
Abstract
Osteogenesis imperfecta (OI) is a heterogeneous heritable skeletal dysplasia characterized by bone fragility and deformity, growth deficiency, and other secondary connective tissue defects. OI is now understood as a collagen-related disorder caused by defects of genes whose protein products interact with collagen for folding, post-translational modification, processing and trafficking, affecting bone mineralization and osteoblast differentiation. This review provides the latest updates on genetics of OI, including new developments in both dominant and rare OI forms, as well as the signaling pathways involved in OI pathophysiology. There is a special emphasis on discoveries of recessive mutations in TENT5A, MESD, KDELR2 and CCDC134 whose causality of OI types XIX, XX, XXI and XXI, respectively, is now established and expends the complexity of mechanisms underlying OI to overlap LRP5/6 and MAPK/ERK pathways. We also review in detail new discoveries connecting the known OI types to each other, which may underlie an eventual understanding of a final common pathway in OI cellular and bone biology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteogenesis imperfecta (OI) or “brittle bone disease” is a genetically and phenotypically heterogeneous heritable skeletal disorder with an incidence of 1:15,000–20,000 live births. OI is primarily characterized by bone fragility and deformity, growth deficiency and highly variable secondary connective tissue findings, including blue sclerae, dentinogenesis imperfecta (DI), cardiovascular and respiratory defects, hearing loss, joint hypermobility. OI inheritance patterns include autosomal dominant and recessive, as well as an X-linked recessive form. The majority of individuals with OI (80–85% in Western countries; about 60% in countries with a high incidence of consanguinity) are caused by structural or quantitative defects in collagen type I genes, COL1A1 and COL1A2, encoding the α1(I) and α2(I) chains of type I collagen, respectively. However, OI is now understood to be a collagen-related disorder, with discovery of OI types caused by defects in genes whose protein products interact with collagen for folding, post-translational modification, processing and trafficking, as well as gene defects that affect primarily bone mineralization and osteoblast differentiation [1, 2].
The Sillence Classification of OI, proposed in 1979, was the first useful nosology. It divided OI into four “types” based on phenotypic (with a strong emphasis on scleral hue) and radiographic characteristics, as well as on the severity of bone fragility. Type I was the mildest group, generally with fractures in childhood but requiring clinical examination and radiographs to distinguish from individuals without OI. Type II was clinically severe, perinatal lethal individuals, who also had undertubulation of long bones on radiographs, generally with prenatal fractures of long bone and ribs and dark blue to blue-grey sclerae. Type III OI represented the severe non-lethal form, called “progressive deforming”. They fracture easily, require surgery and assistive devices for mobility, have striking short stature, barrel chest and scoliosis, and, frequently, relative macrocephaly. Type IV is the moderately severe form. These individuals have childhood fractures that lessen in frequency in adolescence. They have notable short stature and may develop scoliosis but can generally be independent ambulator with placement of intramedullary rods. The Sillence Classification was published before any of the genetic causes of OI were determined and proposed that there would be both dominant and recessive causes, with a greater severity in recessive cases.
In the subsequent decades, structural or quantitative defects in type I collagen were identified in the majority of individuals with OI. It emerged that most individuals with mild type I OI had dominant mutations in COL1A1 causing an effectively null allele and leading to the production of a reduced amount of structurally normal collagen in bone matrix. Mutations in COL1A1 and COL1A2, predominantly substitutions for glycine residues that uniformly occur at every third position of the collagen helical region or splicing defects, cause the full phenotypic range of types II–III–IV OI. Genotype–phenotype correlation studies have shown some alignment of specific collagen mutations with OI type. However, there is considerable variability of clinical outcome of mutations at the same site and molecular overlap of mutations with very different outcome. For these reasons, the Sillence types are still first a clinical classification.
After 2006, various advances led to the identification of multiple other genes causing OI and leading to a Genetic Classification. To date, the Genetic Classification describes 22 OI types. Type I–IV OI are reserved for those individuals with dominant defects in type I collagen genes. Type I OI is reserved for the subset of individuals whose collagen genes lead to a quantitative defect of type I collagen. Types II–III–IV are based on the Sillence types, that is, while these individuals all have collagen structural defects, the OI type assignment is based on the same phenotypic features as in the original Sillence nosology.
Types V and VI OI were defined histologically before their gene defect was known. They are characterized by altered bone mineralization, caused by a recurrent dominant mutation in IFITM5 in type V, recessive null mutations in SERPINF1 in VI, and IFITM5/BRIL p.S40L in atypical VI (aVI), respectively. Recessive defects in CRTAP, P3H1 and PPIB underlie absence of the procollagen prolyl 3-hydroxylation complex components, causing OI types VII, VIII and IX respectively. Recessive mutations in SERPINH1, FKBP10 and BMP1 affecting collagen processing and cross-linking, cause OI types X, XI and XIII, respectively. Impaired osteoblasts differentiation is a common features of OI types XII, XIV, XV, XVI, XVII, XVIII, caused by recessive mutations in SP7, TMEM38B, WNT1, CREB3L1, SPARC and MBTPS2. WNT1 also causes less severe dominantly inherited bone fragility. The recent discoveries about the bone mechanisms of newly established OI types XIX, XX, XXI and XXII have fully validated recessive mutations in TENT5A, MESD, KDELR2 and CCDC134, respectively, as OI-causative within the umbrella of type I collagen-related gene defects.
The Genetic Classification is not simply a list of genes in which defects cause osteogenesis imperfecta. In fact, largely driven by the process of rare disease gene identification, the genes were identified in logical “clusters” by mechanism. For example, when one component of the procollagen prolyl 3-hydroxylation complex was identified in recessive OI, investigators naturally looked quickly to the other two components. Or when a skeletal phenotype in the murine model for CREB3L1/OASIS led to children with rare recessive defects in OASIS, it becomes logical that defects in its processing protease, S2P, encoded by MBTPS2, could also cause OI. Sometimes serendipity intervenes and the related gene is not the next one identified, but their proximity in the gene list leads to their presence in the same “mechanism group”, thus serving both as an aide to memory and a guide to further research. Some (predominantly clinical) investigators wish to fold the new genes into the phenotypic classification based on the Sillence nosology. This is said to have the advantage of being simpler for clinicians and families, and to result in more uniform treatment based on phenotype. However, this simplicity masks some obvious and some latent opportunities for inaccuracy in a purely phenotypic classification. The OI patients in each Sillence phenotypic type will have multiple different genetic etiologies to their bone dysplasia, reflecting different mechanisms and likely/possibly responding differently to the same pharmacological or non-pharmacological treatment. Parents of children with the same rare condition are connected to each other via social media and OI-clinics; they are astute in figuring out which patient is different and are reasonably interested in understanding the differences and their implications. A second factor is the fluidity and variability of phenotype. Individuals, even in the same family, with mutations in the same gene may have sufficient difference in severity to be assigned to a different phenotypic type. Or phenotype my worsen over the decades of a life, changing the assigned type. These factors would confuse patients, families and primary doctors, and potentially muddling treatment protocols. The best of both genetic and phenotypic systems can be obtained by having the Genetic Classification as the primary type, with a secondary designation for the clinical severity of the phenotype which could change with time and circumstance. The genetic classification of OI types is listed in Table 1, with mechanisms and signaling pathways presented in Fig. 1.

Mechanisms and signaling pathways of OI. The two proα1(I) and one proα2(I) chains of the procollagen heterotrimer undergo post-translational modifications of proline and lysine residues by P4H and LH1, respectively, in the ER. During procollagen folding, the ER-residing prolyl 3-OH complex, consisting of P3H1, CRTAP, and PPIB, hydroxylates the P986 residue important for alignment of procollagen chains. HSP47, an ER chaperone, binds to procollagen triple helix to facilitate collagen folding and secretion. FKBP10, another ER chaperone, cooperate with HSP47 in procollagen trafficking from the ER to the Golgi. Another protein that functions closely with HSP47 is KDEL receptor 2, which has a role in intracellular recycling of ER-resident proteins through retrograde transport. Once the procollagen triple helix is secreted into extracellular space, N- and C-propeptides of procollagen are cleaved by ADAMTS-2 and BMP1 enzymes, respectively. Type I collagen is released and incorporated into extracellular matrix. An important regulator of collagen assembly is SPARC, which binds collagen as a matricellular glycoprotein. At the level of transcriptional regulation, Osterix/Sp7, a zinc finger transcriptional factor, interacts with RUNX2, which induces the expression of collagen genes. Intracellular calcium is important for collagen post-translational modification and metabolism. TRIC-B together with IP3R regulate calcium flux from the ER to cytoplasm. In addition, TRIC-B absence impacts the fusion and fission of mitochondria, affecting MFN2 and DRP1 regulators at the ER-mitochondria contact sites. Transmembrane transcriptional factor OASIS is transported from the ER to the Golgi membrane for RIP signaling. OASIS is cleaved by S1P and S2P in the Golgi and the released N-terminal end of OASIS translocates into the nucleus for transcription of genes important for extracellular matrix, such as collagens. Also affecting collagen expression and bone mineralization in osteoblasts is FAM46A, encoded by TENTA, which is involved in the polyadenylation of collagen transcripts and other proteins involved in bone mineralization. BRIL is an important regulator of bone mineralization and impacts SERPINF1 transcription in the nucleus. SERPINF1 encodes the PDEF protein, which binds collagen to activate its own anti-angiogenic function. Wnt signaling, important for bone development, is activated by binding of WNT1 ligand to the Frizzled and LRP5/6 receptors, which inhibits the degradation complex, consisting of Axin, APC, and GSK3, in order to stabilize β-catenin. β-catenin translocates to the nucleus to activate transcription of Wnt target genes. MESD, a chaperone residing in the ER, has a role in Wnt signaling by translocating LRP5/6 receptors, and is also a direct chaperone of pro-α1(I) in osteoblasts. BMP signaling is another important pathway for bone metabolism. BMP binds to BMP receptors and induces phosphorylation of SMAD 1/5/8. Together SMAD 1/5/8, FAM46A, and SMAD4 form a complex that further translocates to the nucleus to induce transcription of BMP target genes. Recently proposed OI-causing gene CCDC134 encodes a secretory protein CCDC134 that inhibits ERK and JNK phosphorylation of MAPK pathway, and impacts osteoblast expression of collagen and mineralization
The clinical presentation of OI ranges from mild to perinatal lethal. Type I OI is the mildest form of OI, which is caused by quantitative defects in collagen and presents clinically with blue sclerae, variable hearing loss, and bone fractures during childhood which decrease in frequency after puberty. The main features of OI include long bone fractures, growth deficiency and skeletal deformities. However, the OI symptoms go beyond bone fragility, including the secondary defects in other connective tissues such as cardiovascular and pulmonary defects, hearing loss, DI, blue sclerae, Wormian bones, and joint hypermobility.
OI types I-IV due to mutations in the helical, N-propeptide, and C-propeptide regions of COL1A1 and COL1A2, and OI Type XIII due to mutations in BMP1.
OI Types I-IV Due to Mutations in the Helical, N-Propeptide, and C-Propeptide Regions of COL1A1 and COL1A2, and OI Type XIII Due to Mutations in BMP1
Introduction
Type I collagen is the most abundant protein in the body and the main constituent of the extracellular matrix (ECM) of bone and skin. Type I procollagen heterotrimer consists of two pro-α1 (I) and one pro-α2 (I) chains, synthesized in the endoplasmic reticulum (ER), that undergo post-translational modifications of proline and lysine residues carried out by enzymes prolyl-4-hydroxylase 1 (P4H1) and lysyl hydroxylase 1 (LH1). ER chaperones facilitate the folding and assembly of the triple helix, which is subsequently transported to Golgi membranes and then to the ECM. Mature type I collagen is formed by cleavage of the N- and C-terminal propeptides via metalloproteinases ADAMTS-2 (ADAM metallopeptidase with thrombospondin type 1 motif 2) and BMP1 (bone morphogenetic protein 1), respectively. Mature collagen can then self-assemble into heterotypic fibrils with types III and V collagen.
Structural and Quantitative Mutations
The most common OI mutations are point mutations which cause the substitution of one of the helical glycines, that occur at every third residue in uninterrupted repeating Gly‐Xaa‐Yaa triplets, by residues with a bulky or charged side chain. Because the glycine residues should normally be accommodated in the sterically restricted internal aspect of the helix, the substitutions cause a slower folding of the collagen, exposing the constituent chains for a longer time to post-translational modification enzymes [3].
Splice site mutations are the second most common type of OI collagen mutations. These may cause exon skipping, intron retention or use of cryptic splice sites. Substitutions of non-glycine residues found at X and Y positions of Gly-Xaa-Yaa triplets are distinct from glycine substitutions. Bone fragility is generally mild, and affected individuals have other connective tissue disorders such as OI/Ehlers-Danlos Syndrome (EDS) or Caffey disease. The most common X or Y position substitution is of an arginine residue by a cysteine, leading to formation of intermolecular disulfide bonds within the collagen helix which cause kinking of the helix and a register shift affecting N-propeptide processing [4,5,6]. A rare mutation consisting of deletion or duplication of one or two Gly-Xaa-Yaa triplets causes severe or lethal OI. These “register shift” mutations induce helical overmodification and disrupt binding sites for non-collagenous proteins along the collagen helix. [7, 8].
Phenotypic variability is one of the major hallmarks of OI and other skeletal dysplasias. It occurs when OI patients with the same mutation, even siblings, have sufficiently different levels of OI clinical severity that they would be given different phenotypic classifications. This phenomenon obscures the therapy of OI patients and further studies are warranted to explore which signaling pathways and modifying factors contribute to this variability.
The Consortium for OI Mutation Genotype–Phenotype analysis [9, 10] showed that substitutions in the α1(I) chain have more severe and lethal phenotype compared to α2(I), clustered in the major ligand binding regions (MLBR) in the collagen triple helix. While the majority of glycine substitutions in the α2(I) chain were not lethal, more lethal splice site mutations occur in this chain [9]. In a recent genotype–phenotype study of a large cohort, 39 of 217 reported glycine substitutions in α1(I) chain were non-lethal, while 58 glycines had at least one lethal case. In α2(I) chain, 63 of 222 reported glycine substitutions were non-lethal and 49 substitutions had a lethal outcome [10]. In another recent population-based study of 166 patients, six pathogenic variants, with four being newly discovered, were identified in the lethal regions of COL1A1 and COL1A2 genes in seven patients with non-lethal OI, adding to the prior reported individuals that indicated not all glycine substitutions located in the lethal domain lead to a fatal outcome [11].
Mutations leading to haploinsufficiency in COL1A1 cause the mildest form of OI, type I. Haploinsufficiency mutations lead to mRNA instability; the mutant allele is thus unable to produce the collagen protein chain, causing quantitative defects of structurally normal collagen. Typical causative mutations introduce a premature stop codon in the coding region of COL1A1 mRNA by either single nucleotide substitutions, insertion and/or deletion of nucleotides, or secondary to alternative splicing when a mutation is located at a splice sites [12]. Haploinsufficiency mutations in COL1A2 result in EDS if the mutation is homozygous or compound heterozygous [13, 14].
