fig. S3E). These data confirmed that HILPDA may be a target downstream of ENPP1. Collectively, changes in the HILPDA expression were consistent with the lipid oxidation level changes observed in vitro and in vivo.
Third, to investigate whether changes in HILPDA expression affect lipid oxidation, we observed that the lipid oxidation was accelerated in TGF-β1-treated HSCs (Fig. 1G–H and Fig. S1M). Surprisingly, when HILPDA was knocked down, which inhibited PPARα and PPARβ expression (Fig. 3M and Fig. S3F), whereas HILPDA overexpression increased the expression of PPARα and PPARβ (Fig. 3N and Fig. S3G). At the same time, the lipid oxidation was enhanced in FFA-treated HSCs (Fig. 3O and Fig. S3H). When HILPDA knocked down, the MDA and ROS levels was inhibited, whereas HILPDA overexpression promoted the MDA and ROS levels (Fig. 3P–Q). In addition, the MDA and ROS levels was enhanced and increased the proliferation of HSCs in FFA-treated HSCs (Fig. 3R and Fig. S3J). When HILPDA was knocked down, the MDA and ROS levels was inhibited, decreased the proliferation rate of HSCs (Fig. 3S and Fig. S3K), whereas HILPDA overexpression promoted the lipid oxidation and increased the proliferation of HSCs (Fig. 3T and Fig. S3I, L). Taken together, these results suggest that ENPP1 regulated lipid oxidation by regulating HILPDA expression.
Fibroblasts specific ENPP1 disruption protects against lipid oxidation and attenuates liver fibrosis
To determine the function of ENPP1 in vivo, we established mouse models of conditional ENPP1 HSCs-specific knockout (ENPP1-cKO mice) (Fig. 4A and Table S1). An overview of the ENPP1 targeting strategy is shown in Fig. 4B. All mice were genotyped via PCR (Fig. 4C and Fig. S4A), and ENPP1 deficiency was verified by RT-qPCR and western blotting (Fig. 4D–E and Fig. S4B). Extract primary HSCs from cKO mice and verify ENPP1 deficiency through RT-qPCR and Western blotting (Fig. S4F-G). As expected, the abundance of ENPP1 and the downstream protein HILPDA was increased in CCl4-induced liver fibrosis mice but was decreased in ENPP1-cKO mice (Fig. 4D–E and Fig. S4B). ENPP1-cKO mice showed marked reduction in collagen deposition, fibrosis, and liver damage (Fig. 4D–E and Fig. S4B). These results validated that ENPP1-deficient ameliorated liver fibrosis.

Fibroblasts Specific ENPP1 Disruption Protects Against Lipid Oxidation and Attenuates Liver Fibrosis. A The schema diagram for Fig. 4. B Diagram of gene targeting scheme to generate the ENPP1 knockout (cKO) mice. C Genotyping of WT and Cre; ENPP1−/− mice by PCRs on WT and cKO liver samples. D The mRNA level of Collagen l, PPARα, PPARβ, α-SMA, HILPDA and ENPP1 were detected in ENPP1−/− mice by RT-qPCR (n = 6). E The protein level of Collagen l, PPARα, PPARβ, α-SMA, HILPDA and ENPP1 were detected in ENPP1−/− mice by western blot (n = 6). F The body weight, liver weight, and liver index (liver weight/body weight) ratio are shown. G The liver TG, and TC level are shown. H The plasma AST and ALT are shown. I The serum TG and TC are shown. J Histopathological study was performed by H&E, Masson, Sirius Red, and Oil Red O staining (n = 6). Scale bars: 50 μm. K The MDA and ROS levels in ENPP1−/− mice is shown (n = 6). L TEM images of lipid droplets (LDs) in ENPP1−/− mice, and the number of LDs in 15.2 μm2 liver tissue was calculated (n = 6). M Fluorescence microscopy images of liver tissue showing the BODIPY 493/503 co-stained with ENPP1 (n = 6), Original scale bars, 50 μm. Colocalization was quantified using ImageJ with JACoP plugin and Manders’s correlation coefficient (MCC). All data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. WT group. ns no significance, two-tailed Student’s t-test
Next, in ENPP1-cKO mice the levels of neutral lipids, such as TC and TG, were found to be decreased, and the fat composition and serum lipids were changed (Fig. 4G–I). The PPARα and PPARβ level was decreased, and the MDA and ROS level was reduced, and these results combined with liver tissue section staining results suggested that abnormal lipid oxidation was significantly attenuated in the Enpp1-cKO mice (Fig. 4D–E, J–K and Fig. S4B–C). Furthermore, we found that the lipid accumulation in the Enpp1-cKO mice had decreased, indicating that ENPP1 may play a key role in lipid oxidation (Fig. 4L–M and Fig. S4D–E). Notably, we found that the rate of LD and ENPP1 colocalization was decreased when ENPP1 was knocked out (Fig. 4M and Fig. S4D). These results indicated that ENPP1 deficiency attenuated lipid oxidation and liver fibrosis.
