
Abstract
Aim/hypothesis
The peroxisome proliferator-activated receptor-γ coactivator α (PGC-1α) plays a critical role in the maintenance of glucose, lipid and energy homeostasis by orchestrating metabolic programs in multiple tissues in response to environmental cues. In skeletal muscles, PGC-1α dysregulation has been associated with insulin resistance and type 2 diabetes but the underlying mechanisms have remained elusive. This research aims to understand the role of TET3, a member of the ten-eleven translocation (TET) family dioxygenases, in PGC-1α dysregulation in skeletal muscles in obesity and diabetes.
Methods
TET expression levels in skeletal muscles were analysed in humans with or without type 2 diabetes, as well as in mouse models of high-fat diet (HFD)-induced or genetically induced (ob/ob) obesity/diabetes. Muscle-specific Tet3 knockout (mKD) mice were generated to study TET3’s role in muscle insulin sensitivity. Genome-wide expression profiling (RNA-seq) of muscle tissues from wild-type (WT) and mKD mice was performed to mine deeper insights into TET3-mediated regulation of muscle insulin sensitivity. The correlation between PGC-1α and TET3 expression levels was investigated using muscle tissues and in vitro-derived myotubes. PGC-1α phosphorylation and degradation were analysed using in vitro assays.
Results
TET3 expression was elevated in skeletal muscles of humans with type 2 diabetes and in HFD-fed and ob/ob mice compared with healthy controls. mKD mice exhibited enhanced glucose tolerance, insulin sensitivity and resilience to HFD-induced insulin resistance. Pathway analysis of RNA-seq identified ‘Mitochondrial Function’ and ‘PPARα Pathway’ to be among the top biological processes regulated by TET3. We observed higher PGC-1α levels (~25%) in muscles of mKD mice vs WT mice, and lower PGC-1α protein levels (~25–60%) in HFD-fed or ob/ob mice compared with their control counterparts. In human and murine myotubes, increased PGC-1α levels following TET3 knockdown contributed to improved mitochondrial respiration and insulin sensitivity. TET3 formed a complex with PGC-1α and interfered with its phosphorylation, leading to its destabilisation.
Conclusions/interpretation
Our results demonstrate an essential role for TET3 in the regulation of skeletal muscle insulin sensitivity and suggest that TET3 may be used as a potential therapeutic target for the metabolic syndrome.
Data availability
Sequences are available from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with accession number of GSE224042.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.

Introduction
The hallmark of type 2 diabetes is persistent hyperglycaemia resulting from a combination of peripheral insulin resistance and inappropriate secretion of insulin and glucagon from the pancreas [1]. Chronic hyperglycaemia in combination with other metabolic aberrations causes damage to organs and blood vessels and substantially increases the risk for various complications, leading to premature disability and death [1, 2]. Skeletal muscle is the major tissue responsible for insulin-stimulated glucose disposal and muscle insulin resistance is a critical component in the pathogenesis of type 2 diabetes [3,4,5]. While the exact causes of muscle insulin resistance remain incompletely understood, available literature points to dysregulation of peroxisome proliferator-activated receptor-γ coactivator α (PGC-1α) as an important mechanism. As a transcriptional coactivator, PGC-1α plays a critical role in the maintenance of glucose, lipid and energy homeostasis by orchestrating metabolic programs in response to numerous environmental stimuli (e.g. nutrients, temperature, exercise) in a tissue-specific manner [6, 7]. Highly expressed in skeletal muscle, PGC-1α has been implicated in the regulation of energy metabolism, myofibre composition, skeletal muscle mass, angiogenesis and neuromuscular junction remodelling [7,8,9]. The best studied role of PGC-1α in myocytes is its ability to powerfully drive the transcriptional program of mitochondrial biogenesis, respiration and fatty acid β-oxidation through co-ordinately activating a wide variety of transcription factors including nuclear respiratory factors, oestrogen related receptors, and peroxisome proliferator-activated receptor (PPAR) family members [7, 8, 10]. An additional role of PGC-1α is to promote fatty acid uptake and lipid biosynthesis by upregulating a set of genes involved in these processes. Given the implication of myocyte overload of lipid/intermediate lipid metabolites in causing insulin resistance, a fine balance between the two actions of PGC-1α (fatty acid uptake/lipid biosynthesis vs fatty acid oxidation) is critical to metabolic health. Indeed, supraphysiological muscle overexpression of PGC-1α paradoxically induces intramuscular lipid accumulation and insulin resistance in transgenic mice [11, 12], whereas modest increases (~25%) in PGC-1α protein abundance in muscle promote insulin sensitisation [9, 13,14,15]. Importantly, repression of the PGC-1α-dependent mitochondrial program has long been documented in skeletal muscles of individuals with type 2 diabetes, and PGC-1α dysregulation and hence mitochondrial insufficiency are widely acknowledged contributors to muscle insulin resistance [9, 16,17,18,19]. However, the causes of PGC-1α dysregulation have remained elusive.
The ten-eleven translocation (TET) family of dioxygenases (TET1, TET2 and TET3) regulate gene expression by oxidising methylated cytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and further oxidised derivatives, leading to DNA demethylation [20, 21]. TET-induced epigenetic regulation can also occur in an enzymatic activity-independent manner [22,23,24,25,26,27,28]. Despite the importance of TETs in development, cancer, stem cells and immunity [20, 24, 25, 29,30,31,32,33], their roles in energy metabolism have just begun to be recognised, as exemplified by a few recent publications [27, 28, 34,35,36]. We were the first to report that TET3 expression is increased in the livers of humans with type 2 diabetes and in mouse models of type 2 diabetes, contributing to hyperglycaemia [34]. Mechanistically, TET3 induces promoter demethylation of the transcription factor, hepatocyte nuclear factor 4α (HNF4α), promoting hepatic glucose production [34]. We later reported that the hepatic TET3/HNF4A regulatory pathway underlies the therapeutic effects of metformin [35]. Recently, a role for TETs in adipose tissues in energy expenditure has also been uncovered. In mouse adipocytes, it was shown that TET1 coordinates with histone deacetylase 1 (HDAC1) to epigenetically suppress thermogenic gene transcription and that adipocyte-specific Tet1 knockout in mice increases energy expenditure and protects against diet-induced obesity and insulin resistance [27]. Likewise, adipose-specific ablation of all three Tet genes enhances β-adrenergic responses, increases energy expenditure and protects against obesity [28]. Mechanistically, TETs suppress the transcription of β3-adrenergic receptor by recruiting histone deacetylases to its promoter [28]. Further, we have recently documented that CRISPR-mediated, agouti-related protein (AgRP) neuron-specific Tet3 ablation induces hyperphagia, systemic insulin resistance, obesity and type 2 diabetes [36]. Mechanistically, we reported a dynamic association of TET3 with the Agrp promoter in response to leptin signalling that induces 5hmC modification and association of a chromatin-modifying complex, leading to transcription inhibition of Agrp. Importantly, this regulation occurs both in mouse models and human cells [36]. In the current work we report that TET3 regulates skeletal muscle insulin sensitivity through a novel mechanism of action.
