
Abstract
The use of chimeric antigen receptor T (CAR-T) cells is a significant therapeutic improvement increasing the prognosis for patients with a variety of hematological malignancies. However, this therapy has also sometimes life-threatening, complications. Therefore, knowledge of the treatment and management of these complications, especially in treatment centers and intensive care units, respectively, is of outstanding importance. This review provides recommendations for the diagnosis, management, and treatment of CAR-T cell-associated complications such as cytokine release syndrome, immune effector cell associated neurotoxicity syndrome, hematotoxicity, hypogammaglobulinemia, and CAR-T cell-induced pseudo-progression amongst others for physicians treating patients with CAR-T cell-associated complications and intensivists.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
This review summarizes the current state of side effects in the growing field of chimeric antigen receptor T (CAR-T) cell therapies. It classifies the most common side effects, such as cytokine release syndrome, immune effector cell-associated neurotoxicity syndrome, hematotoxicity, hypogammaglobulinemia, and CAR-T cell-induced pseudo-progression, among others, and provides a practical guide for orientation in clinical practice. |
Introduction
Chimeric antigen receptor T-cell (CAR-T cell) therapies represent a significant advancement in cancer immunotherapy, offering remarkable response rates and durable remissions in patients with hematological malignancies [1, 2]. While expanding therapeutic options, CAR-T cell therapy introduces a unique set of potential side effects and complications due to its specific immunological mechanisms. These adverse events range from cytokine release syndrome (CRS) [3] to neurotoxicity, following a typical timeline and posing challenges in therapy. Managing these side effects requires a multidisciplinary team, including specialists from stem cell transplantation units, intensive care unit (ICU), neurology amongst others. Despite advancements in treatment strategies, knowledge gaps remain regarding the pathophysiology and management of many complications, emphasizing the need for well-trained intensivists to handle patients with severe complications [4]. This review aims to provide a detailed understanding of CAR-T cell therapy's side effects and management approaches.
Immunological function of CAR-T cell therapy
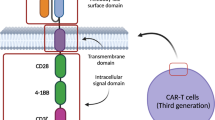
CAR-T cell therapy leverages the immune system to target and destroy cancer cells. T cells are collected and engineered to express a chimeric antigen receptor through viral vectors or gene-editing techniques. CAR-T cells are activated upon binding to cancer cells displaying specific antigens like CD19, B-cell maturation antigen (BCMA), or G protein–coupled receptor class C group 5 member D (GPRC5D) [5, 6], initiating signaling for targeted tumor recognition and CAR-T cell activation [5, 7]. This activation triggers the release of cytotoxic molecules leading to cancer cell death [8], recruits immune components for enhanced response [5, 7], and can differentiate into memory T-cells for long-term surveillance and potential recurrence protection [9, 10]. Co-stimulatory domains like CD28 and 4-1BB in CAR constructs enhance CAR-T cell function and persistence by providing additional signaling cues [11, 12], leading to varied molecular and functional outcomes [13]. CD28 promotes rapid T-cell expansion and 4-1BB enhances cell survival, memory-like phenotype development, and sustained anti-tumor responses [9,10,11]. Differences in these domains affect therapeutic efficacy and persistence of CAR-T cell products [12] and vary the severity of acute side effects, requiring careful consideration in clinical applications [7].
Clinical procedure of CAR-T cell therapy
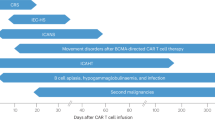
Lymphapheresis leads to extraction of patients lymphocytes, which are then separated, genetically modified, and expanded to create CAR-T cells. The period from apheresis to infusion requires planned bridging therapy, aiming for remission at infusion to enhance outcomes and reduce side effects. CAR-T cell therapy necessitates an immune-modulated environment, making lymphodepletion (LD) essential immediately before CAR-T cell therapy. Lymphodepletion, usually including cyclophosphamide (Cy) and fludarabine (Flu), is critical for CAR-T cell therapy success. It significantly improves response rates [14, 15] and CAR-T cell persistence [9] by altering cytokine profiles and suppressing competing immune cells, thus creating an optimal environment for CAR-T cell expansion [9, 16]. Lymphodepletion predisposes patients to profound bone marrow suppression, leading to adverse events such as severe neutropenia, infection risk, and delayed hematopoietic recovery. These side effects necessitate close monitoring and prompt management to mitigate complications. Lymphodepletion is typically administered from day -5 to day -3, followed by CAR-T cell infusion on day 0. Subsequently, patients are closely monitored for side effects like CRS, various neurotoxicities, hematotoxicity, infections or other adverse events, the extent of which depends on factors such as tumor burden, age, cell dosing, comorbidities, timing and the specific CAR-T cell product used (supplementary Table 1). The period following the end of aplasia is particularly high-risk for neurological complications as well as for CRS. Continuous electrocardiogram (ECG) and vital parameter monitoring in the first 10 days post-therapy are important. Figure 1 illustrates a simplified cartoon of the apheresis, production, and infusion of CAR-T cells.

The process preceding CAR-T cell therapy. 1) First, a lymphapheresis is performed through a large-volume venous access. 2) This is followed by the transduction of a chimeric antigen receptor (CAR) to the collected T-cells and 3) in vitro expansion of CAR-T cells. It is important to note that various generations of CAR-T cell receptors are now available, differing depending on the CAR-T cell product with different approvals. 4) The infusion of the CAR-T cells occurs after a few weeks. It is important to consider that, especially in highly proliferative diseases, other bridging therapies are administered in the time between lymphapheresis and CAR-T cell infusion. Furthermore, the infusion of CAR-T cells only occurs after a lymphodepleting chemotherapy has been applied
Cytokine release syndrome
Cytokine release syndrome (CRS) is a serious complication of CAR-T cell therapy, marked by excessive pro-inflammatory cytokine release, usually within days to a week after infusion, presenting with fever, fatigue, and flu-like symptoms. Triggered by CAR-T cells activated upon target antigen contact it leads to macrophage and other immune cell activation and cytokine release, notably IL-6, causing further immune activation, endothelial cell activation, capillary leakage, and tissue edema [3, 17].