Increased ENPP1 m6A methylation accompanied by WTAP alteration in vivo and vitro
We investigated the mechanism by which ENPP1 promotes liver fibrosis (Fig. 5A). Because m6A methylation is an important epigenetic regulation for gene expression [35] and is related to liver development and considering that ENPP1 can be regulated by epigenetic [36], we examined whether the regulation of the m6A modification was attributable to liver fibrosis. Notably, according to the identification of the sequence obtained with a m6A modification site prediction tool (http://www.cuilab.cn/sramp), ENPP1 mRNA contains multiple potential m6A modification sites, and the three sites with the highest probability scores were found to be at approximately 571–676, 793–899 and 1797–1896 in the ENPP1 mRNA sequence (Fig. 5B). Meanwhile, the m6A modification site of ENPP2-ENPP7 was predicted as Fig. S5A. Notably, the “RRACH” (R = G, U or A; H = U or C) sequence motif was found to be highly enriched in m6A immunoprecipitated RNAs in a bioinformatic analysis performed with SRAMP software (Fig. S5B). To determine the potential function of ENPP1 mRNA m6A methylation in liver fibrosis and in HSCs, we first measured the global level of m6A in mouse liver fibrosis tissues and TGF-β1-induced HSCs via m6A dot blot assays. The results showed that the m6A mRNA abundance was substantially increased in the disease state (Fig. 5C–D and Fig. S5C-D). Therefore, we hypothesized that m6A modification is involved in dysregulated ENPP1 expression, which may lead to lipid oxidation and liver fibrosis.

Increased ENPP1 m6A Methylation Accompanied by WTAP alteration in vivo and vitro. A The schema diagram for cells. B The genomic sequence of a representative ENPP1 transcript (NM_053535.1) was used to predict the m6A modification on website (http://www.cuilab.cn/sramp), and an overview of the result page. C–D Global m6A levels of RNA extracted were measured via m6A dot blot assays (n = 3) from liver tissue induced by CCl4 (C) and HSC cells induced by TGF-β1 (D). E The expressions of WTAP, ENPP1, and HILPDA at the mRNA level in WTAP knockdown HSC were examined by RT-qPCR (n = 6). F The expressions of WTAP, ENPP1, and HILPDA at the protein level in WTAP knockdown HSC were examined by western blotting (n = 6). G Global m6A level of RNA extracted from WTAP knockdown HSC were measured via m6A dot blot assays (n = 3). H RNA decay analysis followed by RT-qPCR of ENPP1 mRNA stability following treatment with actinomycin D in WTAP knockdown HSC (n = 3). I WTAP interacts with ENPP1 by the reciprocal RIP assays in liver tissue induced by CCl4 (n = 6). J WTAP interacts with ENPP1 by the reciprocal RIP assays in LV3-WTAP liver tissue (n = 6). K WTAP interacts with ENPP1 by the reciprocal RIP assays in HSC cells were induced by TGF-β1 (n = 6). L WTAP interacts with ENPP1 by the reciprocal RIP assays in WTAP knockdown HSC (n = 6). M MeRIP analysis followed by RT-qPCR was applied to assess the m6A modification of ENPP1 in TGF-β1 induced HSC (n = 6). N MeRIP analysis followed by RT-qPCR was applied to assess the m6A modification of ENPP1 in WTAP knockdown HSC (n = 6). O The MDA and ROS levels in WTAP knockdown HSC (n = 6). P MTT and Q CCK-8 assays were applied to evaluate the proliferation abilities of WTAP knockdown HSC (n = 6). R Cell populations at different phases were detected by cell cycle analysis in WTAP knockdown HSC (n = 6). S The protein level of Collagen l, PPARα, PPARβ and α-SMA were detected in WTAP knockdown HSC by western blot (n = 6). T The schema diagram for mice. U The protein level of Collagen l, PPARα, PPARβ, α-SMA, WTAP, HILPDA and ENPP1 were detected in LV3-WTAP liver tissues by western blot (n = 6). V Global m6A level of RNA extracted from WTAP knockdown liver tissues were measured via m6A dot blot assays (n = 3). W TEM images of lipid droplets (LDs) in WTAP knockdown liver tissues, and the number of LDs in 15.2 μm2 liver tissue was calculated (n = 6). X The MDA and ROS levels in WTAP knockdown liver tissues is shown (n = 6). All data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Vehicle group, Control group, si-NC group and LV3-NC group. ns no significance, two-tailed Student’s t-test
Second, we evaluated the mRNA and protein expression levels of m6A modification-associated genes, namely, methyltransferases (METTL3, METTL14 and WTAP), and demethylases (FTO and ALKBH5), and found that the expression of WTAP was substantially increased in CCl4-induced liver fibrosis mice and TGF-β1-treated HSCs compared to the level in the corresponding control group (Fig. S5E-H). The expression level of FTO was slightly elevated in vivo but not in vitro (Fig. S5E-H). To identify the effects of WTAP on m6A modification, we transfected siRNA-WTAP into HSCs. Importantly, we observed that downregulation of WTAP marked reduced the abundance of m6A-modified mRNA and the expression of ENPP1 and HILPDA (Fig. 5E–G and Fig. S5I-J). These results indicated that WTAP may be a key element for the mRNA m6A modification in liver fibrosis.
