
Abstract
Aims/hypothesis
Understanding the molecular networks controlling ectopic lipid deposition and insulin responsiveness in skeletal muscle is essential for developing new strategies to treat type 2 diabetes. We recently identified serine/threonine protein kinase 25 (STK25) as a critical regulator of liver steatosis, hepatic lipid metabolism and whole body glucose and insulin homeostasis. Here, we assessed the role of STK25 in control of ectopic fat storage and insulin responsiveness in skeletal muscle.
Methods
Skeletal muscle morphology was studied by histological examination, exercise performance and insulin sensitivity were assessed by treadmill running and euglycaemic–hyperinsulinaemic clamp, respectively, and muscle lipid metabolism was analysed by ex vivo assays in Stk25 transgenic and wild-type mice fed a high-fat diet. Lipid accumulation and mitochondrial function were also studied in rodent myoblasts overexpressing STK25. Global quantitative phosphoproteomics was performed in skeletal muscle of Stk25 transgenic and wild-type mice fed a high-fat diet to identify potential downstream mediators of STK25 action.
Results
We found that overexpression of STK25 in transgenic mice fed a high-fat diet increases intramyocellular lipid accumulation, impairs skeletal muscle mitochondrial function and sarcomeric ultrastructure, and induces perimysial and endomysial fibrosis, thereby reducing endurance exercise capacity and muscle insulin sensitivity. Furthermore, we observed enhanced lipid accumulation and impaired mitochondrial function in rodent myoblasts overexpressing STK25, demonstrating an autonomous action for STK25 within cells. Global phosphoproteomic analysis revealed alterations in the total abundance and phosphorylation status of different target proteins located predominantly to mitochondria and sarcomeric contractile elements in Stk25 transgenic vs wild-type muscle, respectively, providing a possible molecular mechanism for the observed phenotype.
Conclusions/interpretation
STK25 emerges as a new regulator of the complex interplay between lipid storage, mitochondrial energetics and insulin action in skeletal muscle, highlighting the potential of STK25 antagonists for type 2 diabetes treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes is strongly associated with ectopic lipid deposition within non-adipose tissue, which actively contributes to the development of insulin resistance [1–3]. Skeletal muscle plays an important role in the pathophysiology of type 2 diabetes accounting for more than 70% of whole body glucose use [4]. Thus, approaches that can suppress ectopic lipid deposition within the skeletal muscle, and increase the responsiveness of muscle to insulin, offer a potential for the development of new therapies for diabetes.
In the search for novel targets that contribute to the pathogenesis of insulin resistance and type 2 diabetes, we recently described serine/threonine protein kinase 25 (STK25; also referred to as YSK1 or SOK1), a member of the sterile 20 (STE20) kinase superfamily [5], as a central regulator of ectopic lipid accumulation, and whole body glucose and insulin homeostasis [6–11]. STK25 is broadly expressed in mouse, rat and human tissues [10–13]. Previous studies have shown that STK25, present in the Golgi complex, regulates cell polarisation and migration in different cell types [14–17]. It is also reported that in cells subjected to extreme stresses, STK25 enters the nucleus and induces cell death [18, 19]. We found that in mice fed on a high-fat diet, transgenic mice overexpressing STK25 display hyperinsulinaemia and impaired whole body glucose and insulin homeostasis compared with wild-type littermates [10]. Reciprocally, our studies showed that, compared with wild-type littermates, Stk25 knockout mice are protected against systemic glucose intolerance and insulin resistance induced by a high-fat diet [6]. Notably, we found that in both mouse and human liver cells, STK25 is localised on the surface of cytosolic lipid droplets [7, 8]. We observed that increased STK25 abundance in mouse liver and human hepatocytes enhances fat deposition in intrahepatocellular lipid droplets by suppressing lipolytic activity and thereby fatty acid release for β-oxidation and triacylglycerol secretion; the reciprocal effect was seen with STK25 knockdown [7, 8]. Furthermore, we found a significant positive correlation between STK25 mRNA expression and fat content in human liver biopsies [8, 9]. Moreover, STK25 mRNA levels were higher in the skeletal muscle of individuals with type 2 diabetes than in healthy volunteers [11].
On the basis of our previous findings, which reveal a central role of STK25 in control of hepatic fat deposition and systemic insulin sensitivity [6–10], we hypothesised that STK25 is also involved in regulation of ectopic lipid storage and insulin responsiveness in skeletal muscle. Here, we provide the first evidence to support the key cell-specific role of STK25 in the excessive accumulation of intramyocellular lipids in the context of chronic exposure to dietary lipids, which is associated with suppressed mitochondrial function, reduced endurance exercise capacity and exacerbated insulin resistance in skeletal muscle.
Methods
Animals
The generation of Stk25 transgenic mice, where mouse Stk25 expression in the targeting construct is controlled by chicken β-actin promoter, and the subsequent breeding with C57BL/6NCrl mice (Charles River, Sulzfeld, Germany), have been described previously [10]. From the age of 6 weeks, male transgenic mice and wild-type littermates were fed a pelleted high-fat diet (45% kilocalories from fat; D12451; Research Diets, New Brunswick, NJ, USA). At the age of 24 weeks, the mice were killed after 4 h of food withdrawal. Gastrocnemius skeletal muscle samples were collected for histological analysis (see Histology and immunofluorescence) or snap frozen in liquid nitrogen and stored at −80°C for analysis of protein and gene expression (see ESM Fig. 1 for a schematic overview). All animal experiments were performed after approval from the local Ethics Committee for Animal Studies at the Administrative Court of Appeals in Gothenburg, Sweden, and followed appropriate guidelines.