Methods
Animals
All animal work was approved by the Yale University Institutional Animal Care and Use Committee. All mice used in this report were female. Mice were housed at 22–24°C under a 12 h light–dark cycle and were fed with regular chow (RC) (Harlan Teklad no. 2018, IN, USA; 18% energy from fat) or high-fat diet (HFD) (Research Diets, NJ, USA, D12451; 45% energy from fat); water was provided ad libitum. C57BL/6J (Jax, CT, USA, 000664), HAS-Cre79 (Jax, 006149) and Lepob/ob (Jax, 000632) mice were purchased from the Jackson Laboratory. The Tet3fl/fl mice were generous gifts from A. Rao from La Jolla Institute for Immunology (CA, USA). The muscle-specific Tet3 knockout (mKD) mice were created by crossing Tet3fl/fl mice with mice expressing Cre recombinase under the control of human α skeletal muscle actin (HSA) promoter (HSA-Cre79 mice). TET3 floxed littermates (wild-type [WT]) were used as controls. Randomisation was not feasible during group assignment, but results were analysed in a blinded manner whenever possible. Data were not included if values were excluded by outlier test. For information on animal numbers, refer to figure legends. Before experiments, mice were allowed to acclimate for at least 7 days in our animal facility.
Body composition and indirect calorimetry analyses
Body composition was assessed by MRI (EcoMRI; Echo Medical Systems, TX, USA). Food intake, energy expenditure and locomotor activity were measured using an indirect calorimetry chamber (TSE Systems, Germany).
GTT and ITT
The GTT and ITT were conducted as previously described [35]. For the GTT, following a 14 h overnight fast, each mouse received an i.p. injection of 2 g/kg body weight glucose (Sigma-Aldrich, MO, USA, G5767) in sterile saline (154 mmol/l NaCl). Blood glucose concentrations were measured using Contour next blood glucose meter (Ascensia Diabetes Care, NJ, USA) via tail vein bleeding at the indicated time points after injection. The ITT was performed in ad libitum fed mice. Each mouse received an i.p. injection of insulin, 1 U/kg (Humulin R; Eli Lilly, MA, USA). Blood glucose concentrations were measured using Contour next blood glucose via tail vein bleeding at the indicated time points after injection.
Hyperinsulinaemic–euglycaemic clamp studies
The hyperinsulinaemic–euglycaemic clamp studies were performed using previously described methods [34]. Briefly, mice at the age of 12 weeks were catheterised in jugular veins with polyethylene catheters under deep anaesthesia. Mice were singly housed for a 4 day recovery period after surgery. The clamp experiments were performed using conscious and unrestrained mice after 16 h overnight fasting. The protocol consisted of a 120 min basal period (t=−120–0 min) followed by a 115 min clamp period (t=0–115 min). [3-3H]Glucose (185,000 Bq; Perkin Elmer, MA, USA) was given at t=−120 min followed by a 1850 Bq/min infusion for 2 h. During the basal period, at t=−15 min and t=−5 min, blood samples were taken for the assessment of basal glucose level and glucose turnover. The clamp period was begun at t=0 min with primed and continuous infusion of human insulin (8 mU/kg bolus followed by a rate of 2.5 mU kg–1 min–1; Humulin R; Eli Lilly). Blood glucose was measured by glucometer (Breeze 2; Bayer HealthCare, NJ, USA) at 10 min intervals, and 30% glucose was infused at a variable rate in order to maintain euglycaemic levels (6.1–7.2 mmol/l). Blood samples were collected every 10 min from t=70 min to t=115 min and processed to determine glucose specific activity. Following collection of the final blood sample, the mice were euthanised and tissues were harvested, frozen in liquid nitrogen and stored at −80°C until later use.
Myoblast culture and differentiation
Undifferentiated mouse C2C12 myoblasts (Sigma-Aldrich, 91031101-iVL) were maintained in growth medium (DMEM [Gibco, NY, USA, 11965-092] supplemented with 10% (vol./vol.) FBS, heat inactivated, 1% (vol./vol.) penicillin/streptomycin, 1% (vol./vol.) l-glutamine and 1 mmol/l sodium pyruvate). Cells were authenticated and free of mycoplasma. To prepare for differentiation, cells were seeded at a density of 2.0×104 cells/well in growth medium in 24-well plates. Differentiation was initiated 2 days later when cells became confluent by replacing growth medium with differentiation medium containing 2% horse serum (16050-130) in place of 10% FBS. The medium was changed every other day until transfection, which was performed on day 4 or 5 after initiation of differentiation. Cryopreserved primary human skeletal myoblasts (HSkMs) (Thermo Fisher Scientific, MA, USA, A11440) were purchased. To prepare for differentiation, cells were thawed in Gibco HSkM Differentiation Medium (DMEM Basal Medium [11885-084] supplemented with 2% horse serum) and seeded in a 24-well plate at a density of 2.4×105 cells/well in the differentiation medium. Cells were incubated in a tissue culture incubator for 48 h to allow rapid differentiation, followed by siRNA transfection.
siRNA transfection
To prepare siRNA transfection solution for each well of cells in a 24-well plate, 20 pmol of non-targeting control siRNA (NT siRNA) (Ambion, CT, USA, AM4636), siRNA specific for mouse Tet3 (Tet3 siRNA; Ambion, 4390815/s101483), mouse Pgc1a (Pgc1a siRNA; Ambion, 4390771/n253420), human TET3 (TET3 siRNA; Ambion, 4392420/s47239) or human PGC1A (PGC1A siRNA; Ambion, 4392420/s21394) was mixed with 100 μl of OPTI-MEM (Gibco, 31985-070) by gentle pipetting. In parallel, 6 μl of Lipofectamine RNAiMAX (Invitrogen, MA, USA, 13778-150) was mixed with 100 μl of OPTI-MEM by gentle pipetting, then the two were combined. Following 5 min of incubation at room temperature, the resulting 200 μl of transfection solution was added to each well of cells. For treatment with NT siRNA alone, 20 pmol of NT siRNA was used for each well of cells. For treatment with TET3 siRNA alone, 10 pmol of NT siRNA and 10 pmol of TET3 siRNA (or Tet3 siRNA) were used for each well of cells. For TET3/PGC-1α double knockdown, 10 pmol of TET3 siRNA (or Tet3 siRNA) and 10 pmol of PGC1A siRNA (or Pgc1a siRNA) were used for each well of cells. Therefore, the total amount of siRNAs for each well of cells was 20 pmol. After 12 h of incubation at 37°C in a 5% humidified CO2 tissue culture incubator, 300 μl of medium was added and incubation was continued for an additional 24 or 48 h until further analyses.
PGC-1α protein stability assay
C2C12 myoblasts were transfected with NT siRNA or Tet3 siRNA as described above for 48 h, followed by addition of cycloheximide (CHX) (Cell Signaling Technology, MA, USA 2112) at a final concentration of 50 μg/ml. Proteins were isolated at 0, 15, 30 and 45 min later and analysed by western blotting using anti-PGC-1α (dilution 1:1000; Proteintech, IL, USA, 66369-1-Ig) and horseradish peroxide (HRP)-conjugated anti-GAPDH (dilution 1:5000; Proteintech, HRP-60004).