OI Murine Models with Collagen Defects
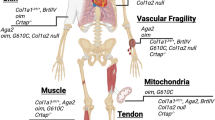
The Brittle mouse model (Brtl+/−) is a model for investigating classical OI. It carries a heterozygous α1(I) Gly349Cys substitution in one of COL1A1 alleles, and presents with symptoms and findings of dominant OI and variability of phenotype. About one-third of newborn mice die on day P1 due to respiratory distress, while living mice have a fairly uniform moderately severe OI phenotype [15]. Affected mice are small and exhibit low bone mass and hypermineralized bone, altered bone biomechanical properties and bone remodeling as well as increased bone fractures [16,17,18].
Another mouse model for studying a classical OI is the G610C OI knock-in mouse, with a Gly610Cys substitution in the COL1A2 gene. G610C, also known as the “Amish mouse”, was created based on the collagen mutation identified in 64 individuals in an Old Order Amish community who had OI type I/IV with considerable variability of phenotype [19]. The mice have low bone mineral density (BMD) and body weight, hypermineralized bone and altered bone biomechanical properties [20].
Oim/oim is an OI mouse model with a spontaneous mutation in Col1a2 gene with a moderate to severe OI phenotype similar to OI type III. Homozygous mice have a guanine deletion at nucleotide 3983 of Col1a1 gene which alters the last 48 amino acids sequence of proα2 (I) propeptide and causes the accumulation of α1 (I) homotrimer collagen. The oim/oim mice have skeletal deformities and fractures, osteopenia, and thin cortical bone [21]. However, this model is atypical for collagen mutations in that the phenotype has recessive inheritance and α1(I) homotrimers are not a normal component of bone matrix.
Aga2 (abnormal gait 2) mouse model, with a dominant frameshift mutation in Col1a1 C-propeptide, is broadly applicable, especially for non-skeletal aspects of dominant OI such as cardiovascular and respiratory defects as well as apoptosis studies [22, 23]. Aga2 mice have two phenotypes, while 34% of newborn mice are lethal due to cardiorespiratory defects, the remaining 66% survive with moderately severe OI with bone fractures, decreased body size and bone mass. Aga2 mice show accumulation of proα1(I) intracellularly that leads to activation of ER-stress-specific unfolded protein response (UPR) and apoptosis of osteoblasts [23].
Col1a1Jrt/+, or the Jrt model, is the first mouse model of combined OI/EDS phenotype which makes it suitable for investigating the OI/EDS pathophysiology [24]. Jrt mice have a dominant mutation in Col1a1 causing skipping of exon 9 and leading to an 18 amino acid deletion in the N-terminal domain of Col1a1. Mice exhibit reduced bone mass and brittle bones susceptible to fractures as OI characteristics, and reduced skin tensile properties and spine curvature as part of EDS phenotype [24].
The Mov13 mouse was the first murine model to confirm that type I collagen mutations caused OI. Mov13 was created in 1983 by the then-prevalent technique of insertion of murine Moloney leukemia virus into the first intron of the Col1a1 gene, preventing initiation of Col1a1 [25]. Homozygous mice produce no type I collagen and die at mid-gestation due to blood vessels rupture, while heterozygous mice survive until adulthood and are a model for type I OI. Heterozygous mice produce 50% less collagen resulting in a 50% decrease of tissue collagen content, which induces bone brittleness [26]. Interestingly, the Mov13 skeleton spontaneously adapted during the first 2 months of life to significantly improved bending strength of long bones [27]. Activation of the provirus at about 4 months of age makes this a difficult mouse to manage, and it is no longer in use.
Generation of a mouse model with a classical collagen glycine substitution in a conditional allele that allows tissue specific expression would be very useful for understanding secondary features of OI.
Defects in Collagen Processing and Propeptides, BMP1
Following synthesis of procollagen chains, they align with each another at the C-propeptide and initiate association and folding toward the N-terminal propeptide. To release mature collagen molecules, N- and C-propeptides are processed by metalloproteinases ADAMTS-2 and BMP1/tolloid-like proteinases, respectively.
A single heterozygous mutation in the signal peptide cleavage site of COL1A1 has been reported in both an infant with lethal type II OI and in a severe terminated fetus [28, 29]. Cells and tissues from the terminated fetus showed delayed collagen secretion and collagen intracellular retention, leading to decreased collagen production. The intracellular collagen appears to be overmodified despite the position of the mutation, while the secreted portion is electrophoretically normal. ER cisternae in cells from the affected fetus are enlarged. The skeletal pathology is attributed to the retained collagen, with putative impaired cleavage and thus impaired release of pre-procollagen from ER-entry sites. This could lead to misfolding and ER stress from both the mutant procollagen chains, a dominant negative effect on entry of normal α1(I) chains, and impaired generalized entry of other proteins to the ER [29].
Mutations in the first approximately 90 residues of the type I collagen α(I) chains interfere with removal of the N-propeptide, although the primary sequence of the N-proteinase cleavage site is intact, and are causative for an OI/EDS phenotype. This phenotype was first described in a group of children presenting with OI with bright blue sclerae, who also had striking hyperextensibility of both large and small joints, reduced collagen fibril diameters and friable tissue [30]. The severity of the OI associated with OI/EDS varied with the specific residue substituted (types III or IV OI) while the EDS symptoms were less striking with further distance from the cleavage site. A subsequent study began with a cohort of patients primarily referred for EDS symptoms, in whom Gly substitutions and exon skipping defects were found in in the amino-end of the COL1A1 or COL1A2 helical region [31]. They shared the delayed N-terminal processing, severe joint hyperlaxity, easy bruising of the cohort presenting predominantly with OI, but some also had signs of vascular fragility, and all had mild OI manifestations with blue sclerae, short stature and few fractures. Thus, mutations in the helical region of either the α1(I) or α2(I) chains adjacent to the N-propeptide cleavage site may present as predominantly OI or EDS.
Substitutions in the type I procollagen C-propeptide cleavage site in COL1A1 or COL1A2 were first reported with a mild dominant OI phenotype in patients with normal or increased DXA z-score and bone mineralization increased higher than in classical OI on BMDD, and impaired C-propeptide processing [32, 33]. Patients have normal growth, light sclerae, straight spine, normal teeth and hearing. Collagen biochemistry was minimally altered with slight backstreaking of α1(I) and α2(I) chains but with normal thermal stability. Histomorphometry yielded normal trabecular bone volume and thick osteoid seams. Tissue fibrils of collagen were thin with a “barbed-wire” appearance [32]. More recently a larger set of patients affected with high bone mass (HBM) OI was reported [33], supporting the predominantly mild phenotype. Although they had fractures of large long bones that persisted into adulthood, fractures were infrequent. Height was in the lower half of the normal curve; again, sclerae were light, teeth and hearing were normal. The mean spinal z-score was + 2.9 SD and qBEI confirmed high levels of bone mineralization. Histomorphometry on two patients confirmed hyperostoidosis. Interestingly, in this older cohort, one-third of subjects older than 15 years had scoliosis.
The C-propeptide itself of type I procollagen is critical for recognition and association of chains to form the triple helix. The C-propeptide is processed in the pericellular space and not incorporated into matrix. Surprisingly, mutations located in the C-propeptide were found to cause mild to lethal OI [34, 35]. The severity of OI phenotype is generally correlated with the structure of the triplet C-propeptide, sometimes compared to a flower, with a base or stem, a transition region, and 4 “petals”, as well as multiple interchain bonds [34]. Mutations that affect areas of interaction between chains or calcium-binding sites lead to a severe consequence. Most but not all mutations in outward facing residues lead to a milder type of OI [34, 35]. These mutations lead to both osteoblast intracellular and matrix dysfunction. Immunofluorescence microscopy indicated that procollagen with C-propeptide defects was mislocalized to the ER lumen, in contrast to the ER membrane localization of normal procollagen and procollagen with a classical collagen helical defect [35]. Slower processing of the mutant C-propeptide leads to pC-collagen incorporation into matrix [35]. Finally, there is a quantitative defect that occurs when nonsense-mediated mRNA decay (NMD) reduces the synthesis of mutant procollagen, leading to a milder OI phenotype [34].
Recessive mutations in BMP1, which encodes metalloprotease BMP1 and its longer isoform mammalian tolloid, cause type XIII OI. BMP1 plus an enhancer protein affects the cleavage of the type I collagen C-propeptide. Type XIII OI is, however, more than the recessive enzymatic counterpart to the dominant structural mutations in the collagen cleavage site causing HBM OI. BMP1 is the C-propeptidase of types I–II–III procollagen and the N-propeptidase of type V collagen, as well as processing non-collagenous ECM components prolysyl oxidase, prodecorin, probiglycan and dentin matrix acidic phosphoprotein 1 (DMP1). Not surprisingly, recessive BMP1 mutations cause a more severe phenotype than HBM OI, ranging from mild to severe OI. Many of these patients have bone hypermineralization by qBEI. Incorporation of pC-collagen into ECM leads to abnormal organization of fibrils and may provide an increased number of sites for mineral nucleation [36,37,38].
All other genes in which mutations directly impact collagen post-translational modification, trafficking, polyadenylation etc. all fall into one of the following (mostly) recessive sets.
Types V, VI, and Atypical VI OI- IFITM5, SERPINF1, IFITM5-p.S40L: Altered Bone Mineralization
In this grouping, dominant mutations in IFITM5/BRIL and recessive mutations in SERPINF1/PDEF both lead to OI with characteristic bone hypermineralization but without alterations in the structure or post-translational modification of type I collagen, although collagen secretion is decreased. Each set of mutations has distinctive histomorphometry focused on lamellar disorganization. A special IFITM5 mutation with a phenotype similar in many respects to that caused by null mutations in PEDF, suggests that their mechanistic pathways are inter-connected and may converge with the biochemical and molecular cascade of events in OI initiated by collagen structural mutations.
A recurrent gain-of-function mutation in the 5’‐UTR of IFITM5 (c.‐14C > T), encoding bone-restricted interferon-inducible transmembrane (IFITM5)-like protein (BRIL), is the cause of almost all instances of OI type V [39, 40]. This IFITM5 mutation generates a new upstream start codon that results in addition of five amino acids (Met-Ala-Leu-Glu-Pro (MALEP)) to the N-terminus of BRIL (MALEP-BRIL) [39].
BRIL was discovered as a protein with increased expression during osteoblast differentiation, especially during the maturation/mineralization phase [41], using a signal trap screening system to detect new genes encoding proteins in bone. This function of BRIL was further corroborated by increased in vitro mineralization by type V osteoblasts derived from patient surgical bone chips [42], indicating a role of BRIL in bone mineralization. Further, MALEP-BRIL showed no differences in cell membrane localization, topology, and synthesis level compared to WT BRIL [43], supporting a gain-of-function mechanism.
Type V OI was defined radiographically and as the first type of OI that is not caused by mutations in collagen genes [44]. Type V OI is a dominantly inherited and moderately severe type of OI, which is highly variable in phenotype [45], characterized by hyperplastic callus formation predominantly in long bones after fractures or surgical interventions, ossification of the forearm interosseous membrane, radial head dislocation, and dense metaphyseal bands in the forearm. Blue sclerae and dentinogenesis imperfecta (DI) are absent. Polarized light microscopy revealed that type V OI Iliac crest bone has an irregular or meshlike appearance of lamellae [44]. Type V OI bone is hypermineralized, with BMDD (bone mineral density distribution) shifted towards increased mineralization, despite having no abnormality of collagen structure or modification [46]. In addition, type V OI bone has an increased osteocyte lacunar density, that, together with irregular bone lamellae, indicates altered bone remodeling [46]. There is currently no surviving murine model for type V OI. Ifitm5-KO mice showed a non-significant bone phenotype compared to patients with type V OI [47]. On the other hand, transgenic mice overexpressing Ifitm5 in osteoblasts were perinatal lethal with skeletal defects, fractures, and delayed mineralization, in addition to impaired in vitro mineralization of osteoblasts [48]. Another mouse model for type V OI, with a knock-in mutation, was perinatal lethal as well, with hypomineralized skull, wavy ribs, shorter long bones and increased hypertrophic chondrocytes in midshaft of long bones. Calvarial osteoblasts from the knock-in mouse showed delayed differentiation and mineralization, and increased expression of inflammatory and adipogenic markers [49].
Type VI OI, an autosomal recessive disorder, is caused by null mutations in serpin family F member 1 (SERPINF1) gene, which encodes the secreted glycoprotein pigment epithelium–derived factor (PEDF) [50, 51]. Type VI OI is generally a severe progressive form of OI, with normal phenotype at birth, and presenting with long bone and vertebral compression fractures in early childhood. Affected individuals have white or faintly blue sclerae and absent DI. Type VI OI was also delineated before the causative gene was identified, based on bone histomorphometry with an irregular fish-scale pattern of lamellae visualized under polarized light, and excessive accumulation of osteoid reflecting increased mineral lag time [52]. This bone is also hypermineralized, although surrounded with areas of low bone mineral content and an increased number of osteocytes [53]. Patients with type VI OI have elevated alkaline phosphatase (ALPL) levels in serum as children [52]. In addition, circulating PEDF is undetectable in their serum [54]. PEDF is a potent endogenous inhibitor of angiogenesis [55], which binds to collagen type I in extracellular matrix. Binding to collagen is critical to activate the anti-angiogenesis function of PEDF; mutation of either the collagen-binding site on PEDF or the PEDF-binding site on collagen abolishes PEDF anti-angiogenic activity [56, 57]. Using a Serpinf1−/− murine model [58], PEDF was shown to have a role in stimulation of osteogenesis and impairing bone vascularization, with TGF-β having the opposite trends [59]. This suggests that PEDF and TGF-β antagonism controls these two processes and contributes to type VI OI pathogenesis. The mechanism of type VI OI remains open for further investigation—competing models focus on the effect of anti-angiogenesis on cartilage ossification [60] or on the collagen-binding properties of PEDF, or even on the capacity of PEDF to induce osteoblast differentiation in cooperation with Wnt3a [61], proper transition to osteocytes and mineralized nodule formation [53].
Ten patients with heterozygous IFITM5, c.119C > T (p.Ser40Leu) mutation have been described with severe progressive OI that is not associated with radiographic or histologic findings of type V OI. These patients develop extremely short stature, vertebral compression fractures, scoliosis, bowed limbs [62,63,64,65,66,67,68,69,70,71]. Intriguingly, their clinical presentation is similar to type VI OI, including elevated unmineralized osteoid and a fish‐scale pattern of lamellation in bone tissue. Serum ALPL levels were elevated in childhood, although the patients do not carry PEDF mutations and the serum PEDF levels are normal. This surprising clinical outcome for an IFITM5 mutation was named atypical type VI OI (aVI OI), connecting types V and VI OI and suggesting synergistic roles of BRIL and PEDF [62] since no evidence has been uncovered for a direct interaction between BRIL and PEDF. While WT BRIL and MALEP-BRIL were shown to attach to the plasma membrane via palmitoylated cysteine residues (C50 and C51), BRIL p.S42L was trapped in the Golgi and poorly palmitoylated, perhaps due to steric hindrance from the proximity of the palmitoylation sites [43]. The newly generated Ifitm5/BRIL p.S42L mouse, a knock-in mouse model for atypical type VI OI, has multiple long bone fractures, and decreased whole body BMD. Mechanical testing demonstrated increased bone brittleness [72]. Additionally, mice showed alterations of bone material properties including hypermineralization of the matrix, increased vascular porosity, elevated osteocyte lacunae density and disordered collagen fibril orientation by second-harmonic generation (SHG) [73].