Histology and immunofluorescence
Gastrocnemius muscle samples were embedded in optimal cutting temperature mounting medium (Histolab Products, Gothenburg, Sweden) and frozen in liquid nitrogen followed by cryosectioning and staining with haematoxylin-eosin (H-E; Histolab Products), Nile Red (Sigma-Aldrich, St Louis, MO, USA) or MitoTracker Red (Thermo Fisher Scientific, Waltham, MA, USA). Enzymatic stainings were performed as previously described [20]. For immunofluorescence, sections were incubated with primary antibodies followed by incubation with secondary antibodies (see ESM Table 1). Gastrocnemius muscle samples were also fixed with 4% formaldehyde in phosphate buffer (Histolab Products), embedded in paraffin, sectioned and stained with Picrosirius Red (Histolab Products) or Periodic acid–Schiff (PAS; Sigma-Aldrich). Ultrastructural analysis of gastrocnemius muscle was performed by transmission electron microscopy (TEM; LEO 912AB; Omega; Carl Zeiss NTS, Oberkochen, Germany) as previously described [21]. Gastrocnemius muscle homogenates were analysed using a Hydroxyproline Colorimetric Assay Kit (Sigma-Aldrich) and a Triglyceride Calorimetric Assay Kit (Biovision, Mountain View, CA, USA).
Cell culture and transient overexpression
L6 myoblasts (Rattus norvegicus, American Type Culture Collection, Manassas, VA, USA) were maintained as described [11] and transfected with pFLAG-Stk25 (cytomegalovirus promoter; GeneCopoeia, Rockville, MD, USA) or an empty control plasmid using Lipofectamine 2000 (Invitrogen, San Diego, CA, USA). Cells were incubated with 50 μmol/l oleic acid for 24 h and stained with Oil Red O or MitoTracker Red as described [8]. Palmitate oxidation was measured as previously described [11]. Cells have been demonstrated to be free of mycoplasma infection by use of the MycoAlert Mycoplasma Detection kit (Lonza, Basel, Switzerland).
Western blot and quantitative real-time PCR
Western blotting was performed as previously described [7] in gastrocnemius muscle of Stk25 transgenic and wild-type mice and/or transfected L6 myoblasts using anti-STK25, anti-adipose triacylglycerol lipase (ATGL), anti-hormone-sensitive lipase (HSL), anti-PLIN2 and anti-actin primary antibodies (see ESM Table 1). The anti-STK25 antibody has been validated by using Stk25-knockout mice [6]. Quantitative real-time PCR was performed in gastrocnemius muscle of Stk25 transgenic and wild-type mice and transfected L6 myoblasts using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) as described [10] (see ESM Table 2).
Ex vivo measurement of lipid metabolism
The oxidation rate of palmitate was measured in quadriceps muscle homogenates as described previously [7]. Oleic acid uptake and triacylglycerol synthesis from [14C]-oleic acid were measured in isolated extensor digitorum longus (EDL) and soleus muscles as described [22, 23].
In vivo assessment of exercise performance and insulin sensitivity
Endurance exercise was assessed by treadmill running until the mice reached fatigue (see ESM Methods). Insulin sensitivity was measured by euglycaemic–hyperinsulinaemic clamp as previously described [10] using an insulin infusion rate of 7 mU/min/kg (see ESM Methods).
Liquid chromatography mass spectrometry analysis
Gastrocnemius muscle samples were heat stabilised and prepared, including tryptic digestion, chemical labelling for relative quantification, enrichment of phosphopeptides and pre-fractionation, as described in ESM Methods. Liquid chromatography mass spectrometry (LC-MS)/MS of these combined tandem mass tagged labelled samples was performed on an Orbitrap Fusion Tribrid MS interfaced to an Easy-nLC 1000 (Thermo Fisher Scientific).
Statistical analysis
Statistical significance between groups was calculated with an unpaired two-tailed Student’s t test or by two-way ANOVA followed by Tukey post hoc test. A p < 0.05 was considered statistically significant.
Results
Evidence for muscle damage in Stk25 transgenic mice, with fibre type composition remaining unaltered
We previously showed that STK25 is highly expressed in mouse, rat and human skeletal muscle [11]. Here we found that endogenous STK25 protein was 2.1 ± 0.3-fold higher in white portions of the gastrocnemius muscle (predominantly type IIb fibres) compared with red portions (predominantly type IIa fibres) in wild-type mice (ESM Fig. 2a). Nonetheless, the endogenous STK25 protein was detected by immunofluorescence in all fibre types (ESM Fig. 2b). STK25 was markedly increased in both red and white portions of Stk25 transgenic vs wild-type muscle (ESM Fig. 2a, b).
Fibre type proportions, as identified by myosin heavy chain (MHC) immunochemistry, were unchanged in Stk25 transgenic compared with wild-type muscle (Fig. 1a); type IIb fibres of transgenic muscle appeared slightly hypertrophic, while no shift in diameter was found in any other fibre type (Fig. 1b–e). Examination of H-E-stained sections revealed evidence of muscle damage in Stk25 transgenic mice as indicated by intracellular inclusions, small angular degenerating fibres, focal necrosis, infiltration of mononuclear inflammatory cells and adipocyte replacement; these features were rarely seen in wild-type muscle (Fig. 1f). TEM showed the presence of well-defined myofibrils and sarcomeric pattern with organised A- and I-bands, Z-discs and M-lines in wild-type muscle (Fig. 1g). In contrast, Stk25 transgenic muscle fibres displayed disorganised myofibril architecture and irregularities of sarcomere elements (Fig. 1g).

