
Abstract
Rationale
Zuranolone is an oral positive allosteric modulator of GABAA receptors. Due to its central nervous system (CNS) activity, zuranolone may impact activities requiring complex cognition, including driving.
Objective
Evaluate the effect of zuranolone on simulated driving performance.
Methods
In this randomized, double-blind, active- and placebo-controlled, four-period crossover study, treatments included once-nightly zuranolone 50 mg on days 1–7, zuranolone 50 mg on days 1–6 and zuranolone 100 mg on day 7, zopiclone 7.5 mg on days 1 and 7, and placebo on days 1–7. Driving was assessed using a validated simulator. Primary endpoint was standard deviation of lateral position (SDLP), evaluated 9 h post-dose on days 2 and 8. Secondary endpoints included additional driving assessments, cognitive tests, pharmacokinetics, and safety.
Results
Healthy adults (N = 67) enrolled and received ≥ 1 dose. Zuranolone 50 mg increased SDLP versus placebo on days 2 (least squares mean difference [LSMD]: 7.4 cm; p < 0.0001) and 8 (LSMD: 4.6 cm; p = 0.0106). Zuranolone 100 mg evoked a larger increase in SDLP versus placebo on day 8 (LSMD 18.9 cm; p < 0.0001). Reduced performance in other driving assessments and cognition were observed with zuranolone 50 mg on day 2; many resolved by day 8. Despite the SDLP observations, most participants judged themselves capable of driving. Frequent adverse events (≥ 20%) were CNS-related; most were mild/moderate.
Conclusion
Zuranolone impaired simulated driving and reduced cognitive function versus placebo 9 h after administration. Although many impairments resolved after 7 days of dosing, driving remained impaired. These results may inform prescriber decision-making.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Postpartum depression (PPD), one of the most common complications of pregnancy, can have substantial impact on the well-being and functioning of the patient as well as the cognitive, behavioral, and emotional development of the child (Frieder et al. 2019). PPD is characterized by a wide range of symptoms, including persistent sadness, poor concentration, feelings of worthlessness and guilt, irritability, anhedonia, insomnia, hypersomnia, recurrent thoughts of death and suicide, and poor bonding with the infant (Suryawanshi and Pajai 2022). Standard-of-care antidepressant therapies (ADTs), approved for the treatment of major depressive disorder, are frequently used off label for the treatment of PPD (Frieder et al. 2019). However, there is limited evidence on the safety and efficacy of these ADTs in PPD, and their delayed onset of action (approximately 4–8 weeks) for some patients underscores a need for new rapid-acting therapeutic options (Kaufman et al. 2022; Machado-Vieira et al. 2008).
Zuranolone is a positive allosteric modulator of synaptic and extrasynaptic GABAA receptors and a neuroactive steroid that is taken once nightly for 14 days. It was approved in 2023 by the US Food and Drug Administration (FDA) as the first oral medication specifically indicated for the treatment of adults with PPD (ZURZUVAE™ PI 2024). In two phase 3 trials, adults with PPD who received zuranolone demonstrated significant improvement in depressive symptoms versus those who received placebo as early as day 3 as measured by the change from baseline in 17-item Hamilton Rating Scale for Depression (HAMD-17) total score (Deligiannidis et al. 2021, 2023). Consistent with its pharmacological action at the GABAA receptor (McKernan et al. 2000), central nervous system (CNS)-depressant effects, including somnolence, dizziness, and sedation, have been reported in zuranolone clinical trials (Deligiannidis et al. 2021, 2023). As CNS depressants can potentially influence activities that require complex cognition and perception, including the ability to operate a motor vehicle, the FDA recommends that psychoactive drugs be evaluated for their impact on driving ability. Such evaluation may include conducting a dedicated “driving study” if accumulated pharmacological and/or clinical data suggest potential for driving impairment (US Food and Drug Administration 2017). According to the guidance, dedicated driving studies may be conducted using actual motor vehicles or a driving simulator and should consider evaluating a dose above the maximum intended dose to account for elevated drug exposure that may occur in some patients, for example, as a result of reduced drug clearance due to organ impairment or a drug-drug interaction (US Food and Drug Administration 2017). Consistent with these recommendations, this study utilized a validated driving simulator platform and assessed the next-day effects of both the approved therapeutic dose of zuranolone (50 mg) and a supratherapeutic dose (100 mg) on simulated driving performance compared to placebo. A positive control was included to confirm that the simulator had sufficient sensitivity to detect impaired driving ability (US Food and Drug Administration 2017).
Methods
Study design, population, and treatment
This was a randomized, phase 1, double-blind, active- and placebo-controlled, four-treatment, four-period crossover study conducted at a clinical research unit (CRU) in Montreal, Quebec, Canada (Fig. 1). Eligible participants were healthy, active drivers (minimum of 6000 km [approximately 3500 miles] per year during the previous 3 years), aged ≥ 21 years, with a valid driver’s license. At screening, participants underwent a driving simulator training session, during which they were required to display acceptable driving performance and demonstrate adequate manual dexterity, vision, hearing, and cognitive ability to reliably perform tasks within the simulator. The acceptable driving performance was defined as maintaining a standard deviation of lateral position (SDLP) within 1 standard deviation (SD) above the mean from a database of healthy adults who previously completed the same driving scenario (data on file – Cognitive Research Corporation). Individuals who displayed signs of motion sickness while performing the simulated drive at screening were excluded from participation. Other eligibility criteria included a minimum body weight of 50 kg, body mass index of 18–35 kg/m2, a regular sleep pattern (usual bedtime between 9 pm and 12 am), and an Epworth Sleepiness Scale (subjective, self-reported measure of daytime sleepiness) score of < 10 at screening, which indicated a normal level of daytime sleepiness (Johns 1991). A positive test result for alcohol or drugs of abuse (including tetrahydrocannabinol-containing products) at any clinic admission was exclusionary. The use of prescription medications was prohibited except for hormonal preparations used for birth control and medications prescribed to participants aged ≥ 65 years for chronic, stable, and well-controlled medical conditions, provided they did not carry a risk for a known drug-drug interaction. Participants who regularly consumed excessive amounts of caffeine, defined as greater than 6 servings (1 serving was approximately equivalent to 120 mg of caffeine) of coffee, tea, cola, or other caffeinated beverages per day, were excluded from the study. Prior experience with the driving simulator was not an exclusion criterion.

