
Abstract
Key message
Association analysis, colocation study with previously reported QTL, and differential expression analyses allowed the identification of the consistent QTLs and main candidate genes controlling seed traits.
Abstract
Common beans show wide seed variations in shape, size, water uptake, and coat proportion. This study aimed to identify consistent genomic regions and candidate genes involved in the genetic control of seed traits by combining association and differential expression analyses. In total, 298 lines from the Spanish Diversity Panel were genotyped with 4,658 SNP and phenotyped for seven seed traits in three seasons. Thirty-eight significant SNP-trait associations were detected, which were grouped into 23 QTL genomic regions with 1,605 predicted genes. The positions of the five QTL regions associated with seed weight were consistent with previously reported QTL. HCPC analysis using the SNP that tagged these five QTL regions revealed three main clusters with significantly different seed weights. This analysis also separated groups that corresponded well with the two gene pools described: Andean and Mesoamerican. Expression analysis was performed on the seeds of the cultivar ‘Xana’ in three seed development stages, and 1,992 differentially expressed genes (DEGs) were detected, mainly when comparing the early and late seed development stages (1,934 DEGs). Overall, 91 DEGs related to cell growth, signaling pathways, and transcriptomic factors underlying these 23 QTL were identified. Twenty-two DEGs were located in the five QTL regions associated with seed weight, suggesting that they are the main set of candidate genes controlling this character. The results confirmed that seed weight is the sum of the effects of a complex network of loci, and contributed to the understanding of seed phenotype control.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Common bean (Phaseolus vulgaris L.) is a diploid and self-pollinated species that is considered the most important legume crop for direct human consumption (FAO 2022). Bean crops are present worldwide, and depending on their genotype, they can be consumed as immature pods (snap beans or green beans) or seeds after rehydration (dry beans). Bean seeds are a valuable source of proteins, carbohydrates, dietary fiber, vitamins, minerals, and bioactive molecules as phenolic components (Hayat et al. 2014). In addition, bean crops provide benefits to the soil, have low carbon and water footprints, and integrate well into sustainable agricultural models (Uebersax et al. 2022).
In the common bean, two main gene pools were found in the analysis of variation in morpho-agronomic traits, seed size, isoenzymes, seed proteins such as phaseolin, and different types of DNA markers in both wild and cultivated populations (Gepts et al. 1986; Singh et al. 1991; Blair et al. 2009): Andean (A) and Mesoamerican (MA). Each gene pool was domesticated independently in parallel domestication events. These two gene pools have also been observed in the European germplasm, although cultivars showing different levels of introgression between both gene pools have also been detected (Santalla et al. 2002; Campa et al. 2018).
Bean seeds exhibit extensive phenotypic variation (e.g., Campa et al. 2018), which can be described by considering a combination of seed shape, seed size, seed coat color, and color distribution. Seed shape is recorded as seed dimensions (length, width, thickness, and area) and the ratios among them, whereas seed size is usually recorded as 100 seed weight. The seed phenotype is an important trait in domestication and is related to consumer acceptability and its potential use as precooking and canned food. Seed size is a yield-related trait, along with the number of pods per plant and the number of seeds per pod (White and González 1990). Seed phenotype is also an important trait in snap bean varieties, which are preferred by white seed color, elongated seed shape, and smaller seed size (Silbernagel et al. 1991). Other relevant characteristics of bean seeds are water absorption and coat proportion because of their relationship with cocking response and consumer acceptability (Berry et al. 2020). Water uptake during soaking has been correlated with cooking time, and there is a relationship between seed size and speed of water absorption (Vidak et al. 2022). The seed coat plays a significant role in the hard-to-cook process of bean hardening before and during storage (de León et al. 1989). The seed coat represents approximately 10% of the seed weight and shows high mineral content (for example, Fe, Ca, and Mg) and antioxidant capacity, as well as many anti-nutrients that affect mineral bioavailability (Blair et al. 2013). The proportion of seed coat is negatively correlated with seed hardness; seeds with a higher percentage of coat tend to have hard shells (Escribano et al. 1997).
Some studies have addressed the inheritance of the traits involved in seed phenotypes. Seed size, shape, water uptake, and coat proportion exhibit quantitative inheritance with moderate-to-high heritability (Moghaddam et al. 2016; Berry et al. 2020). Many quantitative trait loci (QTL) involved in the genetic control of these seed traits have been described (González et al. 2016; Murube et al. 2020), although some QTL for seed size and shape have been collocated in different backgrounds and studies (Murube et al. 2020). These studies were conducted in different environments and used biparental populations, which revealed variation between the involved parents. Genome-wide association studies (GWAS), in which variation is captured among a defined population, have also reported genomic regions associated with seed size, shape, and quality traits (Blair et al. 2009; Schmutz et al. 2014; Cichy et al. 2015; Moghaddam et al. 2016; Giordani et al. 2022; Amongi et al. 2023). All of these genetic studies have described major, minor, and epistatic QTL for seed traits across all 11 common bean chromosomes (Arriagada et al. 2022). However, most of the QTL involved in the inheritance of seed phenotype traits have not been validated in different backgrounds (genotypes and environments) or are not well delimited in the bean genome, an important feature before being used in plant breeding.
