
Abstract
The clinical response to immune checkpoint inhibitors may vary by tumor type and many tumors present with either primary or acquired resistance to immunotherapy. Improved understanding of the molecular and immunologic mechanisms underlying immunotherapy resistance is essential for developing biomarkers and for guiding the optimum approach to selecting treatment regimens and sequencing. This is increasingly important for tumors with primary resistance as effective biomarkers in this setting can guide clinicians about appropriate treatment regimen selection in the first-line setting. Multiple potential biological mechanisms of primary resistance have been proposed but most are yet to be validated in prospective clinical cohorts. Individual biomarkers have poor specificity and sensitivity, and the development of validated and integrated predictive models may guide which patient will benefit from monotherapy versus combination therapy. In this review, we discuss the emerging data identifying the molecular mechanisms of primary resistance to immunotherapy and explore potential therapeutic strategies to target these.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunotherapy with immune checkpoint inhibitors (ICIs) targeting the PD-1/PD-L1 and CTLA4 axis has transformed clinical outcomes for patients across several cancer histologies and has become an integral part of standard treatment regimens. Depending on the tumor type, the response to ICIs varies, and many tumors present with either primary or acquired resistance to immunotherapy. There are emerging data on potential mechanisms underlying primary and/or acquired resistance to immunotherapy which is derived mainly from either preclinical studies or secondary correlative analyses from clinical trials. Improved understanding of the molecular and immunologic mechanisms underlying immunotherapy resistance is essential for developing biomarkers and for guiding the optimum approach to selecting treatment regimens and sequencing. This is increasingly important for tumors with primary resistance as effective biomarkers in this setting can guide clinicians about appropriate treatment regimen selection in the first-line setting. Multiple potential biological mechanisms of primary resistance have been proposed: ineffective priming of a T-cell response, lack of tumor recognition due to defective antigen presentation, inability of T cells to penetrate effectively, and the inability of T cells to eliminate tumor cells due to suppression via other checkpoints such as lymphocyte-activation gene 3 (LAG-3) [1]. Tumor-immune evasion and resistance to PD-(L)1 inhibitors can also be mediated by canonical cancer signaling pathways such as Wnt–β-catenin signaling, cell cycle regulatory signaling, and mitogen-activated protein kinase signaling. In this review, we discuss the emerging data identifying the molecular mechanisms of primary resistance to immunotherapy and explore potential therapeutic strategies to target these.
Defining primary resistance to immunotherapy
The immune evasive measures used by cancer cells are broadly separated into two categories based on the tumor microenvironment: an inflamed T-cell phenotype that suppresses immune activation and a non-inflamed phenotype that passively escapes immune detection [2, 3]. The inflamed phenotype is characterized by tumor infiltration by CD8 + T cells leading to tumor cell cytotoxicity [4]. T-cell activation is a well-regulated process that involves a balance between co-stimulatory and co-inhibitory signals exchanged in the binding of the T-cell receptor (TCR) to the major histocompatibility complex (MHC) peptide complex or the antigen presenting cells (APCs). These antigens, which include tumor-derived neoantigens, are phagocytosed, processed, and presented in the MHC molecules on the cell surface of APCs in recognizable form to train immune cells such as effector T cells, leading to their activation [5]. Two main signals are required for T-cell activation: (I) engagement of MHC-bound antigen on APCs by the TCR, and (II) co-stimulation via CD80/CD86 and CD28 interactions between APCs and T cells. PD-L1/PD-1 interactions counteract this co-stimulation [6, 7]. The PD-1/PD-L1 pathway is a crucial self-tolerance pathway that tumor cells hijack to escape immune elimination and ICIs such as PD-(L)1 inhibitors target this pathway to enable immune mechanisms to target tumor cells [2].