Third, we found that, compared with that in the NC group, the stability of ENPP1 mRNA was decreased in the si-WTAP group (Fig. 5H), confirming that the m6A modification of ENPP1 mRNA expression may have been regulated by WTAP. To further evaluate the relationship between WTAP and ENPP1, we performed RNA immunoprecipitation (RIP) assays and confirmed that ENPP1 interacted with WTAP in liver fibrosis liver tissues (Fig. 5I), but the correlation was reduced after WTAP knockdown (Fig. 5J). Similar results were found with TGF-β1-treated HSCs (Fig. 5K–L).
Hence, we focused on ENPP1 as a target of WTAP action. By conducting methylated RNA immunoprecipitation (MeRIP)-qPCR, we found that ENPP1 mRNA was increased, as indicated by the anti-m6A antibody level, in TGF-β1-treated HSCs (Fig. 5M) and was reduced when WTAP was silenced (Fig. 5N). Subsequently, MeRIP-qPCR showed that WTAP knockdown markedly attenuated the reduction in m6A abundance on ENPP1 mRNA at site 2 (793–899) (Fig. 5M–N). A detailed primary sequence analysis revealed a 5-nucleotide consensus sequence (GAACU) in the ENPP1 mRNA sequence (at site 855) (Fig. S5K). Furthermore, ENPP1 expression was upregulated in CCl4-induced liver fibrosis tissues and TGF-β1-induced HSCs (Fig. 1A–B, G–H and Fig. S1H, M) and was downregulated when WTAP was knocked down (Fig. 5E–F and Fig. S5I). As described above, these results indicated that ENPP1 was a target of WTAP.
We continued to study the effects of WTAP knockdown in HSCs and liver fibrosis (Fig. 5A and T). Surprisingly, when WTAP was knocked down, the level of downstream HILPDA was decreased, which may have inhibited PPARα and PPARβ expression (Fig. 5S and Fig. S5O–P), suggesting that WTAP knockdown significantly alleviated lipid oxidation. Correspondingly, silencing WTAP markedly repressed the level of MDA and ROS (Fig. 5O). Moreover, knockdown of WTAP decreased the proliferative and migratory abilities of HSCs (Fig. 5P–R and Fig. S5L-N). Moreover, we observed that the levels of collagen l, α-SMA were significantly decreased (Fig. 5S, and Fig. S5O–P), demonstrating that WTAP knockdown significantly reduced lipid oxidation, HSCs proliferation and migration.
To determine the therapeutic potential of WTAP in liver fibrosis, mice with CCl4-induced liver fibrosis were injected with LV3-WTAP lentiviral vectors to introduce shRNA for WTAP knockdown (Fig. 5T), and the knockdown efficiency was confirmed (Fig. 5U and Fig. S5Q–R). Importantly, the expression levels of ENPP1 and HILPDA and the m6A modification abundance were reduced when WTAP was knocked down in HSCs (Fig. 5U–V and Fig. S5Q–S). Moreover, we found that WTAP knockdown markedly ameliorated collagen deposition, reduced lipid oxidation, and attenuated liver fibrosis and damage, as indicated by physiological indexes, biochemical indexes, morphological assay and molecular level measurements (Fig. 5W and Fig. S5T–Y). In addition, knocking down WTAP reduced the PPARα and PPARβ expression, the MDA and ROS level (Fig. 5U, X and Fig. S5Q-R), possibly via suppression of the ENPP1/HILPDA axis. In summary, silencing of WTAP consistently reduced lipid oxidation, HSCs proliferation and migration in liver fibrosis.
WTAP increases ENPP1 m6A methylation in a m6A-YTHDF1-dependent manner
The biological function of the m6A modification is mediated by specific m6A-bound reading proteins. Importantly, a protein–protein interaction network showed that WTAP can interact with five classical m6A readers, YTHDF1-3 and YTHDC1-2 (Fig. 6A). RT-qPCR showed that the expression of YTHDF1/2/3 was significantly increased, especially the expression of YTHDF1, in CCl4-induced liver fibrosis mice and TGF-β1-induced HSCs, but the expression of YTHDC1/2 was only slightly increased (Fig. 6B–C). Therefore, we evaluated the role played by YTHDF1/2/3 in ENPP1 mRNA stabilization. Specific siRNAs were specifically designed to target YTHDF1-3, and the knockdown efficiency was confirmed (Fig. 6D). We found that silencing YTHDF1 significantly inhibited ENPP1 mRNA expression, but knocking down YTHDF2 or YTHDF3 exerted a less profound effect (Fig. 6E). Correspondingly, western blotting showed that YTHDF1 expression was significantly upregulated in CCl4-induced liver fibrosis mice and TGF-β1-treated HSCs (Fig. 6F and Fig. S6A), and knocking down YTHDF1 inhibited ENPP1 expression (Fig. 6E, G and Fig. S6B).