Western blot analysis
To extract proteins from cultured cells, myoblasts or myotubes were homogenised in situ using a pipette tip in 2 × SDS sample buffer with 10% β-mercaptoethanol at room temperature in less than 5 s followed by heating at 100°C for 5 min with occasional vortex. To extract proteins from muscle tissues, frozen tissue samples (~50 mg) were homogenised in 200 μl of tissue lysis buffer (15% SDS, 75 mmol/l Tris HCl, pH 7.4, 1× protease inhibitor cocktail [Thermo, 78438], 1× phosphatase inhibitor cocktail [Thermo, 78427], 5% β-mercaptoethanol) using a BeadBug6 Microtube homogeniser (Benchmark) set at speed 3600, 20 s on and 20 s off for six cycles. The lysate was cooled down on ice for 10 min, followed by centrifugation at 12,000 g at 4°C for 7 min to remove insoluble materials. The supernatant fraction was transferred to a new tube and glycerol (final concentration 20%) and bromophenol (for tracking purpose during gel running) were added. Samples were then heated at 100°C for 5 min with occasional vortex, aliquoted, and stored at −80°C until use. Tissue samples were freshly diluted at 1:1 – 1:3 in 2 × SDS sample buffer before loading. Cell and tissue samples were loaded at 5–10 μl per well onto 4–15% gradient SDS gels (Bio-Rad), followed by western blot analysis. The antibodies used were anti-TET3 (for mouse TET3; dilution 1:1000; Active motif, CA, USA, 61395), anti-TET3 (for human TET3; dilution 1:1000; GeneTex, CA, USA, GTX121453), anti-PGC-1α (dilution 1:1000; Proteintech, 66369-1-Ig), anti-phospho-Ser265 (dilution 1:1000), anti-phospho-Thr298 (dilution 1:1000), anti-TET2 (dilution 1:1000, Proteintech, 21207-1-AP) and HRP-conjugated anti-GAPDH (dilution 1:5000; Proteintech, HRP-60004). The secondary antibodies were HRP-linked anti-rabbit IgG (dilution 1:10,000; Rockland, PA, USA, 611-1322) and HRP-linked anti-mouse IgG (dilution 1:10,000; Cell Signalling Technology, 7076S).
RNA extraction and real-time quantitative PCR
For cultured myoblasts and myotubes, total RNA was extracted using PureLink RNA Mini Kit (Ambion, 12183018A). For muscle tissues, total RNA was extracted using RNeasy Fibrous Tissue Mini Kit (Qiagen, MD, USA, 74704). cDNAs were synthesised using PrimeScript RT Reagent Kit (Invitrogen, TAKARA, RR037A) in a 20 μl reaction containing 0.2–0.5 μg of total RNA. Real-time quantitative PCR (RT-qPCR) was performed in a 15 μl reaction containing 0.5–1 μl of cDNA using iQSYBRGreen (Bio-Rad, CA, USA) in a Bio-Rad iCycler. PCR was performed by initial denaturation at 95°C for 5 min, followed by 40 cycles of 30 s at 95°C, 30 s at 60°C and 30 s at 72°C. Specificity was verified by melting curve analysis and agarose gel electrophoresis. The threshold cycle (Ct) values of each sample were used in the post-PCR data analysis. Gene expression levels were normalised against the following housekeeping genes: β-tubulin for muscle tissues and RPLP0 for cultured myoblasts and myotubes. Real-time PCR primers are listed in electronic supplementary material (ESM) Table 1.
Glucose uptake assay
The glucose uptake assay was performed on in vitro differentiated mouse and human myotubes in a 96-well plate using the Glucose Uptake Cell-Based Assay Kit (Cayman Chemical, MI, USA, catalogue no. 600470) according to the manufacturer’s instructions with minor modifications. On the day of the assay, culture media were replaced with 200 μl of glucose-free DMEM (Gibco, catalogue number 11966-025) and incubation was carried out for 2 h. Then, the medium was replaced with 100 μl of new glucose-free DMEM in the presence or absence of 100 nmol/l of insulin for 15–20 min. Subsequently, 100 μl of new glucose-free DMEM containing fluorescent 2-(N-[7-nitrobenz-2-oxa-1,3-diazol-4-yl]amino)-2-deoxyglucose at a final concentration of 150 μg/ml was added. Incubation was carried out in the dark for an additional 15 min in a tissue culture incubator. The medium was then removed and the myotubes were washed once with 200 μl of ice-cold PBS. After adding 100 μl of new ice-cold PBS to the myotubes, fluorescent intensity was immediately determined using the fluorescent plate reader (FilterMax F3&F5 Multi-Mode Microplate Reader; Molecular Devices, CA, USA). Results are presented with NT siRNA-transfected myotubes without insulin stimulation arbitrarily set as 1.
Seahorse analysis
In vitro differentiated mouse and human myotubes in 24-well plates were used. Mitochondrial respiration analyses of myotubes were performed by the Islet, Oxygen consumption, Mass Isotopomer flux Core (IOMIC) at Yale using Seahorse XFPro (https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-pro-software-2007523).
Immunoprecipitation
To prepare antibodies, 50 μl (packed volume) of ChIP grade Dynabeads Protein G (Invitrogen, Thermo Scientific, 10004D) were washed twice with 1 ml of immunoprecipitation buffer (0.5% Triton X-100, 150 mmol/l NaCl, 10 mmol/l Tris HCl at pH 7.5, and 10 mmol/l EDTA), followed by incubation with 5 μg of rabbit polyclonal anti-TET3 (Active Motif, 61395), rabbit polyclonal anti-PGC-1α (Novus, NBP1-04676) or preimmune rabbit IgG in 300 μl of immunoprecipitation buffer at 4°C overnight. Antibody-bound beads were pelleted and kept on ice until use. To prepare lysate from muscle tissues, PBS-washed gastrocnemius muscle (GAS) tissues, freshly isolated from mice, were homogenised using a grinding tube in 1 ml of freshly prepared gentle lysis buffer (0.5% Triton X-100, 10 mmol/l NaCl, 10 mmol/l Tris HCl at pH 7.5, 10 mmol/l EDTA and 1× protease inhibitor cocktail). After centrifugation at 12,000 g at 4°C for 15 min to remove insoluble materials, 5 mol/l of NaCl was added to a final concentration of 200 mmol/l, and the lysate was transferred to a tube containing antibody/preimmune IgG-coated beads (250 μl of lysate per immunoprecipitation). Immunoprecipitation was carried out at 4°C for 4 h. Following this, beads were quickly washed twice with 1 ml of cold immunoprecipitation buffer and washed an additional three times by rotating at 4°C for 5 min each time. After the final wash, residual liquid was completely removed and the beads were eluted with 16 μl of 2 × SDS buffer (containing 1× phosphatase inhibitor cocktail and 1× protease inhibitor cocktail) at 100°C for 5 min. Eluant was loaded (10 μl per gel well) onto a 4–15% gradient SDS gel (Bio-Rad, 456-8086). For western blot analysis, anti-TET3 (Active Motif, 61395) and anti-PGC-1α (Novus, NBP1-04676) were used. The secondary antibodies used were Rabbit IgG TrueBlot (1:1000, Rockland, 18-8816-33). These unique HRP-conjugated monoclonal secondary antibodies enable detection of immunoblotted target proteins without hindrance by interfering immunoprecipitating immunoglobulin heavy and light chains.
RNA-Seq and data analysis
Total RNAs were extracted from GAS tissues of 14-week-old RC-fed WT and mKD mice using RNeasy Fibrous Tissue Mini Kit (Qiagen, 74704). RNA-Seq library preparation and sequencing were conducted at Yale Stem Cell Center Genomics Core facility through poly A enrichment (lllumina TruSeq Stranded mRNA Library Prep Kit). Differential expression analysis between two different groups was performed using Partek Flow software, version 9.0.20.0622 (Partek, St Louis, MO, USA; https://www.partek.com/partek-flow/). Genes with a false discovery rate (FDR) below 0.05 and absolute fold change over 1.0 were analysed with Ingenuity Pathway Analysis using IPA software (Qiagen). Sequences are available from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE224042.