This intense immune activation amplifies the inflammation and contributes to the severity of CRS symptoms, leading to a sepsis-imitating condition with hypotension, tachycardia, and organ dysfunction, with symptoms including fever resistant to antipyretics, hypotension unresponsive to intravenous (IV) fluids, respiratory distress, hypoxia, coagulopathy, renal, and hepatic impairment. The severity of CRS can be graded using various scoring systems [18, 19].
General approach to patients with CRS
Since the molecular pathophysiology of CRS is relatively well understood there is a possibility to directly interrupt the cascade via interleukin (IL)-6-receptor (IL-6-R) inhibition, e.g. through tocilizumab [17, 20]. However, low grade CRS might be treated symptomatically. Whenever patients develop temperatures above 38 °C after CAR-T cell therapy, CRS should be considered, but infections must also be ruled out, especially after lymphodepleting chemotherapy which might induce neutropenia, and therefore cause neutropenic fever [21]. If the focus is unclear, a Pseudomonas-effective antibiotic (piperacillin/tazobactam, imipenem, meropenem, cefepime, or ceftazidime) must be initiated within one hour [21]. Given the complexity of distinguishing between sepsis and CRS in clinical practice, including high cytokine levels [22], and considering that approximately 10% of patients develop sepsis which can mimic CRS [23], it is not uncommon for patients to require empiric treatment for both conditions simultaneously. Following a rapid differential diagnosis for other causes of symptoms such as fever, hypotension, hemodynamic instability, or shortness of breath, but also considering often preceding prodromal symptoms like tachycardia or tachypnea, the (suspected) diagnosis of CRS can be made, allowing for prompt treatment.
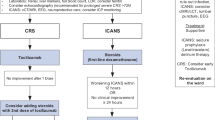
Diagnosis and treatment of patients with CRS according to CRS grade
The ICU should be notified before CAR-T cell therapy to ensure timely intervention and patient safety. The management of CRS after CAR-T cell therapy is stratified by symptom severity into four grades, each dictating specific interventions.
Grade 1 CRS involves mild symptoms like fever without oxygen or vasopressor needs. Management includes supportive care, monitoring vital signs every 2 h and antipyretic medication, e.g. acetaminophen 1000 mg IV. For slight hypotension, IV crystalloid fluid substitution is considered [3].
Grade 2 CRS is characterized by moderate symptoms, such as fever with hypotension or oxygen supplementation (≤ 6L/min). Treatment involves tocilizumab (8 mg/kg, repeatable after 8 h, up to 4 doses daily) and, for axicabtagene-ciloleucel, early steroid intervention with dexamethasone 10 mg IV as a single dose once grade 2 CRS criteria are met [24] or if hypotension persists despite tocilizumab. Supportive care and ICU readiness for rapid progression are crucial; ICU should be informed when CRS progresses from grade 1 to grade 2 to prepare for potential transfer [25].
Grade 3 CRS indicates severe symptoms, requiring ICU transfer for high fever and hypotension that needs vasopressor support or oxygen (> 6L/min). Management includes continuous vital sign monitoring, dexamethasone (10 mg IV every 6 h (q6h), with increasing the dose to 20 mg in case no improvement occurs), and aggressive IV fluids according to the ‘fluid challenge' principle, alongside tocilizumab treatment [26].
Grade 4 CRS represents life-threatening conditions, necessitating multiple vasopressors or intubation for severe hypoxia. Antibiotic therapy should be adjusted according to ICU-specific risks of nosocomial infections [27]. High-dose corticosteroids (1000 mg methylprednisolone daily) and intensive care is mandatory. Anakinra can be used for refractory CRS at doses up to 12 mg/kg/day, especially in grade 4 or high-grade cases unresponsive to other treatments [28]. For refractory to third-line CRS, cyclophosphamide (1.5 g/m [2]) might be administered to irreversibly impair CAR-T cells [29].
Figure 2 depicts the grading, diagnostic measures, and management of CRS.

The various gradings and respective measures for the most common side effect of CAR-T cell therapy—cytokine release syndrome (CRS). For a CRS I°, a systemic infection should be ruled out, and treatment with antipyretics and IV cristalloids should be administered. A CRS II° is defined by a temperature above 38 °C, decreased blood pressure, and/or the need for oxygen therapy with ≤ 6L/min. For a CRS II°, additionally the administration of tocilizumab 8 mg/kg IV should be carried out. In CRS III°, the administration of a vasopressor is necessary and/or oxygen therapy with > 6L/min is required. Transfer to the intensive care unit is necessary and intravenous administration of dexamethasone is performed. For CRS IV°, the administration of > 1 vasopressor is required, and intubation or PAP is often necessary. In addition to the previously mentioned therapy, the administration of intravenous methylprednisolone must be performed
Resolution and recovery of CRS
Appropriate CRS management can resolve symptoms within days to weeks as inflammation diminishes and cytokine levels fall, allowing for patient recovery. During recovery, vital signs should be monitored every 8 h to identify potential complications like neutropenic fever or CRS relapse. Understanding CRS stages is key for prompt management, thus preventing severe complications. Ongoing research aims to identify predictive biomarkers and enhance treatment strategies, improving CAR-T therapy safety.