Types VII, VIII and IX OI-CRTAP, P3H1, PPIB—Procollagen prolyl 3-Hydroxylation Complex
Collagen has a triple-helical tertiary structure and undergoes post-translational modification of proline and lysine residues by prolyl 4-hydroxylase (P4H1) and LH1, an important step for proper folding and stability of the collagen helix [74]. Although the 3-hydroxylation of the α1(I)Pro986 residue had been known for several decades, neither its role nor the identity of the prolyl 3-hydroxylase was known. Isolation of prolyl 3-hydroxylase revealed that an ER-resident complex of 3 proteins was responsible for prolyl 3-hydroxylation of α1(I)Pro986 on each α1(I) chain of collagen types I and II [75, 76], specifically prolyl 3-hydroxylase 1 (P3H1), which accomplishes the enzymatic function, cartilage‑associated protein (CRTAP), the helper protein providing mutual stabilization to P3H1, and cyclophilin B (CyPB), the major collagen PPIase which here functions as a chaperone. The identification of the genes encoding the components of the prolyl 3-hydroxylation complex as the causative genes for 3 severe types of recessive OI demonstrated that the complex and its relatively minor modification have a major role in bone development [77].
The first evidence of an important role of CRTAP in bone was discovered by studies on Crtap-null mice, which developed osteochondrodysplasia with severe osteopenia and decreased osteoid formation [78]. Null and point mutations in CRTAP are responsible for development of type VII OI [78,79,80,81,82], a recessive form with a generally lethal outcome, although there are some very severe surviving patients. The distinctive clinical characteristic of type VII is a rhizomelia, reflecting the role of CRTAP in both cartilage and bone. Other features include fractures at birth, bowing of lower extremities, white or light blue sclerae and osteopenia.
Recessive type VIII OI is caused by null mutations in the P3H1 gene that encodes P3H1, which was independently isolated as an extracellular matrix proteoglycan named leprecan [81, 83,84,85]. As for CRTAP, most individuals with type VIII OI have a perinatal lethal phenotype; severe survivors are more common for Type VIII than for type VII OI and share their rhizomelia. Recently, unusual dental anomalies such as hypodontia, a mesiodens, and single rooted second permanent molars were reported in type VIII OI [86]. Using Crtap−/− murine model, it was shown that lung fibroblasts synthesize type I collagen with altered post-translational modifications, negatively impacting lung function [87].
In vitro studies of fibroblasts from types VII and VIII OI patients showed mutual stabilization of CRTAP and P3H1 in the ER collagen prolyl 3-hydroxylation complex. The studies revealed that both CRTAP and P3H1 proteins were absent or reduced in cells with null mutations in either gene, while rescue studies transfecting fibroblasts with CRTAP or P3H1 constructs restored both CRTAP and P3H1 protein levels [88]. In addition, knock-in mice with an α1(I)P986A substitution that cannot be 3-hydroxylated revealed the role of the 3Hyp substrate site itself in recessive OI as affecting collagen cross-linking and structural organization. Mutant mice had otherwise normal bone phenotype; however, the bone collagen hydroxylysyl pyridinoline (HP) and lysyl pyridinoline (LP) crosslink ratio was doubled, with decreased collagen fibril diameter [89]. Complementary studies of knock-in mice with a single amino acid substitution in the active site of P3h1 showed P3h1 activity was suppressed but it was still able to form a complex with Crtap thus retaining the collagen chaperone function. This mutation also resulted in the absence of prolyl 3-hydroxylation at Pro986 of the α1(I) and α1(II) collagen chains [90].
CyPB, encoded by peptidylprolyl isomerase B (PPIB), is a peptidyl-prolyl cis–trans isomerase which is an ER resident [91]. It is multifunctional, with roles in inflammation, cancer and viral infection [92], as well as in procollagen folding and secretion [93]. Mutations in PPIB cause recessive type IX OI, with moderate to lethal phenotype, similar to CRTAP and P3H1 mutations [94,95,96]. The role of CyPB in the 3-hydroxylation complex was illuminated by studies of a mouse model lacking PPIB expression [97]. Ppib knockout mice had decreased bone volume, bone mechanical properties and increased bone fragility. CypB-deficient osteoblasts had normal CRTAP levels, while levels of P3H1 were reduced by half, resulting in a marked reduction in collagen prolyl 3-hydroxylation and in site-specific changes in collagen lysyl hydroxylation and glycosylation. Intracellular folding of type I collagen in CyPB-null cells was slower than WT and collagen secretion was reduced; interestingly, collagen folding could be reduced further by the addition of cyclosporin A (CsA), suggesting some redundancy exists for the PPIase function required for collagen folding. The reduced lysyl hydroxylation of CypB-null cells is associated with a marked decrease of the HP/LP crosslink ratio, which is the opposite of the crosslink ratio alteration seen in classical OI caused by a collagen structural change [97]. The reduced prolyl 3-hydroxylation, changes in lysyl modification and altered cross-links may alter matrix assembly and ultimately bone stability and strength.
Type X OI-HSP47, Type XI OI-FKBP10, and Type XXI OI—KDELR2-Collagen Folding and Cross-linking
Type X is an extremely rare type of recessive OI, with a severe to lethal phenotype. At birth, patients have severe skeletal defects, including thin ribs with multiple fractures, generalized severe undermineralization, platyspondyly, deformed long bones, hydranencephaly, macrocephaly, blue sclerae, and Wormian bones. Most presented with sudden early death due to respiratory issues [98,99,100]. Because of the rarity of Type X OI, information on the histomorphometry and mineralization of bone tissue has not been reported.
Type X OI is caused by homozygous mutations in serpin family H member 1 (SERPINH1) gene, which encodes heat shock protein 47 (HSP47). HSP47 is a collagen- specific chaperone which resides in the ER and facilitates folding of collagen chains [101]. It binds to fully folded triple helical procollagen in the ER, then dissociates in the cis-Golgi, preventing procollagen aggregation [102]. Hsp47-knockout mice are embryonically lethal [103], with reduced production of mature collagen types I and IV, and accumulation of type I collagen in the ER of Hsp47−/− cells [104]. Collagen secreted from cells with HSP47 defects has increased protease susceptibility, suggesting it is not properly folded [98]. In a recent study, a type X OI child with a homozygous mutation in the collagen interacting surface of HSP47, which reduced its affinity to type I collagen, was shown to have increased post-translational modification of type I procollagen, with significant upregulation of several chaperones (FKBP65, PDI, P4HA1, CypB, P3H1, CRTAP, P3H3, SC65, LH1 and LH3) involved in modification and folding of procollagen as part of a larger compensatory mechanism [100].
Another molecular chaperone, FKBP65, which is a PPIase, also resides in the ER and interacts with collagen [105]. Mutations in the gene that encodes FKBP65, FKBP10, cause either recessive type XI OI, Bruck syndrome or Kuskokwim syndrome. The initial report of homozygous mutations in FKBP10 presented patients with moderately severe OI with progressive kyphoscoliosis, long bones fractures with severe deformities, ligamentous laxity, grayish sclerae and no DI [106]. FKBP65 was shown to have a role in collagen cross-linking since patient cells deficient in FKBP10 had normal folding of type I collagen, but significant reduction of collagen cross-linking and collagen deposition in the matrix [107, 108]. On other hand, mutations in FKBP10 were shown to be a cause of Bruck syndrome [109,110,111], a rare recessive disorder similar to OI, characterized with congenital joint contractures, bone fragility, osteoporosis and short stature. Recent study showed that novel variants in FKBP10 caused Bruck syndrome with orodental changes such as dental malocclusion and enamel hypoplasia [112]. Finally, Kuskokwim syndrome, in which affected individuals have congenital contractures with minimal skeletal manifestations, is caused by an in-frame deletion of a conserved tyrosine in the third PPIase domain of FKBP65 [113].
There is an interesting relationship between the proteins responsible for types X and XI OI. Cells with HSP47 mutations have decreased FKBP65, and procollagen aggregates in ER-adjacent compartments, possibly autophagosomes [114]. While absence of FKBP65 does not affect HSP47 levels, it does result in mis-localization of HSP47 [114]. Apparently, the two chaperones cooperate in procollagen trafficking from the ER to the Golgi.
Type XXI OI, caused by recessive defects in KDELR2, intersects with type X OI, caused by deficiency of HSP47. Patients with type XXI OI have a severe progressive deforming OI [115, 116]. Fewer than ten case, ranging from a preterm fetus to the 5th decade of life have been reported, with multiple fractures, long bone and chest deformities, wheelchair dependence, and short stature. Blue sclerae, dentinogenesis imperfecta and neurodevelopmental delay are present in some individuals.
These individuals have homozygous missense or frameshift mutations in KDELR2, which encodes the KDEL receptor 2. This receptor, a member of the KDEL receptor family, is localized in the cis-Golgi, the ER, and the intermediate ER-Golgi compartment (ERGIC). The receptors are part of the COPI vesicles involved in retrograde transport of ER-resident proteins containing the signal peptide Lys-Glu-Asp-Leu (KDEL) from the cis-Golgi back to the ER in a pH-dependent process [117, 118]. KDELR1 and 3 cannot compensate for loss of KDELR2. Thus, loss-of-function defects in KDELR2 result in secretion of the normally ER-localized type I collagen-specific chaperone HSP47 to the extracellular space. When extracellular HSP47 interacts with collagen molecules in matrix, it binds to monomeric and multimeric collagen and impairs their fibrillogenesis, yielding thin fibrils with excess bound HSP47, but not does alter collagen modification [116]. Interestingly, XXI OI fibroblasts have reduction of both intracellular and secreted type I collagen. Depleted intracellular HSP47 also results in reduction of intracellular FKBP65, overlapping type XXI OI with type XI OI, as well as type X [116]. The Kdelr2−/− murine model has premature lethality; its bone structure appears to model the patient skeletal phenotype, as well has having cleft palate [119].
The remaining sections are about OI types that have an indirect collagen-related mechanism through changes in osteoblast development.
Type XII OI-SP7—Osteoblast Differentiation and UPR Pathway
Homozygous or heterozygous mutations in SP7 are the cause of extremely rare type XII OI, with either with recessive [120,121,122,123] or dominant inheritance [124] and characterized by predominantly moderate bone fragility and deformity, delayed eruption of teeth, normal sclerae and variable dentinogenesis imperfecta. The first reported individual had a homozygous frameshift mutation, moderately severe OI and died early in childhood of respiratory etiology [120]. The same homozygous mutation was reported later in a 17 year old Chinese boy [125]. Recently, a homozygous p.Arg316Cys substitution, which alters the first zinc finger domain in SP7, was identified in independent reports [121, 126]. First, a 13-year old Iraqi boy had short stature, bone fragility fractures, scoliosis, hearing loss and craniofacial anomalies. A bone biopsy showed increased cortical porosity and increased trabecular bone turnover. Second, Kuwaiti male siblings in their early 20 s have moderately severe OI, stature in the normal range, several long bone fractures, dentinogenesis imperfecta, conductive hearing loss and craniofacial anomalies [126].
Intriguingly, there have also been reports of pathological heterozygous mutations in SP7. Siblings with a heterozygous substitution in a highly conserved zinc finger domain of osterix (p.Glu340Ala) share low cortical density with the recessive OI, but have low bone turnover [124] rather than the high bone turnover seen in SP7 recessive OI. Unrelated patients were reported with different phenotypic outcomes of a heterozygous p.Ser309Trp substitution, one patient with cranial hyperostosis, craniosynostosis, long bone fragility, and high bone turnover [127] and one with Juvenile Paget’s Disease [128].
Further investigations of the cellular and biochemical features will be required to provide understanding of the variable outcome of inheritance and phenotype of Type XII OI.
SP7 gene encodes Osterix, a zinc finger transcriptional factor important for osteoblast differentiation and maturation. Osterix interacts with Runx2 to induce COL1A1 gene expression [129]. It also induces expression of antagonists of the Wnt pathway, sclerostin and DKK1 [130]. Conversely, activation of this pathway may also play an important role in classical OI with ER stress, since the IRE1a-XBP1 branch of the UPR promotes SP7 transcription [131].
Mice with an SP7 deletion die shortly after birth with absent bone development due to lack of osteoblast differentiation [132].
Type XIV OI-TMEM38B-Collagen Production and Modification, Cell Energetics and Attachment
Type XIV OI, with recessive inheritance, is caused by mutations in the TMEM38B gene, encoding the ER membrane trimeric intracellular cation channel type B (TRIC-B) [133,134,135,136]. These are predominantly null mutations caused by deletions; other mutations include splice site variant mutations in TMEM38B [137, 138]. Type XIV OI is characterized by a broad phenotypic range, from asymptomatic to severe. The common clinical presentation consists of bone fragility, frequent fractures, bowed limbs and severe osteopenia. Type XIV OI bone is distinctive in remodeling compared to other OI types. Typically, OI bones are hypermineralized and exhibit high bone turnover. However, type XIV OI has low bone turnover with low osteoblast and osteoclast numbers, with mainly normal bone matrix mineralization [139].
TRIC-B protein, a monovalent cation channel, together with inositol 1,4,5-trisphosphate receptor (IP3R), is important for regulating calcium flux from the ER to cytoplasm [140]. Ca2+ signaling is essential for collagen metabolism. TMEM38B-null osteoblasts and fibroblasts have been shown to have impaired ER-Ca2+ flux kinetics, which induces ER stress, thus affecting collagen synthesis and secretion. In addition, although LH1 was increased and calcium-dependent FKBP65 was decreased in TRIC-B cells, absence of the TRIC-B channel caused reduction in hydroxylation of lysines in the collagen helical region and increased telopeptide hydroxylation. Procollagen chain assembly was delayed, causing retention of misfolded collagen in TMEM38B deficient cells and ultimately affecting the collagen assembly in matrix [136]. Recently, it was shown that deletion of TMEM38B in osteoblasts impacts a broader range of calcium-dependent cellular processes, emphasizing a novel role of TRIC-B in osteoblast differentiation and mineralization. The prolonged ER stress resulting from the absence of TRIC-B decreases the expression of mitochondrial fusion/fission markers, resulting in strikingly elongated mitochondria. Furthermore, increased generation of reactive oxygen species (ROS) by mitochondria demonstrates their functional impairment. Contact sites between ER and mitochondria (ER-MCSs) regulate mitochondrial fusion/fission, calcium, and lipid trafficking, important processes for the maintenance of mitochondrial physiology, further emphasizing the indirect but important role of TRIC-B in mitochondria function. This was the first evidence of mitochondria changes in osteoblasts from OI types with recessive inheritance. In addition, deletion of TRIC-B disrupted cell adhesion of osteoblasts, further affecting the cell–cell communication such as gap and tight junctions, cell proliferation and cell cycle [141].
Tric-b knockout mice die shortly after birth due to respiratory distress [142], and have decreased bone mineralization and altered collagen deposition [143]. Recent murine studies in a new Tmem38b knockout mouse model demonstrated that absence of the TRIC-B channel affected skeleton growth, as well as osteoblast differentiation and collagen synthesis, due to reduced calcium calmodulin kinase II-mediated and TGF-β signaling [144].