WTAP increases ENPP1 m6A Methylation in a m6A-YTHDF1-dependent manner. A A protein–protein interaction network direct prediction by String. B The mRNA levels of m6A readers (YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2) were determined in liver tissue induced by CCl4 by real-time PCR (n = 6). C The mRNA levels of m6A readers (YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2) were determined in TGF-β1 induced HSC by real-time PCR (n = 6). D The mRNA levels of YTHDF1, YTHDF2, or YTHDF3 were determined in YTHDF1 knockdown, YTHDF2 knockdown, or YTHDF3 knockdown HSC by real-time PCR (n = 6). E The mRNA levels of ENPP1 were determined in YTHDF1 knockdown, YTHDF2 knockdown, or YTHDF3 knockdown HSC by real-time PCR (n = 6). F The protein level of YTHDF1 were detected in liver tissue induced by CCl4 or in TGF-β1 induced HSC using western blotting (n = 6). G The protein level of ENPP1 and YTHDF1 were detected in YTHDF1 knockdown HSC using western blotting (n = 6). H WTAP interacts with YTHDF1 by the reciprocal Co-IP assays in HSC cells induced by TGF-β1. I WTAP interacts with YTHDF1 by the reciprocal Co-IP assays in YTHDF1 knockdown HSC. J WTAP interacts with YTHDF1 by the reciprocal Co-IP assays in mice liver fibrosis. K RNA decay analysis followed by RT-qPCR of ENPP1 mRNA stability following treatment with actinomycin D in YTHDF1 knockdown HSC (n = 3). L YTHDF1 interacts with ENPP1 by the reciprocal RIP assays in liver tissue induced by CCl4 (n = 6). M YTHDF1 interacts with ENPP1 by the reciprocal RIP assays in LV3- YTHDF1 liver tissue (n = 6). N YTHDF1 interacts with ENPP1 by the reciprocal RIP assays in HSC cells were induced by TGF-β1 (n = 6). O YTHDF1 interacts with ENPP1 by the reciprocal RIP assays in YTHDF1 knockdown HSC (n = 6). P MeRIP analysis followed by RT-qPCR was applied to assess the m6A modification of ENPP1 in YTHDF1 knockdown HSC (n = 6). Q Schematic diagram of the m6A motif (GAACU) in ENPP1 and the mutation. R Immunoblotting of YTHDF1 after RNA pull down assay showed that the binding of YTHDF1 to ENPP1 was decreased by the ENPP1 mutation of the m6A-binding motif. S Binding of YTHDF1 with the ENPP1-WT and ENPP1-Mut were analyzed by RIP assays in HSC (n = 6). T The protein expression of ENPP1 was measured by western blot after transfected with ENPP1-WT and ENPP1-Mut plasmid, HSC were treated with TGF-β1 (n = 6). U Binding of WTAP with the ENPP1-WT and ENPP1-Mut were analyzed by RIP assays in HSC (n = 6). V After pretreatment with TGF-β1, knockdown/overexpression of WTAP, knockdown/overexpression of YTHDF1 or knockdown of ENPP1 were performed to determine the expression of HILPDA protein. All data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. WT or Control or si-NC group. ns no significance, two-tailed Student’s t-test
To better understand the relationship between WTAP and YTHDF1, we performed Co-IP assays and verified that WTAP binds to YTHDF1 in activated HSCs and liver fibrosis tissues and that the interaction rate increases when WTAP knockdown (Fig. 6H–J and Fig. S6C–E). These results suggest that the role played by WTAP in the m6A modification of ENPP1 mRNA is mediated through YTHDF1.
Third, to investigate the role of YTHDF1 in mediating the stability and translation of m6A-modified mRNA, RNA stability experiments showed that YTHDF1 knockdown decreased ENPP1 mRNA stability after actinomycin D (5 g/ml) treatment, indicating that YTHDF1 plays a key role in maintaining ENPP1 mRNA stability (Fig. 6K). To further assess the relationship between YTHDF1 and ENPP1, RIP assays were performed, and the results clearly showed that ENPP1 interacted with YTHDF1 in liver fibrosis tissues (Fig. 6L), but the interaction was modulated by YTHDF1 knockdown (Fig. 6M). Similar results were found in vitro (Fig. 6N–O). As expected, the m6A modification of ENPP1 mRNA was found to be lower after YTHDF1 was knocked down (Fig. 6P). These results suggest that YTHDF1 enhanced ENPP1 mRNA stability in a m6A-dependent manner.