Immune effector cell–associated neurotoxicity syndrome
Immune effector cell–associated neurotoxicity syndrome (ICANS) is an acute reaction usually occurring later than CRS but mostly also within the first 10 days after CAR-T cell therapy [25, 30]. ICANS represents a neurological and/or neuropsychiatric syndrome and can vary in severity. Starting with the CAR-T cell infusion, assessment of the ICE-score, measuring orientation, language, attention, and handwriting, (see below) is recommended. ICANS almost always follows CRS; thus, new neurological complications without preceding CRS necessitate differential diagnosis to rule out other causes, as detailed in the ICANS grading section. ICANS grading, and diagnostic and therapeutic measures are summarized in Fig. 3.

Grading and therapy of CAR-T cell-associated neurotoxicity (ICANS). In ICANS I°, an ICE score of 7–9 out of a total of 10 points is mandatory. This is determined with a simple questionnaire. Grade 1 indicates a mild disturbance of consciousness, where the patient is spontaneously arousable. Close monitoring of the patient should be carried out, and the prophylactic application of antiepileptics can be considered. ICANS II° is characterized by a disturbance of consciousness where the patient is arousable by speech. The application of intravenous dexamethasone must be carried out. Additionally, aspiration prophylaxis should be considered, and medications should no longer be administered orally. Cerebral imaging should be performed, and a lumbar puncture should be discussed. In ICANS III°, the disturbance of consciousness is more severe, and the patient only responds to tactile stimulus. Epileptic seizures may occur but respond to intervention. A focal cerebral edema in imaging may also be present. Transfer to the intensive care unit should take place, and the application of methylprednisolone must be carried out. In ICANS IV°, severe disturbance of consciousness is observed, where the patient is not spontaneously arousable and only responds to repetitive tactile stimuli. Epileptic seizures are life-threatening. Higher degree motor deficits (hemi- or paraparesis) and diffuse cerebral edema in imaging may occur, accompanied by symptoms such as decorticate or decerebrate rigidity, abducens palsy, papilledema, or Cushing’s reflex (increased intracranial pressure, increased blood pressure, decreased heart rate). The placement of intracranial pressure monitoring should and plasmapheresis may be considered. Additionally, further immunosuppressive drug strategies with anakinra IV amongst others may be applied
ICANS grading
The prodromal phase of ICANS occurs within days post-CAR-T cell infusion, presenting with mild neurological symptoms like headache, confusion, and subtle cognitive changes, alongside hypertension and tachycardia. As these symptoms may be overlooked, rigorous testing, including the immune effector cell encephalopathy (ICE)-score is necessary. If organ system failure (e.g., due to CRS) makes ICE-score monitoring unfeasible, brain imaging should exclude cerebral edema, electroencephalograms (EEGs) and the bispectral index (BIS) can monitor the level of consciousness and rule out epileptogenic potentials. Intracranial pressure monitoring should be initiated in sedated patients with unexplained bradycardia, hypertension, or anisocoria. Intracranial pressure monitoring should be initiated in sedated patients with unexplained bradycardia, hypertension, or anisocoria. Grade 1 ICANS symptoms are mild, including slower speech, confusion, difficulty concentrating, and mild headaches, with an ICE-score of 7–9 indicating grade 1. Diagnosis involves neurological exams while treatment emphasizes supportive care, symptom management, and seizure prophylaxis with 750 mg levetiracetam QD [31]. For grade 2 ICANS (ICE-score 3–6), diagnostic imaging, rule-out-EEG are crucial. Lumbar puncture should be discussed in a case-by-case situation. Since patients after CAR T-cell therapy are deeply immunocompromised, infectious differential diagnoses (i.e., infectious meningitis/encephalitis) must be considered. When suspected a rigorous diagnostic methodology should be performed. Thus, initial combinatorial treatment, with dexamethasone (and tocilizumab if CRS co-incides [25]) and treatment of suspected infectious encephalopathy (i.e., meningitis or encephalitis) should be considererd – de-escalation of one treatment path should occur once pending test results eliminate a differential diagnosis or undermine implausibility. Grade 3 and 4 ICANS require intensive diagnostic measures and treatments, including high-dose corticosteroids, intracranial pressure monitoring, ensuring adequate oxygenation, normotension, normovolemia, normocapnia, normoglycemia and normothermia. Intracranial pressure (ICP) should be kept < 20 mmHg, with a target cerebral perfusion pressure (CPP) of 60–70 mmHg, and elevated ICP can be reduced with mannitol or moderate hyperventilation. [32]. Tocilizumab should be discontinued if neurological signs develop due to the risk of IL-6 accumulation in the cerebrospinal fluid (CSF) [33, 34]. In highly proliferative diseases like diffuse large-cell B-cell lymphoma (DLBCL) and B-acute lymphoblastic leukemia (B-ALL) earlier escalation of corticosteroid dosage or switch to methylprednisolone can also prevent severe side effects [35]. For life-threatening grade 4 ICANS, aggressive management in ICU, high-dose methylprednisolone, and antiepileptic treatment for seizures are mandatory [25, 36]. Anakinra should be considered in steroid-refractory high-grade ICANS (1 mg/kg QID with an upper dose of 100 mg) [28, 37]. The use of cyclophosphamide, as described above, could potentially be considered, if there are no other options available. However, there is very limited data about this therapeutic option [29].
Taken together, management of ICANS across grades involves multidisciplinary care and proactive interventions to mitigate severe outcomes (Fig. 4).