Statistical analysis
The number of independent experiments and the statistical analysis for each figure are indicated in the legends. All statistical analyses (except RNA-seq which was performed using DESeq2 software) were performed using GraphPad Prism version 8 for Windows (GraphPad Software, La Jolla California USA; www.graphpad.com) and are presented as mean ± SEM. Two-tailed Student’s t tests (or as otherwise indicated) were used to compare means between groups. p<0.05 was considered significant.
Results
TET3 expression is elevated in skeletal muscles of humans and mice with diabetes
We performed data mining on publicly available datasets (Gene Expression Omnibus). The expression of TET3 (but not TET1 and TET2) mRNA was significantly increased in skeletal muscle tissues of humans with diabetes as compared with non-diabetic control counterparts (Fig. 1a,b) [37, 38]. Increased TET3 expression was also detected in myocytes from individuals with diabetes (Fig. 1c) [39]. Next, we asked whether muscle TET3 expression would change in mice with obesity and diabetes. Thus, we subjected mice to HFD feeding for 12 weeks, starting at the age of 6 weeks. Age-matched mice fed RC were used as a control. Compared with control mice, the HFD-fed mice developed obesity and impaired glucose metabolism, as judged by a significant increase in body weight and fat mass accompanied by a decrease in lean body mass and an increase in fasting glucose level (ESM Fig. 1). To assess TET3 expression, GAS muscle, composed of both slow-twitch type I oxidative and fast-twitch type II glycolytic fibres, was isolated, followed by qPCR and immunoblotting analyses. TET3 expression was significantly increased in muscle of HFD-fed mice as compared with RC-fed mice, at both the mRNA and protein level (Fig. 1d). The specificity of TET3 antibody has been previously validated [36, 40, 41]. Similar observations were made in ob/ob mice, a genetic mouse model of obesity and type 2 diabetes (Fig. 1e). The strong positive correlation between obesity/diabetes and increased muscle TET3 expression in both humans and mice suggests a role for TET3 in regulation of muscle insulin sensitivity.

TET3 expression is increased in muscles of humans with diabetes and muscles of HFD-fed mice. (a) Relative TET1, TET2 and TET3 mRNA levels in muscle tissues from humans with diabetes (age 60.0±4.8 years, BMI 31.8±6.5 kg/m2) or without diabetes (age 59.6±5.0 years; BMI 27.4±5.4 kg/m2) (from the GSE22435 dataset). (b) Relative TET1, TET2 and TET3 mRNA levels in muscle tissues from humans with diabetes (age 51.5±3.6 years, BMI 30.8±2.5 kg/m2) or without diabetes (age 37.8±2.9 years, BMI 25.2±0.8 kg/m2) (from the GSE25462 dataset). (c) Relative TET1, TET2 and TET3 mRNA levels in myocytes from women with diabetes (age 46–63 years, BMI 24–33 kg/m2) or without diabetes (age 41–63 years, BMI 24–35 kg/m2) (from the GSE81965 dataset). (d) qPCR analysis and immunoblot of TET3 in GAS muscles from RC- and HFD-fed mice. The lane of each blot represents an individual mouse; molecular size in kDa is shown. (e) qPCR and immunoblot of TET3 in GAS muscles from control and ob/ob mice at the age of 10 weeks. All data are presented as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 (two-tailed Student’s t test). Ctrl, control; PS, Ponceau S; T2D, type 2 diabetes
Muscle-specific TET3 knockdown enhances insulin sensitivity
To determine whether TET3 regulates muscle insulin sensitivity, we created mice with muscle-specific Tet3 knockout (mKD) by breeding TET3 floxed mice (Tet3fl/fl) with transgenic mice (HSA-Cre79) expressing Cre recombinase under the control of HSA promoter, which has been shown to enable muscle-specific expression of transgenes [42, 43]. The WT TET3 floxed littermates were used as controls.
Muscle TET3 expression was reduced by ~50% at the mRNA level (Fig. 2a) and by ~25% at the protein level (Fig. 2b) in mKD as compared with WT littermates. The residual expression of TET3 in muscle extracts could be attributed to an incomplete deletion of the floxed allele in a fraction of myocytes and/or to the presence of other cell types in the tissue [44]. To exclude the potential for functional compensation of TET3 knockdown, we also examined the expression of other TET family isoforms. While TET1 expression in skeletal muscles was negligible, TET2 expression was abundant and was not altered by TET3 deletion (ESM Fig. 2). Total body weight, fat mass and lean body mass were similar when comparing mKD mice with WT littermates (ESM Fig. 3a,b) and indirect calorimetry experiments showed no differences in food intake, energy expenditure or locomotion (ESM Fig. 3c). However, when subjected to a GTT, the mKD mice showed an enhanced glucose tolerance as compared with WT littermates (Fig. 2c). To determine the cause of alterations in blood glucose more directly, hyperinsulinaemic–euglycaemic clamp studies were performed. Compared with WT littermates, the mKD mice showed significantly higher glucose infusion rate required to maintain euglycaemia (Fig. 2d,e), reflecting increased whole-body insulin sensitivity. This was not due to increased insulin-stimulated suppression of endogenous glucose production (Fig. 2f) but was due to increased insulin-stimulated glucose uptake into skeletal muscle (Fig. 2g). Collectively, these results demonstrate that muscle-specific TET3 knockdown increases muscle and whole-body insulin sensitivity.

Muscle TET3 knockdown enhances insulin sensitivity. (a) qPCR of Tet3 mRNA in GAS from WT and mKD mice at the age of 12 weeks. n=5 mice for each genotype. (b) Immunoblot of TET3 protein in GAS from WT and mKD mice at the age of 12 weeks. Each lane represents an individual mouse, with TET3 protein quantification shown. (c) GTT following 14 h overnight fasting of WT and mKD mice at the age of 10 weeks. n=8 mice in each group. (d–g) Hyperinsulinaemic–euglycaemic clamp studies from WT and mKD mice at the age of 12 weeks. n=8 animals in each group. (h) Per cent body fat of WT (n=8) and mKD (n=10) mice after exposure to HFD for 10 weeks. (i, j) Results of GTT (i) and ITT (j) of WT (n=6) and mKD (n=8) mice after exposure to HFD for 10 weeks. All data are presented as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 (a, b, f, g and h, two-tailed Student’s t test; c–e and j, two-way ANOVA with Sidak post-test). BAT, brown adipose tissue; EGP, endogenous glucose production; EWAT, epididymis white adipose tissue; GIR, glucose infusion rate; PS, Ponceau S
mKD mice are resistant to diet-induced insulin resistance
mKD and WT mice were subjected to HFD for 10 weeks, starting at the age of 6 weeks. The mKD mice gained less body fat than WT mice (Fig. 2h), with no difference in lean mass between the groups (data not shown). In addition, mKD mice were more glucose tolerant and had a better insulin sensitivity (Fig. 2i,j). These results further support the importance of muscle TET3 in modulation of whole-body glucose homeostasis.