Type XV OI-WNT1-Osteocyte-Specific Function
The Wnt/β-catenin pathway is a major pathway regulating the bone development. Wnt 1 is a secreted glycoprotein; palmitoylation of WNT1 at Serine224 is required for intracellular trafficking and full protein activity [145]. Mutations in WNT1 can cause either recessive or dominant type XV OI [146,147,148,149,150,151,152,153,154,155,156,157,158]. Homozygous mutations in WNT1, causing recessive OI, are characterized with fractures, short stature, osteoporosis, vertebral compression, and variable neurological problems, while heterozygous mutations in WNT1, causing dominant OI, present with early-onset osteoporosis. In vitro studies showed that Wnt1 protein with either homozygous or heterozygous mutations could not activate LRP5-mediated WNT-regulated β-catenin signaling [147, 149, 158].
Wnt1-null mice die postnatally due to diminished development of central nervous system [159]. The swaying mouse (Wnt1sw/sw), which carries a spontaneous single nucleotide deletion in Wnt1, was shown to have similar skeletal characteristics as patients with type XV OI [160]. The mice exhibited fractures, osteopenia, reduced bone strength and decreased osteoblast function. Wnt1 expression in bone is surprisingly low, but expression during neurological development is high [159]. The studies on the late-osteoblast-specific and osteocyte-specific WNT1 loss- and gain-of-function mouse models further illuminated the role of osteocyte-specific WNT1 in bone. Wnt1 absence in osteocytes induced low bone mass with fractures, while Wnt1 overexpression increased bone formation, partially mediated through mammalian target of rapamycin complex 1 (mTORC1) signaling [161].
Type XVI OI-CREB3L1, Type XVIII OI-MBTPS2—the RIP Pathway Affects Bone
Type XVI and XVIII OI revealed an unexpected role for Regulated Intramembrane Proteolysis (RIP) in bone development. RIP had previously been extensively studied, including the seminal work of Brown and Goldstein, for its role in cholesterol metabolism. Now defects in Oasis, a substrate for RIP processing, and in S2P, the second of two sequentially acting Golgi membrane-embedded proteases which activate a variety of transcription factors by cleaving them, have pointed to a significant role in bone.
Recessive type XVI OI is caused by mutations in cAMP responsive element binding protein 3-like 1 (CREB3L1) gene, which encodes OASIS (old astrocyte specifically induced-substance). The phenotype of type XVI shows a substantial variability from mild to perinatal lethal [162,163,164,165,166,167,168]. The initial report of type XVI OI involved a Turkish family with two affected children, one with in utero fractures and bowed limbs who died at 9 month and other terminated in utero with thin ribs and fractures revealed by autopsy [162]. Oasis-null mice were shown to develop severe osteopenia and fractures, with reduced type I collagen in bone and osteoblasts, but not in dermal fibroblasts. OASIS is a transmembrane basic leucine zipper (bZIP) transcriptional factor, residing in the ER, which under stress conditions relocates to Golgi membrane and undergoes cleavage by RIP, releasing its N-terminal domain which induces gene transcription in the nucleus (169). OASIS binds to the Col1a1 gene promoter via unfolded protein response element (UPRE)-like sequence and increases its transcription [170]. OASIS also regulates transcription of SEC24D, which is involved in ER-Golgi trafficking [166].
Type XVIII OI, the first X-linked recessive OI, is caused by missense mutations in membrane-bound transcription factor site-2 protease (MBTPS2) gene, encoding S2P. In OMIM (Online Mendelian Inheritance in Man), gene defects in MBTPS2 are listed as type XIX OI (#301014). S2P catalyzes the second of two sequential cleavage reactions in the RIP process, following S1P cleavage, that occur in the Golgi membrane when ER-stress and other lipid-related processes cause the transport of transcription factors from the ER membrane to the Golgi.
Type XVIII OI is characterized by short stature, bone fractures and bowing, barrel chest and vertebral compressions and scoliosis [171]. It is noteworthy that the published and unpublished S2P mutations causing OI are all localized to residues in the NPDG motif that is crucial for zinc ion coordination. Dozens of other missense mutations in S2P occur throughout the remainder of the protein, but especially in the active site, and have been shown to induce cholesterol-related conditions—ichthyosis follicularis, atrichia, and photophobia (IFAP), BRESEK/BRESHECK syndrome, and keratosis follicularis spinulosa decalvans (KFSD) [172,173,174]. It is interesting that recent omics studies of S2P patient skin fibroblasts [175] or fibroblasts from a proband umbilical cord [176] showed upregulation of fatty acid pathways [175]. However, OI Type XVIII patients do not have evidence of cholesterol-related conditions, nor do IFAP/KFSD patients have symptoms of OI. Given the prior demonstration that transcription factors related to lipid, such as SREBPs, are also not well-cleaved in type XVIII OI-affected osteoblasts [171], this may speak to a tissue-specific critical function distal to RIP cleavage of OASIS that makes a specific set of S2P mutations crucial for bone formation. In type XVIII OI, not only is collagen expression reduced, but also hydroxylation of the α1(I) and α 2(I) Lys87 residue that is crucial for collagen cross-linking is reduced by half, weakening bone strength [171].
Furthermore, mutations in S1P, which cause a full absence of S1P/S2P cleavage, have also been reported to affect the skeletal system. A patient reported with a recessive mutation in the MBTPS1 gene, encoding S1P, has features of a skeletal dysplasia, including growth retardation, kyphoscoliosis, and dysmorphic facial features, but intriguingly not OI [177]. Cartilage-specific S1P knockout mice, which die shortly after birth, were shown to develop chondrodysplasia with absent endochondral ossification [178]. Further investigation of these very rare patients and important murine models are needed to understand the full complexity of the RIP system, its substrates and their interactions.
Type XVII OI-SPARC Matrix Protein with Intracellular and Matrix Functions
Homozygous mutations in secreted protein acidic and rich in cysteine (SPARC) are responsible for causing the extremely rare recessive type XVII OI. Patients with type XVII OI usually present normally at birth, developing severe progressive OI in early childhood, with frequent long bones fractures, vertebral compression fractures, muscular hypotonia, and scoliosis [122, 179, 180]. Their bone tissue is hypermineralized, as in classical OI [179]. SPARC protein or osteonectin was identified over 40 years ago as a bone-specific protein important for mineralization [181]. It is a secreted matricellular glycoprotein that binds different ECM proteins including collagen in a calcium-dependent manner [182]. It is interesting that defects in a protein component of matrix have recessive inheritance, which points to its pathology being based on disruption of interactive rather than structural functions. The residues critical for type XVII OI are located in the EC domain of osteonectin and form a salt bridge critical for Ca2+-binding and subsequent collagen binding. Osteonectin has also been proposed to have other intracellular and matrix functions. Cells from type XVII OI patients secrete reduced amounts of collagen, which is partially overmodified, supporting a collagen chaperone function for osteonectin assisting HSP47 [179]. SPARC-null mice exhibited osteopenia and decreased bone formation and bone remodeling [183]. In addition, SPARC was shown to affect murine collagen fibril formation in skin, producing smaller fibrils [184], perhaps via increased activity of transglutaminase for cross-linking of matrix proteins [185].
Type XIX OI—TENT5A/FAM46A Post-Transcriptional Regulation of Collagen Production
A small number of patients with a severe to lethal skeletal dysplasia have been reported to have homozygous or bialleleic missense or frameshift mutations in TENT5A, and designated autosomal recessive type XIX OI [186]. In OMIM, gene defects in TENT5A are listed as type XVIII OI (# 617952). TENT5A encodes FAM46A, a member of the nucleotidyltransferase (NTase)-fold protein family of non-canonical poly(a) polymerases, which are conserved in all known animal genomes [187].
This newer addition to the genetic etiologies of OI fulfills its expected collagen-related function by its involvement in the polyadenylation of transcripts for Col1a1 and Col1a2 in murine studies [188]. However, the collagen in type XIX OI bone matrix is disordered, as well as decreased, suggesting significant indirect contributions to the bone defect. FAM46A mutations causing type XIX OI are located in the conserved DUF1693 domain and include both missense substitutions and stop codons [186]. Distinction between misfunction and loss-of-function cannot yet be made since neither transcript levels nor western blots from patient cells have been reported. In addition, studies of Type XIX OI bone tissue by histomorphometry and BMDD will be critical in the future.
The diagnosis of type XIX OI is made in the first months to years of life, based on typical features such as congenital bowing of limbs, early fractures which accelerate in frequency in childhood, blue sclerae, hyperlaxity and motor delay [186, 189]. Some have short stature and dental abnormalities. Vertebral compressions and Wormian bones are present on radiographs.
Because the children with type XIX OI were first detected by screening a group of patients with Stuve-Wiedmann Syndrome [190], with whom they share congenital bowing and progressive scoliosis, the assignment of this gene as OI-causative was cautiously undertaken. Now, the combination of patient data, murine phenotype and molecular data on polyadenylation of type I collagen transcripts has made the assignment to OI nosology clear.
The Fam46a null mouse, which carries a homozygous null mutation, was generated by ENU mutagenesis before TENT5A was proposed as an OI-causative gene [191]. Its small size, long bone abnormalities, with bent and twisted limbs, compressed rib cage, and thin cranium support the skeletal dysplasia assignment of the gene defect. Long bone fragility, as well as thin cortices and reduced trabecular bone were reported. In mouse, Tent5a is expressed in both newborn and adult bone tissues, suggesting both prenatal roles in bone development and post-natal roles in bone remodeling. Murine calvarial osteoblasts have impaired mineralization in vitro. The murine osteoblast collagen secretion deficiency is reduced to the level of a collagen null allele. One might have expected a rather mild skeletal dysplasia from a straightforward quantitative reduction of normally modified type I collagen. However, the collagen in murine bone tissue is strikingly abnormal, with thinner and disorganized fibrils. This may be related to the broader role of FAM46A, which polyadenylates a set of cytoplasmic mRNAs encoding proteins impacting bone development and which are secreted coordinately with bone mineralization [188]. Fifty-two mRNAs in Tent5a KO osteoblasts had significantly shortened poly-A tails; the transcripts at the top of the list were Col1a1, Col1a2, SerpinF1 and Sparc [188]. Yet another function of FAM46A which may contribute to the bone dysplasia of type XIX OI is its role as a binding partner for SMADs in the TGF-β pathway, physically interacting with Smad1 and Smad4 to stabilize them, and thus promoting BMP target expression [192]. The relative contribution of significantly reduced collagen, reduction of other bone matrix proteins which are related to OI types, and presumptive reduction in BMP signaling has not been investigated.
In addition, the association of FAM46A defects with other conditions remains intriguing. FAM46A was first described as a candidate gene for human retinal disease; expression of TENT5A in retina underlies the recessive retinitis pigmentosa associated with novel VNTRs (variable number tandem repeats) in exon 2 [193]. Elsewhere in the body, TENT5A exon 2 VNTRs are associated with skeletal conditions such as osteoarthritis of hip and knee [194] and adolescent idiopathic scoliosis [195], an assignment made reasonable by the strong expression of TENT5A in human and murine osteoblasts.
Type XX OI—MESD Disrupting the Intersection of Collagen and LRP5/6 and Wnt Signaling
Type XX OI, a severe to lethal recessive skeletal dysplasia, is caused by biallelic or frameshift mutations in exons 2 or 3 of MESD [196,197,198]. Some affected individuals have been stillborn. Common features reported in more than a dozen children who survived have included an initial fracture by age 2 years, with a progressive deforming course. When young, they have a low birth weight and blue sclerae, and radiographic evidence of Wormian bones, gracile ribs and severe overtubulation. By the second decade of life, dental abnormalities, such as disorganized dentition and oligodontia [196, 197], are prominent. They have short stature and rhizomelia or micromelia is prevalent. Progressive scoliosis and kyphosis are associated with vertebral compressions. Type XX OI patients often have intellectual disability [196, 199], a relative rarity among OI types, previously reported in some individuals with type XV OI, due to WNT1 defects, as well as type XXI OI with KDELR2 mutations, in which neurodevelopmental disorders were reported [115].
The MESD gene encodes MESD (mesoderm development candidate 2), which functions as a chaperone of the low-density lipoproteins in the ER [200]. The critical function of MESD for the correct localization of LRP5/6 at the cell surface puts type XX OI at the overlap of pathways leading to OI or to LRP5/6-related conditions. Generally, the mutant MESD protein cannot be retained in the ER, although it is expressed and has similar stability and chaperone function as normal MESD [196, 201]. The mis-localization of LRPs results in their aggregation in the cytoplasm [200, 201], causing loss of normal signaling. For example, the oligodontia that is distinctive to this OI type has been speculated to arise from reduced LRP6 signaling.
While the consequences of overlap of type XX OI and LRP5/6 conditions can be dissected, it is clear that bone with MESD defects is distinctive from most OI types [198]. One of the salient features common to most OI dominant and recessive types, the hypermineralization of bone when assayed by BMDD, is not present. Instead, BMDD of MESD bone matrix shows lower and less homogeneous mineralization than control bone but has islands of hypermineralized cartilage remnants. There are an increased number of irregularly shaped lacunae, connected by broad canaliculi. Though mindful of the distinctiveness of type XX OI, it also fulfills the OI biochemical expectation of being type 1 collagen-related. MESD has recently been shown to be a direct chaperone of pro-α1(I) [201]; consequently, cells with mutant MESD have cytosolic aggregates of type I collagen that block intercellular nanotubes, resulting in significant procollagen intracellular retention. To degrade the collagen aggregates, patient cells increase autophagy, as confirmed by co-localization of aggregates with LC3, an autophagy marker, and impaired collagen degradation in cells treated with bafilomycin A [201]. Failure to fully compensate for the retained aggregates leads to an overall cellular stress. This is notably different from the ER stress response that occurs in OI patient cells with collagen misfolding or retention of overmodified collagen.
It is also interesting that the non-secreted aggregates of type I collagen are mechanistically related to the type XX OI phenotypic feature of dermal aging-related skin laxity. Low co-localization of pro-α1(I) with integrin subunit β1 (ITGB1) and deficiency of paxillin-positive foci in patient but not control cells underlie the weak attachment of proband fibroblasts with matrix surface [201]. It is notable that impaired cell–cell and cell–matrix attachment were also recently described in osteoblasts with type XIV OI, caused by impaired intracellular calcium flux in the absence of the TMEM38B channel [141].
Finally, a connection can be made back to WNT signaling, consistent with the intellectual and neurological aspects of type XX OI. Since MESD is a chaperone for LRP5 and LRP6, their aggregation in patient cells may disrupt WNT signaling. In fact, expression of BMP2 and BMP4 were strikingly downregulated, connecting types XX and XV OI [201]. Thus, the failure of mutant MESD to be retained in the ER leads to its inadequate availability as a chaperone for type 1 procollagen, LRP5, and LRP6, all of which aggregate in the cytoplasm, leading to cell stress, impaired matrix attachment and disruption of WNT signaling.
Type XXII OI—CCDC134, OI Overlap with MAPK/ERK Signaling
Homozygous missense mutations in the first codon of CCDC134 cause a severe recessive skeletal fragility syndrome that has been given the designation OI type XXII [202,203,204]. These defects, which have been reported in only a handful of patients, share pre- and post-natal short stature, multiple fractures and bowing of long bones, low bone density and Wormian bones. Three patients had pseudoarthroses while scleral hue has been variable. Patient bone histomorphometry reveals the decreased BV/TV typically found in OI, associated with atypical increased cortices, with normal mineral apposition rate (MAR) and bone formation rate (BFR), more characteristic of increased extracellular signal-regulated kinase (ERK) signaling [202].