The development of handwriting malfunction during the rise and fall of immune effector cell-associated neurotoxicity syndrome (ICANS). The patient is asked to repeatedly write down the same standard sentence (e.g. here: “my favourite color is green”, in German: “Meine Lieblingsfarbe ist grün”)
Movement and neurocognitive treatment-emergent adverse events
Understanding neurological adverse events in CAR-T cell therapy remains limited. ICANS is a well-documented neurotoxic side effect, but novel CAR-T products, like ciltacabtagene-autoleucel, have been associated with movement and neurocognitive treatment-emergent adverse events (MNTs), as seen in the CARTITUDE-1 studies. These studies noted MNTs post-ICANS, indicating the need to differentiate between ICANS and MNTs based on clinical presentation, including movement and personality changes not typical of ICANS. Safety analysis of CARTITUDE-1 identified non-ICANS neurotoxic symptoms like movement changes and cognitive decline post-ICANS/CRS recovery [38]. Risk factors for MNTs involve high post-therapy CAR-T cell counts, pre-therapy elevated cytokine levels, and links to higher baseline tumor burden and severe CRS or ICANS, necessitating targeted preventive strategies. Treatment for MNTs has included steroids (methylprednisolone), IV immunoglobulins, and/or plasmapheresis, with essential neurology consultations. Differential diagnoses, like viral infections or alternative neurotoxicity causes, must be excluded.
CAR-T cell induced pseudoprogression
Pseudoprogression following CAR-T cell therapy, unlike the documented occurrence in checkpoint-inhibitor therapy (CPI) [39, 40], lacks a comprehensive understanding and description. While CPI-induced pseudoprogression benefits from established toxicity criteria such as RECIST, LUGANO and LYRIC [41], CAR-T cell therapy has seen only a few reported cases of transient progression followed by regression [42, 43]. In most cases reported thus far, CAR‐T cell induced pseudoprogression (CARTiPP) has been shown with clinical and/or radiological findings. Nonetheless, CARTiPP can have serious consequences if the tumor is anatomically located in a highly vulnerable region [43], such as near the central nervous system, large blood vessels, or circumscribed hollow organs. The incidence of CARTiPP and therapeutic strategies remain underexplored. Clinicians are advised to consider the potential impacts of tumor swelling due to pseudoprogression and evaluate bridging therapies like radiotherapy and/or immunochemotherapy pre-CAR-T treatment. For suspected CARTiPP, diagnostic measures should include circumference measurements and imaging to assess tissue blood flow. Treatment options, including corticosteroids, tocilizumab, or cyclophosphamide, require consultation with CAR-T experienced clinicians.
Tumor inflammation-associated neurotoxicity
Tumor inflammation-associated neurotoxicity (TIAN) is identified as a distinct neurotoxicity in patients with cerebral tumor locations, separate from systemic ICANS. It manifests as local complications such as increased intracranial pressure or seizures, often caused by local swelling from pseudoprogression or inflammation-induced changes in the cellular environment enhancing epileptogenicity [44, 45]. Clinically, distinguishing TIAN from CARTiPP and ICANS is challenging, with symptoms like fever, headaches, and neurological disturbances related to tumor location, but neuroimaging can reveal a progression of the original tumor lesion, unlike ICANS, which shows diffuse cerebral edema or other signs of encephalopathy. TIAN severity is classified into four grades, from mild symptoms to life-threatening conditions requiring urgent intervention. Management includes early radiological imaging, neurological consultation, EEG, vital sign monitoring, and treatments similar to ICANS, including dexamethasone, potentially escalated to methylprednisolone, anakinra IV for early stages, and consideration of external ventricular drainage. Anti-epileptics such as levetiracetam are recommended [36, 44]. The TIAN Grading system is summarized in supplementary Table 2.
Aside from the main complications such as CRS and ICANS, CAR-T cell therapy may induce additional harm. Unintended interactions with non-target antigens, referred to as off-target off-tumor (OTOT) toxicities, can lead to immune-related adverse effects. These OTOT toxicities may cause hematological or solid organ damage and provoke secondary complications. Notable examples include hematotoxicity, hypogammaglobulinemia, hemophagocytic lymphohistiocytosis (HLH), and macrophage activation syndrome (MAS), alongside potential organ-specific toxicities.
Immune effector cell–associated hematotoxicity
OTOT toxicities may disrupt hematopoiesis, known as immune effector cell–associated hematotoxicity (ICAHT), with proposed molecular explanations [46]. First identified in 2019 [47,48,49], most evidence pertains to CD19 CAR-induced cytopenias from tisagenlecleucel and axicabtagene-ciloleucel treatments. ICAHT typically exhibits a biphasic pattern, with initial recovery from neutropenia in half of patients and thrombocytopenia in one third, followed by relapsing cytopenia. This pattern correlates with therapy phases, initially post-lymphodepleting therapy and CRS or HLH peak, and a second phase without clear linkage to earlier events. Recovery timelines and severity vary by co-stimulatory domain. CD28 co-stimulatory axicabtagene-ciloleucel treatments show faster cytopenia recovery within 1–2 months, unlike longer hematotoxicity durations with tisagenlecleucel. Additionally, CD28-associated ICAHT cases are often more severe, with with higher occurrence of grade 3 or 4 ICAHT [50].
Rejeski et al. developed the CAR-HEMATOTOX model for clinical use, focusing on hematopoietic reserve (such as platelet count, hemoglobin, absolute neutrophil count [ANC] (> or < 1200/µL)) and baseline inflammation to assess risk for delayed cytopenia post-CAR-T cell therapy. This model highlights the importance of bone marrow reserve and pre-treatment inflammation in managing ICAHT risk [51]. Additionally, tumour necrosis factor (TNF)-α and CRP have been linked to early ICAHT onset [52], emphasizing the necessity for clinicians to evaluate causality, timing, and severity in treatment decisions. An overview of the CAR-HEMATOTOX score and a therapy algorithm is displayed in Table 1 and supplementary Table 3.