TET3 affects mitochondrial pathway gene expression
To gain a mechanistic insight into TET3-mediated regulation of muscle insulin sensitivity, we performed genome-wide expression profiling (RNA-seq) on RNA isolated from GAS muscles of mKD and WT littermates fed on RC. We performed Ingenuity Pathway Analysis on our RNA-seq data, using a cut-off of 1.2-fold change (p<0.05) in gene expression. We acknowledge that many common, complex disorders are characterised by modest but coordinated changes in expression of multiple genes of a biological pathway. Indeed, Gene Set Enrichment Analysis (GSEA) was previously used to successfully identify a subset of genes involved in oxidative phosphorylation whose expression was co-ordinately downregulated (20–50%) in muscles of humans with type 2 diabetes [18]. Importantly, this set of genes are also targets of PGC-1α, underscoring the long-standing notion of repression of the PGC-1α-dependent mitochondrial program in muscles of diabetic individuals [9, 16,17,18,19]. Our RNA-seq studies revealed profound gene expression changes induced by muscle Tet3 knockout as compared with controls (ESM Fig. 4a and ESM Table 2). Pathway analysis identified ‘Mitochondrial Function’ and the ‘PPARα pathway’ to be among the top biological processes affected by TET3 (ESM Fig. 4b). By examining specifically how TET3 influenced the expression of genes in the mitochondrial pathway, we noticed a set of 31 mitochondrial respiration genes that were co-ordinately upregulated by at least 1.2-fold (p<0.05) in mKD vs WT mice (ESM Fig. 4c). As PGC-1α is a master regulator of mitochondrial biogenesis and function, we hypothesised that PGC-1α might be an important downstream target of TET3.
TET3 post-transcriptionally affects PGC-1α expression
As a crucial metabolic node, PGC-1α is subjected to both transcriptional and post-transcriptional regulation [45,46,47]. Our RNA-seq analysis did not detect a significant change in PGC-1α expression at the mRNA level in muscles of mKD vs WT mice (ESM Table 2). However, a change at the protein level could not be excluded. Thus, RNA and protein were isolated from muscles of mKD and WT mice and analysed. Consistent with our RNA-seq data, no significant change was detected in Pgc1a (also known as Ppargc1a) mRNA in muscles of mKD vs WT mice (Fig. 3a). However, there was a modest (~25%) but significant increase in PGC-1α protein in mKD mice (Fig. 3b). These results are in line with previous reports that physiologically relevant increases (~25%) in PGC-1α protein enhance muscle insulin sensitivity [9, 13,14,15], given that mKD mice also show increased insulin sensitivity (Fig. 2). Importantly, when muscles from mice were examined, we observed a ~25% and ~60% decrease in PGC-1α protein in HFD- vs RC-fed and ob/ob vs control mice, respectively, with no changes at the mRNA level (Fig. 3c–f). Collectively, our results suggested a post-transcriptional regulation of PGC-1α expression by TET3 in myocytes.

TET3 negatively regulates PGC-1α expression at the post-transcriptional level. (a) qPCR of Pgc1a mRNA in GAS tissues isolated from WT and mKD mice. (b) Immunoblot of PGC-1α in GAS tissues isolated from WT and mKD mice. (c) qPCR of Pgc1a in GAS tissues isolated from RC- and HFD-fed mice. (d) Immunoblot of PGC-1α in GAS tissues isolated from RC- and HFD-fed mice. (e) qPCR of Pgc1a in GAS tissues isolated from age-matched control and ob/ob mice. (f) Immunoblot of PGC-1α in GAS tissues isolated from age-matched control and ob/ob mice. Data are presented as mean ± SEM, n=5 mice in each group. **p<0.01, ***p<0.001 (two-tailed Student’s t test). Ctrl, control
TET3 affects mitochondrial respiration and insulin sensitivity in myocytes
There is substantial evidence from human and animal studies that mitochondrial respiration is intrinsically coupled to muscle insulin sensitivity [48]. Notably, a ~25% increase of PGC-1α protein in muscle promotes insulin-sensitising effects including enhanced mitochondrial respiration and insulin-stimulated glucose uptake [9, 13,14,15, 18]. Our studies revealed that mKD mice exhibit increased insulin sensitivity (Fig. 2) with a concomitant increase in PGC-1α protein (Fig. 3b). Given the inverse relationship between TET3 and PGC-1α protein levels observed in human and mouse skeletal muscle tissues, we asked whether reducing TET3 protein levels in myocytes using siRNAs specifically targeting TET3 mRNA for degradation would elevate PGC-1α protein level thereby enhancing mitochondrial respiration and insulin-stimulated glucose uptake. The siRNAs (TET3 siRNA and Tet3 siRNA for human and mouse genes, respectively) and the control non-targeting siRNA (NT siRNA) have been previously documented [40]. We also asked whether bringing PGC-1α protein levels back down to basal levels via siRNA-mediated knockdown in TET3 siRNA-treated myocytes would abolish these effects. As expected, the PGC-1α protein level increased in human primary myotubes transfected with TET3 siRNA as compared with NT siRNA (Fig. 4a). Co-transfection with TET3 siRNA and PGC1a siRNA restored PGC-1α protein to basal levels (Fig. 4a). While reducing the TET3 protein level (which led to increased PGC-1α protein level, Fig. 4a) increased mitochondrial maximal respiration and spare respiration capacity, co-transfection with TET3 siRNA and PGC1a siRNA (which restored PGC-1α protein to basal levels) abolished these effects (Fig. 4b). Likewise, insulin-stimulated glucose uptake increased by 1.4- and 3.5-fold in NT siRNA and TET3 siRNA-transfected myotubes, respectively. This insulin-sensitising effect of TET3 knockdown was abrogated when PGC-1α protein was restored to basal levels (Fig. 4c). Similar results were obtained from mouse C2C12 myotubes (Fig. 4d–f). Based on these results we conclude that TET3 negatively regulates mitochondrial respiration and insulin sensitivity in myocytes and that PGC-1α is a major mediator of this regulation.

PGC-1α is required for TET3-mediated regulation of insulin sensitivity. (a) Immunoblot of TET3 and PGC-1α from human primary myotubes transfected with NT siRNA, TET3 siRNA or TET3 siRNA plus PGC1A siRNA. (b) Mitochondrial respiration of human primary myotubes treated as in (a). n=8 in each group. (c) Glucose uptake of human myotubes transfected with NT siRNA, TET3 siRNA or TET3 siRNA plus PGC1A siRNA as in (a) in the absence (−) or presence (+) of insulin at 100 nmol/l. Results are presented as relative glucose uptake with values in the absence of insulin set as 1. n=3 in each group. (d) Immunoblot of TET3 and PGC-1α from mouse C2C12 myotubes transfected with NT siRNA, Tet3 siRNA or Tet3 siRNA plus Pgc1a siRNA. (e) Mitochondrial respiration of mouse C2C12 myotubes treated as in (d). (f) Glucose uptake of mouse C2C12 myotubes transfected with NT siRNA, Tet3 siRNA or Tet3 siRNA plus Pgc1a siRNA as in (d) in the absence (−) or presence (+) of insulin at 100 nmol/l. n=3 in each group. Data are representative of two independent transfection experiments. *p<0.05, **p<0.01, ***p<0.001 (b and e, two-way ANOVA with Sidak post-test; c and f, two-tailed Student’s t test). FCCP, carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone; OCR, oxygen consumption rate
TET3 destabilises PGC-1α protein
Because PGC-1α regulates mitochondrial respiration and insulin sensitivity and because TET3 knockdown affected these processes without altering the mRNA abundance of PGC-1α, we hypothesised that TET3 affects PGC-1α stability. Thus, TET3 was knocked down in C2C12 myoblasts (Fig. 5a), followed by time course analysis of PGC-1α in the presence of CHX, a protein synthesis inhibitor. PGC-1α became more stable in Tet3 siRNA-transfected as compared with NT siRNA-transfected cells (Fig. 5b). The levels of PGC-1α were reduced to ~80% and 20% of steady-state levels in Tet3 siRNA-treated and NT siRNA-treated cells, respectively (Fig. 5c), suggesting that TET3 knockdown leads to stabilisation of PGC-1α.