Resistance to ICIs is complex and can present immediately after treatment initiation (primary resistance) or after initial clinical benefit (secondary or acquired resistance) [8]. As per recommendations from the first meeting of the SITC immunotherapy resistance taskforce, a patient who has disease progression after receiving at least 6 weeks (~ two complete cycles of therapy), but no more than 6 months of ICI therapy is considered to have primary resistant disease [8]. Conversely, any patient who experiences progression after demonstrating initial clinical benefit with PD-(L)1 inhibitor is defined as having secondary or acquired resistance. The biological definition of primary resistance to PD-(L)1 inhibitors is defined as the inability of immune cells to mount an antitumor response on initial drug exposure [9]. The rates of primary resistance to immunotherapy vary and range from 35 to 40% for metastatic melanoma [10, 11] and 39% for advanced NSCLC [12]. To date, the clinical factors that have been associated with primary resistance are elevated levels of serum LDH [13], increased tumor burden [14], and lack of PD-L1 expression [15] but it is unclear whether these measures are surrogates for resistance or have a direct mechanistic role [16]. The tumor microenvironment, particularly the presence and activity of T cells, has also been shown to influence resistance [17] with both tumor-extrinsic as well as tumor-intrinsic molecular mechanisms such as lack of T-cell infiltration [18], insufficient neoantigens [19], or absence of an interferon signature [20] identified as being associated with ICI resistance.
Genomic and molecular mechanisms of primary resistance
Multiple tumor-intrinsic molecular mechanisms have been identified for primary resistance to immunotherapy: lack of T-cell response due to loss of antigen presentation (deletion in beta-2-microglobulin (β2M)), genetic T-cell exclusion (MAPK or PI3K oncogenic signaling, stabilized b-catenin, oncogenic PD-L1 expression), or insensitivity to T cells (mutations in interferon-gamma pathway signaling) [21].
Deletion, mutation, or loss of heterozygosity in β2M
β2M is associated with the heavy chain of MHC I, and thus, β2M mutations impact MHC I antigen presentation. Without β2M on the tumor cell surface, HLA class I molecules are unstable and unable to present antigen to CD8 + T cells. β2M point mutations, deletions, or loss of heterozygosity (LOH) were detected in 29% of patients (n = 17) with metastatic melanoma progressing on ICI treatment [22]. In two independent cohorts of melanoma patients treated with anti-CTLA4 and anti-PD-1, β2M LOH was enriched threefold in non-responders (~ 30%) compared to responders (~ 10%), and loss of both copies of β2M was found only in non-responders [23]. In β2M gene knockout models in human and murine cell lines, lack of antigen presentation, cancer cell recognition, and cytotoxicity by T cells was observed [23]. Notably, β2M loss does not affect response to ICIs in mismatch repair–deficient colorectal cancer and these tumors have been observed to have increased intratumoral infiltration of CD4 + T cells [24]. This indicates that CD4 + T cells may play a role in ICI resistance associated with β2M loss and CD4 + T-cell-based adoptive therapy approach may be evaluated further in this setting [25].
Several approaches are being explored to target tumors without intact β2M. Bempegaldesleukin (NKTR-214), a prodrug of conjugated IL-2 leads to sustained activation of the IL-2 pathway and leads to a systemic expansion of both CD4 + , CD8 + T, and NK cells [26]. Combination therapy with NKTR-214 and ICI may be synergistic, and the administration of NKTR-214 attenuated anti-PD-1 resistance in β2M knockout tumors and prolonged survival in β2M knockout melanoma mice [27]. However, the clinical program with NKTR-214 was discontinued when the primary endpoint was not reached in the phase III PIVOT-09 and PIVOT-10 trials in RCC and urothelial cancers (NCT03729245, NCT03785925). NK cell-based therapy may also be a promising immunotherapy for ICI-resistant melanoma caused by β2M deficiency [28] as NK cells may be activated to recognize “missing self” [29]. Another potential strategy is to use plasmids or adenoviruses to deliver wild-type human β2M gene into tumor cells to restore tumor cell HLA class I antigen expression [28]. An in vivo study using the Ma-Mel-86b tumor xenograft model in nude mice showed that the intratumoral injection of β2M-carrying adenoviral vectors restored regular HLA class I expression [30]. Based on