Therapy of immune effector cell–associated hematotoxicity
Infections are a significant adverse event from ICAHT, necessitating anti-infective prophylaxis. A survey revealed variability in handling high-risk HEMATOTOX scores, with some centers advocating for prophylactic bone marrow aspiration or biopsy [50]. However, investigations have shown increased grade ≥ 2 CRS in case of prophylactic granulocyte colony stimulating factor (G-CSF)- use, suggesting to limit the use as a therapeutic option for ICAHT [53]. Growth factors are the most common treatment across all ICAHT durations, with autologous stem cell boosts as a favored alternative for G-CSF refractory cases, with infused cell dosages ranging from 1.1 to 11.5 × 106 CD34 + /kg [50, 54]. In the absence of stem cell backup, thrombopoietin (TPO) agonists like romiplostim, with doses of 1 to 15 µg/kg weekly, are preferred [55]. For anemia, darbepoetin alfa starting at 2.25 μg/kg weekly is an option, but transfusions are often needed [50]. Early autologous stem cell boosts can improve one-year survival, negatively impacted by prolonged neutropenia [56]. Pulse dosing glucocorticoid therapy and prophylactic autologous stem cell collection for high hematoxicity risk patients are considered, with a collection target of 1 to 1.5 × 106 CD34 + cells/kg. In case of hematotoxicity that could be connected with high amounts of cytokines, like in CRS or infections, the IL-1-receptor antagonist anakinra should be considered, too. As there is no clear dosage recommendation for anakinra in the context of CAR-T cell therapy-associated ICAHT, therapy analogous to the recommended dosages for refractory CRS/ICANS of up to 12 mg/kg/day IV could be a possible option [28, 50]. Clinicians should monitor ICAHT grading closely, consider early G-CSF application, and screen for viremia and rule out HLH. For therapy-resistant ICAHT allogeneic HCT displays the most invasive therapeutic measure acknowledging that it will eliminate CAR-T cells [50].
Hypogammaglobulinemia and infections
Hypogammaglobulinemia (HGGA) emerges as a significant immunological complication following CAR-T cell therapy, with multifactorial pathophysiology largely attributed to the targeted destruction of B-cells by anti-CD-19 CARs. B-cell depletion by CAR-T cells diminishes gamma globulin levels, precipitating HGGA, defined as IgG < 700 mg/dL, IgA < 70 mg/dL, and IgM < 40 mg/dL. Within the initial 90 days post-therapy, up to 67% of patients may develop HGGA. The persistence of CAR-T cells, indicating ongoing activity, can lead to sustained HGGA. Additional factors contributing to B-cell aplasia and subsequent HGGA include systemic inflammation, complications like CRS, pre-CAR-T lymphodepletion, and prior immunochemotherapy-induced B-cell toxicity. The absence of a unified HGGA definition complicates incidence assessment post-CAR-T, yet the direct link between CAR-T cell therapy and HGGA is well documented [57, 58].
Comparing various clinically established CAR-T products, HGGA rates vary: 15%-40% after axicabtagene-ciloleucel, 14%-29% after tisagenleucel, about 32% after lisocabtagene-maraleucel, around 15% after brexucabtagene-autoleucel, and 41% after idecabtagene-vicleucel. Axicabtagene-ciloleucel shows higher HGGA rates compared to tisagenleucel, possibly due to its longer persistence. Multiple myeloma-targeted CAR-T therapies like idecabtagene-vicleucel and ciltacabtagene-autoleucel, targeting BCMA, have heterogenous HGGA rates. Data on CAR-related HGGA after anti-BCMA-CAR-T therapy are scarce, but BCMA-targeted CAR-T might attack long-living plasma cells, depleting polyclonal, healthy and gamma-globulin-producing plasma cells [58]. A retrospective study found no significant difference in total infection rates compared to similar CD19-targeting therapies, yet viral infections were more common in BCMA-CARs, while bacterial infections predominated in CD19-CARs [59].
Age significantly impacts HGGA rates, with pediatric patients showing higher incidences than adults or adolescents due to later development of immunoglobulin subclasses and plasma cell maturation [60]. Patients with HGGA may require immunoglobulin replacement therapy, recommended at 0.2–0.4 g/kg every 3 weeks, though more frequent substitution could be necessary under intensive care due to potentially higher immunoglobulin consumption. It is critical to prevent IgG levels from dropping below 450 mg/dL, with various polyvalent immunoglobulins considered largely equivalent in efficacy. Antiviral, antifungal, and antibiotic prophylaxis should be considered based on the absolute ANC, especially in cases of neutropenia lasting longer than 7 days. Recommendations include acyclovir, PCP prophylaxis with cotrimoxazole, antifungal prophylaxis with fluconazole, and antibiotic prophylaxis with ciprofloxacin might be considered, emphasizing the importance of rapid antibiotic escalation in case of fever to cover a broad spectrum of pathogens [61].
Infections post-CAR-T therapy, heightened by compromised humoral and cellular immune responses from HGGA and lymphodepleting chemotherapy, peak within the first 10 days. Up to 42% of infections are reported in the first month, with about 21% occurring between days 29 and 90. Infections are related to HGGA severity and CRS, with a meta-analysis reporting a 34% rate of severe infections, predominantly respiratory-tract infections. Encapsulated bacteria, such as Streptococcus pneumoniae and Haemophilus influenzae, are particularly incriminated in this setting [62]. Despite a morbidity rate lower than 2%, severe infections develop in 23–34% of patients. Risk factors for infectious complications overlap with those for hematotoxicity and/or HGGA, including previous allogenic HCT, multiple lines of pre-treatment immunochemotherapy (> 3 lines), baseline neutropenia (ANC < 500/µL) before lymphodepleting therapy, and the need for anti-IL-6-R agents and corticosteroids [63, 64].