Concerning the candidate genes controlling seed morphological traits, Schmutz et al. (2014) suggested 15 candidate genes associated with seed weight in a Mesoamerican panel consisting of 280 genotypes, three of which were highlighted by (Moghaddam et al. 2016) for seed weight in the same population (Phvul.006G069300, Phvul.008G013300, and Phvul.010G017600). Other GWAS using multi-environment trials for 4 decades confirmed the involvement of genes located on chromosomes Pv02 and Pv10, and found two additional genes for seed weight, Phvul.002G150600 and Phvul.003G039900 (MacQueen et al. 2020). Additionally, 13 candidate genes for seed shape and size were proposed by Giordani et al. (2022) based on a GWAS conducted on a Brazilian panel of 180 accessions. However, analysis of gene expression in the Negro Jamapa genotype during seed development showed that 10,453 genes modified their expression levels, with the majority (9,701) showing decreased expression (O’Rourke et al. 2014). Many of these genes are transcription factors, although the genes involved in starch biosynthesis (e.g., Phvul.001G082500.1) and sucrose synthesis are highly expressed in developing seeds. Also, the high expression of abscisic acid biosynthesis genes (e.g., Phvul.002G018700.1 and Phvul.005G031500.1) was observed in developing seeds, with expression decreasing as the seeds matured (O’Rourke et al. 2014). In other species, multiple pathways, including G-protein signaling, ubiquitin–proteasome pathway, mitogen-activated protein kinase (MAPK) pathway, BR signaling, transcriptional regulatory factors, and auxin signaling, are involved in the regulation of seed development (Li et al. 2019). In the ubiquitin–proteasome pathway, the ubiquitin receptor DA1 and E3 ubiquitin ligases EOD1/BB and DA2 physically interact to control seed size in Arabidopsis by regulating cell proliferation in integuments (Xia et al. 2013; Li et al. 2019). MAPK pathway consists of the different combinations of MKKK, MKK, and MAPK proteins which the plants use to regulate distinct biological processes, like plant growth, development, and defense response (Xu et al. 2018; Jiang et al. 2022), as well as could be important to regulate the grain size in rice (Xu et al. 2018; Tian et al. 2021; Wu et al. 2022). In addition, a LECTIN RECEPTOR KINASE, LecRK-VIII.2, has been reported to coordinate silique number, seed size, and seed number to determine seed yield in Arabidopsis by acting upstream of the MAPK gene (Xiao et al. 2021). All evidence indicates that the bean seed phenotype is regulated by a complex network of genes; however, there is no evidence that any loci have a higher effect on the expression of this trait.
The bean genome is available (e.g., Schmutz et al. 2014). Therefore, it is necessary to strengthen the connection between phenotype, genotype, and genome to identify annotated genes related to the expression of particular characters. The main goal of this study was to identify the consistent genomic regions and candidate genes involved in the genetic control of common bean seed size, shape, and quality traits, by combining association and differential expression studies. These analyses contribute to the understanding of the complex network of genes involved in seed phenotype control.
Materials and methods
Plant material
The Spanish Diversity Panel (SDP), with wide variation in seed phenotypes, was used in this study (Campa et al. 2018; https://zenodo.org/records/10263706). The SDP has homozygous lines derived from 220 landraces, most of which are from the updated Spanish Core Collection. SDP included 51 snap bean cultivars, 37 lines derived from traditional old cultivars, and well-known breeding lines. In all, 298 homozygous lines of SDP were used in this study.
The genotype ‘Xana’ was used for the analysis of differentially expressed genes (DEG) during seed development (Fig. 1). The cultivar ‘Xana’ was grouped in the Andean gene pool (Campa et al. 2018). This genotype has very large white seeds, determinate growth habits, and is classified as Fabada market class. ‘Xana’ is included in the SDP as line SDP308.

Pods of cv. Xana showing the three different growth stages of seeds used in the differential expression analysis
Genotyping
The genotyping-by-sequencing (GBS) method, as described by Elshire et al. (2011), was performed at BGI-Tech (Copenhagen, Denmark) using ApeKI restriction enzyme (Campa et al. 2018; Table S1). Sequencing reads from different genotypes were aligned using the bean genome G19833 v1.0, https://www.ncbi.nlm.nih.gov/genome/380). Genotypic data were filtered using software Tassel v5 (Bradbury et al. 2007). Lines with more than 50% missing data were removed, and in the remaining genotypes, SNP were filtered using the following criteria: (i) proportion of missing data < 10% and (ii) minor allele frequency (MAF) > 0.05. SNPs were named according to their physical position in the bean genome G19833 v1 (chromosome followed by the physical position in base pairs).
Phenotyping
SDP was grown in a greenhouse in Villaviciosa, Spain (43°29′01′ N, 5°26′11′ W; elevation 6.5 m) for three seasons (2016, 2017, and 2018). The experimental design was a randomized complete design in which there was a replicate with a single 1 m row plot, including 8–10 plants per line. Standard agronomic practices for tillage, irrigation, fertilization, and pest control were followed to ensure adequate plant growth and development. Phenotyping was conducted for seven seed traits: seed shape (area, length, width, and length-to-width ratio), seed weight, and seed quality after rehydration (coat proportion and water absorption) (Table 1). Seed dimensions were digitally recorded and analyzed using SmartGrain software (Tanabata et al. 2012). The trait 25-seed weight was manually recorded. Coat proportions and water absorption traits were recorded manually according to Castellanos et al. (1995).
Statistical analysis seed traits
All statistical analyses were performed using R software version 4.3.0 (R Core Team 2023). Outliers were removed before mean estimation using the mode ‘blup’ in the phenotype package (Piepho et al. 2008). Phenotypic variation in individual traits was visualized using the probability density distribution generated by the ggplot2 package (Wickham 2016). Spearman’s correlation coefficients between the seven traits were calculated using the R corrplot package (Wei and Simko 2021).
Hierarchical clustering on principal components (HCPC) analysis was performed to identify the main clusters in which bean lines could be grouped based on the genotypic data. This analysis was performed using R software with the packages FactoMineR and FactoExtra (Lê et al. 2008). Putative significant differences in seed traits among the clusters established from the HCPC analysis were investigated using ANOVA, followed by a post hoc Tukey test.
GWAS and haplotype block detection
Association analysis was performed using the FASTmrEMMA model (Wen et al. 2018), implemented in the mrMLM package (Zhang et al. 2020) of the R project (R Core Team 2023). Principal component analysis (PCA) and the kinship matrix obtained by the centered-IBS method were considered to account for multiple levels of relatedness within the lines included in the panel. A restricted maximum likelihood (REML) and critical LOD score of 3 were considered critical thresholds of significance for the identification of significant associations trait-SNP (QTN).
Haplotype blocks around QTNs were investigated using the Haploview 4.2 software (Barrett et al. 2005) with default software parameters and algorithms. The identified blocks were named using the prefix ‘Seed,’ chromosome number, and start position in Mb. To complete the characterization of the delimited blocks revealed by GWAS, the genomic positions in the bean genome were compared with those of QTL previously reported in common bean (González et al. 2016; Murube et al. 2020; Berry et al. 2020; Bassett et al. 2021; Giordani et al. 2022; Ugwuanyi et al. 2022; Arriagada et al. 2022). The list of annotated genes under the QTL was established based on those residing within the region delimited by the leftmost and rightmost flanking SNP in the defined haplotype blocks.
RNA-seq
Plants of the genotype ‘Xana’ were grown in pots of 7 litters during the summer of 2022 under greenhouse conditions. Seeds collected at three different growth stages were used to identify differentially expressed genes by RNA-seq (see Fig. 1): D1, the beginning of seed development (seeds with 0.8–1 cm length and green color); D2, intermediate development stage (1.5–2 cm length and green color); and D3, final development stage (2–2.5 cm length and green–white appearance). The experimental design included two biological replicates corresponding to two seed samples from different plants and pods.