Fourth, to confirm that WTAP promotes the ENPP1 level in a m6A-YTHDF1- dependent manner, we mutated the aforementioned m6A motif in ENPP1, which led to decreased binding with YTHDF1, as determined by pull-down assays (Fig. 6Q–R and Fig. S6F). This outcome indicated that the binding of YTHDF1 to ENPP1 depends on the m6A modification. To confirm the roles played by the m6A binding sites in ENPP1, we performed RIP assays, and the results showed that, in the ENPP1-Mut group, the binding of YTHDF1 and ENPP1 mRNA was completely abrogated, in contrast to that in the ENPP1-WT group (Fig. 6S). Furthermore, western blotting indicated that ENPP1-mut clearly impaired the TGF-β1-induced upregulation of ENPP1 (Fig. 6T and Fig. S6G). In addition, after ENPP1 was altered by a point mutation, ENPP1 abundance was significantly reduced in HSCs, as measured with an anti-WTAP antibody assay (Fig. 6U). The rescue experiments results showed that WTAP, YTHDF1 and ENPP1 regulate HILPDA (Fig. 6V and Fig. S6H). Collectively, these data revealed that WTAP elevated ENPP1 expression by promoting its translation in a m6A-YTHDF1-dependent manner.
Upregulated WTAP, YTHDF1 and ENPP1 correlate with lipid oxidation in human liver fibrosis tissue
We confirmed the causal roles of WTAP, YTHDF1, and ENPP1 in human liver fibrosis and their possible clinical significance (Fig. 7A). First, as expected, WTAP, YTHDF1, ENPP1, collagen I, α-SMA, and HILPDA levels were increased in human liver fibrosis samples (Fig. 7B–D and Fig. S7A–B). Moreover, an association study showed that ENPP1 was positively correlated with WTAP and YTHDF1 separately in human liver fibrosis samples (Fig. 7E–F), supporting the notion that WTAP and YTHDF1 play important regulatory roles. Additionally, we confirmed that the global m6A level was increased by performing a dot blot assay (Fig. 7G and Fig. S7C). Meanwhile, transmission electron microscopy (TEM) analysis found that the number of LDs was increased in patient compared with normal (Fig. 7H and Fig. S7E). These results indicate that WTAP or YTHDF1 participates in regulating ENPP1 expression via the m6A modification in human liver fibrosis progression. Second, we identified the baseline characteristics of patients with liver disease status (Fig. S7D). Compared with those of normal patient samples, samples from patients with liver fibrosis exhibited a significant increase in PPARα and PPARβ and MDA, ROS levels (Fig. 7C–D, I and Fig. S7A). In addition, the serum ALT and AST levels were significantly increased in the liver fibrosis patients (Fig. 7J). Furthermore, the fat composition and blood lipid levels in the serum from humans with liver fibrosis were significantly increased (Fig. 7K). Finally, compared with normal liver samples, liver fibrosis tissues exhibited more significant liver damage, lipid accumulation and collagen deposition, as detected by H&E, Masson’s trichrome, Sirius red, and oil red O staining (Fig. 7L and Fig. S7F). Collectively, these data indicate upregulation of WTAP, YTHDF1 and ENPP1 expression in human liver fibrosis tissue and confirm that WTAP is associated with increased lipid oxidation and an increased risk of fibrosis in human liver fibrosis patients.

Upregulated WTAP, YTHDF1 and ENPP1 Correlate with Lipid Oxidation in Human Liver Fibrosis Tissue. A The schema diagram for Fig. 7. B The expression of α-SMA, ENPP1, WTAP, and YTHDF1 were detected by immunohistochemistry (n = 6). C Western blotting was used to determine the protein expression of WTAP, YTHDF1, Collagen l, PPARα, PPARβ, α-SMA, ENPP1, and HILPDA (n = 6). D Real-time PCR was measured to determine the mRNA expression of WTAP, YTHDF1, Collagen l, PPARα, PPARβ, α-SMA, ENPP1, and HILPDA (n = 6). E Pearson correlation analysis of ENPP1 and WTAP. F Pearson correlation analysis of ENPP1 and YTHDF1. G Global m6A level of RNA extracted from human liver tissue was measured via m6A dot blot assays (n = 3). H TEM images of lipid droplets (LDs) from human liver tissue, and the number of LDs in 15.2 μm2 liver tissue was calculated (n = 6). I The MDA and ROS levels in human liver tissues is shown (n = 6). J Determination of aspartate aminotransferase (AST) level in serum (n = 6). K The serum cholesterol and triglycerides are shown. L Histopathological study was performed by H&E, Masson, Sirius Red, and Oil Red O staining (n = 6). Scale bars: 50 μm. All data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Normal group, two-tailed Student’s t-test
Discussion
An increasing number of studies have suggested excessive lipid oxidation in several diseases, such as liver diseases [37], cardiovascular disease [38], and cancers [39]. Nonetheless, we lack comprehensive understanding of the underlying mechanisms of the relationship between lipid oxidation and liver fibrosis. In this study, we demonstrate that WTAP increases lipid oxidation and HSC proliferation and migration by increasing ENPP1 m6A mark abundance in a YTHDF1-dependent manner (Fig. 8).