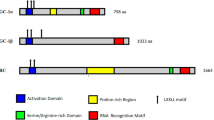
TET3 interacts with and destabilises PGC-1α. (a, b) C2C12 myoblasts were transfected with NT siRNA or Tet3 siRNA. After 48 h, proteins were isolated and TET3 expression was measured by immunoblotting (a). To perform time course analysis, CHX was added at a final concentration of 50 μg/ml and proteins were harvested at the indicated time points, followed by immunoblotting for PGC-1α and GAPDH (b). (c) Quantification of (b); the dotted line indicates the trendline. (d) Schematic of PGC-1α protein domain organisation, showing the activation domain, repression domain, arginine–serine-rich domain (RS) and RNA recognition motif (RRM). Numbers represent amino acids. The blue vertical lines represent phosphorylation at Thr262, Ser265 and Thr298. Not drawn to scale. (e) Mouse GAS tissues were used for immunoprecipitation using preimmune IgG, anti-TET3 or anti-PGC-1α. Representative immunoblots are shown. (f) C2C12 myoblasts were transfected with NT siRNA or Tet3 siRNA as in (a). After 48 h, proteins were isolated and analysed by immunoblotting using antibodies specific for total PGC-1α and PGC-1α phosphorylated at S265 and T298, respectively. (g) Quantification of (f). Data are presented as mean ± SEM. *p<0.05, **p<0.01 (two-tailed Student’s t test). Data are representative of two independent transfection experiments. IB, immunoblotting; IP, immunoprecipitation
PGC-1α is known to be targeted for ubiquitin-mediated proteolysis [49, 50]. Phosphorylation of PGC-1α at Thr262, Ser265 and Thr298 (Fig. 5d) by p38 mitogen-activated protein kinase (MAPK) results in an increase in its activity and protein stability [51,52,53]. We hypothesised that a complex between TET3 and PGC-1α might interfere with phosphorylation on these sites, accelerating PGC-1α degradation. First, we tested whether TET3 forms a complex with PGC-1α. We prepared lysates from mouse GAS muscle tissues, followed by immunoprecipitation using antibodies specific for TET3 [34, 54, 55] and PGC-1α, respectively. When TET3 was immunoprecipitated using anti-TET3 antibodies, we observed an enrichment of PGC-1α in the TET3-containing protein complex (Fig. 5e). Reciprocally, TET3 was readily detected in PGC-1α -containing protein complex (Fig. 5e). These results support complex formation between TET3 and PGC-1α in myocytes. Next, we analysed effects of TET3 knockdown on phosphorylation at S265 and T298 in C2C12 myoblasts using antibodies that specifically recognise these sites [53]. When TET3 was downregulated (Fig. 5a), increased phosphorylation at both sites (pS265 and pT298) was observed (Fig. 5f,g). Based on these results, we conclude that TET3 induces PGC-1α degradation at least in part by inhibiting phosphorylation at both sites.
Discussion
In this work we show that TET3 plays an important role in the regulation of skeletal muscle insulin sensitivity. We identify PGC-1α as a major downstream effector of TET3 in the regulation of mitochondrial respiration and insulin-stimulated glucose uptake in both human and mouse myocytes. PGC-1α has previously been shown to be activated and stabilised by p38 MAPK phosphorylation at Thr262, Ser265 and Thr298 [51,52,53]. We demonstrate that TET3 complexes with PGC-1α and prevents its phosphorylation on Ser265 and Thr298, thereby accelerating protein degradation. This TET3-mediated post-translational regulation of PGC-1α in muscle insulin sensitivity is further supported by our observation that in skeletal muscles of humans with type 2 diabetes and mouse models of type 2 diabetes, there is an increase in TET3 expression and a decrease in PGC-1α protein as compared with non-diabetic controls.
Despite extensive studies of TETs in development, stem cells, malignancies and immunity [21, 56], their roles in metabolic regulation have just begun to be recognised. In mouse adipocytes, TET1 was found to act in concert with histone deacetylase 1 to epigenetically suppress thermogenic gene transcription. Phenotypically, adipocyte-specific Tet1 knockout increased energy expenditure and protected against diet-induced obesity and insulin resistance [27]. Likewise, adipose-specific deletion of all three Tet genes in mice enhanced β-adrenergic responses, increased energy expenditure, and prevented obesity [28]. In pancreas beta cells, eliminating TET2 reduced pathological immune cell activation and beta cell killing during type 1 diabetes [57]. We reported a chronic increase in TET3 expression in the livers of humans and mice with type 2 diabetes and that TET3 induced Hnf4a promoter demethylation leading to heightened hepatic glucose production and hence hyperglycaemia [34, 35]. In addition, we documented that CRISPR-mediated genetic ablation of Tet3 specifically in AgRP neurons in the mouse hypothalamus induced hyperphagia, systemic insulin resistance, obesity and type 2 diabetes [36]. Mechanistically, TET3 deficiency led to AgRP neuron activation and coordinated upregulation of Agrp, Npy and the vesicular γ-aminobutyric acid (GABA) transporter gene Slc32a1. Specifically, we demonstrated a dynamic association of TET3 with the Agrp promoter in response to leptin signalling, which induced 5hmC modification and association of a chromatin-modifying complex leading to transcription inhibition [36]. In the current manuscript we uncover a new role for TET3 in energy homeostasis in yet another major metabolic tissue, the skeletal muscle. However, the model of action of TET3 in this tissue is completely different from that previously defined in other tissues and with other TET family members. Instead of regulating target gene expression at the epigenetic level, TET3 post-translationally targets PGC-1α protein for degradation through inhibiting its phosphorylation. Our data support the notion that TET3 and PGC-1α are in a complex together but do not address whether they directly interact with each other. Nor it is clear how TET3 inhibits PGC-1α phosphorylation, given that TET3 is not a phosphatase. Does PGC-1α contain a binding motif that supports TET3 association? Does association with TET3 lead to altered subcellular localisation of PGC-1α such that it cannot be phosphorylated? A clear understanding of the mechanism of action of TET3 in regulation of PGC-1α stability warrants future in-depth investigation. An additional limitation of our study is that GAPDH is not an optimal loading control for skeletal muscle due to the plasticity in the expression of glycolytic pathway genes.
Increased expression of TETs in peripheral metabolic organs (i.e. liver, adipose tissue, pancreas and skeletal muscle) appears to have adverse effects on metabolic homeostasis [27, 28, 34, 35, 57]. Interestingly, this does not seem to be the case in the central nervous system [36], highlighting tissue/cell-specific functions of TETs. Finally, dysregulation of PGC-1α has been connected to many human diseases, such as Huntington’s disease, amyotrophic lateral sclerosis, heart failure, and Duchenne muscular dystrophy [58,59,60,61,62,63]. Our results demonstrating that TET3 regulates PGC-1α may have a broader impact on the prevention and treatment of other human diseases.
