
Abstract
Purpose
The ATN model represents a research framework used to classify subjects based on the presence or absence of Alzheimer’s disease (AD) pathology through biomarkers for amyloid (A), tau (T), and neurodegeneration (N). The aim of this study was to assess the relationship between ATN profiles defined through imaging and cognitive decline in a memory clinic cohort.
Methods
One hundred-eight patients from the memory clinic of Geneva University Hospitals underwent complete clinical and neuropsychological evaluation at baseline and 23 ± 5 months after inclusion, magnetic resonance imaging, amyloid and tau PET scans. ATN profiles were divided into four groups: normal, AD pathological change (AD-PC: A + T-N-, A + T-N +), AD pathology (AD-P: A + T + N-, A + T + N +), and suspected non-AD pathology (SNAP: A-T + N-, A-T-N + , A-T + N +).
Results
Mini-Mental State Examination (MMSE) scores were significantly different among groups, both at baseline and follow-up, with the normal group having higher average MMSE scores than the other groups. MMSE scores changed significantly after 2 years only in AD-PC and AD-P groups. AD-P profile classification also had the largest number of decliners at follow-up (55%) and the steepest global cognitive decline compared to the normal group. Cox regression showed that participants within the AD-P group had a higher risk of cognitive decline (HR = 6.15, CI = 2.59–14.59), followed by AD-PC (HR = 3.16, CI = 1.17–8.52).
Conclusion
Of the different group classifications, AD-P was found to have the most significant effect on cognitive decline over a period of 2 years, highlighting the value of both amyloid and tau PET molecular imaging as prognostic imaging biomarkers in clinical practice.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The current research framework for the classification of subjects within the Alzheimer’s disease (AD) spectrum is based on the presence or absence of biomarker pathology: extracellular amyloid plaques (A), neurofibrillary tau tangles (T), and neurodegeneration (N) [1]. Each biomarker is considered independent from each other, and subjects can be classified as positive or negative for each of them. Together, the three biomarkers compose the ATN model, where each subject is classified with an unbiased descriptive pathological biomarker profile. Despite being considered independent from each other, a dynamic model for a pathological cascade has been proposed, where the disease starts with A deposits, followed by T, which finally leads to N [2]. Therefore, a temporal evolution of biomarker status is expected with the progression to AD. Indeed, previous studies have shown that the ATN classification is correlated with a clinical progression towards dementia [3, 4], memory worsening [5] and cognitive decline [6,7,8,9,10]. These ATN profiles can be categorised into four larger groups: normal biomarkers, AD pathological change (AD-PC; profiles with partial characteristic AD pathology, i.e., positive for A but not for T pathology), AD pathology (AD-P; profiles with characteristic AD pathology, i.e., positive for A and T pathology), and suspected non-Alzheimer’s disease pathology (SNAP; profiles negative for A but positive for T and/or N).
Positron emission tomography (PET) and magnetic resonance imaging (MRI) are imaging modalities that have been shown to provide good markers for individually predicting disease progression when compared to fluid biomarkers [4, 8, 11, 12]. PET images can be used to show in vivo deposits of A and T, while MRI provides a measure of brain atrophy, which is related to N. The ensuing measurements can be quantified, and subjects can be classified into different ATN profiles based on either visual inspection of the images or specific quantitative thresholds.
As imaging includes expensive techniques to assess biomarkers, other methods for measuring the same biomarkers have been used. However, PET and MRI have been shown to be the most precise approaches to predict disease progression [8]. Furthermore, previous studies have focused on cohorts of patients situated at an early stage of the AD spectrum and in research cohorts. A longitudinal study investigating the effect of ATN profiles, measured only through imaging techniques, in global cognitive decline has not been performed yet. Furthermore, a study of this design using data from subjects with a variety of diagnoses and cognitive statuses at baseline, evaluated at a memory clinic level, is also at need. Therefore, the aim of this study was to assess the relationship between ATN profiles and cognitive decline, measured through a decline in MMSE scores, in a memory clinic population.
Materials and methods
Subjects
A cohort of 108 subjects was selected from an ongoing study at the Geneva Memory Clinic at the Geneva University Hospitals (HUG), Geneva, Switzerland. These subjects were selected from a larger cohort of available subjects since they fitted the inclusion criteria: (1) amyloid and tau PET imaging performed within 12 months of each other (average 3.8 ± 5.1 months), (2) 3D T1 MRI scans performed within a year from the PET image (average 0.4 ± 10.5 months), (3) neuropsychological assessment at baseline was performed within 6 months of PET imaging (average 0.4 ± 5.8 months), (4) a follow-up neuropsychological assessment was performed after an average of 24 months from baseline (average 23.0 ± 11.1 months). The local review board (Cantonal Research Ethics Commission, Geneva, Switzerland) approved the study, which has been conducted in concordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice. All subjects provided written informed consent to have collected data be used in research.