Study design. Participants were randomized 1:1:1:1 to the following treatment sequences A→C→D→B, B→ D→ C→A, C→B→A→ D, or D→A→ B→C
R randomization
After a screening period of up to 28 days, eligible participants were admitted to the CRU on day 1 of the first treatment period and remained housed in the CRU until completion of all assessments on day 9. After a period of 7 to 14 days, participants were readmitted to the CRU on day 1 of subsequent treatment periods. Participants were stratified by age (< 50 years, 50–64 years, ≥ 65 years) and randomized 1:1:1:1 into one of four treatment sequences comprising the following treatments: (A) zuranolone 50 mg once nightly on days 1–7, (B) zuranolone 50 mg on days 1–6 followed by zuranolone 100 mg on day 7, (C) zopiclone 7.5 mg on days 1 and 7, and (D) placebo on days 1–7 (Fig. 1). Zopiclone, a short-acting hypnotic agent with well-established negative effects on driving performances in both on-the-road and simulated driving scenarios, was included as a positive (active) control, (Kay et al. 2017; Leufkens et al. 2009; Mets et al. 2011; Simen et al. 2015; Vermeeren et al. 2016, 2019).
Consecutive treatment periods were separated by a washout period of 7–14 days, after which participants were readmitted to the clinical research unit on day 1 of subsequent treatment periods and received study treatment according to the assigned treatment sequence. Five (± 2) days after discharge from period 4, participants received a follow-up phone call from the clinical research unit to evaluate any adverse events (AEs) and provide any new information.
Study treatments were administered each night (days 1–7) at approximately 11:00 pm, after a meal (800–1000 kcal; approximately 50% total calories from fat) at approximately 8:30 pm and a snack (250 kcal; 25% calories from fat) given 30 min prior to dosing. All treatments were identical in appearance, and placebo capsules were administered as necessary to maintain the blinding for participants and site personnel across the four treatment periods. Participants were awakened the following morning (8 h after dosing) and provided with a low-fat breakfast prior to performing any study-related assessments.
This study was performed in accordance with the Declaration of Helsinki and is consistent with Good Clinical Practice guidelines according to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Harmonized Tripartite Guideline, as well as all applicable regulatory requirements. All participants provided written informed consent.
Simulated driving assessments
Driving was assessed the morning of days 2 and 8 following nighttime administration of the first (day 1) and last (day 7) dose in each treatment period. The Cognitive Research Corporation Driving Simulator (CRCDS) MiniSim was used in this study; this simulator has demonstrated sensitivity to the effects of alcohol and other medications with CNS-depressant effects, including antihistamines, benzodiazepines, and muscle relaxants (Caron et al. 2020; Pearlman et al. 2020). The CRCDS is a computer-based driving simulator featuring three 24-inch LCD front channel monitor displays that provide a 105° field of view, an 18-inch dashboard monitor display, a full-size steering wheel with pedals (ECCI Trackstar 6000), a 2.1 audio system, and a tactile transducer that emulates engine vibration. The Country Vigilance Divided Attention driving scenario was selected to assess a participant’s ability to maintain driving lane position during a 60-minute, monotonous, two-lane highway driving task. Participants were trained to drive the entire scenario at the posted speed limit of 95 km/h (approximately 59 mph) and to maintain a steady position in their lane, with as little weaving as possible. Standardized driving instructions were provided in order to ensure a standardized driving test environment and minimize variability that could confound driving performance evaluation. The SDLP (a measure of lane position control) and additional driving performance assessments such as lane exceedance (including number, maximum, and duration of exceedance), excessive speed count (number of times the vehicle exceeded the speed threshold of 1.2 times the posted speed limit of 95 km/h), average speed and speed deviation, excessive cornering speed threshold (the number of times the lateral acceleration of the vehicle exceeded the cornering threshold of 0.25 G), and total number of collisions (an event where the participant collided with another vehicle or roadway object in the scene, or drove in excess of a predefined distance outside of the lane and, therefore, was presumed to have crashed) were captured electronically.
During the simulated drive, participants were periodically presented with targets (i.e., arrow stimuli) in the periphery and instructed to press a button on the steering wheel or ignore the target depending on its location. The stimuli appeared for 5 s, or disappeared when the response was provided (Simen et al. 2015). Participants’ reaction time and accuracy of response were recorded.
Cognitive assessments
Cognitive assessments were completed before each simulated drive using the CogScreen Symbol Digit Coding (SDC) task, a computer-based analogue of the conventional digit symbol substitution test (Kay 1995). Attention, visual scanning, working memory, and information processing speed were evaluated utilizing the number of correct responses, accuracy of responses, and the SD of reaction time recorded within the SDC task.