Morphology and fibre composition in gastrocnemius muscle of Stk25 transgenic and wild-type mice. (a) Representative immunofluorescence images double-stained with antibodies for MHC type I, IIa, IIx or IIb (green) and laminin (red); nuclei stained with DAPI (blue). White bars, wild-type mice fed high-fat diet; black bars, transgenic mice fed high-fat diet. Scale bar, 50 μm. Histogram shows quantification of fibre types (b–e) Fibre size distribution. White circles, wild-type mice fed high-fat diet; black circles, transgenic mice fed high-fat diet. (f) Representative images stained with H-E showing the presence of intracellular inclusions (black single arrowhead), small angular degenerating fibres (open single arrowheads), focal necrosis (arrows), infiltration of mononuclear inflammatory cells (open double arrowheads) and adipocyte replacement (black double arrowheads). Scale bar, 50 μm. (g) Representative electron micrographs from longitudinal sections showing disrupted sarcomere organisation (open arrowhead). Scale bar, 1 μm. (h) Representative images stained with Picrosirius Red (scale bar, 50 μm) and quantification of Picrosirius Red staining. (i) Hydroxyproline content in the muscle extract. In (a–e, h–i) data are mean ± SEM from 5–8 mice per genotype. *p < 0.05; **p < 0.01. HFD, high-fat diet; TG, transgenic; WT, wild-type
Picrosirius Red staining for collagen revealed that perimysial fibrosis was increased in Stk25 transgenic vs wild-type muscle and endomysial fibrosis, not present in wild-type muscle, was readily observed in transgenic muscle (Fig. 1h). Consistently, hydroxylated proline, a main constituent of collagens, was 1.6 ± 0.2-fold higher in Stk25 transgenic muscle homogenates (Fig. 1i).
STK25 overexpression in mice augments fat storage and impairs mitochondrial function in skeletal muscle
We measured the intramyocellular lipid accumulation in the fibre types with the highest lipid content (type I, IIa and IIx) in gastrocnemius muscle of Stk25 transgenic and wild-type mice fed a high-fat diet. The relative area of these muscle fibres staining with lipophilic dye Nile Red was approximately 1.3- to 1.6-fold higher in Stk25 transgenic mice (Fig. 2a). In contrast, the relative area of these muscle fibres staining with MitoTracker Red, a fluorescent dye that specifically accumulates within respiring mitochondria, was approximately 1.2-fold lower in Stk25 transgenic mice compared with wild-type mice (Fig. 2b). Consistent with a reduced MitoTracker Red signal, histochemical stainings revealed that Stk25 transgenic muscle exhibited repressed pigment retention in enzymatic activity assays for NADH, succinate dehydrogenase (SDH) and cytochrome c oxidase (COX), commonly used as markers of oxidative metabolism (Fig. 2c).

Lipid storage, mitochondrial function and glycogen content in gastrocnemius muscle of Stk25 transgenic and wild-type mice. (a) Representative immunofluorescence images stained with lipophilic Nile Red dye (green). Scale bar, 15 μm. Histograms show quantification of Nile Red staining. (b) Representative immunofluorescence images stained with MitoTracker Red (yellow). Scale bar, 15 μm. Histograms show quantification of MitoTracker staining. (c) Representative histochemical staining for NADH, SDH and COX activities. Scale bar, 50 μm. (d) Representative electron micrographs from cross-sections showing lipid droplets (red arrows) and mitochondria, which are swollen (red arrowheads), display disarrayed cristae and reduced electron density of the matrix (open arrowheads), and internal vesicles (green arrowhead). Scale bar, 2 μm. (e) Representative images stained with PAS (scale bar, 15 μm) and quantification of PAS staining. In (a, b and e) data are mean ± SEM from six mice per genotype. *p < 0.05; **p < 0.01. HFD, high-fat diet; TG, transgenic; WT, wild-type
TEM demonstrated that a significant fraction of mitochondria in Stk25 transgenic, but not wild-type, muscle were structurally distorted and appeared swollen, and displayed disarrayed cristae, reduced electron density of the matrix and/or internal vesicles (Fig. 2d, ESM Fig. 3). TEM also revealed the presence of large lipid droplets in transgenic but not wild-type muscle (Fig. 2d). Mitochondrial DNA (mtDNA) copy number, and the expression of key transcriptional activators mediating mitochondrial biogenesis—PGC1α (Ppargc1a), PGC1β (Ppargc1b) and nuclear respiratory factor 1 (Nrf1)—were similar in Stk25 transgenic vs wild-type muscle (ESM Figs 4, 5).
Our previous studies have shown that the glycogen content was similar in gastrocnemius muscle homogenates of Stk25 transgenic and wild-type mice [10]. Consistent with these findings, PAS staining in glycogen-rich type IIa and IIx fibres was similar between the genotypes (Fig. 2e).
Overexpression of STK25 induces lipid accumulation and represses mitochondrial function in myoblasts
The global overexpression of STK25 in transgenic mice does not allow us to address whether the impact of STK25 on skeletal muscle lipid metabolism is direct or secondary to the action of STK25 in tissues other than muscle. To study the cell-specific role of STK25 in muscle cells, we transiently transfected the rat myoblast cell line L6 with the STK25 expression plasmid or an empty control plasmid (mock; Fig. 3a). Subsequent to transfection, the cells were exposed to oleic acid, which efficiently induces steatosis in vitro and thereby mimics the dietary challenge in mice. To analyse lipid deposition, cells were stained with Oil Red O. STK25 overexpression increased lipid accumulation 1.8 ± 0.1-fold based on quantification of Oil Red O staining (Fig. 3b). Notably, staining with MitoTracker Red was 2.8 ± 0.05-fold lower in cells overexpressing STK25 suggesting an impairment of mitochondrial function (Fig. 3c). Consistent with the reduced MitoTracker Red signal, there was a tendency (p = 0.09) for a lower rate of β-oxidation in cells overexpressing STK25 (ESM Fig. 6a).