This memory clinic cohort included subjects with a variety of diagnoses at baseline: healthy control individuals (HC), and patients with subjective cognitive decline (SCD), mild cognitive impairment (MCI), or dementia. Diagnosis was based on a clinical assessment combined with the results of the neuropsychological assessment. Subjects with SCD were evaluated at the Geneva Memory Clinic with a self-experience of deterioration in cognitive abilities but did not present objective cognitive impairment through formal neuropsychological testing [13]. MCI patients presented objective cognitive impairment and no functional impairing in everyday life [14]. Individuals were diagnosed with dementia if they matched MCI requirement but differ from MCI subjects for the impairment in everyday life [15]. Dementia patients were further diagnosed based on probable aetiology: two suspected non-AD dementia, one dementia due to stroke, one Parkinson’s dementia, and 11 AD dementia. Diagnosis was used to characterise the included population, but it was not included as a significant variable in the assessment of cognitive decline of each profile. Neuropsychological assessment also included a Mini-Mental State Examination (MMSE), which was used in this study as a measure of global cognition and to assess cognitive decline [16]. Cognitive decline was defined by a decrease of one MMSE point per year [17]. A subset of the cohort underwent other neuropsychological tests for verbal episodic memory (free and cued selective remining test — RLRI16 in French), attention (trail making test — TMT), and verbal fluency (categorical and phonemic).
Imaging acquisition and processing
MRI exams were performed at the Division of Radiology Department of HUG. 3D T1 images were acquired using a 3 Tesla Siemens Magnetom Skyra scanner (Siemens Healthineers, Erlangen, Germany) equipped with a 64-channel head coil and were acquired in close accordance with IMI pharmacog WP5/European ADNI sequences and published procedures [18]. In summary, a field of view of 256 mm, 0.9–1 mm slice thickness, 1819–1930 ms repetition time, 2.19–2.4 ms echo time, 8° flip angle, and no fat suppression were used.
PET imaging was performed at the Nuclear Medicine and Molecular Imaging Division of the HUG. For amyloid imaging, 42 subjects were injected with 204 ± 23 MBq of [18F]florbetapir, and images were acquired 50 min after intravenous administration of the radiotracer (3 × 5 min image frames that were averaged into a single image). The remaining 66 subjects were scanned using 171 ± 19 MBq of [18F]flutemetamol, and images were acquired 90 min after intravenous administration of the radiotracer (4 × 5 min image frames that were averaged into a single image). For tau images, subjects were injected with 200.70 ± 18.98 MBq of [18F]flortaucipir, and images were acquired 75 min after tracer injection (6 × 5 min image frames that were averaged into a single image). All images were acquired using a Siemens Biograph PET scanner (Siemens Medical Solutions, USA), reconstructed using a 3D OSEM algorithm (4 iterations, 8 subsets), a 2 mm Gaussian convolution kernel, corrected for dead time, normalisation, attenuation, and sensitivity. All commercially available radiotracers were synthesised at radiopharmaceutical Good Manufacturing Practice laboratories.
All images were processed at the Geneva Memory Centre of HUG, Geneva, Switzerland, using SPM12 (Wellcome Trust Centre for Neuroimaging, London, UK) and MATLAB R2018b version 9.5 (MathWorks Inc., Sherborn, USA). Firstly, 3D T1 MRI images were aligned to the anterior commissure. Then, they were normalised to the Montreal Neurologic Institute (MNI) space using tissue probability maps [19]. PET images were aligned to the subject’s respective MRI images and then, using the MRI transformation matrix, they were transformed into the MNI space.
Cortical reconstruction and volumetric segmentation of T1 images were performed using Freesurfer (v7, recon-all [20]). Right and left hippocampal volumes were extracted, averaged, and normalised to the total intracranial volume. Amyloid PET data were converted into the centiloid scale [21], so that data from the different radiotracers could be equally compared, and global centiloid values were used in this study. For the tau PET images, standardised uptake value rations (SUVR) were generated using the cerebellar crus as a reference tissue [22, 23], and data were extracted using the automated anatomic labelling atlas 3 [24, 25]. Regional data were combined in a weighted average according to the Simplified Temporal-Occipital Classification (STOC) into four main regions: medial temporal lobe (MTL; hippocampus, amygdala, parahippocampus, and fusiform gyrus), lateral temporal lobe (LTL; Heschl, temporal inferior and middle gyrus), superior temporal gyrus (STG), and the primary visual cortex (PVC) [23]. Furthermore, a global tau SUVR value was calculated based on a combination of the amygdala, parahippocampus, middle occipital gyrus, and inferior temporal gyrus [26] for further analyses. As a supplementary analysis, N was also estimated using the cortical thickness of an AD-specific region of interest (weighted average cortical thickness in the entorhinal, inferior temporal, middle temporal, and fusiform) [27], setting a threshold of 2.57 for discriminating between N + and N − subjects [28].