WTAP increases lipid oxidation and HSC proliferation and migration by increasing ENPP1 m.6A mark abundance in a YTHDF1-dependent manner (Created with BioRender.com)
Several of our findings support our hypothesis. First, WTAP and YTHDF1 expression was significantly elevated in the fibrotic liver tissue of both liver fibrosis humans and mice. The upregulation of WTAP was further associated with increased m6A marks on the RNA in these tissues, corresponding with previous research suggesting that WTAP expression and m6A modification may be necessary for liver fibrosis.
Second, m6A modification is key to control cellular processes such as proliferation, migration, and lipid oxidation. Indeed, the link between m6A-induced regulation and liver fibrosis was demonstrated. Based on our findings, knocking down WTAP or YTHDF1 contributes to a reduced level collagen and lipid deposition and suppresses lipid oxidation to ameliorate liver fibrosis. The findings suggest that WTAP and YTHDF1 are closely associated with lipid oxidation, cell proliferation and migration.
Third, the increasing evidence is demonstrating that ENPP1 regulates cell proliferation and migration, energy metabolism and lipid metabolism. Importantly, ENPP1 is regulated by epigenetic factors. Our results showed that after TGF-β1 stimulation, differentiated HSCs displayed increased levels of WTAP and YTHDF1 mRNA and protein levels, with a corresponding increase in ENPP1 expression. Through our bioinformatics analysis, mutation analysis, and MeRIP-qPCR assay, ENPP1 was confirmed to be modified by WTAP-related m6A modification. Knocking down WTAP resulted in a significant reduction in the m6A modification of ENPP1 mRNA, leading to a decrease in ENPP1 level. m6A readers consistently recognize m6A marks, resulting in different fates for the marked mRNA, including changes in RNA stability [40]. YTHDF1, a m6A reader, induces the targeted translation of a tumor suppressor protein that accelerates tumor progression, with its regulatory function catalyzed by the WTAP-induced m6A modification. We have provided evidence of direct binding of YTHDF1 and WTAP. Notably, knocking down YTHDF1 significantly counteracted the effects of increased WTAP on the expression of ENPP1. MeRIP-qPCR confirmed that the m6A modification of ENPP1 was specifically recognized by YTHDF1; following YTHDF1 knockdown, a significant reduction in the m6A modification of ENPP1, which inhibited ENPP1 expression, was identified. These results further support the theory that WTAP catalyzes the m6A modification of ENPP1 and the subsequent binding of YTHDF1 to ENPP1, increasing the expression of the latter.
Fourth, we demonstrated that the augmentation of lipid oxidation is necessary to promote HSC proliferation and migration, similar to findings reported in studies on pulmonary hypertension. In mechanistic findings, the Co-IP and pull-down assay confirmed that the downstream target of ENPP1 is HILPDA, which has been confirmed to regulate lipid oxidation [34], as evidenced by our results. Crucially, overexpression of ENPP1 led to a profound increased HILPDA, PPARα and PPARβ, α-SMA and collagen l expression in HSCs, promoting lipid oxidation, as well as HSC proliferation and migration, while knocking down ENPP1 led to the opposite results. Although the later experiment has the verification efficiency, the current technical means have low efficiency in liver knockout.
The findings of our current study clarified the mechanism underlying the role of excessive lipid oxidation in HSCs proliferation, migration, and fibrosis. This study is the first to document that WTAP boosts HILPDA-mediated lipid oxidation and HSCs proliferation and migration by increasing ENPP1 mRNA methylation via m6A in a YTHDF1-dependent manner. Overall, our research offers a better understanding of the roles of lipid oxidation and m6A regulation in liver fibrosis.
Data and materials availability
All data are available in the main text or the supplementary materials.
Data sharing statement
The original RNA-seq for this study have been deposited at the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/) with the accession numbers PRJNA922741 and PRJNA922743.