Abbreviations
- AgRP:
-
Agouti-related protein
- CHX:
-
Cycloheximide
- GAS:
-
Gastrocnemius (muscle)
- HFD:
-
High-fat diet
- HNF4α:
-
Hepatocyte nuclear factor 4α
- HRP:
-
Horseradish peroxide
- HSA:
-
Human α skeletal muscle actin
- HSkM:
-
Human skeletal myoblasts
- MAPK:
-
Mitogen-activated protein kinase
- mKD:
-
Muscle-specific Tet3 knockout
- NT siRNA:
-
Non-targeting control siRNA
- PGC-1α:
-
Peroxisome proliferator-activated receptor-γ coactivator α
- RC:
-
Regular chow
- RT-qPCR:
-
Real-time quantitative PCR
- TET:
-
Ten-eleven translocation
- WT:
-
Wild-type
References
DeFronzo RA, Ferrannini E, Groop L et al (2015) Type 2 diabetes mellitus. Nat Rev Dis Primers 1:15019. https://doi.org/10.1038/nrdp.2015.19
Bluher M (2019) Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol 15(5):288–298. https://doi.org/10.1038/s41574-019-0176-8
Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn CR (1992) Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet 340(8825):925–929. https://doi.org/10.1016/0140-6736(92)92814-v
Rothman DL, Magnusson I, Cline G et al (1995) Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci U S A 92(4):983–987. https://doi.org/10.1073/pnas.92.4.983
Petersen MC, Shulman GI (2018) Mechanisms of insulin action and insulin resistance. Physiol Rev 98(4):2133–2223. https://doi.org/10.1152/physrev.00063.2017
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92(6):829–839. https://doi.org/10.1016/s0092-8674(00)81410-5
Lin J, Handschin C, Spiegelman BM (2005) Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 1(6):361–370. https://doi.org/10.1016/j.cmet.2005.05.004
Chan MC, Arany Z (2014) The many roles of PGC-1alpha in muscle–recent developments. Metabolism 63(4):441–451. https://doi.org/10.1016/j.metabol.2014.01.006
Lukaszuk B, Kurek K, Miklosz A, Zendzian-Piotrowska M, Chabowski A (2015) The role of PGC-1alpha in the development of insulin resistance in skeletal muscle - revisited. Cell Physiol Biochem 37(6):2288–2296. https://doi.org/10.1159/000438584
Benton CR, Wright DC, Bonen A (2008) PGC-1alpha-mediated regulation of gene expression and metabolism: implications for nutrition and exercise prescriptions. Appl Physiol Nutr Metab 33(5):843–862. https://doi.org/10.1139/H08-074
Miura S, Kai Y, Ono M, Ezaki O (2003) Overexpression of peroxisome proliferator-activated receptor gamma coactivator-1alpha down-regulates GLUT4 mRNA in skeletal muscles. J Biol Chem 278(33):31385–31390. https://doi.org/10.1074/jbc.M304312200
Choi CS, Befroy DE, Codella R et al (2008) Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc Natl Acad Sci U S A 105(50):19926–19931. https://doi.org/10.1073/pnas.0810339105
Benton CR, Nickerson JG, Lally J et al (2008) Modest PGC-1alpha overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in subsarcolemmal, not intermyofibrillar, mitochondria. J Biol Chem 283(7):4228–4240. https://doi.org/10.1074/jbc.M704332200
Mathai AS, Bonen A, Benton CR, Robinson DL, Graham TE (2008) Rapid exercise-induced changes in PGC-1alpha mRNA and protein in human skeletal muscle. J Appl Physiol (1985) 105(4):1098–1105. https://doi.org/10.1152/japplphysiol.00847.2007
Benton CR, Holloway GP, Han XX et al (2010) Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1alpha) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia 53(9):2008–2019. https://doi.org/10.1007/s00125-010-1773-1
Patti ME, Butte AJ, Crunkhorn S et al (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100(14):8466–8471. https://doi.org/10.1073/pnas.1032913100
Cheng Z, Tseng Y, White MF (2010) Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab 21(10):589–598. https://doi.org/10.1016/j.tem.2010.06.005
Mootha VK, Lindgren CM, Eriksson KF et al (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34(3):267–273. https://doi.org/10.1038/ng1180
Szendroedi J, Phielix E, Roden M (2011) The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 8(2):92–103. https://doi.org/10.1038/nrendo.2011.138
Wu X, Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18(9):517–534. https://doi.org/10.1038/nrg.2017.33
Lio CJ, Yue X, Lopez-Moyado IF, Tahiliani M, Aravind L, Rao A (2020) TET methylcytosine oxidases: new insights from a decade of research. J Biosci 45:21. https://doi.org/10.1007/s12038-019-9973-4
Chen Q, Chen Y, Bian C, Fujiki R, Yu X (2013) TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493(7433):561–564. https://doi.org/10.1038/nature11742
Vella P, Scelfo A, Jammula S et al (2013) Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell 49(4):645–656. https://doi.org/10.1016/j.molcel.2012.12.019
Zhang Q, Zhao K, Shen Q et al (2015) Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525(7569):389–393. https://doi.org/10.1038/nature15252
Xue S, Liu C, Sun X et al (2016) TET3 inhibits type I IFN production independent of DNA demethylation. Cell Rep 16(4):1096–1105. https://doi.org/10.1016/j.celrep.2016.06.068
Guan W, Guyot R, Samarut J, Flamant F, Wong J, Gauthier KC (2017) Methylcytosine dioxygenase TET3 interacts with thyroid hormone nuclear receptors and stabilizes their association to chromatin. Proc Natl Acad Sci U S A 114(31):8229–8234. https://doi.org/10.1073/pnas.1702192114
Damal Villivalam S, You D, Kim J et al (2020) TET1 is a beige adipocyte-selective epigenetic suppressor of thermogenesis. Nat Commun 11(1):4313. https://doi.org/10.1038/s41467-020-18054-y
Byun S, Lee CH, Jeong H et al (2022) Loss of adipose TET proteins enhances beta-adrenergic responses and protects against obesity by epigenetic regulation of beta3-AR expression. Proc Natl Acad Sci U S A 119(26):e2205626119. https://doi.org/10.1073/pnas.2205626119
Pan W, Zhu S, Qu K et al (2017) The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity 47(2):284-297 e285. https://doi.org/10.1016/j.immuni.2017.07.020
Montalban-Loro R, Lozano-Urena A, Ito M et al (2019) TET3 prevents terminal differentiation of adult NSCs by a non-catalytic action at Snrpn. Nat Commun 10(1):1726. https://doi.org/10.1038/s41467-019-09665-1
Tanaka S, Ise W, Inoue T et al (2020) Tet2 and Tet3 in B cells are required to repress CD86 and prevent autoimmunity. Nat Immunol 21(8):950–961. https://doi.org/10.1038/s41590-020-0700-y
Wang H, Huang Y, Yu M et al (2021) Muscle regeneration controlled by a designated DNA dioxygenase. Cell Death Dis 12(6):535. https://doi.org/10.1038/s41419-021-03817-2
Zhang H, Wang S, Zhou Q et al (2022) Disturbance of calcium homeostasis and myogenesis caused by TET2 deletion in muscle stem cells. Cell Death Discov 8(1):236. https://doi.org/10.1038/s41420-022-01041-1
Li D, Cao T, Sun X et al (2020) Hepatic TET3 contributes to type-2 diabetes by inducing the HNF4a fetal isoform. Nat Commun 11(1):342. https://doi.org/10.1038/s41467-019-14185-z
Xie D, Chen F, Zhang Y et al (2022) Let-7 underlies metformin-induced inhibition of hepatic glucose production. Proc Natl Acad Sci U S A 119(14):e2122217119. https://doi.org/10.1073/pnas.2122217119