ATN classification
Positivity for each biomarker was defined based on previously published thresholds. Subjects were considered A + when the centiloid value was above 12 [29]. Tau status was assessed based on the STOC model [23]. Each region from the STOC classification, described on the previous section (MTL, LTL, STG, and PVC) was considered positive if its SUVR value was above 1.28, and tau STOC stage was defined based on these positivises. Stages 0 (all negative) and 1 (MTL positive only) were considered T-, while stages 2 (LTL or MTL + LTL positive) 3 (stage 2 + STG), and 4 (stage 3 + PVC) were considered T + . Neurodegeneration positivity was decided based on each subject’s ratio of hippocampal volume to total intracranial volume, a variable called from here on as ‘hippocampal ratio’. If this value was below 0.00215, subjects were considered N + [27]. Combining biomarker positivity information, subjects were then classified into ATN profiles. Due to the small number of subjects in some of the ATN classifications, profiles were then combined into larger groups that comprise of more than one profile (with the exception of one classification): normal (A-T-N-), AD-PC (A + T-N-, A + T-N +), AD-P (A + T + N-, A + T + N +), and SNAP (A-T + N-, A-T-N + , A-T + N +).
Statistical analysis
Kruskal-Wallis test and Dunn tests for multiple corrections using false discovery rate (FDR) were performed to explore differences in age, years of education, MMSE (both at baseline and follow up), centiloid, global tau SUVR, and hippocampal ratio between groups. Significant differences between baseline and follow-up MMSE scores were assessed using paired Wilcoxon tests for each group individually.
Linear mixed models were used to estimate the association between ATN profiles at baseline and changes in MMSE scores in a 2-year period considering the normal group as the reference. Cox proportional hazards analysis was performed to evaluate the association between each group (normal biomarker group as reference) and cognitive decline, defined as a decrease of one MMSE point per year [16]. Linear mixed models were also used for the association between A, T, and N biomarkers individually at baseline and changes in MMSE scores at follow-up using negative biomarkers as reference, and cognitive decline, using the same criterium.
Neuropsychological tests (RLRI16, TMT, and verbal fluency) were analysed using the same methods as for the MMSE: a Kruskal-Wallis test and Dunn test for multiple correction using FDG to compare between ATN groups, and a paired Wilcoxon test to compare between baseline and follow up scores.
A p-value of 0.05 was considered the significance threshold for all analyses, which were performed using RStudio (version 2022.07.1, R version 4.2.1). For the Kruskal-Wallis and Dunn tests, the FSA R package (version 0.9.3) was used. For the linear mixed models, the lme4 package (version 1.1-29) was used. The survival package (version 3.4-0) was used for the Cox proportional hazards estimations and the survminer package (version 0.4.9) was used for plotting Kaplan-Meyer curves.
Results
Population
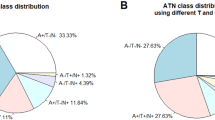
Figure 1 shows the distribution of ATN profiles and groups. Table 1 shows the demographic, clinical, cognitive, and imaging characteristics of each ATN group at baseline. Education was not significantly different between the groups. Age was significantly different between normal and AD-PC groups, with the latter having a significantly higher age in comparison to the former. MMSE was significantly higher in the normal group when compared to the AD-P and SNAP groups, while in the AD-PC group it was significantly higher than in the AD-P group. Centiloid values were significantly higher in the AD-PC and AD-P groups when compared to normal and SNAP subjects, but not between each other. Global tau SUVR was higher in the AD-P group when compared to all others. Finally, hippocampal ratio was significantly higher in the normal individuals when compared to all other groups. A total of 4 subjects were included as HC, 26 were diagnosed as SCD, 63 as MCI, and 15 as Dementia at baseline. The characteristics of each group are summarised in Supplementary Table 1. No significant differences in population demographics were found when using cortical thickness to define N (Supplementary Table 2).

Pie chart illustrating the distribution of ATN profiles and their percentage. In green, the normal group (negative for all biomarkers); in shades of orange, the profiles included in the AD-PC (A + T −) group; in shades of red, the profiles included in the AD-P (A + T +) group; and in blue, the profiles included in the SNAP group
Cognitive follow-up
Table 2 shows MMSE scores per group at baseline and follow-up. Scores were significantly different between groups at both time points, with the normal group having higher MMSE values in comparison to AD-PC and SNAP groups, and the AD-PC group also having higher MMSE values when compared to the AD-P group. However, scores were significantly different between time points in the AD-PC and AD-P groups alone. Furthermore, the AD-P group had the largest percentage of decliners (55%). Figure 2 presents a spaghetti plot of MMSE changes between baseline and follow-up by group, with red lines denoting subjects with significant decline.

Individual MMSE scores per subject by ATN group. Lines connect points that belong to the same subject, expressing evolutionary trajectory. Each column represents a different ATN group. Red colour represents subjects that significantly declined, and black colour, stable subjects
For the memory (RLRI16), attention (TMT), and verbal fluency (categorical fruits) tests, no significant differences between groups were found at baseline, but groups were significantly different at follow-up. However, only the AD-P group showed differences in scores between baseline and follow-up in the sum of free recalls, sum of total recalls and delayed total recall, TMTA tests. The phonemic verbal fluency test showed no significant differences between groups neither at baseline nor at follow-up. Furthermore, no significant differences between baseline and follow-up were found in each of the groups. Results for the neuropsychological assessments of the specific domains are presented in Supplementary Table 3 and Supplementary Fig. 1.