The missense mutation responsible for type XXII OI alters the start codon of CCDC134, resulting in a loss of function of this secreted protein. CCDC134 contains a coiled-coil domain and is involved in some mitogen-activated protein kinase (MAPK) signaling pathways, as well as being responsible for the inhibition of phosphorylation of ERK and C-Jun N terminal kinase (JNK) [205]. Thus, primary osteoblasts derived from individuals with type XXII OI have increased ERK1/2 phosphorylation, reduced expression of osteopontin and COL1A1, and reduced mineralization in vitro [202]. This makes type XXII OI another OI form which contains elements of both OI and another set of skeletal dysplasias, in this case, bone dysplasias with dysregulation of the MAPK/ERK pathways [206], including Noonan syndrome, neurofibromatosis type 1, and cardiofaciocutaneous syndrome. ERK1/2 activation and decreased osteoblast differentiation have also been demonstrated in the classic “dripping candle wax” form of melorheostosis, although melorheostosis is caused by MAP2K1 somatic mosaicism [207]. Given that ERK activation is important for osteoblast differentiation and maturation, the situation in type XXII OI appears more complex, with reduced osteoblast collagen and mineralization. Investigation of these pathways in OI XXII will be important to clarify how CCDC134 mutations result in different outcomes, perhaps with overlapping pathways. Ccdc134−/− mice have not been informative, since they die at E12.5–E14.5 of cerebral hemorrhages and their bone properties cannot be studied [208]. For the present, diagnostically grouping these mutations with recessive OI is the most appropriate course.
Intracellular and Matrix Consequences of Different OI Mutations
The trafficking of collagen molecules is a complex process, consisting of anterograde and retrograde transport, which is dependent on the proper functioning of chaperone molecules to facilitate the collagen transport. After post-translational modifications and folding of the triple helix are completed, procollagen is transported anterograde from the ER to the Golgi through COPII vesicles [209]. For the transport of large molecules such as collagen, COPII vesicles require the transmembrane protein transport and Golgi organization 1 (TANGO1); null mutations of MIA3/TANGO result in procollagen retention in the ER [210]. Conversely, retrograde transport returns certain proteins back to the ER to fulfill their function. KDEL receptors, among others, function to mediate the sorting of ER proteins by binding COPI vesicles [209]. Studies of KDELR2 mutations, which cause severe progressive deforming type XXI OI, highlight its crucial role in collagen transport [116].
Misfolded collagen chains, often with increased post-translational modification, are either secreted slowly from cells or retained in the ER, causing ER stress and activation of UPR. UPR facilitates structure restoration of retained proteins. However, if the repair is not possible, the misfolded proteins are degraded either through ER-associated degradation (ERAD), which consists of ubiquitination and degradation by the proteasome, or through autophagy, a lysosomal self-digestion. The induction of ER stress and the subsequent activation of UPR pathways were reported in several OI types, both dominant and recessive. Some but not all mutations in COL1A1 and COL1A2 caused ER accumulation of collagen, increased expression of binding immunoglobulin protein (BiP), a chaperone that directly binds to collagen to support its folding and assembly, as well as UPR activation, autophagy and apoptosis in OI fibroblasts [211]. The same findings were reported in OI patient fibroblasts with mutations in CRTAP, P3H1 and PPIB impairing prolyl 3-hydroxylation [212]. Both studies showed that the application of chaperone 4-phenylbutyrate (4-PBA) decreased UPR and ameliorated the cellular homeostasis. Recessive type XIV OI, caused by null mutations in the TMEM38B/TRIC-B channel, causes significant collagen accumulation in both osteoblasts and fibroblasts in the ER, inducing ER stress and UPR with a significant increase of BiP [136]. In recessive OI types involving RIP, there is a distruption of the process by which transcription factors such as OASIS are translocated to the Golgi membrane upon ER stress, cleaved by S1P/S2P, and transported to the nucleus for transcription of genes for ER-stress response or UPR pathways [169]. Thus, to which extent ER stress and UPR contribute to the OI phenotype, versus helping to mitigate it, remains unresolved.
One of the most intriguing recent findings is mitochondrial involvement in OI pathophysiology. Mitochondrial dysfunction was first observed in muscle tissue of OI murine models such as oim/oim, with evident reduction of respiration rates, biogenesis markers, mitophagy and electron transport chain components [213]. In a transcriptomics analysis of muscle in Jrt and oim/oim mice, Mss51, a mitochondrial metabolic stress inducible factor, was shown to be downregulated [214]. Recent studies of osteoblasts from dominant COL1A2 G610C OI mice, showed that integrated stress response (ISR), induced by ER collagen accumulation, was regulated by expression changes in mitochondrial Hspa9/HSP70 (mt-HSP70) [215]. Recently, it was found for the first time that mitochondrial dysfunction is a contributing factor to OI osteoblast pathology. Deletion of TMEM38B/TRIC-B in type XIV OI osteoblasts induced striking changes in mitochondrial morphology, which became very elongated, developed crystolysis and increased production of superoxide [141]. Expression of mitochondrial fission/fusion markers was decreased, consistent with mitochondrial morphology and with transmission of ER stress through the contact sites between ER and mitochondria (ER-MCSs). The contact sites regulate various mitochondrial functions, including mitochondrial fusion/fission, calcium and lipid trafficking [216]. Fission and fusion components form hotspots and colocalize at ER-MCSs [217], which are enriched with IP3Rs, implicating the role of contact sites in regulation of mitochondrial calcium levels. Investigation of the role of mitochondria in OI bone pathology is likely to yield further exciting findings.
The stability of collagen in matrix is dependent on intermolecular cross-links that are formed by hydroxylation of lysine residues, carried out by lysyl hydroxylases (LH1, LH2, and LH3), encoded by PLOD1, PLOD2, and PLOD3, respectively. Abnormal collagen cross-linking is reported in OI and Bruck syndrome (skeletal disorder similar to OI with congenital joint contractures) via mechanisms of underhydroxylation and overhydroxylation of collagen telopeptide lysines [218, 219].
The main cause of increased fragility of OI bone is abnormal bone material properties, especially hypermineralization of bone matrix. The increased mineralization of bone matrix occurs in most types of OI and is one of the unifying feature of OI bone. Quantitative backscattered electron imaging (qBEI) is used to directly determine the bone mineralization density distribution (BMDD). qBEI studies showed increased bone mineral content in patient with dominant [32, 46, 220, 221], as well as in recessive OI types [53, 222,223,224]. Only exception to this rule are types XIV (TMEM38B) and XV (WNT1), with mostly normal mineralization, and XX (MESD) in which patients exhibit reduced bone mineralization [139, 225, 226].
OI bone has been shown to contain a higher number of osteocytes [227] and the resulting increase in porosity also contributes to bone fragility. This finding was consistent with investigations of the Crtap−/− recessive mouse model, which demonstrated increased TGF-β signaling [228]. Furthermore, the osteocyte transcription was found to be significantly dysregulated in the Crtap−/− mouse, with major changes in both WNT and TGF-β signaling [229], suggesting an important role of osteocytes in OI pathology. See sections on these types below.
The major cause of mortality and morbidity of patients with OI is respiratory dysfunction [230]. It was thought that impaired pulmonary function was extrinsic to the impaired collagen, caused by presence of scoliosis in OI patients [231]. However, studies showed that lung disease and some cardiac issues develop independently of scoliosis [232]. Pulmonary function was shown to decline in children with classical OI who did not have scoliosis [22]. More recently, the lung tissue abnormalities in OI were shown to have an intrinsic etiology, which can be exacerbated by scoliosis [233]. In this study of children and young adults with classical OI, all patients with types III and half with type IV had lung restriction, and many had parenchymal defects such as atelectasis and reticulation. As a result of functional defects caused directly by collagen abnormalities, 90% of patients in the study group had reduced gas exchange. Further, decreased forced expiratory flow (FEF) 25–75% and significant thickening of small bronchi wall indicate a critical role for small airways, the mechanism of which is currently not known [233].
Aortic root dilatation and valvular dysfunction in OI patients has been extensively investigated [234,235,236,237]. Patients with OI have a higher risk of cardiovascular abnormalities, such as valvular heart disease, heart failure, and aortic root dilation [238]. A single study reported on atrial fibrillation and hypertension in patients with OI [239]. Further studies are needed to understand the underlying mechanism of cardiovascular abnormalities in OI patients.
Conclusions
The genes responsible for OI have now largely been defined. While several rare types may emerge, they will not pertain to very many individuals and will most likely be related to pathways containing the known genes. The identification of genetic defects causing OI and their mechanism are important. Further research on mechanism of the types and their interrelatedness will hopefully lead to a more encompassing conceptualization of OI pathophysiology, and ultimately to broad therapies.
Nosology, on the other hand, is not the equivalent of scientific data. It is an organization of the information at hand at the time, and, since that information changes with time, so will nosology. A particular nosology will be utilized if it represents a convenient organization for understanding the status quo and moving research forward. While it is possible that different nosology might be more useful to researchers versus clinicians and families, we propose that a dual or combination nosology might well serve both communities and even facilitate cross-talk. For example, the use of a genetic type in combination with an OI severity label based on both skeletal and non-skeletal features, would keep the distinctions of genetics in the forefront and clarify phenotypic variability. The shared goal is the use of genetics to understand the cause and change the outcome.
References
Marini JC, Cabral WA (2018) Osteogenesis imperfecta. Genet Bone Biol Skeletal Dis, pp 397–420
Jovanovic M, Guterman-Ram G, Marini JC (2022) Osteogenesis imperfecta: mechanisms and signaling pathways connecting classical and rare OI types. Endocr Rev 43(1):61–90
Raghunath M, Bruckner P, Steinmann B (1994) Delayed triple helix formation of mutant collagen from patient with osteogenesis imperfecta. J Mol Biol 236(3):940–949
Malfait F, Symoens S, De Backer J, Hermanns-Lê T, Sakalihasan N, Lapière CM et al (2007) Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat 28(4):387–395
Cabral WA, Makareeva E, Letocha AD, Scribanu N, Fertala A, Steplewski A, et al (2007) Y‐position cysteine substitution in type I collagen (α1 (I) R888C/p. R1066C) is associated with osteogenesis imperfecta/Ehlers‐Danlos syndrome phenotype. Human Mutation 28(4):396–405
Gensure RC, Mäkitie O, Barclay C, Chan C, DePalma SR, Bastepe M et al (2005) A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Investig 115(5):1250–1257
Cabral WA, Mertts MV, Makareeva E, Colige A, Tekin M, Pandya A et al (2003) Type I collagen triplet duplication mutation in lethal osteogenesis imperfecta shifts register of α chains throughout the Helix and Disrupts incorporation of mutant helices into fibrils and extracellular matrix. J Biol Chem 278(12):10006–10012
Pace JM, Atkinson M, Willing MC, Wallis G, Byers PH (2001) Deletions and duplications of Gly-Xaa-Yaa triplet repeats in the triple helical domains of type I collagen chains disrupt helix formation and result in several types of osteogenesis imperfecta. Hum Mutat 18(4):319–326
Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S et al (2007) Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 28(3):209–221
Garibaldi N, Besio R, Dalgleish R, Villani S, Barnes AM, Marini JC et al (2022) Dissecting the phenotypic variability of osteogenesis imperfecta. Dis Models Mech 15(5):dmm049398
Sałacińska K, Pinkier I, Rutkowska L, Chlebna-Sokół D, Jakubowska-Pietkiewicz E, Michałus I et al (2021) Novel mutations within collagen alpha1 (I) and alpha2 (I) ligand-binding sites, broadening the spectrum of osteogenesis imperfecta–current insights into collagen type I lethal regions. Front Genet 12:692978
Willing MC, Deschenes SP, Scott DA, Byers PH, Slayton RL, Pitts SH et al (1994) Osteogenesis imperfecta type I: molecular heterogeneity for COL1A1 null alleles of type I collagen. Am J Hum Genet 55(4):638
Pihlajaniemi T, Dickson L, Pope F, Korhonen V, Nicholls A, Prockop D et al (1984) Osteogenesis imperfecta: cloning of a pro-alpha 2 (I) collagen gene with a frameshift mutation. J Biol Chem 259(21):12941–12944
Schwarze U, Hata R-I, McKusick VA, Shinkai H, Hoyme HE, Pyeritz RE et al (2004) Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Human Genet 74(5):917–930
Forlino A, Porter FD, Lee EJ, Westphal H, Marini JC (1999) Use of the Cre/lox recombination system to develop a non-lethal knock-in murine model for osteogenesis imperfecta with an α1 (I) G349C substitution: variability in phenotype in BrtlIV mice. J Biol Chem 274(53):37923–37931
Kozloff KM, Carden A, Bergwitz C, Forlino A, Uveges TE, Morris MD et al (2004) Brittle IV mouse model for osteogenesis imperfecta IV demonstrates postpubertal adaptations to improve whole bone strength. J Bone Mineral Res 19(4):614–622
Uveges TE, Collin-Osdoby P, Cabral WA, Ledgard F, Goldberg L, Bergwitz C et al (2008) Cellular mechanism of decreased bone in Brtl mouse model of OI: imbalance of decreased osteoblast function and increased osteoclasts and their precursors. J Bone Miner Res 23(12):1983–1994