Abbreviations
- α-SMA:
-
Alpha-smooth muscle actin
- ABCC6:
-
Tissue nonspecific alkaline phosphatase
- ALKBH5:
-
AlkB Homolog 5
- ALT:
-
Alanine aminotransferase
- AMP:
-
Adenosine monophosphate
- ANK:
-
Ankylosis protein
- AST:
-
Aspartate aminotransferase
- ATP:
-
Adenosine Triphosphate
- BCA:
-
Bicinchoninic acid
- ENPP1:
-
Ecto-nucleotide pyrophosphatase/phosphodiesterase
- CCl4 :
-
Carbon tetrachloride
- CCK-8:
-
Cell counting kit-8
- Co-IP:
-
Co-immunoprecipitation
- Collagen I:
-
Type I of collagen
- DEPC:
-
Diethylpyrocarbonate
- DMEM:
-
Dulbecco’s modified eagle medium
- DMSO:
-
Dimethyl sulfoxide
- ECL:
-
Electrochemiluminescence
- ECM:
-
Extracellular matrix
- FBS:
-
Fetal bovine serum
- FFA:
-
Free fatty acid
- FTO:
-
Fat mass and obesity-associated protein
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- HILPDA:
-
Hypoxia-inducible lipid droplet-associated
- HRP:
-
Horseradish peroxidase
- HSC:
-
Hepatic stellate cell
- IGF2BP1/2/3:
-
Insulin-like growth factor 2 mRNA-binding proteins family 1/2/3
- LV3:
-
Lentivirus-3
- LDs:
-
Lipid droplets
- m6A:
-
N6-methyladenosine
- MDA:
-
Malonydialdehyde
- METTL14:
-
Methyltransferase like 14
- METTL3:
-
Methyltransferase like 3
- MTT:
-
Methyl Thiazolyl Tetrazolium
- NC:
-
Negative control
- OCR:
-
Oxygen consumption rate
- PBS:
-
Phosphate buffer saline
- PC-1:
-
Plasma cell membrane glycoprotein
- PMSF:
-
Phenylmethylsulfonyl fluoride
- PPARα:
-
Peroxisome proliferator activated receptor alpha
- PPARβ:
-
Peroxisome proliferator activated receptor beta
- PPi:
-
Pyrophosphate
- RIPA:
-
Radio immunoprecipitation assay lysis buffer
- ROS:
-
Reactive oxygen species
- TC:
-
Total cholesterol
- TG:
-
Triglyceride
- TGF-β1:
-
Transforming growth factor-β1
- TNAP:
-
Tissue nonspecific alkaline phosphatase
- WT:
-
Wild-type
- WTAP:
-
Wilms tumor 1 associated protein
- YTH:
-
YT521-B homology
- YTHDC1/2:
-
YTH domain-containing proteins 1/2
- YTHDF1/2/3:
-
YTH domain family proteins 1/2/3
References
Gao J, Wei B, de Assuncao TM, Liu Z, Hu X, Ibrahim S et al (2020) Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol 73(5):1144–1154
Zhang Z, Yuan Y, Hu L, Tang J, Meng Z, Dai L et al (2023) ANGPTL8 accelerates liver fibrosis mediated by HFD-induced inflammatory activity via LILRB2/ERK signaling pathways. J Adv Res 47:41–56
Zhong L, Zhao J, Huang L, Liu Y, Pang X, Zhan K et al (2023) Runx2 activates hepatic stellate cells to promote liver fibrosis via transcriptionally regulating Itgav expression. Clin Transl Med 13(7):e1316
Fondevila MF, Fernandez U, Heras V, Parracho T, Gonzalez-Rellan MJ, Novoa E et al (2022) Inhibition of carnitine palmitoyltransferase 1A in hepatic stellate cells protects against fibrosis. J Hepatol 77(1):15–28
Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ (2018) Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24(7):908–922
Hou W, Syn WK (2018) Role of metabolism in hepatic stellate cell activation and fibrogenesis. Front Cell Dev Biol 6:150
Wang F, Okamoto Y, Inoki I, Yoshioka K, Du W, Qi X et al (2010) Sphingosine-1-phosphate receptor-2 deficiency leads to inhibition of macrophage proinflammatory activities and atherosclerosis in apoE-deficient mice. J Clin Invest 120(11):3979–3995
Ramosaj M, Madsen S, Maillard V, Scandella V, Sudria-Lopez D, Yuizumi N et al (2021) Lipid droplet availability affects neural stem/progenitor cell metabolism and proliferation. Nat Commun 12(1):7362
Suh JM, Zeve D, McKay R, Seo J, Salo Z, Li R et al (2007) Adipose is a conserved dosage-sensitive antiobesity gene. Cell Metab 6(3):195–207
Bellanti F, Villani R, Facciorusso A, Vendemiale G, Serviddio G (2017) Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radic Biol Med 111:173–185
Houten SM, Wanders RJA, Ranea-Robles P (2020) Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim Biophys Acta Mol Basis Dis 1866(5):165720
Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW et al (2002) Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA 99(14):9445–9449
Jiang S, Zhang C, Lu Y, Yuan F (2022) The molecular mechanism research of cartilage calcification induced by osteoarthritis. Bioengineered 13(5):13082–13088
Li Y-Y (2012) ENPP1 K121Q polymorphism and type 2 diabetes mellitus in the Chinese population: a meta-analysis including 11,855 subjects. Metabolism 61(5):625–633
Ruiz-Fernández de Córdoba B, Moreno H, Valencia K, Perurena N, Ruedas P, Walle T et al (2022) Tumor ENPP1 (CD203a)/haptoglobin axis exploits myeloid-derived suppressor cells to promote post-radiotherapy local recurrence in breast cancer. Cancer Discov 12(5):1356–1377
Li S, Yokota T, Wang P, Ten Hoeve J, Ma F, Le TM et al (2022) Cardiomyocytes disrupt pyrimidine biosynthesis in nonmyocytes to regulate heart repair. J Clin Invest. https://doi.org/10.1172/JCI149711
Meyre D, Bouatia-Naji N, Tounian A, Samson C, Lecoeur C, Vatin V et al (2005) Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat Genet 37(8):863–867