ATN prediction of cognitive decline
A linear mixed model was used to estimate the association between groups and global cognitive status measured through MMSE scores corrected for the effect of time. Table 3 shows estimated baseline MMSE scores and change in 2 years by ATN group. Significant associations were found only in the AD-P group (β = − 3.81, p < 0.01). Furthermore, the same group had significant interactions with time when measuring cognitive decline through MMSE scores, showing a consistent effect (β = − 3.21, p < 0.01). Correcting the model for age, gender, and education did not significantly change the results.
Supplementary Table 4 shows the results of the linear mixed models used to estimate the association between A, T, and N biomarkers separately and cognitive decline measure through MMSE scores corrected for the effect of time. Significant associations were found for the T biomarker at follow-up (β = − 1.96, p = 0.04). Correcting the model for age, gender, and education did not significantly change the results.
Supplementary Table 5 shows the results of the linear mixed model used to estimate the association between groups and global cognitive status measured though MMSE scores corrected for the effect of time with N measured through cortical thickness. As for N measured through hippocampal ratio, significant association were only found in the AD-P group (β = − 3.22, p < 0.01). Furthermore, the same group had significant interactions with time when measuring cognitive decline through MMSE scores, showing a consistent effect (β = − 3.07, p < 0.01).
ATN profile risk of cognitive decline
Table 2 shows the complete results from the Cox proportional hazard analysis. This analysis showed that compared to the normal group, subjects in the AD-PC and AD-P groups were at increased risk of cognitive decline with an incremental increase in hazard ration (HR) (HR 3.16 [1.17–8.52], and HR 6.15 [2.59–14.59], respectively). The SNAP group did not show a significant increase in risk of cognitive decline. Figure 3 shows the Kaplan-Meyer curves illustrating cognitive decline per ATN group.

Kaplan-Meyer curves illustrate cognitive decline defined as an MMSE loss of 1 point per year. Separate lines represent each of the ATN groups. Numbers at risk at every 2-month time point are depicted in the risk table below the graph
Discussion
The aim of this study was to assess the prognostic value of different ATN groups in cognitive decline in a memory clinic population. To this end, subjects with PET and MRI scans, and two independent cognitive assessments conducted on average 2 years apart were selected from a prospective cohort. While 33% of the included subjects presented a normal biomarker profile (A-T-N-), 22% of the subjects presented an A + T- profile and were classified as being within the AD-PC group, and 39% with an A + T + profile which were considered within the AD-P group. The use of ATN stratification has the potential to offer complementary knowledge to clinical and genetic information to predict cognitive decline. Different profiles are expected to progress differently over time because of additive effects of accumulated pathological proteins. Moreover, it is expected that the more pronounced the underlying pathological process is, the faster a patient will decline over time. This study showed that positivity for an AD biomarker in itself is sufficient to predict short-time cognitive decline. Independently of classification, all groups showed an average MMSE score that decreased at follow-up, except for the normal group. However, only A + individuals within the AD-PC and AD-P groups, had significantly different MMSE scores at follow-up when compared to baseline (Table 2).
Most of AD drug trials are focused on amyloid interventions, and include amyloid status information alone. Indeed, it was observed that subjects within the AD continuum presented significant short-time changes in MMSE scores, in comparison to baseline measures (Table 2), supporting the hypothesis that amyloid positivity is a risk factor for cognitive decline. However, there is a significant interplay between different biomarkers that affects disease progression. AD-P subjects had not only MMSE scores significantly lower than AD-PC at baseline and follow-up but also a larger number of subjects who progressed within a 2-year time frame, highlighting the added value of T in assessing cognitive impairment and cognitive decline. Although amyloid positivity is a risk factor for AD and is correlated to disease progression, there is a significant number of A + subjects that do not decline [30]. Furthermore, studies have shown that other factors might help in avoiding cognitive decline even in the presence of amyloid and tau pathologies, such as cognitive reserve [31], diet [32], education [33], lifestyle [31, 34], and others [35, 36]. The identification of patients’ ATN profiles could be used to assess possible therapeutic interventions as profiles decline cognitively at different rates. The urgency of treatment can also be assessed based on the profile, as patients positive for amyloid and tau deposition tend to progress at a faster rate than compared to negative profiles.
AD progression and AD patients’ cognitive decline has been shown to be largely influenced by positivity for the ε4 allele of the apolipoprotein E gene (APOE4) [37]. However, not all subjects of the cohort used in this study had information regarding the presence of the APOE4 allele. Nonetheless, previous studies on the decline of subjects stratified into ATN profiles have shown that short-time progression is independent of APOE4 presence [5, 10, 11]. Therefore, although the inclusion of APOE4 status might improve model prediction, it is unlikely that it would have significantly changed the results found in this study.
Differently from previous results, the only profiles able to predict global cognitive decline in this study were those of the AD-PC and AD-P groups. Previous studies have found that subjects without amyloidosis but with tau pathology significantly declined in a 2-year follow-up period [3]. This study could not confirm this finding in this cohort possibly due to the small sample of subjects in the SNAP group, but also related to the different approaches to assess cognitive decline. Few published works measured cognitive decline using MMSE scores, whereas different domains of cognition individually instead of global cognition were more frequently assessed. Furthermore, while 2 years is a long enough time to state short-time cognitive decline, most studies focused on longer follow-up periods. In addition, it is important to notice that the large variance in the estimation in the linear mixed models applied in this study could also be related to the smaller samples of subjects in some of the groups.