Xie D, Stutz B, Li F et al (2022) TET3 epigenetically controls feeding and stress response behaviors via AGRP neurons. J Clin Invest 132(19):e162365. https://doi.org/10.1172/JCI162365
Pihlajamaki J, Lerin C, Itkonen P et al (2011) Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab 14(2):208–218. https://doi.org/10.1016/j.cmet.2011.06.007
Jin W, Goldfine AB, Boes T et al (2011) Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J Clin Invest 121(3):918–929. https://doi.org/10.1172/JCI41940
Varemo L, Henriksen TI, Scheele C et al (2017) Type 2 diabetes and obesity induce similar transcriptional reprogramming in human myocytes. Genome Med 9(1):47. https://doi.org/10.1186/s13073-017-0432-2
Song J, Lv H, Liu B et al (2023) Let-7 suppresses liver fibrosis by inhibiting hepatocyte apoptosis and TGF-beta production. Mol Metab 78:101828. https://doi.org/10.1016/j.molmet.2023.101828
Lv H, Catarino J, Li D et al (2023) A small-molecule degrader of TET3 as treatment for anorexia nervosa in an animal model. Proc Natl Acad Sci U S A 120(16):e2300015120. https://doi.org/10.1073/pnas.2300015120
Miniou P, Tiziano D, Frugier T, Roblot N, Le Meur M, Melki J (1999) Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res 27(19):e27. https://doi.org/10.1093/nar/27.19.e27
Wang G, Yu Y, Cai W et al (2020) Muscle-specific insulin receptor overexpression protects mice from diet-induced glucose intolerance but leads to postreceptor insulin resistance. Diabetes 69(11):2294–2309. https://doi.org/10.2337/db20-0439
Long YC, Cheng Z, Copps KD, White MF (2011) Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol Cell Biol 31(3):430–441. https://doi.org/10.1128/MCB.00983-10
Fernandez-Marcos PJ, Auwerx J (2011) Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93(4):884S – 890. https://doi.org/10.3945/ajcn.110.001917
Luo X, Liao C, Quan J et al (2019) Posttranslational regulation of PGC-1alpha and its implication in cancer metabolism. Int J Cancer 145(6):1475–1483. https://doi.org/10.1002/ijc.32253
Ruan HB, Han X, Li MD et al (2012) O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab 16(2):226–237. https://doi.org/10.1016/j.cmet.2012.07.006
Genders AJ, Holloway GP, Bishop DJ (2020) Are alterations in skeletal muscle mitochondria a cause or consequence of insulin resistance? Int J Mol Sci 21(18):6948. https://doi.org/10.3390/ijms21186948
Olson BL, Hock MB, Ekholm-Reed S et al (2008) SCFCdc4 acts antagonistically to the PGC-1alpha transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev 22(2):252–264. https://doi.org/10.1101/gad.1624208
Trausch-Azar J, Leone TC, Kelly DP, Schwartz AL (2010) Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1{alpha} via the N-terminal pathway. J Biol Chem 285(51):40192–40200. https://doi.org/10.1074/jbc.M110.131615
Puigserver P, Rhee J, Lin J et al (2001) Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell 8(5):971–982. https://doi.org/10.1016/s1097-2765(01)00390-2
Fan M, Rhee J, St-Pierre J et al (2004) Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev 18(3):278–289. https://doi.org/10.1101/gad.1152204
Roth RJ, Le AM, Zhang L et al (2009) MAPK phosphatase-1 facilitates the loss of oxidative myofibers associated with obesity in mice. J Clin Invest 119(12):3817–3829. https://doi.org/10.1172/JCI39054
Cao T, Jiang Y, Wang Z et al (2019) H19 lncRNA identified as a master regulator of genes that drive uterine leiomyomas. Oncogene 38(27):5356–5366. https://doi.org/10.1038/s41388-019-0808-4
Xu Y, Sun X, Zhang R et al (2020) A positive feedback loop of TET3 and TGF-beta1 promotes liver fibrosis. Cell Rep 30(5):1310-1318 e1315. https://doi.org/10.1016/j.celrep.2019.12.092
Gerecke C, Egea Rodrigues C, Homann T, Kleuser B (2022) The role of ten-eleven translocation proteins in inflammation. Front Immunol 13:861351. https://doi.org/10.3389/fimmu.2022.861351
Rui J, Deng S, Perdigoto AL et al (2021) Tet2 controls the responses of beta cells to inflammation in autoimmune diabetes. Nat Commun 12(1):5074. https://doi.org/10.1038/s41467-021-25367-z
Eschbach J, Schwalenstocker B, Soyal SM et al (2013) PGC-1alpha is a male-specific disease modifier of human and experimental amyotrophic lateral sclerosis. Hum Mol Genet 22(17):3477–3484. https://doi.org/10.1093/hmg/ddt202
Weydt P, Pineda VV, Torrence AE et al (2006) Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab 4(5):349–362. https://doi.org/10.1016/j.cmet.2006.10.004
Arany Z, He H, Lin J et al (2005) Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 1(4):259–271. https://doi.org/10.1016/j.cmet.2005.03.002
Lin J, Wu PH, Tarr PT et al (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119(1):121–135. https://doi.org/10.1016/j.cell.2004.09.013
Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM (2007) PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev 21(7):770–783. https://doi.org/10.1101/gad.1525107
Arany Z (2008) PGC-1 coactivators and skeletal muscle adaptations in health and disease. Curr Opin Genet Dev 18(5):426–434. https://doi.org/10.1016/j.gde.2008.07.018
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Acknowledgements
We thank A. Rao from La Jolla Institute for Immunology for the gift of Tet3fl/fl mice, M. Zhong from Yale Stem Cell Center Genomics Core for assistance with RNA sequencing, and R. Cardone at the Yale IOMIC for mitochondrial function analyses (Seahorse) of human and mouse myotubes.
Data availability
Sequences are available from the Gene Expression Omnibus with accession number of GSE224042. All other study data are included in the article and/or ESM.
Funding
This work was supported by a grant from the National Institute of Health (R01 DK124321 to YH). The study funder was not involved in the design of the research; the collection, analysis and interpretation of data; writing the report; and did not impose any restrictions regarding the publication of the report.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
YH conceived and directed the studies and wrote the manuscript. BL, DX, XH and YD performed the experiments, analysed the data and helped with manuscript preparation. SJ and SD designed and performed the hyperinsulinaemic–euglycaemic clamp studies, performed data analyses and critically reviewed the manuscript. XS and DL carried out bioinformatic analyses and critically reviewed the manuscript. AMB provided the phospho-specific antibodies to Ser265 and Thr298 of PGC-1α, performed data interpretation and critically read the manuscript. All authors have read and approved the final version of the manuscript. YH is responsible for the integrity of the work as a whole.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, B., Xie, D., Huang, X. et al. Skeletal muscle TET3 promotes insulin resistance through destabilisation of PGC-1α. Diabetologia 67, 724–737 (2024). https://doi.org/10.1007/s00125-023-06073-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-023-06073-5