Analysis of lipid accumulation and mitochondrial function in rodent myoblasts overexpressing STK25. L6 cells were transiently transfected with Stk25 expression plasmid or vector control (mock) and incubated with oleic acid for 24 h. (a) Representative western blot with anti-STK25 antibodies; actin was used as a loading control (endogenous STK25 48 kDa, FLAG-tagged STK25 51 kDa). (b) Representative cell images stained with Oil Red O (scale bar, 50 μm) and quantification of Oil Red O staining. (c) Representative cell images stained with MitoTracker Red (scale bar, 20 μm) and quantification of MitoTracker staining. Data are mean ± SEM from 3–5 wells. **p < 0.01
As with the results obtained in Stk25 transgenic vs wild-type muscle (see above), mtDNA copy number and the expression of key transcriptional activators mediating mitochondrial biogenesis (PGC1α, PGC1β and Nrf1) were largely similar in cells transfected with the STK25 expression plasmid and vector control (ESM Fig. 6b, c). Our previous studies have shown that in skeletal muscle of Stk25 transgenic mice fed a high-fat diet, the mRNA expression of carnitine palmitoyltransferase 1 (CPT1), the rate-limiting enzyme in fatty acid oxidation in mitochondria, was significantly reduced [10]. Surprisingly, Cpt1 mRNA levels were markedly increased in STK25-overexpressing L6 cells (ESM Fig. 6c); while the significance of this observation remains unclear, in view of other data suggesting repressed mitochondrial function.
Overexpression of STK25 in mice reduces skeletal muscle β-oxidation, while lipid uptake and synthesis remain unaltered
We further characterised lipid metabolism in skeletal muscle of Stk25 transgenic and wild-type mice fed a high-fat diet ex vivo. Consistent with the reduced MitoTracker Red staining and repressed activity of oxidative metabolism markers, the muscle homogenates of Stk25 transgenic mice displayed a lower β-oxidation rate (75% of the capacity of wild-type; Fig. 4a). Notably, no significant difference in the level of acylcarnitines, the by-products of incomplete fatty acid oxidation and markers of skeletal muscle insulin resistance, was observed in muscle homogenates comparing the genotypes (ESM Fig. 7).

Assessment of lipid metabolism in Stk25 transgenic and wild-type mice. (a) β-oxidation in quadriceps muscle extract. (b) Oleic acid uptake and (c) triacylglycerol synthesis in isolated EDL and soleus muscle. White bars, wild-type mice fed high-fat diet; black bars, transgenic mice fed high-fat diet. Data are mean ± SEM from 16–18 (a) or 11–12 (b, c) mice per genotype. *p < 0.05. HFD, high-fat diet; OA, oleic acid; TAG, triacylglycerol; TG, transgenic; WT, wild-type
In addition, we incubated isolated skeletal muscle from Stk25 transgenic and wild-type mice with radiolabelled oleate and observed that cell-associated radioactivity was not significantly altered between the genotypes, indicating similar fatty acid influx (Fig. 4b). Furthermore, skeletal muscles isolated from both genotypes displayed similar incorporation of oleate into triacylglycerol (Fig. 4c).
Stk25 transgenic mice have reduced running performance
To assess the impact of the disorganised myofibril architecture and fibrosis observed in Stk25 transgenic muscle on endurance exercise capacity, we next compared the responses of Stk25 transgenic and wild-type mice fed a high-fat diet to treadmill running. A markedly reduced exercise performance was found in Stk25 transgenic mice compared with wild-type littermates both in terms of running time and distance to fatigue (Fig. 5a, b). The post-exercise concentration of plasma lactate, a by-product of anaerobic glycolysis, was comparable between the genotypes (Fig. 5c).

Assessment of endurance running capacity and insulin sensitivity in Stk25 transgenic and wild-type mice. (a) Time and (b) distance to fatigue during a treadmill run. (c) Plasma lactate levels measured directly after exercise using an l-Lactate Assay Kit (Abcam, Cambridge, UK). (d) Insulin-stimulated glucose uptake in individual tissues determined during a euglycaemic–hyperinsulinaemic clamp. White bars, wild-type mice fed high-fat diet; black bars, transgenic mice fed high-fat diet. Data are mean ± SEM from 8–9 mice per genotype. *p < 0.05. BAT, brown adipose tissue; eWAT, epididymal white adipose tissue; Gas, gastrocnemius muscle; HFD, high-fat diet; Quad, quadriceps muscle; Sol, soleus muscle; sWAT, subcutaneous white adipose tissue; TG, transgenic; WT, wild-type
Stk25 transgenic mice display reduced in vivo insulin-stimulated glucose uptake in skeletal muscle
The observation that STK25 overexpression increased intramyocellular lipid levels and favoured fat storage rather than oxidation in the muscle prompted us to investigate whether these changes would affect skeletal muscle insulin sensitivity. To this end, euglycaemic–hyperinsulinaemic clamp experiments with a glucose tracer were performed in Stk25 transgenic and wild-type mice fed a high-fat diet. Insulin infusion significantly increased plasma insulin concentration at the end of the clamp for both genotypes (ESM Fig. 8a). There was no difference in glucose infusion rate or blood glucose level at steady state of the clamp comparing the two genotypes (ESM Fig. 8b). Insulin-stimulated glucose uptake was 1.5 ± 0.1-fold and 1.6 ± 0.1-fold lower in gastrocnemius and quadriceps muscles of Stk25 transgenic mice, respectively, with a similar tendency seen in EDL and soleus muscles (Fig. 5d).