The seeds were extracted from the pods, flash-frozen in liquid nitrogen, and stored at − 80 °C before RNA extraction. Total RNA from two biological replicates per growth stage was isolated using the RNeasy Plant Mini Kit, following the manufacturer’s instructions (Qiagen, Germany). RNA was quantified using fluorometric methods, and the quantity was determined using a 2100 Bioanalyzer Instrument (Agilent Technologies, UK). RNA libraries were prepared using the TruSeq Stranded mRNA Sample Preparation Kit (Illumina), and sequencing was performed on the Illumina platform (Macrogen, Korea).
The reads were mapped to the reference genome G19833 v1.0 (Schmutz et al. 2014) using HISAT2 splice-aware aligner (Kim et al. 2015). Expression profiles were represented as read counts and normalized by calculating the Trimmed Mean of M-values (TMM). Genes with expression levels less than 33% were removed. A principal component analysis (PCA) was performed to detect the possible sources of noise in the results. The NOISeqBIO function of the NOISeq package in R (Tarazona et al. 2015) was used to identify differentially expressed genes (DEG) through comparisons at different growth stages: D3 versus D1, D2 versus D1, and D3 versus D2 (see Fig. 1). DEGs were identified using q > 0.99. Specific and common DEGs among the three comparisons were detected and visualized using Venn diagrams constructed using the package ggVenn/ggplot2 (Wickham 2016).
Gene ontology enrichment analysis
Gene ontology (GO) annotation was done using the ‘Phaseolus vulgaris’ organism database in AnnotationHub resource (Morgan and Shepherd 2023) considering the three categories: biological process (BP), molecular function (MF), and cellular components (CC) to investigate the functional groups of the observed DEGs. Overrepresentation analysis (ORA) of candidate genes was performed using the R package clusterProfiler (Wu et al. 2021) based on the hypergeometric test (p value) and Benjamin–Hochberg method for controlling the false discovery rate (q value).
Approach to candidate genes
The identification of potential candidate genes was focused on considering the following criteria: (i) The QTL regions revealed by this study and colocalized with previously reported QTL for the same traits; (ii) DEGs located in the genomic regions delimited in this study; (iii) match with the candidate genes for domestication events previously proposed by Schmutz et al. (2014); (iv) match with the reported DEGs during seed development from the common bean expression atlas (O’Rourke et al. 2014).
Complete genomic sequences of the selected putative candidate genes were obtained from the Phytozome v12 database (Mesoamerican genomes UI111 v1.1, Labor Ovalle1.1 and 5 593v1.1). The sequences obtained were analyzed by alignment (BLASTn) with sequences of the reference genome G19833 v1.1, using default parameters. The polymorphisms identified were nucleotide variation, insertions, deletions, and the number of predicted genes.
Results
Genotyping of the SDP
Genotyping of 298 lines included in the Spanish Diversity Panel (SDP) was filtered considering homozygous sites, missing values (< 10%), and minor allele frequency (> 0.05), resulting in 4,658 SNPs distributed across the 11 bean chromosomes (Table S1; Figure S1). The number of SNPs per chromosome ranged between 298 (Pv10) and 619 (Pv02) SNPs.
Phenotypic variation
Table 2 shows the observed variation in the seven traits evaluated. Phenotypic evaluation of the SDP panel revealed a wide variation in all cases (see Figure S2). For instance, SL and SW ranged from 8.68 mm (observed in SDP262) to 22.41 mm (SDP308), and from 3.9 g (SDP009) to 27.59 g (SDP308), respectively (see Table S2). Similarly, WA showed wide variation in this panel, ranging from 39.3% (SDP083) to 63.3% (SDP106). All traits showed a good fit to a normal distribution (Kolmogorov–Smirnov test), except for LWR (Figure S2). Significant correlations were detected in most cases, except for SW, LWR, WA, SA, SW, and CP (Fig. 2). Correlations (r) ranged from − 0.07 to 0.94 and were positive in most cases. Overall, 11 of the 21 trait combinations showed moderately positive correlations (r > 0.4).

Spearman’s correlation coefficient between the evaluated seed traits. Black crosses represent not significantly associated (p > 0.05)
Genome-wide association study
Association analysis revealed 38 SNP-trait-significant associations (QTN), although 11 SNPs were associated with more than one character. The 27 unique SNPs were located on 10 chromosomes (all except for chromosome Pv06). The distribution and characteristics of QTN are presented in Table 3 (see Figure S3). For instance, the six QTN for SW were located on chromosomes Pv01, Pv03, Pv04, Pv07, Pv08, and Pv10, whereas those for SL were located on chromosomes Pv04, Pv07, Pv08 (two regions), Pv10, and Pv11.
Linkage disequilibrium (LD) analysis showed that the 27 SNP were organized in 23 genomic regions (blocks) ranging between 5 bp (Seed02_39.8) and 8.4 Mbp (Seed09_23.4) (Table 4). The QTL region Seed11_1.5 was tagged with an SNP. The positions of the genomic regions were compared to previously reported QTL regions associated with seed traits. Seven studies on biparental populations and diversity panels were considered, and 34 QTL associated with the genetic control of seed traits were found, revealing 13 overlapping regions on chromosomes Pv01, Pv02, Pv03, Pv04, Pv05, Pv08, Pv09, and Pv10 (Table 4). Interestingly, eight regions (most of which were associated with SW) were detected in more than one study: Seed01_50.7, Seed01_51.9, Seed02_28.1, Seed03_45.6, Seed08_55.3, Seed09_10.1, Seed09_23.4, Seed10_39.1, and Seed10_40.3 (see Fig. 3).

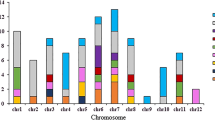
Chromosomal positions of genomic regions associated with seven seed traits identified by GWAS (green boxes). * Regions colocated with previously reported QTL for seed traits. Genes differentially expressed during seed development underlying those genomic regions revealed by RNA-seq analysis are shown at the right of each chromosome
Seed weight QTL dissection
Five QTL associated with SW have been consistently identified in other studies: Seed01_51.9, Seed03_45.6, Seed07_0.62, Seed08_55.3, and Seed10_39.1. A total of 102 SNPs that tagged these five QTL were selected, and hierarchical clustering on principal components (HCPC) analysis was performed with these SNPs revealing two main dimensions that explained 47.3% of the variance and led to the establishment of three main clusters with the SDP lines (Fig. 4; Table S2).