Davis MS, Kovacic BL, Marini JC, Shih AJ, Kozloff KM (2012) Increased susceptibility to microdamage in Brtl/+ mouse model for osteogenesis imperfecta. Bone 50(3):784–791
Daley E, Streeten EA, Sorkin JD, Kuznetsova N, Shapses SA, Carleton SM et al (2010) Variable bone fragility associated with an Amish COL1A2 variant and a knock-in mouse model. J Bone Miner Res 25(2):247–261
Kohler R, Tastad CA, Creecy A, Wallace JM (2021) Morphological and mechanical characterization of bone phenotypes in the Amish G610C murine model of osteogenesis imperfecta. PLoS ONE 16(8):e0255315
Chipman SD, Sweet HO, McBride DJ Jr, Davisson MT, Marks SC Jr, Shuldiner AR et al (1993) Defective pro alpha 2 (I) collagen synthesis in a recessive mutation in mice: a model of human osteogenesis imperfecta. Proc Natl Acad Sci 90(5):1701–1705
Thiele F, Cohrs CM, Flor A, Lisse TS, Przemeck GK, Horsch M et al (2012) Cardiopulmonary dysfunction in the Osteogenesis imperfecta mouse model Aga2 and human patients are caused by bone-independent mechanisms. Hum Mol Genet 21(16):3535–3545
Lisse TS, Thiele F, Fuchs H, Hans W, Przemeck GKH, Abe K et al (2008) ER stress-mediated apoptosis in a new mouse model of osteogenesis imperfecta. PLoS Genet 4(2):e7
Chen F, Guo R, Itoh S, Moreno L, Rosenthal E, Zappitelli T et al (2014) First mouse model for combined osteogenesis imperfecta and Ehlers-Danlos syndrome. J Bone Miner Res 29(6):1412–1423
Schnieke A, Harbers K, Jaenisch R (1983) Embryonic lethal mutation in mice induced by retrovirus insertion into the α 1 (I) collagen gene. Nature 304(5924):315–320
Bonadio J, Saunders TL, Tsai E, Goldstein SA, Morris-Wiman J, Brinkley L et al (1990) Transgenic mouse model of the mild dominant form of osteogenesis imperfecta. Proc Natl Acad Sci 87(18):7145–7149
Bonadio J, Jepsen K, Mansoura M, Jaenisch R, Kuhn J, Goldstein S (1993) A murine skeletal adaptation that significantly increases cortical bone mechanical properties. Implications for human skeletal fragility. J Clin Investig 92(4):1697–705
Pollitt R, McMahon R, Nunn J, Bamford R, Afifi A, Bishop N et al (2006) Mutation analysis of COL1A1 and COL1A2 in patients diagnosed with osteogenesis imperfecta type I‐IV. Hum Mutat 27(7):716
Lindert U, Gnoli M, Maioli M, Bedeschi M, Sangiorgi L, Rohrbach M et al (2018) Insight into the pathology of a COL1A1 signal peptide heterozygous mutation leading to severe osteogenesis imperfecta. Calcif Tissue Int 102:373–379
Cabral WA, Makareeva E, Colige A, Letocha AD, Ty JM, Yeowell HN et al (2005) Mutations near amino end of α1 (I) collagen cause combined osteogenesis imperfecta/Ehlers-Danlos syndrome by interference with N-propeptide processing. J Biol Chem 280(19):19259–19269
Malfait F, Symoens S, Goemans N, Gyftodimou Y, Holmberg E, López-González V et al (2013) Helical mutations in type I collagen that affect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers-Danlos Syndrome overlap syndrome. Orphanet J Rare Dis 8:1–10
Lindahl K, Barnes AM, Fratzl-Zelman N, Whyte MP, Hefferan TE, Makareeva E et al (2011) COL1 C-propeptide cleavage site mutations cause high bone mass osteogenesis imperfecta. Hum Mutat 32(6):598–609
Cundy T, Dray M, Delahunt J, Hald JD, Langdahl B, Li C et al (2018) Mutations that alter the carboxy-terminal-propeptide cleavage site of the chains of type I procollagen are associated with a unique osteogenesis imperfecta phenotype. J Bone Miner Res 33(7):1260–1271
Symoens S, Hulmes DJ, Bourhis JM, Coucke PJ, De Paepe A, Malfait F (2014) Type I procollagen C-propeptide defects: study of genotype–phenotype correlation and predictive role of crystal structure. Hum Mutat 35(11):1330–1341
Barnes AM, Ashok A, Makareeva EN, Brusel M, Cabral WA, Weis M, et al (1865) COL1A1 C-propeptide mutations cause ER mislocalization of procollagen and impair C-terminal procollagen processing. Biochim Biophys Acta (BBA)-Molecular Basis Dis 1865(9):2210–2223
Martínez-Glez V, Valencia M, Caparrós-Martín JA, Aglan M, Temtamy S, Tenorio J et al (2012) Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat 33(2):343–350
Valencia M, Caparrós-Martin JA, Sirerol-Piquer MS, García-Verdugo JM, Martínez-Glez V, Lapunzina P et al (2014) Report of a newly indentified patient with mutations in BMP1 and underlying pathogenetic aspects. Am J Med Genet A 164(5):1143–1150
Pollitt RC, Saraff V, Dalton A, Webb EA, Shaw NJ, Sobey GJ et al (2016) Phenotypic variability in patients with osteogenesis imperfecta caused by BMP1 mutations. Am J Med Genet A 170(12):3150–3156
Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J et al (2012) A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet 91(2):349–357
Cho T-J, Lee K-E, Lee S-K, Song SJ, Kim KJ, Jeon D et al (2012) A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet 91(2):343–348
Moffatt P, Gaumond MH, Salois P, Sellin K, Bessette MC, Godin É et al (2008) Bril: a novel bone-specific modulator of mineralization. J Bone Miner Res 23(9):1497–1508
Reich A, Bae AS, Barnes AM, Cabral WA, Hinek A, Stimec J et al (2015) Type V OI primary osteoblasts display increased mineralization despite decreased COL1A1 expression. J Clin Endocrinol Metab 100(2):E325–E332
Patoine A, Gaumond MH, Jaiswal PK, Fassier F, Rauch F, Moffatt P (2014) Topological mapping of BRIL reveals a type II orientation and effects of osteogenesis imperfecta mutations on its cellular destination. J Bone Miner Res 29(9):2004–2016
Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P et al (2000) Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res 15(9):1650–1658
Rauch F, Moffatt P, Cheung M, Roughley P, Lalic L, Lund AM, et al (2013) Osteogenesis imperfecta type V: marked phenotypic variability despite the presence of the IFITM5 c.− 14C> T mutation in all patients. J Med Genet 50(1):21–24
Blouin S, Fratzl-Zelman N, Glorieux FH, Roschger P, Klaushofer K, Marini JC et al (2017) Hypermineralization and high osteocyte lacunar density in osteogenesis imperfecta type V bone indicate exuberant primary bone formation. J Bone Miner Res 32(9):1884–1892
Hanagata N, Li X, Morita H, Takemura T, Li J, Minowa T (2011) Characterization of the osteoblast-specific transmembrane protein IFITM5 and analysis of IFITM5-deficient mice. J Bone Miner Metabol 29:279–290
Lietman CD, Marom R, Munivez E, Bertin TK, Jiang MM, Chen Y et al (2015) A transgenic mouse model of OI type V supports a neomorphic mechanism of the IFITM5 mutation. J Bone Miner Res 30(3):489–498
Rauch F, Geng Y, Lamplugh L, Hekmatnejad B, Gaumond M-H, Penney J et al (2018) Crispr-Cas9 engineered osteogenesis imperfecta type V leads to severe skeletal deformities and perinatal lethality in mice. Bone 107:131–142
Becker J, Semler O, Gilissen C, Li Y, Bolz HJ, Giunta C et al (2011) Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet 88(3):362–371
Homan EP, Rauch F, Grafe I, Lietman C, Doll JA, Dawson B et al (2011) Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J Bone Mineral Res 26(12):2798–2803
Glorieux FH, Ward LM, Rauch F, Lalic L, Roughley PJ, Travers R (2002) Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J Bone Miner Res 17(1):30–38
Fratzl-Zelman N, Schmidt I, Roschger P, Roschger A, Glorieux FH, Klaushofer K et al (2015) Unique micro-and nano-scale mineralization pattern of human osteogenesis imperfecta type VI bone. Bone 73:233–241
Rauch F, Husseini A, Roughley P, Glorieux FH, Moffatt P (2012) Lack of circulating pigment epithelium-derived factor is a marker of osteogenesis imperfecta type VI. J Clin Endocrinol Metab 97(8):E1550–E1556
Dawson D, Volpert O, Gillis P, Crawford S, Xu H-J, Benedict W et al (1999) Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 285(5425):245–248
Meyer C, Notari L, Becerra SP (2002) Mapping the type I collagen-binding site on pigment epithelium-derived factor: Implications for its antiangiogenic activity. J Biol Chem 277(47):45400–45407
Hosomichi J, Yasui N, Koide T, Soma K, Morita I (2005) Involvement of the collagen I-binding motif in the anti-angiogenic activity of pigment epithelium-derived factor. Biochem Biophys Res Commun 335(3):756–761
Bogan R, Riddle RC, Li Z, Kumar S, Nandal A, Faugere MC, Boskey A, Crawford SE, Clemens TL (2013) A mouse model for human osteogenesis imperfecta type VI. J Bone Miner Res 28(7):1531–1536
Kang H, Aryal Ac S, Barnes AM, Martin A, David V, Crawford SE et al (2020) Antagonism Between PEDF and TGF-β Contributes to Type VI Osteogenesis Imperfecta Bone and Vascular Pathogenesis. J Bone Miner Res 37(5):925–937
Mackie E, Ahmed Y, Tatarczuch L, Chen K-S, Mirams M (2008) Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol 40(1):46–62
Belinsky GS, Sreekumar B, Andrejecsk JW, Saltzman WM, Gong J, Herzog RI et al (2016) Pigment epithelium–derived factor restoration increases bone mass and improves bone plasticity in a model of osteogenesis imperfecta type VI via Wnt3a blockade. FASEB J 30(8):2837
Farber CR, Reich A, Barnes AM, Becerra P, Rauch F, Cabral WA et al (2014) A novel IFITM5 mutation in severe atypical osteogenesis imperfecta type VI impairs osteoblast production of pigment epithelium-derived factor. J Bone Mineral Res 29(6):1402–1411
Lindsay SE, Nicol LE, Gamayo AC, Raney EM (2021) An unusual presentation of osteogenesis imperfecta: a case report. JBJS Case Connector 11(4):e21
Grebennikova TA, Gavrilova AO, Tiulpakov AN, Tarbaeva NV, Belaya ZE, Melnichenko GA (2020) First description of a type v osteogenesis imperfecta clinical case with severe skeletal deformities caused by a mutation p. 119C> T in IFITM5 gene in Russia. Osteoporosis Bone Dis 22(2):32–37
Hoyer-Kuhn H, Semler O, Garbes L, Zimmermann K, Becker J, Wollnik B, Schoenau E, Netzer C (2014) A nonclassical IFITM5 mutation located in the coding region causes severe osteogenesis imperfecta with prenatal onset. J Bone Miner Res 29(6):1387–1391
Guillén-Navarro E, Ballesta-Martínez MJ, Valencia M, Bueno AM, Martinez-Glez V, López-González V et al (2014) Two mutations in IFITM5 causing distinct forms of osteogenesis imperfecta. Am J Med Genet A 164(5):1136–1142
Rodriguez Celin M, Moosa S, Fano V (2018) Uncommon IFITM5 mutation associated with severe skeletal deformity in osteogenesis imperfecta. Ann Hum Genet 82(6):477–481
Liu Y, Asan, Ma D, Lv F, Xu X, Wang J, et al (2017) Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporosis Int 28:2985–2995
Dagdeviren D, Tamimi F, Lee B, Sutton R, Rauch F, Retrouvey JM (2019) Dental and craniofacial characteristics caused by the p. Ser40Leu mutation in IFITM5. Am J Med Genet A 179(1):65–70
Chandler N, Best S, Hayward J, Faravelli F, Mansour S, Kivuva E et al (2018) Rapid prenatal diagnosis using targeted exome sequencing: a cohort study to assess feasibility and potential impact on prenatal counseling and pregnancy management. Genet Med 20(11):1430–1437
He Y, Yan J-M, Liu Y-H, Han J, Li D-Z (2017) A prenatal case of osteogenesis imperfecta diagnosed with next-generation sequencing. J Obstet Gynaecol 37(6):809–810
Guterman-Ram G, Hedjazi G, Stephan C, Blouin S, Zwerina J, Kozloff KM, Fratzl-Zelman N, Marini JC (2021) Atypical type VI Osteogenesis Imperfecta mouse models the intersection of IFITM5 and SERPINF1 pathways in patients. Bone Rep 14:100805
Hedjazi G, Guterman-Ram G, Blouin S, Schemenz V, Wagermaier W, Fratzl P, et al (2022) Alterations of bone material properties in growing Ifitm5/BRIL p. S42 knock-in mice, a new model for atypical type VI osteogenesis imperfecta. Bone 162:116451
Myllyharju J, Kivirikko KI (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 20(1):33–43
Vranka JA, Sakai LY, Bächinger HP (2004) Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J Biol Chem 279(22):23615–23621
Tryggvason K, Majamaa K, Risteli J, Kivirikko KI (1979) Partial purification and characterization of chick-embryo prolyl 3-hydroxylase. Biochem J 183(2):303–307
Marini JC, Cabral WA, Barnes AM, Chang W (2007) Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle 6(14):1675–1681
Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M et al (2006) CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127(2):291–304
Ward L, Rauch F, Travers R, Chabot G, Azouz E, Lalic L et al (2002) Osteogenesis imperfecta type VII: an autosomal recessive form of brittle bone disease. Bone 31(1):12–18
Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR et al (2006) Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med 355(26):2757–2764
Baldridge D, Schwarze U, Morello R, Lennington J, Bertin TK, Pace JM et al (2008) CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum Mutat 29(12):1435–1442
Marini JC, Cabral WA, Barnes AM (2010) Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res 339:59–70
Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S et al (2007) Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 39(3):359–365
Willaert A, Malfait F, Symoens S, Gevaert K, Kayserili H, Megarbane A et al (2009) Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J Med Genet 46(4):233–241
Wassenhove-McCarthy DJ, McCarthy KJ (1999) Molecular characterization of a novel basement membrane-associated proteoglycan, leprecan. J Biol Chem 274(35):25004–25017
Kantaputra PN, Dejkhamron P, Intachai W, Ngamphiw C, Cairns JRK, Kawasaki K et al (2021) A novel P3H1 mutation is associated with osteogenesis imperfecta type VIII and dental anomalies. Oral Surg Oral Med Oral Pathol Oral Radiol 132(6):e198–e207
Dimori M, Heard-Lipsmeyer ME, Byrum SD, Mackintosh SG, Kurten RC, Carroll JL et al (2020) Respiratory defects in the Crtap KO mouse model of osteogenesis imperfecta. Am J Physiol-Lung Cell Molec Physiol 318(4):L592–L605
Chang W, Barnes AM, Cabral WA, Bodurtha JN, Marini JC (2010) Prolyl 3-hydroxylase 1 and CRTAP are mutually stabilizing in the endoplasmic reticulum collagen prolyl 3-hydroxylation complex. Hum Mol Genet 19(2):223–234
Cabral WA, Fratzl-Zelman N, Weis M, Perosky JE, Alimasa A, Harris R et al (2020) Substitution of murine type I collagen A1 3-hydroxylation site alters matrix structure but does not recapitulate osteogenesis imperfecta bone dysplasia. Matrix Biol 90:20–39
Homan EP, Lietman C, Grafe I, Lennington J, Morello R, Napierala D et al (2014) Differential effects of collagen prolyl 3-hydroxylation on skeletal tissues. PLoS Genet 10(1):e1004121
Price ER, Zydowsky LD, Jin M, Baker CH, McKeon FD, Walsh CT (1991) Human cyclophilin B: a second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc Natl Acad Sci 88(5):1903–1907