Subjective assessments
Prior to each simulated drive, participants rated their sleepiness level using the Karolinska Sleepiness Scale (KSS), a 9-point categorial scale with scores ranging from 1 = “extremely alert” to 9 = “very sleepy, great effort to keep awake, fighting sleep” (Akerstedt et al. 2014), and answered a question about whether they feel ready to drive (yes or no). Upon completion of the drive, each participant’s motivation to drive was assessed by asking them the following question: “How motivated did you feel to drive at your best during the last 60 minutes of driving?” Participants recorded their response on a 100-mm visual analogue scale (VAS) ranging from “Not motivated” (0) to “Motivated” (100). Each participant also provided a self-appraisal of their driving performance using a VAS ranging from “Not satisfactory” (0) to “Satisfactory” (100).
Pharmacokinetics
Blood samples for zuranolone plasma concentrations and correlations to SDLP were collected prior to driving assessments on days 2 and 8 and prior to dosing on day 7.
Endpoints
The primary endpoint of this study was SDLP after single and multiple nightly doses of zuranolone. Secondary endpoints included additional driving measures, cognitive function tests, subjective assessments, pharmacokinetics, and safety evaluations such as AEs, vital signs, clinical laboratory data, electrocardiogram (ECG) parameters, and suicidal ideation and behavior using the Columbia Suicide Severity Rating Scale (C-SSRS).
Statistical methods
The primary endpoint was analyzed using a mixed model with fixed effects for sequence, period, and treatment, with repeated periods for participants. An unstructured covariance structure and Kenward-Roger degrees of freedom were used. Pairwise comparisons of differences in means and two-sided 95% confidence intervals (CIs) on differences were calculated for all active treatments versus placebo. Formal statistical tests were two-sided and tested at the alpha = 0.05 level of significance. To address multiplicity, a predefined sequential testing procedure maintained the overall type I error rate at 0.05. Doses of zuranolone were interpreted in a sequential manner, starting with zuranolone 50 mg multiple doses (day 8) compared with placebo, followed by zuranolone 50 mg single dose (day 2) compared with placebo, and then by zuranolone 100 mg (i.e., supratherapeutic dose) compared with placebo. This procedure evaluated each comparison in a hierarchical manner, progressing to the next comparison only after the previous comparison was statistically significant, thereby limiting the family-wise error rate (Dmitrienko and D’Agostino 2018). A clinically relevant effect in SDLP was ruled out if the upper bound of the 95% CI for the mean difference in SDLP between active treatment and placebo was < 4.4 cm. This threshold represents the least squares mean (LSM) difference in SDLP between a blood alcohol concentration of 0.05% and placebo determined previously using the same driving simulator and driving scenario (data on file – Cognitive Research Corporation; Online Resource 1), and is consistent with the finding from the National Highway Traffic Safety Administration indicating that a blood alcohol concentration of 0.05% is associated with an elevated risk of traffic accidents (Compton and Berning 2015).
As a supportive analysis, pairwise within-participant differences in SDLP were tested for symmetry around zero using the maximally selected McNemar test (Laska et al. 2012). Symmetry around zero was rejected if the maximum McNemar’s test statistic exceeded the critical value of 7.2. If there was no significant difference between placebo and zuranolone and if the distribution of paired differences was symmetrical around zero (critical value was less than 7.2), it could be concluded that zuranolone did not impair driving. For both primary and secondary endpoints, day 2 and day 8 comparisons utilized data from both treatment groups (zuranolone 50 mg once nightly on days 1–7, and zuranolone 50 mg on days 1–6 followed by zuranolone 100 mg on day 7).
The secondary endpoints of driving performance, VAS, KSS, and SDC, were evaluated using a mixed model for repeated measures similar to that used for the primary endpoint. Lane exceedance was log-transformed (ln[x + 1]) prior to analysis. The number of collisions for each pairwise comparison was evaluated using a Wilcoxon signed-rank test. No adjustment to alpha levels was made for secondary endpoints. The pharmacokinetic/pharmacodynamic relationship between plasma zuranolone concentrations and SDLP was assessed by correlational analyses. All safety endpoints were summarized by treatment group based on the safety set, defined as all participants who received at least one dose of any drug.
All statistical analyses were completed using SAS, version 9.4 (SAS Institute Inc, Cary, NC, USA).
Results
Participants
Sixty-seven participants enrolled and received at least one dose of study treatment, and 58 (86.6%) completed the study. Among nine participants who did not complete the study, five withdrew from the study, two terminated the study early due to treatment-emergent AEs (TEAEs), one terminated early due to noncompliance with protocol, and one was terminated from the study due to heightened stress that impacted demeanor, as decided by the investigator (Online Resource 2). The median (range) age of the population was 45.0 (22–81) years, with seven (10.4%) participants aged ≥ 65 years. A slightly greater proportion of participants were male (56.7%) than female (43.3%); the majority (88.1%) were White (Table 1).
Primary endpoint: SDLP
After treatment with zopiclone, mean SDLP increased compared with placebo on day 2 (LSM difference [95% CI], 3.931 cm [1.176–6.686]; p = 0.0056; Fig. 2) and on day 8 (LSM difference [95% CI], 3.236 cm [− 0.138–6.610]; p = 0.0600; Fig. 2). The upper limit of the 95% CI exceeded the pre-established noninferiority criterion of 4.4 cm for both days, which was consistent with impaired driving performance, thus, confirming sensitivity of the simulator (Table 2).