Mărginean C, Mărginean CO, Iancu M, Meliț LE, Tripon F, Bănescu C (2019) MC4R and ENPP1 gene polymorphisms and their implication in maternal and neonatal risk for obesity. Sci Rep 9(1):10858
Wei J, Yu X, Yang L, Liu X, Gao B, Huang B et al (2022) FTO mediates LINE1 mA demethylation and chromatin regulation in mESCs and mouse development. Science 376(6596):968–973
Frye M, Harada BT, Behm M, He C (2018) RNA modifications modulate gene expression during development. Science 361(6409):1346–1349
Fu Y, Dominissini D, Rechavi G, He C (2014) Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet 15(5):293–306
Shi H, Wei J, He C (2019) Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 74(4):640–650
Chelmicki T, Roger E, Teissandier A, Dura M, Bonneville L, Rucli S et al (2021) m(6)A RNA methylation regulates the fate of endogenous retroviruses. Nature 591(7849):312–316
Zaccara S, Ries RJ, Jaffrey SR (2019) Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20(10):608–624
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D et al (2014) N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505(7481):117–120
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H et al (2015) N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161(6):1388–1399
Caillon L, Nieto V, Gehan P, Omrane M, Rodriguez N, Monticelli L et al (2020) Triacylglycerols sequester monotopic membrane proteins to lipid droplets. Nat Commun 11(1):3944
Stefan C, Jansen S, Bollen M (2005) NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem Sci 30(10):542–550
Lee S-Y, Müller CE (2017) Nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1) and its inhibitors. Medchemcomm 8(5):823–840
Valli-Jaakola K, Suviolahti E, Schalin-Jäntti C, Ripatti S, Silander K, Oksanen L et al (2008) Further evidence for the role of ENPP1 in obesity: association with morbid obesity in Finns. Obesity (Silver Spring) 16(9):2113–2119
Jenkinson CP, Coletta DK, Flechtner-Mors M, Hu SL, Fourcaudot MJ, Rodriguez LM et al (2008) Association of genetic variation in ENPP1 with obesity-related phenotypes. Obesity (Silver Spring) 16(7):1708–1713
Du M (2020) Targeting NFATc4 attenuates non-alcoholic steatohepatitis in mice. J Hepatol 73(6):1333–1346
Tsuchida T, Friedman SL (2017) Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 14(7):397–411
Mattijssen F, Georgiadi A, Andasarie T, Szalowska E, Zota A, Krones-Herzig A et al (2014) Hypoxia-inducible lipid droplet-associated (HILPDA) is a novel peroxisome proliferator-activated receptor (PPAR) target involved in hepatic triglyceride secretion. J Biol Chem 289(28):19279–19293
Li Y, Kang X, Zhou Z, Pan L, Chen H, Liang X et al (2022) The m6A methyltransferase Mettl3 deficiency attenuates hepatic stellate cell activation and liver fibrosis. Mol Ther 30(12):3714–3728
Dongiovanni P, Valenti L, Rametta R, Daly AK, Nobili V, Mozzi E et al (2010) Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 59(2):267–273
Li S, Tan HY, Wang N, Zhang ZJ, Lao L, Wong CW et al (2015) The role of oxidative stress and antioxidants in liver diseases. Int J Mol Sci 16(11):26087–26124
McIntyre TM, Hazen SL (2010) Lipid oxidation and cardiovascular disease: introduction to a review series. Circ Res 107(10):1167–1169
Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L et al (2017) Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer 16(1):76
Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF et al (2019) m(6)A enhances the phase separation potential of mRNA. Nature 571(7765):424–428
Funding
This project was supported by the National Natural Science Foundation of China (81600477), the National Natural Science Foundation Incubation Program of the Second Affiliated Hospital of Anhui Medical University (2019GMFY07), the Natural science research project of Anhui Province (2208085MH215), and the Natural science research project of Anhui Province (KJ2020A0181), Research Fund of Anhui Institute of Translational Medicine (2021zhyx-C64).
Author information
Authors and Affiliations
Contributions
Chao Lu and Hui Tao designed the study. Jing-Jing Yang reviewed the manuscript. Lin Jia and Juan Wang performed molecular docking and molecular dynamics simulation experiments. Feng Sun, Qi-Qi Dong and Juan Wang performed animal and cell experiments. Yang Yang, Jing-Jing Yang and Juan Wang analyzed and interpreted data. Hui Tao and Wei Hu helped revise the manuscript. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
Authors declare that they have no competing interests.
Ethical approval and consent to participate
Studies involving human samples have been approved by the Ethics Committee of Anhui Medical University.
Consent for publication
All authors agree to publish this manuscript. The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jing-Jing Yang is Lead contact.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, F., Wang, J., Yang, Y. et al. Epitranscriptomic regulation of lipid oxidation and liver fibrosis via ENPP1 mRNA m6A modification. Cell. Mol. Life Sci. 81, 387 (2024). https://doi.org/10.1007/s00018-024-05420-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05420-y