Biplots showing the results of hierarchical clustering on principal components using 102 SNP tagging the five consistent QTL regions associated with SW
Cluster 1 is formed by 85 lines, including typical Mesoamerican genotypes such as Sanilac (SDP290), Cornell49242 (SDP225), IVT7214 (SDP248), and AB136 (SDP005). The group had an average weight of 10.54 g per 25 seeds and contained 16 lines classified in the intermediate population and 1 classified in the Andean population (Campa et al. 2018).
Cluster 2 is formed by 43 lines, 38 of them classified as intermediate between both gene pool. The group had an average weight of 10.37 g per 25 seeds and did not differ significantly from cluster 1. Many of these lines are snap bean cultivars such as Fin de Bagnols (SDP232), Triomphe de Farcy (SDP293), Gloire De Saumur (SDP242), and Manteca de los Mercados (SDP247).
Cluster 3 consisted of 170 lines, of which 141 were classified in the Andean gene pool. This group included the typical Andean cultivars Tendergreen (SDP295), MDRK (SDP256), Perry Marrow (SDP276), and G19833 (SDP238), one of the bean genomes available (Schmutz et al. 2014). Cluster 3 included 28 lines classified as intermediate between both gene pools, and a line grouped in the Mesoamerican gene pool (SDP080). The mean seed weight was 14.06 g in this group, which differed significantly from that of clusters 1 and 2.
An analysis was also carried out for each QTL region to evaluate the effect of each region on seed weight. HCPC analysis showed three groups for each QTL region (Figure S4). The percentage of explained variance varied between 52.3% (Seed07_0.62) and 89% (Seed01_51.9). Significant differences in SW were detected between the groups of lines established for each QTL (Table 5). The QTL regions Seed01_51.9 and Seed10_39.1 showed the greatest differences between the two groups with extreme mean values (6.63 and 6.47 g, respectively) and significant differences in the means of the three groups.
Differentially expressed genes
The read counts for the expression levels at each seed development stage (D1, D2, and D3) and the replicates per locus are shown in Table S3. The reads from all samples were used for transcriptome assembly, and an average of 91.2% of the reads were mapped to the reference genome. The mapped reads were normalized by calculating the TMM-normalized reads, which revealed two components that explained 67% of the variation (Figure S5a). The obtained plot shows the grouped samples, except in the case of a sample derived from the D1 development stage (Figure S5b). Differentially expressed genes (DEGs) were identified by comparing three stages of seed development. (D2-D1; D3-D1; D3-D2; see Fig. 1). In total, 2,085 differentially expressed genes involving 1992 unique genes were identified in this analysis (Table S4). The majority of DEGs were up-regulated (1,888) (Fig. 5), and most of them were detected when the comparison was made between stages D3 and D1. Down-regulated DEGs were not detected in the comparison between the development stages, and up-regulated DEGs common among the three stages were also not detected.

Venn diagrams showing DEGs detected in each comparison (D2 vs. D1, D3 vs. D1, D3 vs. D2). a Up-regulated genes. b Down-regulated genes
GO enrichment analysis of DEGs
Gene ontology (GO) enrichment analysis was performed with 1,934 genes, corresponding to the DEGs in D3 compared to D1. The biological process (BP) and cellular component (CC) categories showed enrichment terms, whereas the molecular function (MF) category did not (Table S5). For BP, 19 GO terms were enriched, with more important terms related to intracellular cell establishment and functions, such as protein transport, protein and macromolecule localization, and compound metabolic processes (Figure S6a). For CC, 34 terms were enriched, most of which were implicated in functions related to endoplasmic reticulum or coated COPI-coated vesicles (Figure S6b).
Putative candidate genes for seed traits
The 23 QTL contained 1,605 annotated genes in the G19833 genome, and 91 genes were differentially expressed in RNA-seq analysis during seed development (Table S6). These 91 differentially expressed genes were located in only 16 QTL regions (Table S7). The number of DEGs per region ranged between one (Seed10_39.1) and 27 (Seed09_23.4). Six of these 91 genes were described in the gene expression atlas during seed development: PHAVU_003G249200g, PHAVU_009G175800g, PHAVU_009G177500g, PHAVU_009G189600g, PHAVU_009G202000g, and PHAVU_011G026700g (O’Rourke et al. 2014; Table S6). In addition, 15 of these 91 genes have been reported as relevant genes in the domestication process by Schmutz et al. (2014). Functional annotation of these 91 DEG revealed molecular functions and biological processes already reported in seed development (see Table S7).
The reference bean genome (G19833) contained 22 DEGs located in five consistent QTL regions associated with SW. The genomes of Labor Ovalle, UI111, and 5-593 had predicted homologous genes for each of these DEGs, except for PHAVU_010G123100g, which had two genes (Table S8). The respective sequences of these genes were aligned, and different types of variation were observed, compared to the reference genome: mismatches, insertions, deletions, and duplications. The genome of 5-593 presented less variation than that of Labor Ovalle and UI111 compared to the genome of G19833. However, the levels of variation were not the same for the 22 DEGs. Thirteen of the 15 predicted genes located on chromosome 3 showed a very low variation with G19833. In contracts, eight of the 22 genes showed high variation in the three genomes in the respective alignments with G19883: PHAVU_001G267600g, PHAVU_001G268900g, PHAVU_003G241500g, PHAVU_007G009100g, PHAVU_008G239600g, PHAVU_008G240600g, PHAVU_008G242800g, and PHAVU_010G123100g. For example, the three homologous genes of PHAVU_008G240600g had an insertion of 46 bp.
Discussion
Seed phenotype is a relevant characteristic of both dry and snap beans. While many studies have focused on seed coat color inheritance, there is limited research on the genetic control of seed size, seed shape, and seed quality traits, as well as limited data on the candidate genes involved. In this study, association and differential expression analyses were combined to identify consistent genomic regions and candidate genes controlling those seed traits. For this purpose, the seed variation for seven characters was evaluated in a diverse panel, the Spanish Diversity Panel (Campa et al. 2018). The results confirmed a continuous distribution of the seven traits, which is in agreement with the results of previous studies. In addition, most evaluated characters were positively and significantly correlated, suggesting the involvement of common loci in genetic control. The exception was WA, which did not show significant correlations with SA, SW, and CP (see Fig. 2). Seed weight, a yield-related trait, was positively correlated with shape traits (SA, SL, and SWI) and the seed quality trait CP.