Summary of SDLP (cm) difference from placebo on day 2 and day 8
*Statistically significant, p < 0.05; p value tests null hypothesis that difference in LSM = 0 versus the alternative hypothesis that difference in LSM ≠ 0. Day 2 zuranolone 50 mg LSM difference is estimated as the average of two zuranolone treatment groups versus placebo. The dashed horizontal line at 4.4 cm depicts the LSM difference in SDLP between a blood alcohol concentration of 0.05% and placebo using the same driving simulator and driving scenario
CI confidence interval, LSM least squares mean, SDLP standard deviation of lateral position
On day 2, approximately 9 h after the initial nighttime dose, SDLP significantly increased following administration of zuranolone 50 mg compared with placebo (LSM difference [95% CI], 7.416 cm [4.730–10.103]; p < 0.0001); the upper limit of the 95% CI (10.103 cm) exceeded the noninferiority criterion of 4.4 cm (Fig. 2; Table 2). On day 8, under steady-state conditions after nightly dosing on days 1–7, the SDLP increase with zuranolone 50 mg was numerically less than on day 2 but remained statistically significant compared with placebo (LSM difference [95% CI], 4.602 cm [1.091–8.113]; p = 0.0106), with the upper limit of the 95% CI (8.113 cm) exceeding the noninferiority criterion. The effect of the supratherapeutic dose, zuranolone 100 mg on day 7 after 50 mg on days 1–6, on SDLP was greater in magnitude on day 8 (LSM difference [95% CI], 18.941 cm [15.326–22.556]; p < 0.0001) relative to that observed with zuranolone 50 mg when both were compared with placebo (Table 2). Within-participant differences on days 2 and 8 were not symmetric around zero (maximum McNemar test statistics of 34.783 [day 2, zuranolone 50 mg], 32.818 [day 8; zuranolone 50 mg], and 24.029 [day 8; zuranolone 100 mg]; all exceeded the critical value of 7.2 required for the McNemar test statistic for symmetry).
On day 2, approximately half of the participants treated with zuranolone 50 mg (35 [56.5%] receiving zuranolone 50 mg on days 1–7 and 32 [54.2%] receiving zuranolone 50 mg on days 1–6 and zuranolone 100 mg on day 7) exceeded the 4.4 cm threshold for within-participant SDLP difference from placebo 9 h after the initial dose administered the previous night. On day 8, 27 participants (43.5%) treated with zuranolone 50 mg and 43 (78.2%) treated with zuranolone 100 mg exceeded the 4.4 cm threshold (Online Resource 3).
Additional driving and cognitive endpoints
Selected secondary endpoints relating to driving performance and cognitive assessments are shown in Fig. 3. The endpoints presented are known to be sensitive to sedative effects (Kay et al. 2013). In consideration of the different scales and measurement outcomes utilized across various endpoints, the data were standardized by dividing each LSM difference and its upper and lower CI by the SD. Observations for secondary outcomes were consistent with the primary outcome of SDLP. Reduction in performance was typically observed after the first dose of zuranolone 50 mg, which decreased in magnitude on day 8 after repeat dosing and increased in magnitude on day 8 following administration of zuranolone 100 mg.

Forest plot of secondary driving endpoints and SDC. Data shown represent LSM difference from placebo with 95% CI. To account for different scales across the endpoints, data were standardized by dividing each LSM difference and its upper and lower CI by the SD
CI confidence interval, DA directed attention, LSM least squares mean, SD standard deviation, SDC symbol digit coding
On day 2, statistically significant differences from placebo were observed following administration of zuranolone 50 mg for all endpoints except excessive cornering speed threshold (Fig. 3). After repeated dosing of 50 mg, the magnitude of the effects diminished and, with the exception of number of lane exceedances and SDC, the 95% CI for the difference from placebo included zero. Administration of zuranolone 100 mg increased the magnitude of effect for all endpoints.
Collisions
The majority of participants had no collisions during the simulated drive; however, the number of collisions was greater following treatment with zuranolone than with placebo. The number of collisions was significantly increased with zuranolone 50 mg compared with placebo on day 2 (mean [SD] = 0.34 [2.040]; p = 0.0260), but not on day 8 (mean [SD] = 0.15 [1.171]; p = 0.7500). On day 8, administration of zuranolone 100 mg was associated with an increased number of collisions compared with placebo (mean [SD] = 2.00 [4.615]; p = 0.0002). Nine participants (16.1%) experienced three or more simulated collisions following treatment with zuranolone 100 mg compared with one participant (1.6%) following repeated administration of zuranolone 50 mg after 8 days.
Self-reported measures
On average, participants reported more subjective sleepiness on the KSS following zuranolone 50 mg compared with placebo on day 2 (LSM difference, 2.0; p < 0.0001) and day 8 (LSM difference, 1.3; p < 0.0001). The increase in subjective sleepiness was most pronounced following treatment with zuranolone 100 mg (LSM difference, 3.5; p < 0.0001) compared with placebo.
Throughout the study, participants on average reported less motivation to drive after they received zuranolone versus placebo, with LSM differences of − 19.1 (p < 0.0001), − 10.5 (p = 0.0117), and − 23.4 (p < 0.0001) for zuranolone 50 mg on day 2, zuranolone 50 mg on day 8, and zuranolone 100 mg on day 8, respectively. Additionally, participants reported a negative self-appraisal of driving performance after administration of zuranolone 50 mg compared with placebo (day 2 LSM difference, − 27.0; p < 0.0001). Participants continued to report a negative self-appraisal of driving performance at day 8 after administration of zuranolone 50 mg and 100 mg (LSM differences, −7.9 [p = 0.0725] and −36.0 [p < 0.0001], respectively).
With respect to evaluating their own ability to drive, on day 2, 74.6% of participants reported feeling ready to drive after receiving zuranolone 50 mg. By comparison, 95.3% of participants who received placebo reported feeling ready to drive. On day 8, 69.8% of participants reported feeling ready to drive after repeated dosing with zuranolone 50 mg, and 49.1% of participants reported feeling ready to drive after dosing with zuranolone 50 mg on days 1–6 and 100 mg on day 7, whereas the percent of participants who felt ready to drive after 7 days of placebo-treatment remained at 95.3%.