An interesting point to further explore regarding cognitive decline in ATN groups could be the changes of individual biomarker profiles over time. However, most of the subjects included in this study did not undergo follow-up PET and MRI scans. Further studies focusing on the temporal evolution of ATN positivity are still needed. Moreover, imaging techniques also offer the option of staging disease progression [38,39,40,41], which would provide different subclassifications within each profile using non-dichotomised A, T, and N biomarker values. Regional uptake in AD-specific regions could provide a better prediction of disease progression. For A, frontal, parietal and occipital regions have been found to be of relevance for identifying specific amyloid deposits [42]. However, their applicability to a standardised general measure as the centiloid scale and the comparability across different amyloid radiotracers should be specifically tested. For T, uptake in regions related to Braak stages [38], or larger and more easily atlas-defined regions such as the medial temporal lobe, lateral temporal lobe, superior temporal gyrus, and the primary visual cortex as defined by the STOC model [23] could be suggested. Finally, N can be expressed as a measure not only of relative hippocampal volume [27], but also hippocampal cortical thickness [43] or through some automated score measurement of AD pattern expression in FDG PET scans [44, 45].
Neurodegeneration is considered, in the ATN framework, as a non-specific marker for AD, and it can be defined not only through MRI, as in this study, but also through [18F]Fluorodeoxyglucose (FDG) [1]. Using a different approach to estimate N might lead to different profile classifications due to the fundamental differences between techniques [46]. In this study, the definition of the N threshold for positivity classification was done based on the ratio of the hippocampal volume relative to the total intracranial volume: a measure of hippocampal atrophy. In comparison, FDG PET assessment for N classification is based in a wider assessment of brain regions that present a hypometabolic pattern [47,48,49]. Although these measurements are correlated [50], they do not measure the same aspect of neurodegeneration and, therefore, different results of profile classifications and a higher sensitivity could be expected using FDG as a biomarker for N instead of MRI [51]. Finally, due to the non-specific nature of N as a biomarker in the ATN framework, the classification of profile subgroups was mostly based on A and T positivity, while N was mostly used to differentiate SNAP patients from normal subjects.
The use of an imaging approach to assess ATN groups, the approach that has been shown to be better correlated to disease progression [8], is the strongest point of this study. In addition to this, the use of a population recruited directly from a memory clinic results in findings that are implementable in clinical practice, in contrary to research populations. Although this study was not focused on subjects’ clinical status, it is interesting to notice that the SCD group was mostly characterised with a normal profile, which is in line with a previous publication that focused on SCD individuals alone [3]. Meanwhile, MCI patients were mostly classified as AD-PC or AD-P. The definition of MCI was solely based on cognitive complaints and did not consider the underlying cause for such complaints. However, as the ATN research framework was created with the purpose of classifying AD, and AD is the most prevalent form of dementia [52], it was expected for MCI population to be classified mostly as AD-PC or AD-P.
Neuroimaging modalities provide reliable biomarkers for the classification of ATN profiles and to assess longitudinal cognitive decline. However, imaging can be an expensive and burdensome technique for the patients. Cerebral spinal fluid is more commonly used in clinical practice, and also provides markers for ATN classification that are related to cognitive decline [7, 53, 54]. Furthermore, in the recent years, a great development of techniques for the assessment of plasma-based biomarkers has been achieved. These markers would be even less invasive for patients as they could easily be implemented in routine blood examens. Studies assessing ATN classification and the prognostic value of this technique to assess ATN biomarkers have shown promising results [10, 55]. Yet, the measurement of plasma-based biomarker levels is still under development and requires validation in different settings for it to be available in clinical routine [56]. Finally, it is important to point out that only neuroimaging techniques provide a visualisation of pathology deposition that allow for disease staging and regional uptake assessment that, as previously discussed, could provide a more precise evaluation of individual prognosis.
This study has a number of limitations worth discussing. Firstly, the population included in this study was a memory clinic population and although the majority of the patients that are referred to the clinic are within the AD spectrum, not all subjects included in this study were classified as dementia due to AD but included other diagnoses. However, given the lower prevalence of non-AD diagnoses, our study did not allow to stratify the prognostic analysis by diagnosis. Secondly, the classification of subjects in the different subcategories of the ATN framework led to small subgroups that were then pooled into larger categories (AD-PC, AD-P, and SNAP subjects), preventing firm conclusions on each specific combination. The ATN framework includes specific markers for AD pathology and a non-specific marker of neurodegeneration (N). Properly investigated, N markers (MRI and FDG PET when available) could also provide specific information on non-AD degenerative disturbances, such as investigating frontal abnormalities. However, given the lower prevalence of these conditions, this study did not use markers for N other than targeting AD and, thus, cannot conclude on their prognostic value in this cohort. Furthermore, a 2-year follow-up period was used, which is not a sensitive measure for slow and gradual decline, namely in healthy individuals, and only allows to identify subjects at a higher risk of declining. Finally, only a minority of the included subjects had a complete neuropsychological evaluation at both time points and for this reason mainly the MMSE as a global measure of cognition was used to measure decline. In particular, the small number of SNAP and AD-PC subjects might have limited the ability of this study to detect significant changes between these groups.