this design, Allovectin-7 (velimogene aliplasmid) is a bicistronic plasmid DNA encoding two transgene proteins, HLA-B7 and β2M [31]. In a phase I trial, Allovectin-7 enabled the synthesis and expression of intact MHC class I complexes on the tumor cell surface and stimulated T-cell-based immune responses to transfected cells and foreign antigens [32, 33]. In a phase II trial of Allovectin-7 among 133 patients with advanced melanoma [34], the overall response rate (ORR) was 11.8% and the safety profile was acceptable. Subsequently, the phase III Allovectin immunotherapy for metastatic melanoma (AIMM) trial was conducted but reported that responders to Allovectin-7 had significantly shorter overall survival (OS; 18.8 months versus 24.1 months, P = 0.491) [35]. This phase III trial included all patients with melanoma and was not limited to tumors with B2M loss or other biomarkers of MHC1 function. The further development of this agent was discontinued. Another approach to target ICI resistance is using radiation therapy (RT). In lung cancer in vivo studies, Wang et al. have demonstrated that localized RT can induce IFN-β production which in turn increases MHC I expression in PD-1 resistant tumor cells and may target PD-1 resistance [36]. Conversely, this radio-sensitization to ICIs is observed only when type I IFN signaling is intact [36].
Mitogen-activated protein kinase (MAPK) signaling pathway
MAPK directs interactions between tumor cells and the surrounding T-cell infiltrate, downregulates T-cell co-stimulatory molecules, and suppresses the expression of negative immune checkpoints such as PD-L1 and CTLA4 in several cancers [37]. In breast cancer, dysregulation of MAPK pathway has been linked to an immune-silent phenotype associated with poor outcome and treatment resistance [38]. These aberrations include mutations of MAP3K1 and MAP2K4, amplification of KRAS, BRAF, and RAF1, and truncations of NF1 [38]. Activation of the Ras-MAPK pathway correlates with reduced tumor-infiltrating leukocytes (TILs) in a subset of triple negative breast cancer patients who failed to achieve pathologic complete response (CR) after neoadjuvant therapy [39]. The combination of MEK inhibitors and PD-(L)1 inhibitors has been investigated to overcome this resistance. A phase 1 trial (NCT02027961) investigated the combination of durvalumab (anti-PD-L1) with dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) in patients with BRAF-mutated melanoma as well as durvalumab and trametinib given concurrently or sequentially in patients with BRAF wild-type melanoma. ORR was 69.2%, 20.0%, and 31.8%, respectively, with improved tumor-immune infiltration in available biopsy samples [40]. Additional trials are currently ongoing. The sequence in which immunotherapy is used with MEK inhibitor may be significant as well. In a phase III trial in metastatic melanoma, immunotherapy with nivolumab and ipilimumab was associated with an ORR of 46%. Conversely, when nivolumab and ipilimumab combination therapy was administered in second line setting in patients who had already received prior therapy with dabrafenib/trametinib, the ORR was lower at 29.6% [41].
PI3K signaling or loss of PTEN
Signaling through the PI3K/AKT/mTOR pathway contributes to tumorigenesis through several processes such as apoptosis, proliferation, motility, and metabolism. A common way to activate this pathway is through loss of expression of the tumor suppressor PTEN, a lipid phosphatase suppressing the activity of PI3K signaling. Additionally, PTEN represses the expression of immunosuppressive cytokines IL-10, IL-16, and VEGF, by blocking the PI3K pathway [42]. PTEN deletion promotes AKT phosphorylation, thereby promoting PI3K/AKT pathway activation, and ultimately inactivation of T cells.
Peng et al. [43] reported for the first time that loss of PTEN may lead to primary resistance to anti-PD-(L)1 therapy. In preclinical melanoma models, loss of PTEN reduced CD8 + T cells tumor infiltration and impeded T-cell-mediated tumor killing. In an analysis of 135 resected advanced melanoma with regional metastases, tumors with loss of PTEN had lower CD8 + T-cell tumor infiltration as compared to PTEN intact tumors. The loss of PTEN significantly up-regulated the expression of immunosuppressive cytokines VEGF and CCL2, leading to reduced T-cell infiltration in tumors, and suppressed autophagy, thus reducing T-cell-mediated cell death. Loss of PTEN has also been reported to be associated with ICI resistance in uterine leiomyosarcoma [44], as well as in glioblastoma [45].