Pharmacokinetics
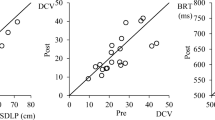
Following administration of zuranolone 50 mg the previous night, the median zuranolone plasma concentration prior to the driving assessment was 77.7 ng/mL (range, < 1.0–147 ng/mL) on day 2 and 108 ng/mL (range, 57.5–203 ng/mL) on day 8. On day 8, the median concentration before the drive following administration of zuranolone 100 mg the previous night was 158 ng/mL (range, 60.4–253 ng/mL). A significant correlation between zuranolone plasma concentration and SDLP on day 2 was evident for participants who received zuranolone 50 mg (Spearman correlation coefficient = 0.3127; p = 0.0133). However, the correlation between zuranolone plasma concentration and SDLP on day 8 was not significant for participants who received nightly dosing of 50 mg through day 7 (Spearman correlation coefficient = 0.2273; p = 0.0781; Online Resource 4). In contrast, a significant correlation between plasma concentration and SDLP on day 8 was evident in participants who received zuranolone 100 mg on day 7 (Spearman correlation coefficient = 0.3425; p = 0.0113; Online Resource 4).
Safety
The overall incidence of TEAEs was 89.1% among participants who received zuranolone 50 mg on days 1–7, 95.2% among those who received zuranolone 50 mg on days 1–6 followed by zuranolone 100 mg on day 7, and 67.8% when participants received zopiclone. The incidence of TEAEs when participants received placebo was 56.3%.
When participants received zuranolone 50 mg on days 1–6 followed by zuranolone 100 mg on day 7, overall TEAE incidence was greater after the 100 mg dose (90.3%) than before the 100 mg dose (77.8%). In this treatment group, the incidence of nervous system and psychiatric TEAEs was 58.7% and 15.9%, respectively, prior to receiving the 100 mg dose of zuranolone on day 7. When participants received the 100 mg dose of zuranolone on day 7, these incidences increased to 75.8% and 35.5%, respectively.
The most frequently reported TEAEs (occurring in ≥ 10% of participants) who received zuranolone 50 mg days 1–7 were dizziness, somnolence, fatigue, tremor, headache, asthenia, nausea, insomnia, speech disorder, and constipation (Table 3). In several instances, the AE frequency was higher in the treatment group in which participants received zuranolone 50 mg on days 1–6 followed by zuranolone 100 mg on day 7. Most participants with TEAEs experienced mild or moderate events, which resolved by the end of the study. Seven (10.4%) participants (n = 6, zuranolone; n = 1, zopiclone) had at least one severe TEAE (zuranolone 50 mg: diarrhea, n = 2; constipation, n = 3; balance disorder, abnormal coordination, and somnolence, n = 1; and zopiclone: constipation, n = 1). These severe TEAEs were considered not related to the study product by the investigator in six participants. One participant who had balance disorder, abnormal coordination, and somnolence also had another severe TEAE (altered state of consciousness) on day 8 after zuranolone 100 mg, which was determined to be related to study treatment.
After receiving zuranolone 100 mg on day 7, two participants had one or more TEAEs (dysarthria, malaise, and somnolence in one participant; altered state of consciousness in one participant) that led to treatment discontinuation and withdrawal from the study. In both cases, the TEAEs leading to discontinuation were considered related to the study treatment by the investigator and resolved within 1 day. There were no clinically relevant mean changes from baseline in clinical laboratory evaluations, vital signs, ECGs, or C-SSRS responses. No deaths or serious AEs were reported.
Additional details of AEs that occurred on and off treatment in the trial are in the supplement (Online Resource 5).
Discussion
In this study, participants who received zuranolone experienced adverse effects on simulated driving ability and tasks related to cognitive performance when evaluated in the morning approximately 9 h after nighttime administration. The effects were noted after a single 50 mg dose, after repeat dosing, and after the supratherapeutic 100 mg dose. While many of these effects diminished upon repeated dosing, the effect on the primary endpoint of SDLP (an index of control of lane position) remained statistically significant the morning after 7 nights of dosing. Following administration of a supratherapeutic dose of 100 mg after steady-state dosing of zuranolone 50 mg, the impairment in SDLP was more pronounced and cognitive performance was further reduced.
The sensitivity of the driving simulator was confirmed by performance assessments following administration of zopiclone 7.5 mg (positive control). While the mean placebo-adjusted SDLP for zopiclone was not statistically different from zero on day 8 (p = 0.0600), the upper bound of the 95% CI (6.6 cm) exceeded the predefined 4.4 cm threshold associated with a clinically relevant effect. Zopiclone has been shown to be associated with significant and meaningful driving impairment compared with placebo in numerous on-the-road driving tests (Mets et al. 2011; Vermeeren et al. 2015, 2019; Verster et al. 2011). Additionally, the zopiclone-related driving impairments in this study (day 2 LSM difference, 3.931 cm; day 8 LSM difference, 3.236 cm) are consistent with previous driving studies that utilized the same simulator scenario, where LSM differences in SDLP following administration of 7.5 mg of zopiclone compared with placebo ranged from 2.62 to 3.518 cm (Kay et al. 2017; Simen et al. 2015).