Yao Q, Li M, Yang H, Chai H, Fisher W, Chen C (2005) Roles of cyclophilins in cancers and other organ systems. World J Surg 29:276–280
Smith T, Ferreira LR, Hebert C, Norris K, Sauk JJ (1995) Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles: a role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J Biol Chem 270(31):18323–18328
van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA et al (2009) PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet 85(4):521–527
Barnes AM, Carter EM, Cabral WA, Weis M, Chang W, Makareeva E et al (2010) Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N Engl J Med 362(6):521–528
Pyott SM, Schwarze U, Christiansen HE, Pepin MG, Leistritz DF, Dineen R et al (2011) Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum Mol Genet 20(8):1595–1609
Cabral WA, Perdivara I, Weis M, Terajima M, Blissett AR, Chang W et al (2014) Abnormal type I collagen post-translational modification and crosslinking in a cyclophilin B KO mouse model of recessive osteogenesis imperfecta. PLoS Genet 10(6):e1004465
Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S et al (2010) Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet 86(3):389–398
Marshall C, Lopez J, Crookes L, Pollitt RC, Balasubramanian M (2016) A novel homozygous variant in SERPINH1 associated with a severe, lethal presentation of osteogenesis imperfecta with hydranencephaly. Gene 595(1):49–52
Syx D, Ishikawa Y, Gebauer J, Boudko SP, Guillemyn B, Van Damme T et al (2021) Aberrant binding of mutant HSP47 affects posttranslational modification of type I collagen and leads to osteogenesis imperfecta. PLoS Genet 17(2):e1009339
Macdonald JR, Bächinger HP (2001) HSP47 binds cooperatively to triple helical type I collagen but has little effect on the thermal stability or rate of refolding. J Biol Chem 276(27):25399–25403
Bonfanti L, Mironov AA, Martínez-Menárguez JA, Martella O, Fusella A, Baldassarre M et al (1998) Procollagen traverses the Golgi stack without leaving the lumen of cisternae: evidence for cisternal maturation. Cell 95(7):993–1003
Nagai N, Hosokawa M, Itohara S, Adachi E, Matsushita T, Hosokawa N et al (2000) Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J Cell Biol 150(6):1499–1506
Ishida Y, Kubota H, Yamamoto A, Kitamura A, Bächinger HP, Nagata K (2006) Type I collagen in Hsp47-null cells is aggregated in endoplasmic reticulum and deficient in N-propeptide processing and fibrillogenesis. Mol Biol Cell 17(5):2346–2355
Ishikawa Y, Vranka J, Wirz J, Nagata K, Bächinger HP (2008) The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J Biol Chem 283(46):31584–31590
Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D et al (2010) Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet 86(4):551–559
Barnes AM, Cabral WA, Weis M, Makareeva E, Mertz EL, Leikin S et al (2012) Absence of FKBP10 in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum Mutat 33(11):1589–1598
Schwarze U, Cundy T, Pyott SM, Christiansen HE, Hegde MR, Bank RA et al (2013) Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum Mol Genet 22(1):1–17
Kelley BP, Malfait F, Bonafe L, Baldridge D, Homan E, Symoens S et al (2011) Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res 26(3):666–672
Shaheen R, Al-Owain M, Sakati N, Alzayed ZS, Alkuraya FS (2010) FKBP10 and Bruck syndrome: phenotypic heterogeneity or call for reclassification? Am J Hum Gene 87(2):306–307
Shaheen R, Al-Owain M, Faqeih E, Al-Hashmi N, Awaji A, Al-Zayed Z et al (2011) Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am J Med Genet A 155(6):1448–1452
Otaify GA, Abdel-Hamid MS, Hassib NF, Elhossini RM, Abdel-Ghafar SF, Aglan MS (2022) Bruck syndrome in 13 new patients: Identification of five novel FKBP10 and PLOD2 variants and further expansion of the phenotypic spectrum. Am J Med Genet A 188(6):1815–1825
Barnes AM, Duncan G, Weis M, Paton W, Cabral WA, Mertz EL et al (2013) Kuskokwim syndrome, a recessive congenital contracture disorder, extends the phenotype of FKBP10 mutations. Hum Mutat 34(9):1279–1288
Duran I, Nevarez L, Sarukhanov A, Wu S, Lee K, Krejci P et al (2015) HSP47 and FKBP65 cooperate in the synthesis of type I procollagen. Hum Mol Genet 24(7):1918–1928
Efthymiou S, Herman I, Rahman F, Anwar N, Maroofian R, Yip J et al (2021) Two novel bi-allelic KDELR2 missense variants cause osteogenesis imperfecta with neurodevelopmental features. Am J Med Genet A 185(7):2241–2249
Van Dijk FS, Semler O, Etich J, Köhler A, Jimenez-Estrada JA, Bravenboer N et al (2020) Interaction between KDELR2 and HSP47 as a key determinant in osteogenesis imperfecta caused by bi-allelic variants in KDELR2. Am J Hum Genet 107(5):989–999
Capitani M, Sallese M (2009) The KDEL receptor: new functions for an old protein. FEBS Lett 583(23):3863–3871
Bräuer P, Parker JL, Gerondopoulos A, Zimmermann I, Seeger MA, Barr FA et al (2019) Structural basis for pH-dependent retrieval of ER proteins from the Golgi by the KDEL receptor. Science 363(6431):1103–1107
Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK et al (2016) High-throughput discovery of novel developmental phenotypes. Nature 537(7621):508–514
Lapunzina P, Aglan M, Temtamy S, Caparrós-Martín JA, Valencia M, Letón R et al (2010) Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet 87(1):110–114
Fiscaletti M, Biggin A, Bennetts B, Wong K, Briody J, Pacey V et al (2018) Novel variant in Sp7/Osx associated with recessive osteogenesis imperfecta with bone fragility and hearing impairment. Bone 110:66–75
Hayat A, Hussain S, Bilal M, Kausar M, Almuzzaini B, Abbas S et al (2020) Biallelic variants in four genes underlying recessive osteogenesis imperfecta. Eur J Med Genet 63(8):103954
Harrington J, AlSubaihin A, Dupuis L, Kannu P, Mendoza-Londono R, Howard A (2021) Diagnostic utility of next-generation sequence genetic panel testing in children presenting with a clinically significant fracture history. Arch Osteoporos 16(1):88
Ludwig K, Ward LM, Khan N, Robinson M-E, Miranda V, Bardai G et al (2022) Dominant osteogenesis imperfecta with low bone turnover caused by a heterozygous SP7 variant. Bone 160:116400
Tung JY-l, Ho JL-i, Wong R, Fung S-c (2022) Dental phenotype in an adolescent with osteogenesis imperfecta type XII. BMJ Case Reports CP 15(4):e246554
Al-Mutairi DA, Jarragh AA, Alsabah BH, Wein MN, Mohammed W, Alkharafi L (2024) A homozygous SP7/OSX mutation causes osteogenesis and dentinogenesis imperfecta with craniofacial anomalies. JBMR Plus 8(5):ziae026
Lui JC, Raimann A, Hojo H, Dong L, Roschger P, Kikani B et al (2022) A neomorphic variant in SP7 alters sequence specificity and causes a high-turnover bone disorder. Nat Commun 13(1):700
Whyte MP, Campeau PM, McAlister WH, Roodman GD, Kurihara N, Nenninger A et al (2020) Juvenile Paget’s disease from heterozygous mutation of SP7 encoding Osterix (specificity protein 7, transcription factor SP7). Bone 137:115364
Ortuño MJ, Susperregui AR, Artigas N, Rosa JL, Ventura F (2013) Osterix induces Col1a1 gene expression through binding to Sp1 sites in the bone enhancer and proximal promoter regions. Bone 52(2):548–556
Cao Z, Liu R, Zhang H, Liao H, Zhang Y, Hinton RJ et al (2015) Osterix controls cementoblast differentiation through downregulation of Wnt-signaling via enhancing DKK1 expression. Int J Biol Sci 11(3):335
Tohmonda T, Miyauchi Y, Ghosh R, Yoda M, Uchikawa S, Takito J et al (2011) The IRE1α–XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep 12(5):451–457
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR et al (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108(1):17–29
Volodarsky M, Markus B, Cohen I, Staretz-Chacham O, Flusser H, Landau D et al (2013) A deletion mutation in TMEM 38 B associated with autosomal recessive Osteogenesis imperfecta. Hum Mutat 34(4):582–586
Shaheen R, Alazami AM, Alshammari MJ, Faqeih E, Alhashmi N, Mousa N et al (2012) Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J Med Genet 49(10):630–635
Rubinato E, Morgan A, D’Eustacchio A, Pecile V, Gortani G, Gasparini P et al (2014) A novel deletion mutation involving TMEM38B in a patient with autosomal recessive osteogenesis imperfecta. Gene 545(2):290–292
Cabral WA, Ishikawa M, Garten M, Makareeva EN, Sargent BM, Weis M et al (2016) Absence of the ER cation channel TMEM38B/TRIC-B disrupts intracellular calcium homeostasis and dysregulates collagen synthesis in recessive osteogenesis imperfecta. PLoS Genet 12(7):e1006156
Lv F, Xu X-j, Wang J-y, Liu Y, Wang J-w, Song L-j et al (2016) Two novel mutations in TMEM38B result in rare autosomal recessive osteogenesis imperfecta. J Hum Genet 61(6):539–545
Kodama Y, Meiri S, Asada T, Matsuyama M, Makino S, Iwai M et al (2023) Novel splice site variant of TMEM38B in osteogenesis imperfecta type XIV. Human Genome Variation 10(1):25
Webb EA, Balasubramanian M, Fratzl-Zelman N, Cabral WA, Titheradge H, Alsaedi A et al (2017) Phenotypic spectrum in osteogenesis imperfecta due to mutations in TMEM38B: unraveling a complex cellular defect. J Clin Endocrinol Metab 102(6):2019–2028
Yazawa M, Ferrante C, Feng J, Mio K, Ogura T, Zhang M et al (2007) TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 448(7149):78–82
Jovanovic M, Mitra A, Besio R, Contento BM, Wong KW, Derkyi A et al (2023) Absence of TRIC-B from type XIV Osteogenesis Imperfecta osteoblasts alters cell adhesion and mitochondrial function–a multi-omics study. Matrix Biol 121:127–148
Yamazaki D, Komazaki S, Nakanishi H, Mishima A, Nishi M, Yazawa M, Yamazaki T, Taguchi R, Takeshima H (2009) Essential role of the TRIC-B channel in Ca2+ handling of alveolar epithelial cells and in perinatal lung maturation. Development 136(14):2355–2361
Zhao C, Ichimura A, Qian N, Iida T, Yamazaki D, Noma N et al (2016) Mice lacking the intracellular cation channel TRIC-B have compromised collagen production and impaired bone mineralization. Sci Signal 9(428):ra49-ra
Besio R, Contento BM, Garibaldi N, Filibian M, Sonntag S, Shmerling D et al (2023) CaMKII inhibition due to TRIC-B loss-of-function dysregulates SMAD signaling in osteogenesis imperfecta. Matrix Biol 120:43–59
Doubravska L, Krausova M, Gradl D, Vojtechova M, Tumova L, Lukas J et al (2011) Fatty acid modification of Wnt1 and Wnt3a at serine is prerequisite for lipidation at cysteine and is essential for Wnt signalling. Cell Signal 23(5):837–848
Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F (2013) Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet 50(5):345–348
Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O et al (2013) Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet 92(4):565–574
Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT et al (2013) WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet 92(4):590–597
Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M et al (2013) WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med 368(19):1809–1816
Stephen J, Girisha KM, Dalal A, Shukla A, Shah H, Srivastava P et al (2015) Mutations in patients with osteogenesis imperfecta from consanguineous Indian families. Eur J Med Genet 58(1):21–27
Umair M, Alhaddad B, Rafique A, Jan A, Haack TB, Graf E et al (2017) Exome sequencing reveals a novel homozygous splice site variant in the WNT1 gene underlying osteogenesis imperfecta type 3. Pediatr Res 82(5):753–758
Kausar M, Siddiqi S, Yaqoob M, Mansoor S, Makitie O, Mir A et al (2018) Novel mutation G324C in WNT1 mapped in a large Pakistani family with severe recessively inherited Osteogenesis Imperfecta. J Biomed Sci 25:1–10
Lu Y, Ren X, Wang Y, Bardai G, Sturm M, Dai Y et al (2018) Novel WNT1 mutations in children with osteogenesis imperfecta: clinical and functional characterization. Bone 114:144–149
Shang L, Shi W, Xu Y, Nong T, Li X, Li Z et al (2024) A novel compound heterozygous variation in the FKBP10 gene causes Bruck syndrome without congenital contractures: a case report. Heliyon 10(7)
Kantaputra PN, Sirirungruangsarn Y, Visrutaratna P, Petcharunpaisan S, Carlson BM, Intachai W et al (2019) WNT1-associated osteogenesis imperfecta with atrophic frontal lobes and arachnoid cysts. J Hum Genet 64(4):291–296
Nampoothiri S, Guillemyn B, Elcioglu N, Jagadeesh S, Yesodharan D, Suresh B et al (2019) Ptosis as a unique hallmark for autosomal recessive WNT1-associated osteogenesis imperfecta. Am J Med Genet A 179(6):908–914
Mäkitie RE, Haanpää M, Valta H, Pekkinen M, Laine CM, Lehesjoki AE et al (2016) Skeletal characteristics of WNT1 osteoporosis in children and young adults. J Bone Miner Res 31(9):1734–1742
Alhamdi S, Lee YC, Chowdhury S, Byers PH, Gottschalk M, Taft RJ et al (2018) Heterozygous WNT1 variant causing a variable bone phenotype. Am J Med Genet A 176(11):2419–2424
McMahon AP, Bradley A (1990) The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell 62(6):1073–1085
Joeng KS, Lee Y-C, Jiang M-M, Bertin TK, Chen Y, Abraham AM et al (2014) The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Hum Mol Genet 23(15):4035–4042
Joeng KS, Lee Y-C, Lim J, Chen Y, Jiang M-M, Munivez E et al (2017) Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Investig 127(7):2678–2688
Symoens S, Malfait F, D’hondt S, Callewaert B, Dheedene A, Steyaert W et al (2013) Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis 8:1–6
Lindahl K, Åström E, Dragomir A, Symoens S, Coucke P, Larsson S et al (2018) Homozygosity for CREB3L1 premature stop codon in first case of recessive osteogenesis imperfecta associated with OASIS-deficiency to survive infancy. Bone 114:268–277
Keller RB, Tran TT, Pyott SM, Pepin MG, Savarirayan R, McGillivray G et al (2018) Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively. Genet Med 20(4):411–419
Cayami FK, Maugeri A, Treurniet S, Setijowati ED, Teunissen BP, Eekhoff EM et al (2019) The first family with adult osteogenesis imperfecta caused by a novel homozygous mutation in CREB3L1. Mol Genet Genomic Med 7(8):e823
Guillemyn B, Kayserili H, Demuynck L, Sips P, De Paepe A, Syx D et al (2019) A homozygous pathogenic missense variant broadens the phenotypic and mutational spectrum of CREB3L1-related osteogenesis imperfecta. Hum Mol Genet 28(11):1801–1809
Andersson K, Malmgren B, Åström E, Nordgren A, Taylan F, Dahllöf G (2020) Mutations in COL1A1/A2 and CREB3L1 are associated with oligodontia in osteogenesis imperfecta. Orphanet J Rare Dis 15:1–12
DeMasters DP, Paulus AO, Scott JN (2023) Osteogenesis imperfecta diagnosed in an active duty female due to CREB3L1 heterozygosity. Mil Med 188(7–8):e2802–e2804