GWAS and LD analysis identified 23 regions that were associated with seven seed traits. Eighteen regions were associated with seed size and shape traits on ten chromosomes while five regions were only associated with quality traits (two for CP and three for WA). Well-characterized QTL, consistent among backgrounds, well delimited and tagged, and with a significant contribution to the expression of the trait are desirable for use in breeding. A few studies have reported QTL for seed size and shape (González et al. 2016; Murube et al. 2020; Bassett et al. 2021; Giordani et al. 2022; Ugwuanyi et al. 2022; Arriagada et al. 2022) and seed quality traits (Berry et al. 2020; Bassett et al. 2021). The results allowed the identification of 13 consistent genomic regions associated with QTL that were previously reported in different genetic backgrounds and environments, indicating the relevance of such regions in the control of seed characters. The SNP s8_55946412 tagging the QTL region Seed08_55.3 was also associated with seed area (Giordani et al. 2022). Two of these QTL regions (Seed08_55.3 and Seed10_39.1) were associated with more than three traits, which agreed with the observed correlations. The QTL regions Seed05_3.2 and Seed10_32.5 were associated with water absorption on chromosomes Pv05 and Pv10. A QTL for WA was previously described by Berry et al. (2020) at the position of Pv10. QTL Seed09_23.4, tagged by 86 SNPs and associated LWR, was the largest QTL region identified. The size of QTL can depend on factors such as variation among genomes, density of markers, recombination of the region, or number of genes that contribute to the characteristics. The QTL region Seed09_23.4 was also involved in SW and SWI (Ugwuanyi et al. 2022; Arriagada et al. 2022). Interestingly, this delimited region annotated the genes Phvul.009G190100 and Phvul.009G202100, which had a significant effect on yield in a set of 42 common bean genotypes evaluated at two locations (Reinprecht et al. 2021). Moreover, seven of the 115 genes previously related to seed weight and domestication by Schmutz et al. (2014) were located in seven regions associated with seed size and shape traits.
Seed size plays a crucial role in the domestication of the common beans. The wild type has smaller seeds than domesticated ones, and domesticated Mesoamerican populations are smaller in size (Singh et al. 1991; Chacón‑Sánchez 2018). Choosing the 102 SNP underlaying the five consistent QTL regions for SW (Seed01_51.9, Seed03_45.6, Seed07_0.62, Seed08_55.3, and Seed10_39.1), three groups could be established from the HCPC analysis, two of which corresponded well with the Andean and Mesoamerican gene pools, and a third with intermediate material between both gene pools (Campa et al. 2018). The results indicated that the lines included in the Andean group had significantly heavier seeds than those included in the Mesoamerican group. In addition, the effects of each of the five QTL on SW were investigated. QTL dissection showed a significant effect on seed weight in the five consistent QTL regions. HCPC analysis based on the SNP genotype of each QTL region revealed three main groups, which significantly differed in SW. Remarkably, differences were established by the QTL regions Seed01_51.9 and Seed10_39.1, 6.63 g (Clusters 2 and 3) and 6.47 g (Clusters 1 and 2) between the two extreme groups (see Table 5). Both regions were found to be associated with seed weight in several QTL analyses (Bassett et al. 2021; Ugwuanyi et al. 2022; Arriagada et al. 2022); therefore, they are of special interest for breeding. Interestingly, the end of chromosome Pv01 is important for bean domestication and adaptation because it contains genes for determinacy (Fin locus) and sensitivity to photoperiod (Ppd locus; Weller et al. 2019).
The WA and CP of bean seeds are highly correlated with cooking time (Elia et al. 1997; Cichy et al. 2015). Seed cooking time in common beans is an important trait for consumer preference, with implications for nutrition, health, and the environment; the long cooking time of common beans is a major hindrance to its widespread consumption (Diaz et al. 2020; Haman et al. 2020). The Seed01_50.7 region, associated with CP in this study, was described as a QTL for seed content protein by Ugwuanyi et al. (2022), and the genes PHAVU_001G251000g and PHAVU_001G252200g were annotated at this position. The gene PHAVU_001G251000g encodes an inositol/myoinositol phosphatase synthase, a molecule precursor to a large variety of compounds and has been implicated in seed development in other legumes (Hegeman et al. 2001; Chiera and Grabau 2007). The gene PHAVU_001G252200g encodes an asparagine synthase, which is an amino acid positively correlated with protein concentration in soybean seeds (Pandurangan et al. 2012). The Seed10_39.1 region, which is also associated with CP, contains the gene PHAVU_010G123100g, which encodes a pectinesterase inhibitor involved in seed coat development in Arabidopsis. WA rate is also related to the emergence and germination of plants (Powell et al. 1986; Vidak et al. 2022), and some putative QTLs that control this trait have been reported (Cichy et al. 2015; Diaz et al. 2020; Berry et al. 2020; Bassett et al. 2021). The region Seed05_37.1, found in this work, associated with WA was reported by Berry et al. (2020); also, the region Seed10_32.5 was previously described as WA associated by Bassett et al. (2021).