Conclusion
The stratification of subjects into ATN groups allows for an accurate estimate of the risk of cognitive decline. In our cohort, the AD-P group had the largest change in MMSE scores and at the highest risk of significant cognitive decline over a short follow-up period.
Data availability
Datasets generated and/or analyzed during the current study are available with the corresponding author upon reasonable request.
Code availability
Not applicable.
References
Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s & Dementia [Internet]. 2018;14:535–62. Available from: https://doi.org/10.1016/j.jalz.2018.02.018.
Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28.
Ebenau JL, Timmers T, Wesselman LMP, Verberk IMW, Verfaillie SCJ, Slot RER, et al. ATN classification and clinical progression in subjective cognitive decline. Neurology [Internet]. 2020;95:e46–58. Available from: https://doi.org/10.1212/WNL.0000000000009724.
Ebenau JL, Pelkmans W, Verberk IMW, Verfaillie S, van den Bosch KA, van Leeuwenstijn M, et al. Association of CSF, plasma, and imaging markers of neurodegeneration with clinical progression in people with subjective cognitive decline. Neurology [Internet]. 2022;0:https://doi.org/10.1212/WNL.0000000000200035. Available from: https://doi.org/10.1212/WNL.0000000000200035.
Jack CR, Wiste HJ, Therneau TM, Weigand SD, Knopman DS, Mielke MM, et al. Associations of amyloid, Tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA [Internet]. 2019;321:2316. Available from: https://doi.org/10.1001/jama.2019.7437.
Soldan A, Pettigrew C, Fagan AM, Schindler SE, Moghekar A, Fowler C, et al. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology [Internet]. 2019;92:e1567–79. Available from: https://doi.org/10.1212/WNL.0000000000007248.
Delmotte K, Schaeverbeke J, Poesen K, Vandenberghe R. Prognostic value of amyloid/tau/neurodegeneration (ATN) classification based on diagnostic cerebrospinal fluid samples for Alzheimer’s disease. Alzheimers Res Ther [Internet]. 2021;13:84. Available from: https://doi.org/10.1186/s13195-021-00817-4.
Bucci M, Chiotis K, Nordberg A. Alzheimer’s disease profiled by fluid and imaging markers: tau PET best predicts cognitive decline. Mol Psychiatry [Internet]. 2021;26:5888–98. Available from: https://doi.org/10.1038/s41380-021-01263-2.
van de Beek M, Ooms FAH, Ebenau JL, Barkhof F, Scheltens P, Teunissen CE, et al. Association of the ATN research framework with clinical profile, cognitive decline, and mortality in patients with dementia with Lewy bodies. Neurology [Internet]. 2022;98:e1262–72. Available from: https://doi.org/10.1212/WNL.0000000000200048.
Cullen NC, Leuzy A, Janelidze S, Palmqvist S, Svenningsson AL, Stomrud E, et al. Plasma biomarkers of Alzheimer’s disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat Commun [Internet]. 2021;12:3555. Available from: https://doi.org/10.1038/s41467-021-23746-0.
Ebenau JL, Visser D, Kroeze LA, van Leeuwenstijn MSSA, van Harten AC, Windhorst AD, et al. Longitudinal change in ATN biomarkers in cognitively normal individuals. Alzheimers Res Ther [Internet]. 2022;14:124. Available from: https://doi.org/10.1186/s13195-022-01069-6..
Young PNE, Estarellas M, Coomans E, Srikrishna M, Beaumont H, Maass A, et al. Imaging biomarkers in neurodegeneration: current and future practices. Alzheimers Res Ther. 2020;12:49.
Ribaldi F, Chicherio C, Altomare D, Martins M, Tomczyk S, Jelescu I, et al. Brain connectivity and metacognition in persons with subjective cognitive decline (COSCODE): rationale and study design. Alzheimers Res Ther. 2021;13:105.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Focus (Madison) [Internet]. 2013;11:96–106. Available from: https://doi.org/10.1176/appi.focus.11.1.96.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia. 2011;7:263–9.
Pangman VC, Sloan J, Guse L. An examination of psychometric properties of the Mini-Mental State Examination and the Standardized Mini-Mental State Examination: implications for clinical practice. Applied Nursing Research [Internet]. 2000;13:209–13. Available from: https://doi.org/10.1053/apnr.2000.9231.
Schneider LS, Kennedy RE, Wang G, Cutter GR. Differences in Alzheimer disease clinical trial outcomes based on age of the participants. Neurology. 2015;84:1121–7.
Jovicich J, Marizzoni M, Sala-Llonch R, Bosch B, Bartrés-Faz D, Arnold J, et al. Brain morphometry reproducibility in multi-center 3T MRI studies: a comparison of cross-sectional and longitudinal segmentations. Neuroimage [Internet]. 2013;83:472–84. Available from: https://doi.org/10.1016/j.neuroimage.2013.05.007.