Hemophagocytic lymphohistiocytosis
Hemophagocytic lymphohistiocytosis and macrophage activation syndrome are inflammatory conditions potentially triggered by CAR-T cell therapy. While full-blown HLH is rare, HLH-like symptoms contributing to hematotoxicity are more common [65]. CRS progression can lead to HLH/MAS through excessive cytokine release, activating T-cells and macrophages [66]. Hyperactivation of CAR-T cells exacerbates this, creating a cycle of immune dysregulation. Additionally, impaired regulatory T-cell function, vital for immune balance, can further promote HLH/MAS development. To differentiate HLH/MAS from infections, specific diagnostic criteria must be met, requiring at least 5 of the following 8 factors: fever, splenomegaly, cytopenia in ≥ 2 compartments, hypertriglyceridemia and/or hypofibrinogenemia, elevated ferritin, elevated sCD25, decreased natural killer cell activity, and hemophagocytosis in bone marrow, CSF, or lymph nodes [67] (Supplementary Table 4).
Classical HLH/MAS treatment involves immunosuppression, starting with high-dose corticosteroids like dexamethasone, combined with ciclosporin A [68]. In CAR-T-induced HLH/MAS, anti-cytokine agents like the IL-6-R inhibitor tocilizumab and for severe cases, anakinra are recommended. Initial therapy typically includes tocilizumab, possibly with corticosteroids while further treatment should be coordinated with a specialist.
If immunosuppressive therapy is insufficient, agents like ciclosporine A, etoposide, or cyclophosphamide may be considered, though the latter two will impair or even abrogate CAR-T cell therapy. Immunomodulatory agents like alemtuzumab or anti-thymocyte globulin might be options for refractory cases. Supportive care, including monitoring vital signs, organ function and managing complications, is critical for patient management [65, 66, 68].
Organ specific toxicity
Depending on the target antigen expression profile, CAR-T cell therapy can lead to damage in specific organs. For example, if the target antigen is expressed in the lung tissue, patients may develop pulmonary toxicity, which can present as dyspnoea, cough, and infiltrates on imaging. Similarly, if the target antigen is expressed in the liver, hepatotoxicity can occur, resulting in elevated liver enzymes and potential liver dysfunction.
Cardiotoxicity
Cardiotoxicity is recognized as an organ-specific side effect following CAR-T cell therapy, occurring in 10 to 36% of cases [69]. It involves various pathophysiological mechanisms, including damage from CRS resulting from macrophage stimulation by IFN-γ, leading to IL-6 release, which can impair cardiac microcirculation and worsen perfusion [70]. On a macroscopic level, IL-6 overactivation can cause volume depletion associated with CRS, reducing cardiac preload and leading to decreased left ventricular ejection fraction, arrhythmias, and ST segment changes on ECG, especially in patients with existing cardiac conditions [69,70,71]. The clinical approach to CAR-T cell-associated cardiotoxicity mirrors the treatment for similar cardiac symptoms, including IL-6-R blockade with tocilizumab at 8 mg/kg, analogous to CRS therapy. In the ICU, caution for arrhythmias, optimal volume, electrolyte, and anti-inflammatory management is crucial.
Future perspectives and summary
CAR-T cell therapy holds immense potential, though it presents complexities and challenges, particularly with managing associated toxicities. One significant adverse effect is OTOT, hindering its establishment in solid tumor therapy [72]. Innovative strategies, such as combining CAR-T cells with engineered hematopoietic stem cell transplantation that lack the CAR-T target antigen (e.g., CD33), aim to improve the safety profile while maintaining efficacy [73].
However, emerging CAR-T products might heighten side effects. For instance, the CARTITUDE-2 trial showed that ciltacabtagene-autoleucel had better efficacy but higher neurotoxicity risk compared to idecabtagen-vicleucel [74]. Allogeneic “off-the-shelf” CAR-T cells offer faster treatment options but could introduce graft-versus-host effects.
Comprehensive investigations into toxicity mechanisms and novel therapeutic interventions are crucial for improving patient outcomes. Rigorous monitoring, early detection, and prompt management of toxicities are essential. As CAR-T therapy expands to more indications, knowledge of toxicity management in specialized centers must be ensured. Continued collaborative efforts and translational research will be pivotal in maximizing CAR-T therapy’s potential while ensuring patient safety, thus facilitating its curative potential.
Data availability
Data sharing is not applicable to this narrative review as no new data were created or analyzed in this review.
References
Couzin-Frankel J (2013) Breakthrough of the year 2013. Cancer Immunotherap Sci 342(6165):1432–1433
June CH, Sadelain M (2018) Chimeric antigen receptor therapy. N Engl J Med 379(1):64–73
Shimabukuro-Vornhagen A, Gödel P, Subklewe M et al (2018) Cytokine release syndrome. J Immunother Cancer 6(1):56–70
Azoulay É, Castro P, Maamar A et al (2021) Outcomes in patients treated with chimeric antigen receptor T-cell therapy who were admitted to intensive care (CARTTAS): an international, multicentre, observational cohort study. Lancet Haematol 8(5):e355–e364
Sterner RC, Sterner RM (2021) CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J 11(4):69
Mailankody S, Devlin SM, Landa J et al (2022) GPRC5D-targeted CAR T cells for myeloma. N Engl J Med 387(13):1196–1206
Garcia Borrega J, Gödel P, Rüger MA et al (2019) In the Eye of the Storm: Immune-mediated Toxicities Associated With CAR-T Cell Therapy. Hemasphere 3(2):191–200
Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int J Mol Sci. 2019;20(6).
Pietrobon V, Todd LA, Goswami A, Stefanson O, Yang Z, Marincola F. Improving CAR T-Cell Persistence. Int J Mol Sci. 2021;22(19).
Honikel MM, Olejniczak SH. Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules. 2022;12(9).