Seed phenotype is the result of seed development. Two distinct phases during seed development have been described in legumes: The first phase involves cell division in the embryo, followed by a second phase, which regulates seed thickness via cell expansion and is highly influenced by the environment (Domoney et al. 2006). Changes in the transcriptomic profile of the ‘Xana’ cultivar during seed development were studied and 1,992 DEGs were detected. Many DEGs were found in the comparison between development stages D3 and D1. GO enrichment analysis revealed GO enrichment only in the categories of biological processes and cellular components. The enriched GO terms have important functions in plant development processes, such as those related to the Golgi apparatus, endoplasmic reticulum, and coated vesicles, which are essential for plant growth (Ahn et al. 2015). In all, 91 of 1,992 DEGs were located under the 23 QTL regions and 15 of them (PHAVU_001G251800g; PHAVU_001G252100g; PHAVU_001G267600g; PHAVU_009G072100g; PHAVU_003G241200g; PHAVU_003G241500g; PHAVU_003G248500g; PHAVU_003G249200g; PHAVU_007G009100g; PHAVU_009G058600g; PHAVU_009G072100g; PHAVU_009G07700g; PHAVU_009G185100g; PHAVU_009G189600g; PHAVU_009G197300g) were considered associated to domestication events by Schmutz et al. (2014). On the other hand, six DEG identified in this study located underlying some QTL regions were also identified as DEG in seed development by O’Rourke et al. (2014): PHAVU_003G249200g, PHAVU_009G175800g, PHAVU_009G177500g, PHAVU_009G189600g, PHAVU_009G202000g, and PHAVU_011G026700g. Identification of the same genes in different studies consolidates their involvement in controlling seed phenotypes. The annotated function of these 91 DEG in the QTL region agreed with functions already reported in seed development. For example, functions related to ubiquitin activities are known to determine seed size in Arabidopsis and rice (Li and Li 2014); the gene PHAVU_002G148100g encodes a ubiquitin hydrolase; the gene PHAVU_003G250800g encodes a ubiquitin receptor DA1, which in Arabidopsis thaliana controls seed and organ growth by restricting cell proliferation (Li and Li 2014); and the gene PHAVU_005G142800g encodes a ubiquitin ligase similar to the E3 ligase EOD1/BB identified as a negative regulator of seed size (Li et al. 2008). Across these 91 genes, we also found functions important in seed development in other species, such as AFP1-RELATED protein (PHAVU_009G202000g), expressed in embryos during the latest stages of seed maturation of Arabidopsis, and PPR protein (PHAVU_009G175100g), which play important roles in seed development in higher plants (Li et al. 2021). MAPKs control signaling cascades that play essential roles in plant growth, development, and defense response (Jiang et al. 2022), and are involved in regulating seed size in rice (Tian et al. 2021; Wu et al. 2022). The gene PHAVU_009G062400g, which encodes a MAP3K3/MEKK3, could be part of this network. Acting upstream of the MAPK gene in Arabidopsis can be found LecRK-VIII.2 that coordinates silique number, seed size, and seed number to determine seed yield (Xiao et al. 2021). A homologous of this gene is a DEG located under the QTL Seed08_55.3, PHAVU_008G239600g which encodes a LECTIN RECEPTOR KINASE VIII.1.
Four bean genomes with predicted genes are available: one from the Andean gene pool (G19833) and three closely related to the Mesoamerican gene pool (UI111, Labor Ovalle, and 5-593). Genotype G19833 has seeds larger than the other three genotypes. The 22 DEGs located in the consistent QTL associated with SW showed high variation when the respective sequences were aligned with the genes predicted in G19833, which may be a consequence of evolutionary differentiation and may contribute to phenotypic differentiation for SW. This variation was not homogeneous among the three MA genotypes, and 12 predicted genes in genotype 5-593 were very similar to those in G19833, suggesting that this observed variation should not be relevant to explicating the phenotypic variation between both gene pools for SW. In contrast, 10 genes were highly variable to those of G19833 in the three MA genotypes (see Table S8), suggesting that they could be relevant for phenotypic variation between both gene pools for seed weight. Interestingly three of them (PHAVU_001G267600g, PHAVU_001G268900g, PHAVU_010G123100g) were located in the QTLs that provided the greatest differences in SW: Seed01_51.9 and Seed10_39.1. From the sequences of these genes, functional markers tagging both genes and QTL regions can be developed and used in marker-assisted selection in breeding programs where SW is a trait considered. However, evidence suggests that SW is not the result of variation in a few loci; rather, it is the consequence of variation in many loci and their interactions with the environment in which the plants develop. Thus, these markers can help to enrich segregating populations in certain SW phenotypes.
Finally, the combination of GWAS and RNA-seq analyses helped elucidate QTL regions and candidate genes that control seed size, shape, and quality traits. GWAS revealed 23 QTL regions that were significantly associated with the evaluated traits, 13 of which were consistent with the regions reported in previous studies. These QTL regions contained 1,605 annotated genes in the G19833 bean genome, of which 91 genes were differentially expressed during seed development in the cultivar ‘Xana.’ DEGs were only found in 16 QTL, and 22 DEGs were located in five consistent QTL regions associated with SW. These regions and DEGs constitute a priority set for future genetic studies focused on SW control, their identification increases our knowledge of the genetic architecture of this trait, and a marker can be use as indirect selection tool, which is a relevant characteristic in many breeding programs.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials. An image of the seeds of each line included in the Spanish Diversity Panel (SDP) was deposited in the Zenodo repository (https://zenodo.org/records/10263706).
References
Ahn H-K, Kang YW, Lim HM et al (2015) Physiological functions of the COPI complex in higher plants. Mol Cells 38:866–875. https://doi.org/10.14348/molcells.2015.0115
Amongi W, Nkalubo ST, Ochwo-Ssemakula M et al (2023) Phenotype based clustering, and diversity of common bean genotypes in seed iron concentration and cooking time. PLoS ONE 18:e0284976. https://doi.org/10.1371/journal.pone.0284976
Arriagada O, Arévalo B, Cabeza RA et al (2022) Meta-QTL analysis for yield components in common bean (Phaseolus vulgaris L.). Plants. https://doi.org/10.3390/plants12010117