Ashburner J, Friston KJ. Unified segmentation. Neuroimage [Internet]. 2005;26:839–51. Available from: https://doi.org/10.1016/j.neuroimage.2005.02.018.
Fischl B. FreeSurfer Neuroimage. 2012;62:774–81.
Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD, Jagust WJ, et al. The centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s & Dementia [Internet]. 2015;11:1. https://doi.org/10.1016/j.jalz.2014.07.003.
Joachim CL, Morris JH, Selkoe DJ. Diffuse senile plaques occur commonly in the cerebellum in Alzheimer’s disease. Am J Pathol [Internet]. 1989;135:309–19. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2675616.
Schwarz AJ, Shcherbinin S, Slieker LJ, Risacher SL, Charil A, Irizarry MC, et al. Topographic staging of tau positron emission tomography images. DADM. 2018;10:221–31.
Rolls ET, Huang C-C, Lin C-P, Feng J, Joliot M. Automated anatomical labelling atlas 3. Neuroimage. 2020;206: 116189.
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage [Internet]. 2002;15:273–89. Available from: https://doi.org/10.1006/nimg.2001.0978.
Mishra S, Gordon BA, Su Y, Christensen J, Friedrichsen K, Jackson K, et al. AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. Neuroimage. 2017;161:171–8.
Mattsson-Carlgren N, Leuzy A, Janelidze S, Palmqvist S, Stomrud E, Strandberg O, et al. The implications of different approaches to define AT(N) in Alzheimer disease. Neurology. 2020;94:e2233–44.
Jack CR, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimer’s Dementia. 2017;13:205–16.
Salvadó G, Molinuevo JL, Brugulat-Serrat A, Falcon C, Grau-Rivera O, Suárez-Calvet M, et al. Centiloid cut-off values for optimal agreement between PET and CSF core AD biomarkers. Alzheimers Res Ther. 2019;11:1–12.
Crary J. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromed. 2016;1:53–7.
Scarmeas N, Stern Y. Cognitive reserve and lifestyle. J Clin Exp Neuropsychol. 2003;25:625–33.
Luchsinger JA, Mayeux R. Dietary factors and Alzheimer’s disease. Lancet Neurol. 2004;3:579–87.
Wilson RS, Li Y, Aggarwal NT, Barnes LL, McCann JJ, Gilley DW, et al. Education and the course of cognitive decline in Alzheimer disease. Neurology. 2004;63:1198–202.
Pope SK, Shue VM, Beck C. Will a healthy lifestyle help prevent Alzheimer’s disease? Annu Rev Public Health. 2003;24:111–32.
Crous-Bou M, Minguillón C, Gramunt N, Molinuevo JL. Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimers Res Ther. 2017;9:71.
de Godoy LL, Alves CAPF, Saavedra JSM, Studart-Neto A, Nitrini R, da Costa LC, et al. Understanding brain resilience in superagers: a systematic review. Neuroradiology. 2021;63:663–83.
Frisoni GB, Altomare D, Thal DR, Ribaldi F, van der Kant R, Ossenkoppele R, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci. 2022;23:53–66.
Therriault J, Pascoal TA, Lussier FZ, Tissot C, Chamoun M, Bezgin G, et al. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat Aging [Internet]. 2022; Available from: https://doi.org/10.1038/s43587-022-00204-0.
Grothe MJ, Barthel H, Sepulcre J, Dyrba M, Sabri O, Teipel SJ. In vivo staging of regional amyloid deposition. Neurology [Internet]. 2017;89:2031–8. Available from: https://doi.org/10.1212/WNL.0000000000004643.
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404.
Biel D, Brendel M, Rubinski A, Buerger K, Janowitz D, Dichgans M, et al. Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers Res Ther [Internet]. 2021;13:137. Available from: https://doi.org/10.1186/s13195-021-00880-x.
Collij LE, Salvadó G, Wottschel V, Mastenbroek SE, Schoenmakers P, Heeman F, et al. Spatial-temporal patterns of amyloid-β accumulation: a subtype and stage inference model analysis. Neurology. Apr2022;98(17):e1692–703. https://doi.org/10.1212/WNL.0000000000200148.
Jack CR, Wiste HJ, Weigand SD, Therneau TM, Knopman DS, Lowe V, et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. Lancet Neurol [Internet]. 2017;16:435–44. Available from: https://doi.org/10.1016/S1474-4422(17)30077-7.
Peretti DE, Vállez García D, Renken RJ, Reesink FE, Doorduin J, de Jong BM, et al. Alzheimer’s disease pattern derived from relative cerebral flow as an alternative for the metabolic pattern using SSM/PCA. EJNMMI Res [Internet]. 2022;12:37. Available from: https://doi.org/10.1186/s13550-022-00909-8.
Herholz K, Salmon E, Perani D, Baron JC, Holthoff V, Frölich L, et al. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage. 2002;17:302–16.
Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003;2:605–13.
Meles SK, Pagani M, Arnaldi D, De Carli F, Dessi B, Morbelli S, et al. The Alzheimer’s disease metabolic brain pattern in mild cognitive impairment. J Cereb Blood Flow Metab. 2017;37:3643–8.