Global phosphoproteomic analysis of skeletal muscle in Stk25 transgenic and wild-type mice
To identify potential downstream mediators of STK25 action in skeletal muscle metabolism in an unbiased manner, global quantitative phosphoproteomic analysis was performed in the gastrocnemius muscle of Stk25 transgenic and wild-type mice fed a high-fat diet with multiplexed isobaric labelling and phosphopeptide enrichment coupled to tandem MS (MS/MS) (Fig. 6a). The analysis also included quantification of non-phosphorylated peptides to determine possible changes in total protein abundance (Fig. 6a). A total of 4918 distinct peptides and 129 phosphopeptides, corresponding to 943 and 80 unique proteins, respectively, were quantified (Fig. 6b, c). We observed that the abundance of 39 peptides representing 28 unique proteins was differentially regulated, by a factor of 1.15-fold or more, in Stk25 transgenic muscle relative to the wild-type controls (Fig. 6b, Table 1). Furthermore, we found that the phosphorylation level of 26 peptides derived from 21 proteins was differentially regulated, by a factor of 1.15-fold or more, comparing the genotypes (Fig. 6c, Table 2). No difference in the abundance of the total proteins corresponding to the altered phosphorylation sites was observed, indicating that changes in phosphopeptide levels were a direct result of alterations in their phosphorylation status.

Global quantitative phosphoproteomic analysis in gastrocnemius muscle of Stk25 transgenic and wild-type mice. (a) Experimental design. Summary of the quantified (blue bars) and differentially regulated (red bars) (b) proteome and (c) phosphoproteome. The subcellular location of the (d) differentially expressed and (e) differentially phosphorylated proteins was annotated according to the Gene Ontology database [50], NCBI OMIM and/or PubMed. Up- or downregulation is indicated by green and red arrows, respectively
Assessment of the known cellular localisation of the differentially expressed proteins revealed a marked enrichment of the targets located to mitochondria (Fig. 6d, Table 1). Among these candidates, carbonyl reductase 4 (CBR4), an enzyme in the mitochondrial fatty acid synthesis pathway [24], reticulon 4 interacting protein 1 (RTN4IP1), enoyl coenzyme A hydratase, short chain, 1 (ECHS1), and NADH dehydrogenase (ubiquinone) 1 beta subcomplex 7 (NDUFB7)—all involved in mitochondrial oxidation [25–27]—as well as two mitochondrial aldehyde dehydrogenases (ALDH2 and ALDH6A1 [28, 29]) were upregulated in transgenic muscle. In addition, mitochondrial fission factor (MFF) and microtubule-associated protein 1S (MAP1S), which control mitochondrial morphology by regulating dynamic fission and fusion process [30, 31], were differentially expressed comparing the genotypes. Apart from these candidates with relatively well-known functions, several additional proteins with poorly described roles in mitochondria were differentially expressed in transgenic muscle, including von Willebrand factor A domain-containing protein 8 (VWA8) and glyoxalase domain-containing 4 (GLOD4).
Analysis of proteins containing differentially regulated phosphosites, on the other hand, revealed an enrichment of the targets with known location in the Z-disk, where actin-containing thin filaments from neighbouring sarcomeres overlap cross-linked by alpha-actinin, and in the myosin-containing A-band of the sarcomere (Fig. 6e, Table 2). These myofibril-associated candidates included proteins with well-characterised roles in regulation of contractile properties of the skeletal muscle, such as titin (TTN), myosin regulatory light chain (MYLPF), creatine kinase (CKM), tropomyosin 1, alpha (TPM1), actinin alpha 3 (ACTN3) and myosin heavy chain II beta (MYH4 or MHCIIB), as well as proteins with less defined roles, such as myozenin 1 and 2 (MYOZ1 and 2) and LIM domain binding 3 (LDB3). Interestingly, the phosphorylation of the three enzymes in the glycolytic pathway—aldolase A, fructose-bisphosphate (ALDOA), enolase 3, beta muscle (ENO3) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)—was altered in Stk25 transgenic compared with wild-type muscle. Of the 26 differentially regulated phosphosites, 24 were annotated in PhosphoSitePlus [32]. However, the functional implication of the phosphorylation at these sites has only been described for TPM1, where phosphorylation at a single Ser-283 residue has been associated with increased Ca2+ activated ATPase activity [33] and regulation of Ca2+ sensitivity [34].
Notably, our previous studies have shown that overexpression of STK25 repressed lipolysis in mouse and human hepatocytes, which probably contributed to the increased lipid storage observed in these cells [7, 8]. The global phosphoproteomic analysis by MS/MS did not detect any lipases above the level of quantification in the skeletal muscle. However, we also measured the skeletal muscle mRNA and protein levels of two lipases (ATGL and HSL) by quantitative real-time PCR and western blot, respectively, and observed no differences between the genotypes (ESM Fig. 9). Recently, the levels of the lipid droplet binding proteins Perilipin 2 (PLIN2 also known as adipose differentiation-related protein [ADRP]) and Perilipin 3 (PLIN3), as well as the activity of AMP-activated protein kinase (AMPK), have been implicated in the regulation of lipid content in muscle cells [35, 36]. PLIN3 was quantified by MS/MS analysis without any alterations in total protein abundance observed between the genotypes, while PLIN2 and AMPK were below the level of quantification. However, we found that the skeletal muscle mRNA of Plin2 and Plin3, and protein abundance of PLIN2 measured by western blot, were not changed when comparing the genotypes (ESM Fig. 9). Furthermore, our previous studies have shown that AMPK activity, measured by AMPKα Thr172 phosphorylation, was similar when comparing the skeletal muscle of Stk25 transgenic and wild-type mice [10].