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265. https://doi.org/10.1093/bioinformatics/bth457
Bassett A, Katuuramu DN, Song Q, Cichy K (2021) QTL mapping of seed quality traits including cooking time, flavor, and texture in a yellow dry bean (Phaseolus vulgaris L.) population. Front Plant Sci 12:670284. https://doi.org/10.3389/fpls.2021.670284
Berry M, Izquierdo P, Jeffery H et al (2020) QTL analysis of cooking time and quality traits in dry bean (Phaseolus vulgaris L.). Theor Appl Genet 133:2291–2305. https://doi.org/10.1007/s00122-020-03598-w
Blair MW, Díaz LM, Buendía HF, Duque MC (2009) Genetic diversity, seed size associations and population structure of a core collection of common beans (Phaseolus vulgaris L.). Theor Appl Genet 119:955–972. https://doi.org/10.1007/s00122-009-1064-8
Blair MW, Izquierdo P, Astudillo C, Grusak MA (2013) A legume biofortification quandary: variability and genetic control of seed coat micronutrient accumulation in common beans. Front Plant Sci 4:275. https://doi.org/10.3389/fpls.2013.00275
Bradbury PJ, Zhang Z, Kroon DE et al (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Campa A, Murube E, Ferreira JJ (2018) Genetic diversity, population structure, and linkage disequilibrium in a spanish common bean diversity panel revealed through genotyping-by-sequencing. Genes. https://doi.org/10.3390/genes9110518
Castellanos JZ, Guzmán-Maldonado H, Acosta-Gallegos JA, Kelly JD (1995) Effects of hardshell character on cooking time of common beans grown in the semiarid highlands of Mexico. J Sci Food Agric 69:437–443. https://doi.org/10.1002/jsfa.2740690406
Chacón-Sánchez MI (2018) The domestication syndrome in phaseolus crop plants: a review of two key domestication traits. In: Pontarotti P (ed) Origin and evolution of biodiversity. Springer, Cham, pp 37–59
Chiera J, Grabau E (2007) Localization of myo-inositol phosphate synthase (GmMIPS-1) during the early stages of soybean seed development. J Exp Bot 58:2261–2268. https://doi.org/10.1093/jxb/erm101
Cichy KA, Wiesinger JA, Mendoza FA (2015) Genetic diversity and genome-wide association analysis of cooking time in dry bean (Phaseolus vulgaris L.). Theor Appl Genet 128:1555–1567. https://doi.org/10.1007/s00122-015-2531-z
de León LF, Bressani R, Elías LG (1989) Effect of the seed coat on the hardening of common beans (Phaseolus vulgaris). Arch Latinoam Nutr 39:405–418
Diaz S, Ariza-Suarez D, Ramdeen R et al (2020) Genetic architecture and genomic prediction of cooking time in common bean (Phaseolus vulgaris L.). Front Plant Sci 11:622213. https://doi.org/10.3389/fpls.2020.622213
Domoney C, Duc G, Ellis THN et al (2006) Genetic and genomic analysis of legume flowers and seeds. Curr Opin Plant Biol 9:133–141. https://doi.org/10.1016/j.pbi.2006.01.014
Elia FM, Hosfield GL, Kelly JD, Uebersax MA (1997) Genetic analysis and interrelationships between traits for cooking time, water absorption, and protein and tannin content of andean dry beans. J Am Soc Hortic Sci 122:512–518. https://doi.org/10.21273/JASHS.122.4.512
Elshire RJ, Glaubitz JC, Sun Q et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. https://doi.org/10.1371/journal.pone.0019379
Escribano MR, Santalla M, de Ron AM (1997) Genetic diversity in pod and seed quality traits of common bean populations from northwestern Spain. Euphytica 93:71–81
FAO (2022) Statistical database. Food and Agriculture Organization of the United Nations. https://www.fao.org/faostat/en/#home. Accessed 2 Nov 2022
Gepts P, Osborn TC, Rashka K, Bliss FA (1986) Phaseolin-protein variability in wild forms and landraces of the common bean (Phaseolus vulgaris): evidence for multiple centers of domestication. Econ Bot 40:451–468. https://doi.org/10.1007/BF02859659
Giordani W, Gama HC, Chiorato AF et al (2022) Genome-wide association studies dissect the genetic architecture of seed shape and size in common bean. G3 (bethesda). https://doi.org/10.1093/g3journal/jkac048
González AM, Yuste-Lisbona FJ, Saburido S et al (2016) Major contribution of flowering time and vegetative growth to plant production in common bean as deduced from a comparative genetic mapping. Front Plant Sci 7:1940. https://doi.org/10.3389/fpls.2016.01940
Haman Y, Abatchoua A, Tchiagam N, Ibrahima (2020) Genetic analysis of cooking time in five common bean (Phaseolus vulgaris L.) cultivars grown in Cameroon. EAS J Biotechnol Genet 2:58–66
Hayat I, Ahmad A, Masud T et al (2014) Nutritional and health perspectives of beans (Phaseolus vulgaris L.): an overview. Crit Rev Food Sci Nutr 54:580–592. https://doi.org/10.1080/10408398.2011.596639
Hegeman CE, Good LL, Grabau EA (2001) Expression of D-myo-inositol-3-phosphate synthase in soybean. Implications for phytic acid biosynthesis. Plant Physiol 125:1941–1948. https://doi.org/10.1104/pp.125.4.1941
Jiang M, Zhang Y, Li P et al (2022) Mitogen-activated protein kinase and substrate identification in plant growth and development. Int J Mol Sci. https://doi.org/10.3390/ijms23052744
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360. https://doi.org/10.1038/nmeth.3317
Lê S, Josse J, Husson F (2008) FactoMineR: an R package for multivariate analysis. J Stat Softw. https://doi.org/10.18637/jss.v025.i01
Li N, Li Y (2014) Ubiquitin-mediated control of seed size in plants. Front Plant Sci 5:332. https://doi.org/10.3389/fpls.2014.00332
Li Y, Zheng L, Corke F et al (2008) Control of final seed and organ size by the DA1 gene family in Arabidopsis thaliana. Genes Dev 22:1331–1336. https://doi.org/10.1101/gad.463608
Li N, Xu R, Li Y (2019) Molecular networks of seed size control in plants. Annu Rev Plant Biol 70:435–463. https://doi.org/10.1146/annurev-arplant-050718-095851
Li X, Sun M, Liu S et al (2021) Functions of PPR proteins in plant growth and development. Int J Mol Sci. https://doi.org/10.3390/ijms222011274
MacQueen AH, White JW, Lee R et al (2020) Genetic associations in four decades of multienvironment trials reveal agronomic trait evolution in common bean. Genetics 215:267–284. https://doi.org/10.1534/genetics.120.303038
Moghaddam SM, Mamidi S, Osorno JM et al (2016) Genome-wide association study identifies candidate loci underlying agronomic traits in a middle American diversity panel of common bean. Plant Genome. https://doi.org/10.3835/plantgenome2016.02.0012
Morgan M, Shepherd L (2023) AnnotationHub: client to access AnnotationHub resources. Version 3.8.0. R package
Murube E, Campa A, Song Q et al (2020) Toward validation of QTLs associated with pod and seed size in common bean using two nested recombinant inbred line populations. Mol Breed 40:7. https://doi.org/10.1007/s11032-019-1085-1