Approximately 75% of participants indicated they felt “ready to drive” the morning after the initial dose of zuranolone 50 mg. However, SDLP endpoints demonstrated that, on average, driving ability was impaired with zuranolone 50 mg. On day 2, more than 50% of participants treated with zuranolone 50 mg had an SDLP difference from placebo that was greater than 4.4 cm, a difference that is comparable to a blood alcohol concentration of 0.05% (data on file – Cognitive Research Corporation; Online Resource 1). On day 8, the number of participants receiving nightly dosing of 50 mg of zuranolone who felt “ready to drive” decreased slightly to approximately 70%.
Following zuranolone 100 mg dosing on day 8, approximately half (50.9%) of participants reported they did not feel ready to drive, demonstrating greater awareness of the potential adverse effects on driving at this dose of zuranolone. This was reflected in the finding that 27 participants (43.5%) treated with zuranolone 50 mg and 43 participants (78.2%) treated with zuranolone 100 mg exceeded the 4.4 cm threshold on day 8. The apparent disconnect between subjective assessments (feeling ready to drive) and objective observations has been reported in prior driving studies (Pearlman et al. 2020; Verster and Roth 2012; Verster et al. 2011); the results from the present study indicate that patients treated with zuranolone may be unable to adequately judge their ability to drive. Based on the results of the present study, the FDA prescribing information for zuranolone for the treatment of adults with PPD includes a boxed warning for impaired ability to drive due to CNS-depressant effects and notes that individuals may not correctly assess the impact of their impairment (ZURZUVAE™ PI 2024).
Plasma concentrations of zuranolone were in the expected range for the doses administered in this study. The elimination half-life of zuranolone is approximately 19.7 to 24.6 h, and steady state is achieved in 3 to 5 days (ZURZUVAE™ PI 2024). Thus, the 7-day dosing period in this study was sufficient to achieve steady-state conditions. In patients with PPD, a 14-day treatment course of once-daily zuranolone demonstrated rapid improvements in depressive symptoms observed as early as day 3 (Deligiannidis et al. 2021, 2023); therefore, assessing driving effects early in the treatment course was important for the design of this study. Although no statistically significant correlation was observed between plasma concentrations of zuranolone and SDLP performance for zuranolone 50 mg on day 8, a significant and stronger trend was evident for zuranolone 100 mg on day 8. Per the US prescribing information, patients receiving zuranolone should not drive for at least 12 h post-dose (ZURZUVAE™ PI 2024).
The results of the present study were consistent with worsened driving ability observed with a sedative antidepressant, mirtazapine. In a previous study, healthy participants receiving an evening dose of mirtazapine (days 1–7, 30 mg; days 8–15, 45 mg) were assessed on the Road Tracking Test (1-hour, on-the-road, 100 km) on days 2, 9, and 16 (Wingen et al. 2005). Treatment with mirtazapine led to a mean (standard error of the mean) SDLP of 21.8 (1.36) cm on day 2 compared with 17.9 (0.72) cm with placebo (p < 0.001). The effect on SDLP subsided upon repeated dosing on days 9 and 16. Furthermore, a review of on-the-road and simulator studies concluded that worsened driving ability after a single dose of mirtazapine 15 or 30 mg was comparable to a blood alcohol concentration of 0.05% (Verster et al. 2015).
The present study evaluated cognitive performance using the SDC task, which measures attention, visual scanning, working memory, and information processing speed. Results indicated worsened SDC task performance on day 2 following the initial dose of zuranolone 50 mg, a milder deficit on day 8 after repeat dosing, and a more pronounced impairment on day 8 following administration of zuranolone 100 mg. These findings are consistent with the primary outcome of the effect of zuranolone on SDLP and corroborate recent studies that suggest a link between cognitive abilities and driving performance (Depestele et al. 2020; Tapia and Dunabeitia 2023).
This study has some limitations. First, driving was evaluated at only a single time point, 9 h post-dose, and the dosing duration was limited to 7 days; therefore, driving ability evaluated at later time points or after longer post-dose periods may yield different results. Importantly, driving is a multifaceted activity, and this study did not evaluate complex driving tasks that assessed risk-taking, which might influence driving performance. In addition, this study included both male and female participants (including women of non-childbearing age), so the overall results may not translate directly to women of childbearing age. Another limitation is that a participant’s driving performance or adverse event profile may have had an unblinding effect. Finally, this study was conducted in healthy, experienced drivers, and the results may not generalize to adults with PPD who could respond differently when treated with zuranolone; for example, greater variability in driving test results has been observed in patients with anxiety compared with healthy volunteers (Verster et al. 2005). Similarly, the effects may not generalize to individuals who did not fulfill the study’s inclusion and exclusion criteria, including those who did not meet the acceptable driving performance criterion.
In summary, results from this study showed that participants who received zuranolone 50 mg demonstrated significantly impaired simulated driving measures and cognitive performance after single-dose administration. These effects diminished after repeated dosing, although the impairment in lane position control remained significant after 7 days of dosing. Although this study was not limited to female participants of childbearing age, these results may inform prescriber decision-making as well as prescriber-patient conversations.
Data availability or code availability
Any data will be made available upon reasonable request to the corresponding author.