Discussion
Insulin resistance in skeletal muscle is a major and early feature in the pathogenesis of type 2 diabetes [37]. In this study, we provide compelling evidence that overexpression of protein kinase STK25 in transgenic mice challenged with a high-fat diet increases myocellular lipid storage and impairs skeletal muscle mitochondrial function as well as sarcomeric ultrastructure, thereby reducing endurance exercise capacity and repressing muscle insulin responsiveness (Fig. 7). Furthermore, we found enhanced lipid accumulation and impaired mitochondrial function in rodent myoblasts overexpressing STK25, demonstrating an autonomous action of STK25 in muscle cells. These results are consistent with our previous studies showing that Stk25 transgenic mice fed a high-fat diet develop aggravated whole body glucose intolerance and insulin resistance compared with wild-type littermates [10].

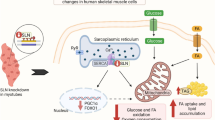
Schematic illustration of metabolic responses at the whole body level as well as in skeletal muscle fibres of Stk25 transgenic mice vs wild-type littermates. Up- or downregulation is indicated by green and red arrows, respectively
The main finding of this study is the substantially increased presence of ultrastructural abnormalities of both subsarcolemmal and intermyofibrillar mitochondria in Stk25 transgenic skeletal muscle, which was associated with reduced MitoTracker Red staining, lower abundance of oxidative metabolism markers and suppressed β-oxidation. Previous studies using TEM have demonstrated alterations in mitochondrial morphology in skeletal muscle of humans and rodent models with insulin resistance and type 2 diabetes [38–40]. In addition, reduced in vivo mitochondrial oxidative capacity has been reported in the skeletal muscle of patients with type 2 diabetes [41–43]. Furthermore, studies in healthy elderly individuals and insulin-resistant offspring of parents with type 2 diabetes have demonstrated that repressed mitochondrial function may predispose these individuals to intramyocellular lipid accumulation and insulin resistance [44]. Moreover, coordinated reduction in the expression of genes involved in mitochondrial function and oxidative phosphorylation was reported in skeletal muscle from type 2 diabetes patients, but also from individuals with insulin resistance but normal glucose tolerance [45, 46]. Based on this evidence, impaired mitochondrial function observed in Stk25 transgenic muscle probably contributed to increased myocellular lipid storage and development of skeletal muscle insulin resistance.
We further explored the underlying mechanisms involved in the regulation of the mitochondrial structure and activity by STK25. We failed to detect any accumulation of STK25 protein in skeletal muscle mitochondria (ESM Fig. 2c), and therefore, STK25 is likely to control mitochondrial function indirectly through the regulation of abundance and/or phosphorylation pattern of, as yet, unknown mitochondrial target proteins. Interestingly, global phosphoproteomic analysis revealed that the two key regulators of mitochondrial fusion and fission process were differentially expressed, with MFF protein levels decreased and MIP1S protein levels increased, in Stk25 transgenic vs wild-type muscle. MFF knockdown in mammalian cells resulted in an interconnected tubular network of mitochondria, whereas MFF overexpression stimulated mitochondrial fission [31, 47]. Accumulation of MAP1S, on the other hand, was associated with irreversible aggregation of mitochondria [48]. Based on this evidence, we speculate that altered protein abundance of MFF and MAP1S may be one underlying mechanism for impaired mitochondrial morphology in Stk25 transgenic muscle.
We also found disorganised myofibril architecture and pronounced fibrosis in Stk25 transgenic muscle, which probably contributed to reduced endurance exercise capacity. This was not observed in wild-type muscle. Notably, global phosphoproteomic analysis revealed a marked enrichment of the differentially phosphorylated proteins located in the myofibril, in particular to the Z-disk and A-band of the sarcomere, in Stk25 transgenic vs wild-type muscle. However, at present it is not known whether these changes have contributed to the disrupted sarcomere organisation in transgenic muscle. Interestingly, recent evidence shows that perturbations in skeletal muscle sarcomere ultrastructure in individuals with heart failure and type 2 diabetes can be improved by stimulating mitochondrial function [49], which supports a close functional connection between mitochondrial alterations and muscle damage, and suggests that mitochondrial abnormalities observed in Stk25 transgenic muscle may have contributed to altered myofibril architecture.
The metabolic changes in the skeletal muscle of Stk25 transgenic mice are consistent with our previous observations in mouse and human liver cells, where STK25 overexpression in conditions of excess dietary fuels increased intrahepatocellular lipid deposition while repressing mitochondrial function and insulin sensitivity [7–9]. Taken together, these findings suggest that STK25 may regulate the shift in the metabolic balance from lipid use to lipid storage in several tissues prone to diabetic damage, contributing to the pathogenesis of whole body insulin resistance and type 2 diabetes.
In this study, we characterised the skeletal muscle phenotype of Stk25 transgenic and wild-type mice challenged with a high-fat diet in order to mimic conditions in high-risk individuals. Notably, our previous investigations have shown that liver lipid deposition and whole body insulin sensitivity were not significantly altered comparing Stk25 transgenic vs wild-type mice fed regular chow [9, 10]. Moreover, we found that skeletal muscle triacylglycerol content and fibrosis were not increased in Stk25 transgenic mice fed regular chow compared with corresponding wild-type littermates (ESM Fig. 10). These data suggest that overexpression of STK25 leads to significant metabolic alterations in mice only after a dietary challenge.