Cappell KM, Kochenderfer JN (2021) A comparison of chimeric antigen receptors containing CD28 versus 4–1BB costimulatory domains. Nat Rev Clin Oncol 18(11):715–727
Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA (2016) CD28 Costimulation: From Mechanism to Therapy. Immunity 44(5):973–988
Zhang X, Zhu L, Zhang H, Chen S, Xiao Y (2022) CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front Immunol 13:927153
Cameron J Turtle L-AH, Carolina Berger, Daniel Sommermeyer, Barbara Pender, Emily M Robinson, Katherine Melville, Tanya M Budiarto, Natalia N Steevens, Colette Chaney, Sindhu Cherian, Brent L Wood, Lorinda Soma, Xueyan Chen, Shelly Heimfeld, Michael C Jensen, Stanley R. Riddell, David G Maloney. Addition of Fludarabine to Cyclophosphamide Lymphodepletion Improves In Vivo Expansion of CD19 Chimeric Antigen Receptor-Modified T Cells and Clinical Outcome in Adults with B Cell Acute Lymphoblastic Leukemia. Blood. 2015;Volume 126(Issue 23):Page 3773.
Bracci L, Moschella F, Sestili P et al (2007) Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res 13(2 Pt 1):644–653
Hay KA, Gauthier J, Hirayama AV et al (2019) Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 133(15):1652–1663
Spooren A, Kolmus K, Laureys G et al (2011) Interleukin-6, a mental cytokine. Brain Res Rev 67(1–2):157–183
Schuster SJ, Maziarz RT, Rusch ES et al (2020) Grading and management of cytokine release syndrome in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv 4(7):1432–1439
Lee DW, Santomasso BD, Locke FL et al (2019) ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant 25(4):625–638
Kotch C, Barrett D, Teachey DT (2019) Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev Clin Immunol 15(8):813–822
Heinz WJ, Buchheidt D, Christopeit M et al (2017) Diagnosis and empirical treatment of fever of unknown origin (FUO) in adult neutropenic patients: guidelines of the Infectious Diseases Working Party (AGIHO) of the German Society of Hematology and Medical Oncology (DGHO). Ann Hematol 96(11):1775–1792
Wang L, Lv Y, Zhou L et al (2024) Cytokine-based models for efficient differentiation between infection and cytokine release syndrome in patients with hematological malignancies. Exp Hematol Oncol 13(1):1–5
Luo H, Wang N, Huang L et al (2019) Inflammatory signatures for quick diagnosis of life-threatening infection during the CAR T-cell therapy. J Immunother Cancer 7(1):271
Topp M, Van Meerten T, Houot R, et al. Earlier Steroid Use with Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory Large B Cell Lymphoma. Blood. 2019;134(Supplement_1):243–243.
Jain MD, Smith M, Shah NN (2023) How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood 141(20):2430–2442
Cecconi M, Parsons AK, Rhodes A (2011) What is a fluid challenge? Curr Opin Crit Care 17(3):290–295
Vincent JL, Bihari DJ, Suter PM, et al. The prevalence of nosocomial infection in intensive care units in Europe. Results of the European Prevalence of Infection in Intensive Care (EPIC) Study. EPIC International Advisory Committee. Jama. 1995;274(8):639–644.
Gazeau N, Liang EC, Wu QV et al (2023) Anakinra for Refractory Cytokine Release Syndrome or Immune Effector Cell-Associated Neurotoxicity Syndrome after Chimeric Antigen Receptor T Cell Therapy. Transplant Cell Ther 29(7):430–437
Garfall AL, Lancaster E, Stadtmauer EA et al (2016) Posterior Reversible Encephalopathy Syndrome (PRES) after Infusion of Anti-Bcma CAR T Cells (CART-BCMA) for Multiple Myeloma: Successful Treatment with Cyclophosphamide. Blood 128(22):5702–5703
Gust J, Taraseviciute A, Turtle CJ (2018) Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 32(12):1091–1101
Rees JH. Part IV: Clinical Management of Patients Treated with CAR-T Cells. In: Kröger N. The EBMT/EHA CAR-T Cell Handbook.2022.
Patel S, Maria-Rios J, Parikh A, Okorie ON (2023) Diagnosis and management of elevated intracranial pressure in the emergency department. Int J Emerg Med 16(1):72–91
Möhn N, Bonda V, Grote-Levi L et al (2022) Neurological management and work-up of neurotoxicity associated with CAR T cell therapy. Neurol Res Pract 4(1):1
Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T (2008) Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 112(10):3959–3964
Lemoine J, Vic S, Houot R (2022) Disease-specific outcomes after chimeric antigen receptor T-cell therapy. Eur J Cancer 160:235–242
Farrokh S, Tahsili-Fahadan P, Ritzl EK, Lewin JJ 3rd, Mirski MA (2018) Antiepileptic drugs in critically ill patients. Crit Care 22(1):153–165
Wehrli M, Gallagher K, Chen YB, et al. Single-center experience using anakinra for steroid-refractory immune effector cell-associated neurotoxicity syndrome (ICANS). J Immunother Cancer. 2022;10(1).