Murakami T, Kondo S, Ogata M, Kanemoto S, Saito A, Wanaka A et al (2006) Cleavage of the membrane-bound transcription factor OASIS in response to endoplasmic reticulum stress. J Neurochem 96(4):1090–1100
Murakami T, Saito A, Hino S-i, Kondo S, Kanemoto S, Chihara K, et al (2009) Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nature Cell Biol 11(10):1205–1211
Lindert U, Cabral WA, Ausavarat S, Tongkobpetch S, Ludin K, Barnes AM et al (2016) MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun 7(1):11920
Oeffner F, Fischer G, Happle R, König A, Betz RC, Bornholdt D et al (2009) IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. Am J Hum Genet 84(4):459–467
Naiki M, Mizuno S, Yamada K, Yamada Y, Kimura R, Oshiro M et al (2012) MBTPS2 mutation causes BRESEK/BRESHECK syndrome. Am J Med Genet A 158(1):97–102
Aten E, Brasz LC, Bornholdt D, Hooijkaas IB, Porteous ME, Sybert VP et al (2010) Keratosis follicularis spinulosa decalvans is caused by mutations in MBTPS2. Hum Mutat 31(10):1125–1133
Lim PJ, Marfurt S, Lindert U, Opitz L, Ndarugendamwo T, Srikanthan P et al (2021) Omics profiling of S2P mutant fibroblasts as a mean to unravel the pathomechanism and molecular signatures of X-linked MBTPS2 osteogenesis imperfecta. Front Genet 12:662751
Lim PJ, Marcionelli G, Srikanthan P, Ndarugendamwo T, Pinner J, Rohrbach M et al (2023) Perturbations in fatty acid metabolism and collagen production infer pathogenicity of a novel MBTPS2 variant in Osteogenesis imperfecta. Front Endocrinol 14:1195704
Kondo Y, Fu J, Wang H, Hoover C, McDaniel JM, Steet R et al (2018) Site-1 protease deficiency causes human skeletal dysplasia due to defective inter-organelle protein trafficking. JCI Insight 3(14)
Patra D, Xing X, Davies S, Bryan J, Franz C, Hunziker EB et al (2007) Site-1 protease is essential for endochondral bone formation in mice. J Cell Biol 179(4):687–700
Mendoza-Londono R, Fahiminiya S, Majewski J, Tétreault M, Nadaf J, Kannu P et al (2015) Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet 96(6):979–985
Durkin A, DeVile C, Arundel P, Bull M, Walsh J, Bishop NJ et al (2022) Expanding the phenotype of SPARC-related osteogenesis imperfecta: clinical findings in two patients with pathogenic variants in SPARC and literature review. J Med Genet 59(8):810–816
Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML, Martin GR (1981) Osteonectin, a bone-specific protein linking mineral to collagen. Cell 26(1):99–105
MAYER U, Aumailley M, Mann K, Timpl R, Engel J (1991) Calcium‐dependent binding of basement membrane protein BM‐40 (osteonectin, SPARC) to basement membrane collagen type IV. Euro J Biochem 198(1):141–150
Delany A, Amling M, Priemel M, Howe C, Baron R, Canalis E (2000) Osteopenia and decreased bone formation in osteonectin-deficient mice. J Clin Investig 105(7):915–923
Bradshaw AD, Puolakkainen P, Wight TN, Sage EH, Dasgupta J, Davidson JM (2003) SPARC-null mice display abnormalities in the dermis characterized by decreased collagen fibril diameter and reduced tensile strength. J Investig Dermatol 120(6):949–955
Trombetta-eSilva J, Rosset EA, Hepfer RG, Wright GJ, Baicu C, Yao H et al (2015) Decreased mechanical strength and collagen content in SPARC-null periodontal ligament is reversed by inhibition of transglutaminase activity. J Bone Miner Res 30(10):1914–1924
Doyard M, Bacrot S, Huber C, Di Rocco M, Goldenberg A, Aglan MS et al (2018) FAM46A mutations are responsible for autosomal recessive osteogenesis imperfecta. J Med Genet 55(4):278–284
Kuchta K, Muszewska A, Knizewski L, Steczkiewicz K, Wyrwicz LS, Pawlowski K et al (2016) FAM46 proteins are novel eukaryotic non-canonical poly (A) polymerases. Nucleic Acids Res 44(8):3534–3548
Gewartowska O, Aranaz-Novaliches G, Krawczyk PS, Mroczek S, Kusio-Kobiałka M, Tarkowski B et al (2021) Cytoplasmic polyadenylation by TENT5A is required for proper bone formation. Cell Reports 35(3)
Tüysüz B, Elkanova L, Alkaya DU, Güleç Ç, Toksoy G, Güneş N et al (2022) Osteogenesis imperfecta in 140 Turkish families: molecular spectrum and comparison of long-term clinical outcome of those with COL1A1/A2 and biallelic variants. Bone 155:116293
Cormier-Daire V, Munnich A, Lyonnet S, Rustin P, Delezoide A-L, Maroteaux P et al (1998) Presentation of six cases of Stüve-Wiedemann syndrome. Pediatr Radiol 28:776–780
Diener S, Bayer S, Sabrautzki S, Wieland T, Mentrup B, Przemeck GK et al (2016) Exome sequencing identifies a nonsense mutation in Fam46a associated with bone abnormalities in a new mouse model for skeletal dysplasia. Mamm Genome 27:111–121
Colland F, Jacq X, Trouplin V, Mougin C, Groizeleau C, Hamburger A et al (2004) Functional proteomics mapping of a human signaling pathway. Genome Res 14(7):1324–1332
Barragan I, Borrego S, Abd El-Aziz M, El-Ashry M, Abu-Safieh L, Bhattacharya S et al (2008) Genetic analysis of FAM46A in Spanish families with autosomal recessive retinitis pigmentosa: characterisation of novel VNTRs. Ann Hum Genet 72(1):26–34
Etokebe GE, Jotanovic Z, Mihelic R, Mulac-Jericevic B, Nikolic T, Balen S et al (2015) Susceptibility to large-joint osteoarthritis (hip and knee) is associated with BAG6 rs3117582 SNP and the VNTR polymorphism in the second exon of the FAM46A gene on chromosome 6. J Orthop Res 33(1):56–62
Min K, Li Y, Wu Z, Dai Z, Feng Z, Qian Z et al (2023) A genetic variant of FAM46A is associated with the development of adolescent idiopathic scoliosis in the Chinese Population. Spine 48(17):1253–1258
Moosa S, Yamamoto GL, Garbes L, Keupp K, Beleza-Meireles A, Moreno CA et al (2019) Autosomal-recessive mutations in MESD cause osteogenesis imperfecta. Am J Hum Genet 105(4):836–843
Tran TT, Keller RB, Guillemyn B, Pepin M, Corteville JE, Khatib S et al (2021) Biallelic variants in MESD, which encodes a WNT-signaling-related protein, in four new families with recessively inherited osteogenesis imperfecta. Human Genet Genom Adv 2(4)
Stürznickel J, Rolvien T, Delsmann A, Butscheidt S, Barvencik F, Mundlos S et al (2020) Clinical phenotype and relevance of LRP5 and LRP6 variants in patients with early-onset osteoporosis (EOOP). J Bone Miner Res 36(2):271–282
Uludağ Alkaya D, Uyguner ZO, Güneş N, Tüysüz B (2022) Long-term follow-up findings in a Turkish girl with osteogenesis imperfecta type XX caused by a homozygous MESD variant. Am J Med Genet A 188(5):1639–1646
Hsieh J-C, Lee L, Zhang L, Wefer S, Brown K, DeRossi C et al (2003) Mesd encodes an LRP5/6 chaperone essential for specification of mouse embryonic polarity. Cell 112(3):355–367
Ghosh DK, Udupa P, Shrikondawar AN, Bhavani GS, Shah H, Ranjan A et al (2023) Mutant MESD links cellular stress to type I collagen aggregation in osteogenesis imperfecta type XX. Matrix Biol 115:81–106
Dubail J, Brunelle P, Baujat G, Huber C, Doyard M, Michot C et al (2020) Homozygous loss-of-function mutations in CCDC134 are responsible for a severe form of osteogenesis imperfecta. J Bone Miner Res 35(8):1470–1480
Holick MF, Shirvani A, Charoenngam N (2021) Fetal fractures in an infant with maternal Ehlers-Danlos syndrome, CCDC134 pathogenic mutation and a negative genetic test for osteogenesis imperfecta. Children 8(6):512
Ali TM, Linnenkamp BD, Yamamoto GL, Honjo RS, Cabral de Menezes Filho H, Kim CA et al (2022) The recurrent homozygous translation start site variant in CCDC134 in an individual with severe osteogenesis imperfecta of non‐Morrocan ancestry. Am J Med Genet A 188(5):1545–1549
Huang J, Shi T, Ma T, Zhang Y, Ma X, Lu Y et al (2008) CCDC134, a novel secretory protein, inhibits activation of ERK and JNK, but not p38 MAPK. Cell Mol Life Sci 65:338–349
Kim J-M, Yang Y-S, Park KH, Oh H, Greenblatt MB, Shim J-H (2019) The ERK MAPK pathway is essential for skeletal development and homeostasis. Int J Mol Sci 20(8):1803
Kang H, Jha S, Deng Z, Fratzl-Zelman N, Cabral WA, Ivovic A et al (2018) Somatic activating mutations in MAP2K1 cause melorheostosis. Nat Commun 9(1):1390
Yu B, Zhang T, Xia P, Gong X, Qiu X, Huang J (2018) CCDC134 serves a crucial role in embryonic development. Int J Mol Med 41(1):381–390
Claeys L, Storoni S, Eekhoff M, Elting M, Wisse L, Pals G et al (2021) Collagen transport and related pathways in Osteogenesis Imperfecta. Hum Genet 140(8):1121–1141
Lekszas C, Foresti O, Raote I, Liedtke D, König E-M, Nanda I et al (2020) Biallelic TANGO1 mutations cause a novel syndromal disease due to hampered cellular collagen secretion. Elife 9:e51319
Besio R, Iula G, Garibaldi N, Cipolla L, Sabbioneda S, Biggiogera M, et al (2018) 4-PBA ameliorates cellular homeostasis in fibroblasts from osteogenesis imperfecta patients by enhancing autophagy and stimulating protein secretion. Biochim Biophys Acta (BBA)-Molecular Basis Dis 1864(5):1642–1652
Besio R, Garibaldi N, Leoni L, Cipolla L, Sabbioneda S, Biggiogera M et al (2019) Cellular stress due to impairment of collagen prolyl hydroxylation complex is rescued by the chaperone 4-phenylbutyrate. Dis Models Mech 12(6):dmm038521
Gremminger VL, Jeong Y, Cunningham RP, Meers GM, Rector RS, Phillips CL (2019) Compromised exercise capacity and mitochondrial dysfunction in the osteogenesis imperfecta murine (oim) mouse model. J Bone Miner Res 34(9):1646–1659
Moffatt P, Boraschi-Diaz I, Bardai G, Rauch F (2021) Muscle transcriptome in mouse models of osteogenesis imperfecta. Bone 148:115940
Gorrell L, Makareeva E, Omari S, Otsuru S, Leikin S (2022) ER, Mitochondria, and ISR regulation by mt-HSP70 and ATF5 upon procollagen misfolding in osteoblasts. Adv Sci 9(29):2201273
Prinz WA, Toulmay A, Balla T (2020) The functional universe of membrane contact sites. Nat Rev Mol Cell Biol 21(1):7–24
Abrisch RG, Gumbin SC, Wisniewski BT, Lackner LL, Voeltz GK (2020) Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J Cell Biol 219(4):e201911122
Lim J, Grafe I, Alexander S, Lee B (2017) Genetic causes and mechanisms of osteogenesis imperfecta. Bone 102:40–49
Ha-Vinh R, Alanay Y, Bank RA, Campos-Xavier AB, Zankl A, Superti-Furga A et al (2004) Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet A 131(2):115–120
Roschger P, Fratzl-Zelman N, Misof BM, Glorieux FH, Klaushofer K, Rauch F (2008) Evidence that abnormal high bone mineralization in growing children with osteogenesis imperfecta is not associated with specific collagen mutations. Calcif Tissue Int 82:263–270
Weber M, Roschger P, Fratzl-Zelman N, Schöberl T, Rauch F, Glorieux FH et al (2006) Pamidronate does not adversely affect bone intrinsic material properties in children with osteogenesis imperfecta. Bone 39(3):616–622
Fratzl-Zelman N, Morello R, Lee B, Rauch F, Glorieux F, Misof B et al (2010) CRTAP deficiency leads to abnormally high bone matrix mineralization in a murine model and in children with osteogenesis imperfecta type VII. Bone 46(3):820–826
Marini JC, Reich A, Smith SM (2014) Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation. Curr Opin Pediatr 26(4):500–507
Hoyer-Kuhn H, Semler O, Schoenau E, Roschger P, Klaushofer K, Rauch F (2013) Hyperosteoidosis and hypermineralization in the same bone: bone tissue analyses in a boy with a homozygous BMP1 mutation. Calcif Tissue Int 93:565–570
Palomo T, Al-Jallad H, Moffatt P, Glorieux FH, Lentle B, Roschger P et al (2014) Skeletal characteristics associated with homozygous and heterozygous WNT1 mutations. Bone 67:63–70
Stürznickel J, Jähn-Rickert K, Zustin J, Hennig F, Delsmann MM, Schoner K et al (2020) Compound heterozygous frameshift mutations in MESD cause a lethal syndrome suggestive of osteogenesis imperfecta type XX. J Bone Miner Res 36(6):1077–1087
Sarathchandra P, Pope F, Kayser M, Ali S (2000) A light and electron microscopic study of osteogenesis imperfecta bone samples, with reference to collagen chemistry and clinical phenotype. J Pathol 192(3):385–395
Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM et al (2014) Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat Med 20(6):670–675
Zimmerman SM, Dimori M, Heard Lipsmeyer ME, Morello R (2019) The osteocyte transcriptome is extensively dysregulated in mouse models of osteogenesis imperfecta. J Bone Miner Res Plus 3(7):e10171
Folkestad L, Hald JD, Canudas-Romo V, Gram J, Hermann AP, Langdahl B et al (2016) Mortality and causes of death in patients with osteogenesis imperfecta: a register-based nationwide cohort study. J Bone Miner Res 31(12):2159–2166
Widmann RF, Bitan FD, Laplaza FJ, Burke SW, DiMaio MF, Schneider R (1999) Spinal deformity, pulmonary compromise, and quality of life in osteogenesis imperfecta. Spine 24(16):1673
Khan SI, Yonko EA, Carter EM, Dyer D, Sandhaus RA, Raggio CL (2020) Cardiopulmonary status in adults with osteogenesis imperfecta: intrinsic lung disease may contribute more than scoliosis. Clin Orthop Relat Res 478(12):2833–2843
Gochuico BR, Hossain M, Talvacchio SK, Zuo MXG, Barton M, Do AND et al (2023) Pulmonary function and structure abnormalities in children and young adults with osteogenesis imperfecta point to intrinsic and extrinsic lung abnormalities. J Med Genet 60(11):1067–1075
Hortop J, Tsipouras P, Hanley J, Maron B, Shapiro J (1986) Cardiovascular involvement in osteogenesis imperfecta. Circulation 73(1):54–61
Bonita RE, Cohen IS, Berko BA (2010) Valvular heart disease in osteogenesis imperfecta: presentation of a case and review of the literature. Echocardiography 27(1):69–73
Lamanna A, Fayers T, Clarke S, Parsonage W (2013) Valvular and aortic diseases in osteogenesis imperfecta. Heart Lung Circ 22(10):801–810
Rush ET, Li L, Goodwin JL, Kreikemeier RM, Craft M, Danford DA et al (2017) Echocardiographic phenotype in osteogenesis imperfecta varies with disease severity. Heart 103(6):443–448
Verdonk SJ, Storoni S, Micha D, van den Aardweg JG, Versacci P, Celli L, et al (2024) Is Osteogenesis imperfecta associated with cardiovascular abnormalities? A systematic review of the literature. Calcified Tissue Int, pp 1–12
Folkestad L, Hald JD, Gram J, Langdahl BL, Hermann AP, Diederichsen AC et al (2016) Cardiovascular disease in patients with osteogenesis imperfecta—a nationwide, register-based cohort study. Int J Cardiol 225:250–257
Funding
Open access funding provided by the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors has any conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jovanovic, M., Marini, J.C. Update on the Genetics of Osteogenesis Imperfecta. Calcif Tissue Int (2024). https://doi.org/10.1007/s00223-024-01266-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00223-024-01266-5