To date, it remains unknown whether any physiological situations occur in which the STK25 protein abundance is enhanced to a level observed in the Stk25 transgenic muscle, which is a limitation of the animal model. Notably, our findings in Stk25 transgenic muscle are reciprocal to our previous observations of increased β-oxidation and improved insulin action in STK25-deficient myoblasts [11] as well as reduced lipid accumulation and enhanced insulin sensitivity in the skeletal muscle of Stk25 -/- mice fed a high-fat diet [6], reinforcing the physiological validity of the results.
In light of the current epidemic of type 2 diabetes, research aimed at understanding the interplay between intramyocellular lipid storage, mitochondrial energetics, and insulin action in skeletal muscle is of utmost importance for the development of new therapeutic strategies. This study provides several layers of evidence that STK25 is an interesting new mediator in the interconnected metabolic network controlling skeletal muscle insulin sensitivity, and that the development of STK25 antagonists for therapeutic applications in type 2 diabetes and related metabolic disease is warranted.
Abbreviations
- COX:
-
Cytochrome c oxidase
- EDL:
-
Extensor digitorum longus
- H-E:
-
Haematoxylin-eosin
- HSL:
-
Hormone-sensitive lipase
- LC:
-
Liquid chromatography
- MAP1S:
-
Microtubule-associated protein 1S
- MFF:
-
Mitochondrial fission factor
- MHC:
-
Myosin heavy chain
- PAS:
-
Periodic acid–Schiff
- SDH:
-
Succinate dehydrogenase
- STK25:
-
Serine/threonine protein kinase 25
- TEM:
-
Transmission electron microscopy
References
Anstee QM, Targher G, Day CP (2013) Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 10:330–344
Perseghin G, Scifo P, De Cobelli F et al (1999) Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 48:1600–1606
Goodpaster BH, Wolf D (2004) Skeletal muscle lipid accumulation in obesity, insulin resistance, and type 2 diabetes. Pediatr Diabetes 5:219–226
DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP (1981) The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30:1000–1007
Thompson BJ, Sahai E (2015) MST kinases in development and disease. J Cell Biol 210:871–882
Amrutkar M, Cansby E, Chursa U et al (2015) Genetic disruption of protein kinase STK25 ameliorates metabolic defects in a diet-induced type 2 diabetes model. Diabetes 64:2791–2804
Amrutkar M, Cansby E, Nunez-Duran E et al (2015) Protein kinase STK25 regulates hepatic lipid partitioning and progression of liver steatosis and NASH. FASEB J 29:1564–1576
Amrutkar M, Kern M, Nunez-Duran E et al (2016) Protein kinase STK25 controls lipid partitioning in hepatocytes and correlates with liver fat content in humans. Diabetologia 59:341–353
Amrutkar M, Chursa U, Kern M et al (2016) STK25 is a critical determinant in nonalcoholic steatohepatitis. FASEB J 30:3628–3643
Cansby E, Amrutkar M, Manneras Holm L et al (2013) Increased expression of STK25 leads to impaired glucose utilization and insulin sensitivity in mice challenged with a high-fat diet. FASEB J 27:3660–3671
Nerstedt A, Cansby E, Andersson CX et al (2012) Serine/threonine protein kinase 25 (STK25): a novel negative regulator of lipid and glucose metabolism in rodent and human skeletal muscle. Diabetologia 55:1797–1807
Osada S, Izawa M, Saito R et al (1997) YSK1, a novel mammalian protein kinase structurally related to Ste20 and SPS1, but is not involved in the known MAPK pathways. Oncogene 14:2047–2057
Pombo CM, Bonventre JV, Molnar A, Kyriakis J, Force T (1996) Activation of a human Ste20-like kinase by oxidant stress defines a novel stress response pathway. EMBO J 15:4537–4546
Preisinger C, Short B, De Corte V et al (2004) YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14-3-3zeta. J Cell Biol 164:1009–1020
Voss K, Stahl S, Schleider E et al (2007) CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8:249–256
Matsuki T, Matthews RT, Cooper JA et al (2010) Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell 143:826–836
Fidalgo M, Fraile M, Pires A, Force T, Pombo C, Zalvide J (2010) CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation. J Cell Sci 123:1274–1284
Nogueira E, Fidalgo M, Molnar A et al (2008) SOK1 translocates from the Golgi to the nucleus upon chemical anoxia and induces apoptotic cell death. J Biol Chem 283:16,248–16,258
Zhou J, Shao Z, Kerkela R et al (2009) Serine 58 of 14-3-3zeta is a molecular switch regulating ASK1 and oxidant stress-induced cell death. Mol Cell Biol 29:4167–4176
De Paepe B, De Bleecker JL, Van Coster R (2009) Histochemical methods for the diagnosis of mitochondrial diseases. Curr Protoc Hum Genet 63:19.12.1–19.12.19
Anderberg C, Cunha SI, Zhai Z et al (2013) Deficiency for endoglin in tumor vasculature weakens the endothelial barrier to metastatic dissemination. J Exp Med 210:563–579
Wu Q, Ortegon AM, Tsang B, Doege H, Feingold KR, Stahl A (2006) FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol Cell Biol 26:3455–3467
Bruce CR, Brolin C, Turner N et al (2007) Overexpression of carnitine palmitoyltransferase I in skeletal muscle in vivo increases fatty acid oxidation and reduces triacylglycerol esterification. Am J Physiol Endocrinol Metab 292:E1231–E1237
Chen Z, Kastaniotis AJ, Miinalainen IJ, Rajaram V, Wierenga RK, Hiltunen JK (2009) 17beta-hydroxysteroid dehydrogenase type 8 and carbonyl reductase type 4 assemble as a ketoacyl reductase of human mitochondrial FAS. FASEB J 23:3682–3691
Angebault C, Guichet PO, Talmat-Amar Y et al (2015) Recessive mutations in RTN4IP1 cause isolated and syndromic optic neuropathies. Am J Hum Genet 97:754–760
Sakai C, Yamaguchi S, Sasaki M, Miyamoto Y, Matsushima Y, Goto Y (2015) ECHS1 mutations cause combined respiratory chain deficiency resulting in Leigh syndrome. Hum Mutat 36:232–239