Cohen AD, Parekh S, Santomasso BD et al (2022) Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J 12(2):32
Wang Q, Gao J, Wu X (2018) Pseudoprogression and hyperprogression after checkpoint blockade. Int Immunopharmacol 58:125–135
Frelaut M, du Rusquec P, de Moura A, Le Tourneau C, Borcoman E (2020) Pseudoprogression and Hyperprogression as New Forms of Response to Immunotherapy. BioDrugs 34(4):463–476
Seymour L, Bogaerts J, Perrone A et al (2017) iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 18(3):e143–e152
Boursier C, Perrin M, Bordonne M, Campidelli A, Verger A (2022) Early 18F-FDG PET Flare-up Phenomenon After CAR T-Cell Therapy in Lymphoma. Clin Nucl Med 47(2):e152–e153
Denton CC, Gange WS, Abdel-Azim H et al (2020) Bilateral retinal detachment after chimeric antigen receptor T-cell therapy. Blood Adv 4(10):2158–2162
Mahdi J, Dietrich J, Straathof K et al (2023) Tumor inflammation-associated neurotoxicity. Nat Med 29(4):803–810
Majzner RG, Ramakrishna S, Yeom KW et al (2022) GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 603(7903):934–941
Dunleavy K, Hakim F, Kim HK et al (2005) B-cell recovery following rituximab-based therapy is associated with perturbations in stromal derived factor-1 and granulocyte homeostasis. Blood 106(3):795–802
Fried S, Avigdor A, Bielorai B et al (2019) Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant 54(10):1643–1650
Schuster SJ, Bishop MR, Tam CS et al (2019) Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 380(1):45–56
Locke FL, Ghobadi A, Jacobson CA et al (2019) Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol 20(1):31–42
Rejeski K, Subklewe M, Aljurf M et al (2023) Immune effector cell-associated hematotoxicity: EHA/EBMT consensus grading and best practice recommendations. Blood 142(10):865–877
Rejeski K, Perez A, Sesques P et al (2021) CAR-HEMATOTOX: a model for CAR T-cell-related hematologic toxicity in relapsed/refractory large B-cell lymphoma. Blood 138(24):2499–2513
Wang Y, Song Z, Geng Y et al (2022) The risk factors and early predictive model of hematotoxicity after CD19 chimeric antigen receptor T cell therapy. Front Oncol 12:987965
Miller KC, Johnson PC, Abramson JS et al (2022) Effect of granulocyte colony-stimulating factor on toxicities after CAR T cell therapy for lymphoma and myeloma. Blood Cancer J 12(10):146
Davis JA, Sborov DW, Wesson W et al (2023) Efficacy and Safety of CD34+ Stem Cell Boost for Delayed Hematopoietic Recovery After BCMA Directed CAR T-cell Therapy. Transplant Cell Ther 29(9):567–571
Gilreath J, Lo M, Bubalo J (2021) Thrombopoietin Receptor Agonists (TPO-RAs): Drug Class Considerations for Pharmacists. Drugs 81(11):1285–1305
Gagelmann N, Wulf GG, Duell J et al (2023) Hematopoietic stem cell boost for persistent neutropenia after CAR T-cell therapy: a GLA/DRST study. Blood Adv 7(4):555–559
Wat J, Barmettler S (2022) Hypogammaglobulinemia After Chimeric Antigen Receptor (CAR) T-Cell Therapy: Characteristics, Management, and Future Directions. J Allergy Clin Immunol Pract 10(2):460–466
Kampouri E, Walti CS, Gauthier J, Hill JA (2022) Managing hypogammaglobulinemia in patients treated with CAR-T-cell therapy: key points for clinicians. Expert Rev Hematol 15(4):305–320
Wudhikarn K, Palomba ML, Pennisi M et al (2020) Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J 10(8):79
Bhoj VG, Arhontoulis D, Wertheim G et al (2016) Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood 128(3):360–370
Hill JA, Seo SK (2020) How I prevent infections in patients receiving CD19-targeted chimeric antigen receptor T cells for B-cell malignancies. Blood 136(8):925–935
Topp M, Feuchtinger T. Part IV Clinical Management of Patients Treated with CAR-T Cells. In: Kröger N, Gribben J, Chabannon C, Yakoub-Agha I, Einsele H. The EBMT/EHA CAR-T Cell Handbook2022.
Hill JA, Li D, Hay KA et al (2018) Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood 131(1):121–130
Park JH, Romero FA, Taur Y et al (2018) Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin Infect Dis 67(4):533–540
Hines MR, Knight TE, McNerney KO et al (2023) Immune Effector Cell-Associated Hemophagocytic Lymphohistiocytosis-Like Syndrome. Transplant Cell Ther 29(7):438.e431-438.e416
Sandler RD, Tattersall RS, Schoemans H et al (2020) Diagnosis and Management of Secondary HLH/MAS Following HSCT and CAR-T Cell Therapy in Adults; A Review of the Literature and a Survey of Practice Within EBMT Centres on Behalf of the Autoimmune Diseases Working Party (ADWP) and Transplant Complications Working Party (TCWP). Front Immunol 11:524
Henter JI, Horne A, Aricó M et al (2007) HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48(2):124–131
Bergsten E, Horne A, Aricó M et al (2017) Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood 130(25):2728–2738
Marar RI, Abbasi MA, Prathivadhi-Bhayankaram S et al (2023) Cardiotoxicities of Novel Therapies in Hematologic Malignancies: Chimeric Antigen Receptor T-Cell Therapy and Bispecific T-Cell Engager Therapy. JCO Oncol Pract 19(6):331–342
Nenna A, Carpenito M, Chello C, et al. Cardiotoxicity of Chimeric Antigen Receptor T-Cell (CAR-T) Therapy: Pathophysiology, Clinical Implications, and Echocardiographic Assessment. Int J Mol Sci. 2022;23(15).
Ganatra S, Dani SS, Yang EH, Zaha VG, Nohria A (2022) Cardiotoxicity of T-Cell Antineoplastic Therapies: JACC: CardioOncology Primer. JACC CardioOncol 4(5):616–623
Flugel CL, Majzner RG, Krenciute G et al (2023) Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol 20(1):49–62
Young RM, Engel NW, Uslu U, Wellhausen N, June CH (2022) Next-Generation CAR T-cell Therapies. Cancer Discov 12(7):1625–1633
Cohen AD, Mateos MV, Cohen YC et al (2023) Efficacy and safety of cilta-cel in patients with progressive multiple myeloma after exposure to other BCMA-targeting agents. Blood 141(3):219–230
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no known competing fincancial interests or persolal relationships that could have appeared to influence the work reported in this review.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Schroeder, T., Martens, T., Fransecky, L. et al. Management of chimeric antigen receptor T (CAR-T) cell-associated toxicities. Intensive Care Med 50, 1459–1469 (2024). https://doi.org/10.1007/s00134-024-07576-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-024-07576-4