Pagani M, Nobili F, Morbelli S, Arnaldi D, Giuliani A, Öberg J, et al. Early identification of MCI converting to AD: a FDG PET study. Eur J Nucl Med Mol Imaging. 2017;44:2042–52.
Morbelli S, Bauckneht M, Arnaldi D, Picco A, Pardini M, Brugnolo A, et al. 18F–FDG PET diagnostic and prognostic patterns do not overlap in Alzheimer’s disease (AD) patients at the mild cognitive impairment (MCI) stage. Eur J Nucl Med Mol Imaging [Internet]. 2017;44:2073–83. Available from: https://doi.org/10.1007/s00259-017-3790-5.
Albrecht F, Ballarini T, Neumann J, Schroeter ML. FDG-PET hypometabolism is more sensitive than MRI atrophy in Parkinson’s disease: a whole-brain multimodal imaging meta-analysis. Neuroimage Clin. 2019;21: 101594.
Boccalini C, Ribaldi F, Hristovska I, Arnone A, Peretti D, Mu L, et al. The impact of tau deposition and hypometabolism on cognitive impairment and longitudinal cognitive decline. Accepted for publication at Alzheimer’s & Dementia. 2023.
Gustavsson A, Norton N, Fast T, Frölich L, Georges J, Holzapfel D, et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimer’s Dement. 2023;19:658–70.
Bartels C, Kögel A, Schweda M, Wiltfang J, Pentzek M, Schicktanz S, et al. Use of cerebrospinal fluid biomarkers of Alzheimer’s disease risk in mild cognitive impairment and subjective cognitive decline in routine clinical care in Germany. Journal of Alzheimer’s Disease [Internet]. 2020;78:1137–48. Available from: https://doi.org/10.3233/JAD-200794.
Ebenau JL, Pelkmans W, Verberk IMW, Verfaillie SCJ, van den Bosch KA, van Leeuwenstijn M, et al. Association of CSF, plasma, and imaging markers of neurodegeneration with clinical progression in people with subjective cognitive decline. Neurology [Internet]. 2022;98:e1315–26. Available from: https://doi.org/10.1212/WNL.0000000000200035.
Kivisäkk P, Magdamo C, Trombetta BA, Noori A, Kuo Y kai E, Chibnik LB, et al. Plasma biomarkers for prognosis of cognitive decline in patients with mild cognitive impairment. Brain Communications, 2022;4(4). https://doi.org/10.1093/braincomms/fcac155.
Altomare D, Stampacchia S, Ribaldi F, Tomczyk S, Chevalier C, Poulain G, et al. Plasma biomarkers for Alzheimer’s disease: a field-test in a memory clinic. J Neurol Neurosurg Psychiatry. 2023.
Acknowledgements
The Clinical Research Centre, at Geneva University Hospital and Faculty of Medicine provides valuable support for regulatory submissions and data management, and the Biobank at Geneva University Hospital for biofluid processing and storage. We thank the Centre for Radiopharmaceutical Sciences of ETH and USZ for providing the PET tracer for tau imaging. We acknowledge Avid radiopharmaceuticals for providing the precursor for the tau radiotracer and emphasise that Avid was not involved in the analysis or interpretation.
Funding
Open access funding provided by University of Geneva The Centre de la mémoire is funded by the following private donors under the supervision of the Private Foundation of Geneva University Hospitals: A.P.R.A.—Association Suisse pour la Recherche sur la Maladie d’Alzheimer, Genève; Fondation Segré, Genève; Race Against Dementia Foundation, London, UK; Fondation Child Care, Genève; Fondation Edmond J. Safra, Genève; Fondation Minkoff, Genève; Fondazione Agusta, Lugano; McCall Macbain Foundation, Canada; Nicole et René Keller, Genève; Fondation AETAS, Genève.
Competitive research projects have been funded by: H2020 (projects n. 667375), Innovative Medicines Initiative (IMI contract n. 115736 and 115952), IMI2, Swiss National Science Foundation (projects n.320030_182772, n.320030_185028, and n. 320030_169876), VELUX Foundation, Schmidheiny foundation, Aetas foundation.
Author information
Authors and Affiliations
Contributions
DEP contributed to the study design, image processing, data analysis, and writing and revision of the manuscript. FR contributed to the study design, image processing, and revision of the manuscript. MS and CC contributed to subject inclusion, data acquisition, and revision of the manuscript. GBF and VG contributed to the study design, coordination of the study, patient inclusion, data acquisition, and revision of the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The local review board (Cantonal Research Ethics Commission, Geneva, Switzerland) approved the study, which has been conducted in concordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice. All subjects provided written informed consent to have collected data be used in research.
Consent for publication
All patients provided signed informed consent for participation in the study and for publication.
Competing interests
VG received financial support for research and/or speaker fees through her institution from Siemens Healthineers, GE Healthcare, and Novo Nordisk. Other authors declare that they have no competing interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peretti, D.E., Ribaldi, F., Scheffler, M. et al. Prognostic value of imaging-based ATN profiles in a memory clinic cohort. Eur J Nucl Med Mol Imaging 50, 3313–3323 (2023). https://doi.org/10.1007/s00259-023-06311-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-023-06311-3