References
Akerstedt T, Anund A, Axelsson J, Kecklund G (2014) Subjective sleepiness is a sensitive indicator of insufficient sleep and impaired waking function. J Sleep Res 23:240–252. https://doi.org/10.1111/jsr.12158
Caron J, Kaye R, Wessel T, Halseth A, Kay G (2020) An assessment of the centrally acting muscle relaxant tolperisone on driving ability and cognitive effects compared to placebo and cyclobenzaprine. J Clin Pharm Ther 45:774–782. https://doi.org/10.1111/jcpt.13165
Compton R, Berning A (2015) Drug and Alcohol Crash Risk [Traffic Safety Facts]: Research Note. National Highway Traffic Safety Administration. https://rosap.ntl.bts.gov/view/dot/1993
Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, Doherty J, Jonas J, Li S, Sankoh AJ, Silber C, Campbell AD, Werneburg B, Kanes SJ, Lasser R (2021) Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry 78:951–959. https://doi.org/10.1001/jamapsychiatry.2021.1559
Deligiannidis KM, Meltzer-Brody S, Maximos B, Peeper EQ, Freeman M, Lasser R, Bullock A, Kotecha M, Li S, Forrestal F, Rana N, Garcia M, Leclair B, Doherty J (2023) Zuranolone for the treatment of postpartum depression. Am J Psychiatry 180:668–675. https://doi.org/10.1176/appi.ajp.20220785
Depestele S, Ross V, Verstraelen S, Brijs K, Brijs T, van Dun K, Meesen R (2020) The impact of cognitive functioning on driving performance of older persons in comparison to younger age groups: a systematic review. Transp Res Part F: Traffic Psychol Behav 73:433–452. https://doi.org/10.1016/j.trf.2020.07.009
Dmitrienko A, D’Agostino RB, Sr (2018) Multiplicity considerations in clinical trials. N Engl J Med 378:2115–2122. https://doi.org/10.1056/NEJMra1709701
Frieder A, Fersh M, Hainline R, Deligiannidis KM (2019) Pharmacotherapy of postpartum depression: current approaches and novel drug development. CNS Drugs 33:265–282. https://doi.org/10.1007/s40263-019-00605-7
Johns MW (1991) A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 14:540–545. https://doi.org/10.1093/sleep/14.6.540
Kaufman Y, Carlini SV, Deligiannidis KM (2022) Advances in pharmacotherapy for postpartum depression: a structured review of standard-of-care antidepressants and novel neuroactive steroid antidepressants. Ther Adv Psychopharmacol 12:20451253211065859. https://doi.org/10.1177/20451253211065859
Kay G (1995) CogScreen Overview. https://cogscreen.com/Overview.aspx
Kay G, Ahmad O, Brown T, Veit A (2013) Comparison of the MINISIM and STISIM driving simulators for the detection of impairment: an alcohol validation study. University of Iowa. https://pubs.lib.uiowa.edu/driving/article/28488/galley/136780/view/
Kay GG, Hochadel T, Sicard E, Natarajan KK, Kim NN (2017) Next-day residual effects of flibanserin on simulated driving performance in premenopausal women. Hum Psychopharmacol 32. https://doi.org/10.1002/hup.2603
Laska E, Meisner M, Wanderling J (2012) A maximally selected test of symmetry about zero. Stat Med 31:3178–3191. https://doi.org/10.1002/sim.5384
Leufkens TR, Lund JS, Vermeeren A (2009) Highway driving performance and cognitive functioning the morning after bedtime and middle-of-the-night use of gaboxadol, zopiclone and zolpidem. J Sleep Res 18:387–396. https://doi.org/10.1111/j.1365-2869.2009.00746.x
Machado-Vieira R, Salvadore G, Luckenbaugh DA, Manji HK, Zarate CA Jr (2008) Rapid onset of antidepressant action: a new paradigm in the research and treatment of major depressive disorder. J Clin Psychiatry 69:946–958. https://doi.org/10.4088/jcp.v69n0610
McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, Garrett L, Bristow L, Marshall G, Macaulay A, Brown N, Howell O, Moore KW, Carling RW, Street LJ, Castro JL, Ragan CI, Dawson GR, Whiting PJ (2000) Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci 3:587–592. https://doi.org/10.1038/75761
Mets MA, de Vries JM, de Senerpont Domis LM, Volkerts ER, Olivier B, Verster JC (2011) Next-day effects of ramelteon (8 mg), zopiclone (7.5 mg), and placebo on highway driving performance, memory functioning, psychomotor performance, and mood in healthy adult subjects. Sleep 34:1327–1334. https://doi.org/10.5665/SLEEP.1272
Pearlman EM, Wilbraham D, Dennehy EB, Berg PH, Tsai M, Doty EG, Kay GG (2020) Effects of Lasmiditan on simulated driving performance: results of two randomized, blinded, crossover studies with placebo and active controls. Hum Psychopharmacol 35:e2732. https://doi.org/10.1002/hup.2732
Simen AA, Gargano C, Cha JH, Drexel M, Bautmans A, Heirman I, Laethem T, Hochadel T, Gheyle L, Bleys K, Beals C, Stoch A, Kay GG, Struyk A (2015) A randomized, crossover, placebo-controlled clinical trial to assess the sensitivity of the CRCDS Mini-sim to the next-day residual effects of zopiclone. Ther Adv Drug Saf 6:86–97. https://doi.org/10.1177/2042098615579314
Suryawanshi Ot, Pajai S (2022) A comprehensive review on postpartum depression. Cureus 14:e32745. https://doi.org/10.7759/cureus.32745
Tapia JL, Dunabeitia JA (2023) Driving safety: investigating the cognitive foundations of accident prevention. Heliyon 9:e21355. https://doi.org/10.1016/j.heliyon.2023.e21355
US Food and Drug Administration (2017) Guidance for industry: evaluating drug effects on the ability to operate a motor vehicle. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/evaluating-drug-effects-ability-operate-motor-vehicle