O’Rourke JA, Iniguez LP, Fu F et al (2014) An RNA-Seq based gene expression atlas of the common bean. BMC Genom 15:866. https://doi.org/10.1186/1471-2164-15-866
Pandurangan S, Pajak A, Molnar SJ et al (2012) Relationship between asparagine metabolism and protein concentration in soybean seed. J Exp Bot 63:3173–3184. https://doi.org/10.1093/jxb/ers039
Piepho HP, Möhring J, Melchinger AE, Büchse A (2008) BLUP for phenotypic selection in plant breeding and variety testing. Euphytica 161:209–228. https://doi.org/10.1007/s10681-007-9449-8
Powell AA, Oliveira MDA, Matthews S (1986) The role of imbibition damage in determining the vigour of white and coloured seed lots of dwarf French beans ( Phaseolus vulgaris). J Exp Bot 37:716–722. https://doi.org/10.1093/jxb/37.5.716
Reinprecht Y, Qi Y, Shahmir F et al (2021) Yield and antiyield genes in common bean (Phaseolus vulgaris L.). Legume Sci. https://doi.org/10.1002/leg3.91
R Core Team (2023) R: a language and environment for statistical computing. Version 4.3.1. R Foundation for Statistical Computing
Santalla M, Rodiño P, De Ron M (2002) Allozyme evidence supporting southwestern Europe as a secondary center of genetic diversity for the common bean. Theor Appl Genet 104:934–944. https://doi.org/10.1007/s00122-001-0844-6
Schmutz J, McClean PE, Mamidi S et al (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46:707–713. https://doi.org/10.1038/ng.3008
Silbernagel MJ, Janssen W, Davis JHC, Montes de Oca G (1991) Snap bean production in the tropics: implications for genetic improvement. In: van Schoonhoven A, Voysest O (eds) Common beans: research for crop improvement. Centro Internacional de Agricultura Tropical (CIAT), Cali, CO, pp 835–862
Singh SP, Gepts P, Debouck DG (1991) Races of common bean (Phaseolus vulgaris, Fabaceae). Econ Bot 45:379–396. https://doi.org/10.1007/BF02887079
Tanabata T, Shibaya T, Hori K et al (2012) SmartGrain: high-throughput phenotyping software for measuring seed shape through image analysis. Plant Physiol 160:1871–1880. https://doi.org/10.1104/pp.112.205120
Tarazona S, Furió-Tarí P, Turrà D et al (2015) Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res 43:e140. https://doi.org/10.1093/nar/gkv711
Tian X, He M, Mei E et al (2021) WRKY53 integrates classic brassinosteroid signaling and the mitogen-activated protein kinase pathway to regulate rice architecture and seed size. Plant Cell 33:2753–2775. https://doi.org/10.1093/plcell/koab137
Uebersax MA, Cichy KA, Gomez FE (et al2022) Dry beans (Phaseolus vulgaris L.) as a vital component of sustainable agriculture and food security—a review. Legume Sci. https://doi.org/10.1002/leg3.155
Ugwuanyi S, Udengwu OS, Snowdon RJ, Obermeier C (2022) Novel candidate loci for morpho-agronomic and seed quality traits detected by targeted genotyping-by-sequencing in common bean. Front Plant Sci 13:1014282. https://doi.org/10.3389/fpls.2022.1014282
Vidak M, Lazarević B, Javornik T et al (2022) Seed water absorption, germination, emergence and seedling phenotypic characterization of the common bean landraces differing in seed size and color. Seeds 1:324–339. https://doi.org/10.3390/seeds1040027
Wei T, Simko V (2021) R package “corrplot”: visualization of a correlation matrix. Version 0.92. R core
Weller JL, Vander Schoor JK, Perez-Wright EC et al (2019) Parallel origins of photoperiod adaptation following dual domestications of common bean. J Exp Bot 70:1209–1219. https://doi.org/10.1093/jxb/ery455
Wen Y-J, Zhang H, Ni Y-L et al (2018) Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief Bioinform 19:700–712. https://doi.org/10.1093/bib/bbw145
White JW, González A (1990) Characterization of the negative association between seed yield and seed size among genotypes of common bean. Field Crops Res 23:159–175. https://doi.org/10.1016/0378-4290(90)90052-D
Wickham H (2016) ggplot2: elegant graphics for data analysis (use R!), 2nd edn. Springer, Cham
Wu T, Hu E, Xu S et al (2021) clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (camb) 2:100141. https://doi.org/10.1016/j.xinn.2021.100141
Wu X, Cai X, Zhang B et al (2022) ERECTA regulates seed size independently of its intracellular domain via MAPK-DA1-UBP15 signaling. Plant Cell 34:3773–3789. https://doi.org/10.1093/plcell/koac194
Xia T, Li N, Dumenil J et al (2013) The ubiquitin receptor DA1 interacts with the E3 ubiquitin ligase DA2 to regulate seed and organ size in Arabidopsis. Plant Cell 25:3347–3359. https://doi.org/10.1105/tpc.113.115063
Xiao W, Hu S, Zou X et al (2021) Lectin receptor-like kinase LecRK-VIII.2 is a missing link in MAPK signaling-mediated yield control. Plant Physiol 187:303–320. https://doi.org/10.1093/plphys/kiab241
Xu R, Duan P, Yu H et al (2018) Control of grain size and weight by the OsMKKK10-OsMKK4-OsMAPK6 signaling pathway in rice. Mol Plant 11:860–873. https://doi.org/10.1016/j.molp.2018.04.004
Zhang Y-W, Tamba CL, Wen Y-J et al (2020) mrMLM v4.0.2: an R platform for multi-locus genome-wide association studies. Genom Proteom Bioinform 18:481–487. https://doi.org/10.1016/j.gpb.2020.06.006
Acknowledgements
The authors thank M Bueno, F Díaz, and JA Poladura (SERIDA) for the technical assistance provided in this study.
Funding
This work was supported by Grant PID2021-123919OB-100 financed by MCIN/AEI/https://doi.org/10.13039/501100011033 and FEDER funds ‘Una manera de hacer Europa.’ MJ is supported by the Grant PRE2019-091249 funded by MCIN/AEI/https://doi.org/10.13039/501100011033 and, as appropriate, by ‘ESF Investing in your future’ or by ‘European Union Next GenerationEU/PRTR.’
Author information
Authors and Affiliations
Contributions
MJ performed part of the association, transcriptomic, and statistical analyses, and contributed to writing the manuscript. AC performed genotyping and contributed to transcriptomic analysis, review, and editing. CG-F performed part of the phenotyping and contributed to association analysis, review, and editing. JJF conceived the work, prepared the material, performed the statistical analysis, reviewed and edited the manuscript, and contributed to writing the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Communicated by Elena Bitocchi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jurado, M., García-Fernández, C., Campa, A. et al. Identification of consistent QTL and candidate genes associated with seed traits in common bean by combining GWAS and RNA-Seq. Theor Appl Genet 137, 143 (2024). https://doi.org/10.1007/s00122-024-04638-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-024-04638-5