Szklarczyk R, Wanschers BF, Nabuurs SB, Nouws J, Nijtmans LG, Huynen MA (2011) NDUFB7 and NDUFA8 are located at the intermembrane surface of complex I. FEBS Lett 585:737–743
Nakashima Y, Ohsawa I, Nishimaki K et al (2014) Preventive effects of chlorella on skeletal muscle atrophy in muscle-specific mitochondrial aldehyde dehydrogenase 2 activity-deficient mice. BMC Complement Altern Med 14:390
Marcadier JL, Smith AM, Pohl D et al (2013) Mutations in ALDH6A1 encoding methylmalonate semialdehyde dehydrogenase are associated with dysmyelination and transient methylmalonic aciduria. Orphanet J Rare Dis 8:98
Xie R, Nguyen S, McKeehan K, Wang F, McKeehan WL, Liu L (2011) Microtubule-associated protein 1S (MAP1S) bridges autophagic components with microtubules and mitochondria to affect autophagosomal biogenesis and degradation. J Biol Chem 286:10,367–10,377
Otera H, Wang C, Cleland MM et al (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191:1141–1158
Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43:D512–D520
Nixon BR, Liu B, Scellini B et al (2013) Tropomyosin Ser-283 pseudo-phosphorylation slows myofibril relaxation. Arch Biochem Biophys 535:30–38
Lu X, Heeley DH, Smillie LB, Kawai M (2010) The role of tropomyosin isoforms and phosphorylation in force generation in thin-filament reconstituted bovine cardiac muscle fibres. J Muscle Res Cell Motil 31:93–109
Kleinert M, Parker BL, Chaudhuri R et al (2016) mTORC2 and AMPK differentially regulate muscle triglyceride content via Perilipin 3. Mol Metab 5:646–655
Bosma M, Hesselink MK, Sparks LM et al (2012) Perilipin 2 improves insulin sensitivity in skeletal muscle despite elevated intramuscular lipid levels. Diabetes 61:2679–2690
DeFronzo RA, Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(Suppl 2):S157–S163
Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE (2005) Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54:8–14
Kelley DE, He J, Menshikova EV, Ritov VB (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51:2944–2950
Liu Y, Turdi S, Park T et al (2013) Adiponectin corrects high-fat diet-induced disturbances in muscle metabolomic profile and whole-body glucose homeostasis. Diabetes 62:743–752
Phielix E, Schrauwen-Hinderling VB, Mensink M et al (2008) Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 57:2943–2949
Schrauwen-Hinderling VB, Kooi ME, Hesselink MK et al (2007) Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia 50:113–120
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350:664–671
Morino K, Petersen KF, Shulman GI (2006) Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 55(Suppl 2):S9–S15
Mootha VK, Lindgren CM, Eriksson KF et al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273
Patti ME, Butte AJ, Crunkhorn S et al (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100:8466–8471
Toyama EQ, Herzig S, Courchet J et al (2016) Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351:275–281
Liu L, Xie R, Yang C, McKeehan WL (2009) Dual function microtubule- and mitochondria-associated proteins mediate mitotic cell death. Cell Oncol 31:393–405
Taub PR, Ramirez-Sanchez I, Ciaraldi TP et al (2013) Perturbations in skeletal muscle sarcomere structure in patients with heart failure and type 2 diabetes: restorative effects of (-)-epicatechin-rich cocoa. Clin Sci (Lond) 125:383–389
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Funding
This work was supported by grants from the Swedish Research Council, the Novo Nordisk Foundation, the Swedish Heart and Lung Foundation, the Diabetes Wellness Network Sweden, the Estonian Research Council, the Swedish Diabetes Foundation, the Royal Society of Arts and Sciences in Gothenburg, the M. Bergvalls Foundation, the Wiberg Foundation, the Adlerbert Research Foundation, the I. Hultman Foundation, the S. and E. Goljes Foundation, the West Sweden ALF Program, the F. Neubergh Foundation, the I.-B. and A. Lundbergs Research Foundation, W. and M. Lundgrens Foundation and the European Foundation for the Study of Diabetes and Novo Nordisk Partnership for Diabetes Research in Europe. The study sponsor was not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; or the decision to submit the report for publication.
Duality of interest
FB is co-founder and shareholder in Metabogen AB. FB has participated on advisory boards and received honoraria from Unilever, Boehringer and Merck. The authors declare that there is no other duality of interest associated with this article.
Author contribution
All the authors made substantial contributions to conception and design, and/or analysis and interpretation of data. UC and EN-D generated the bulk of the results. EC, MA, SS, MEJ, FB and MM contributed to the research data. MS and JB performed the analysis of acylcarnitines. B-MO and CS conducted the primary global phosphoproteomic analysis. BRJ conducted the TEM studies. All the authors revised the article critically for important intellectual content and approved the final version of the article to be published. MM directed the project, designed the study, interpreted the data and wrote the manuscript. MM is the guarantor of this work.
Additional information
Urszula Chursa and Esther Nuñez-Durán contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1700 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Chursa, U., Nuñez-Durán, E., Cansby, E. et al. Overexpression of protein kinase STK25 in mice exacerbates ectopic lipid accumulation, mitochondrial dysfunction and insulin resistance in skeletal muscle. Diabetologia 60, 553–567 (2017). https://doi.org/10.1007/s00125-016-4171-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4171-5