Vermeeren A, Sun H, Vuurman EF, Jongen S, Van Leeuwen CJ, Van Oers AC, Palcza J, Li X, Laethem T, Heirman I, Bautmans A, Troyer MD, Wrishko R, McCrea J (2015) On-the-road driving performance the morning after bedtime use of suvorexant 20 and 40 mg: a study in non-elderly healthy volunteers. Sleep 38:1803–1813. https://doi.org/10.5665/sleep.5168
Vermeeren A, Vets E, Vuurman EF, Van Oers AC, Jongen S, Laethem T, Heirman I, Bautmans A, Palcza J, Li X, Troyer MD, Wrishko R, McCrea J, Sun H (2016) On-the-road driving performance the morning after bedtime use of suvorexant 15 and 30 mg in healthy elderly. Psychopharmacology 233:3341–3351. https://doi.org/10.1007/s00213-016-4375-x
Vermeeren A, Jongen S, Murphy P, Moline M, Filippov G, Pinner K, Perdomo C, Landry I, Majid O, Van Oers ACM, Van Leeuwen CJ, Ramaekers JG, Vuurman E (2019) On-the-road driving performance the morning after bedtime administration of lemborexant in healthy adult and elderly volunteers. Sleep 42. https://doi.org/10.1093/sleep/zsy260
Verster JC, Roth T (2012) Drivers can poorly predict their own driving impairment: a comparison between measurements of subjective and objective driving quality. Psychopharmacology 219:775–781. https://doi.org/10.1007/s00213-011-2400-7
Verster JC, Veldhuijzen DS, Volkerts ER (2005) Is it safe to drive a car when treated with anxiolytics? Evidence from on-the-road driving studies during normal traffic. Curr Psychiatr Rev 1:215–225. https://doi.org/10.2174/1573400054065613
Verster JC, Spence DW, Shahid A, Pandi-Perumal SR, Roth T (2011) Zopiclone as positive control in studies examining the residual effects of hypnotic drugs on driving ability. Curr Drug Saf 6:209–218. https://doi.org/10.2174/157488611798280933
Verster JC, van de Loo AJ, Roth T (2015) Mirtazapine as positive control drug in studies examining the effects of antidepressants on driving ability. Eur J Pharmacol 753:252–256. https://doi.org/10.1016/j.ejphar.2014.10.032
Wingen M, Bothmer J, Langer S, Ramaekers JG (2005) Actual driving performance and psychomotor function in healthy subjects after acute and subchronic treatment with escitalopram, mirtazapine, and placebo: a crossover trial. J Clin Psychiatry 66:436–443. https://doi.org/10.4088/jcp.v66n0405
ZURZUVAE™ PI (2024) Full prescribing information. Sage Therapeutics, Inc. and Biogen Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/217369s001lbl.pdf
Acknowledgements
This study was funded by Sage Therapeutics, Inc., and Biogen Inc. Medical writing and editorial support were provided by Shivani Vaidya, PharmD, of Parexel, and Johnson Ying, PhD, of Red Nucleus, and funded by Sage Therapeutics, Inc., and Biogen Inc.
Funding
This study was funded by Sage Therapeutics, Inc. and Biogen Inc. Medical writing and editorial support were provided by Shivani Vaidya, PharmD, of Parexel, and Johnson Ying, PhD, of Red Nucleus, and funded by Sage Therapeutics, Inc. and Biogen Inc.
Author information
Authors and Affiliations
Contributions
All authors participated in the study design, data collection, and conceptualization, review, and editing of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
This study was performed in accordance with the Declaration of Helsinki and is consistent with Good Clinical Practice guidelines according to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Harmonized Tripartite Guideline, as well as all applicable regulatory requirements.
Consent to participate
All participants provided written informed consent.
Competing interests
Joi Dunbar, Carrie Vaudreuil, Indrani Nandy, and Victor Ona are employees of Sage Therapeutics, Inc., and may hold stock and/or stock options. Gaetano Morelli has no conflicts of interest to disclose. Rakesh Jain reports research funding from AbbVie, Lilly, Lundbeck, Otsuka, Pfizer, Shire, and Takeda and participation on advisory boards for Adamas Pharmaceuticals; Alkermes; Corium; Eisai; Janssen; Lilly; Lundbeck; Merck; Neos Therapeutics; Neurocrine Biosciences; Otsuka; Pamlab; Pfizer; Sage Therapeutics, Inc.; Shire; Sunovion; Supernus; Takeda; Teva Pharmaceuticals; and Usona. Rakesh Jain also reports honoraria for speakers’ bureaus from AbbVie, Alkermes, Almatica, Axsome, Corium, Eisai, Intra-Cellular Therapies, Ironshore, Janssen, Lilly, Lundbeck, Merck, Neos Therapeutics, Otsuka, Pamlab, Pfizer, Shire, Sumitomo Pharma, Sunovion, Takeda, Tris Pharma, and Viatris and consulting fees from AbbVie; Acadia; Adamas Pharmaceuticals; Alfasigma; Alkermes; Almatica; Axsome; Biogen Inc.; Boehringer Ingelheim; Cingulate; Corium; Eisai; Evidera; Impel Pharmaceuticals; Janssen; Lilly; Lundbeck; Merck; Neos Therapeutics; Neurocrine Biosciences; Osmotica Pharmaceuticals; Otsuka; Pamlab; Pfizer; Sage Therapeutics, Inc.; Shire; Sumitomo Pharma; Sunovion; Supernus; Takeda; Teva Pharmaceuticals; Transcend Therapeutics; and Viatris. Margaret K. Moseley and Seth Levin are employees of Biogen Inc. and may hold stock. Gary Kay is an employee of Cognitive Research Corporation and may hold stock.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dunbar, J., Morelli, G., Jain, R. et al. Effects of zuranolone on next-day simulated driving in healthy adults. Psychopharmacology (2024). https://doi.org/10.1007/s00213-024-06687-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00213-024-06687-6