To target the PI3K-AKT pathway, one study combined PD-1 and PI3K inhibitors in a mouse model of head and neck squamous cell carcinoma [46]. It observed increased survival due to an activated immunostimulatory transcriptional program, enhanced expression of proinflammatory cytokines, and enhanced T-cell cytotoxicity [46]. Another preclinical study with triple CTLA4, PD-1, and PI3K blockade reshaped the tumor microenvironment, enhancing T-cell–mediated tumor regression [47]. Several trials are evaluating the combination of PD-(L)1 inhibitors with PI3K inhibitors (NCT02646748).
Wnt/β-catenin signaling pathway
Wnt/β-catenin signaling is involved in several cell processes. In melanoma cells in vivo, Wnt/β-catenin signaling prevents the priming of antitumor responses by disrupting the recruitment of dendritic cells expressing basic leucine zipper transcriptional factor ATF-like 3 (BATF3) [48]. In several cancers such as colon cancer, mutations in the destruction complex components (APC, AXIN2, and FAM123B/WTX) or regulators of the receptors/ligand (RNF43/ZNRF3, RSPO2, or RSPO3) components can lead to unchecked Wnt signaling [49]. Furthermore, the conversion of tryptophan into kynurenine is catalyzed by IDO1, which is a transcriptional target downstream of Wnt5A-induced signaling and this metabolic shift promotes the development of regulatory T cells while suppressing effector T-cell activity [50]. Inhibition of this metabolic shift augments the efficacy of anti-PD-1 immunotherapy in a model of BRAF V600E/PTEN − / − mouse melanoma [51].
Spranger et al. [52] used mouse melanoma models to describe how tumor-intrinsic active β-catenin signaling results in T-cell exclusion and ICI resistance. There was also a correlation between activation of the Wnt/β-catenin signaling pathway and absence of a T-cell gene expression signature in tumor samples from patients with metastatic melanoma. Another study analyzing samples from gastric cancer reported that a high β-catenin expression was associated with an absence of CD8 + T-cell infiltration [53]. A large analysis The Cancer Genome Atlas (TCGA) reported Wnt/β-catenin signaling genes were significantly mutated in all colorectal cancer subtypes, and activated Wnt/β-catenin was correlated with the absence of T-cell infiltration [54].
Several drugs inhibiting constitutive Wnt/β-catenin pathway signaling are in clinical development and can be combined with anti–PD-(L)1 therapy to overcome this mode of primary resistance. One study identified serine/threonine-protein kinase PAK4, a Wnt signaling mediator, to be enriched in immunologically cold tumors from patients with melanoma not responsive to anti-PD-1 immune checkpoint blockade [55]. In mouse models, deletion or pharmacological inhibition of PAK4 resulted in reversal of resistance to anti-PD-1 therapy. DKN-01 is a monoclonal antibody neutralizing DKK1, an immune-suppressive protein produced by tumors with Wnt/β catenin activation. A recent phase IIa trial exploring the effect of DKN-01 in combination with chemoimmunotherapy showed encouraging results in patients with advanced gastroesophageal adenocarcinoma [56]. DKB-01 is also under investigation in a phase I/II trial (NCT03645980) as a potential treatment of hepatocellular carcinoma with Wnt/β catenin activation.
Oncogenic PD-L1 expression
Intrinsic or constitutive PD-L1 overexpression is associated with the presence of multiple molecular mechanisms such as genetic amplification of chromosome 9, which contains the locus of PD-L1 and PD-L2, MYC overexpression, and EGFR or ALK alterations observed in NSCLC [57]. While constitutive PD-L1 signaling exerts a tumor-promoting function through the activation of oncogenic pathways, these tumors are associated with reduced TILs thereby making them “immune-cold” and unlikely to respond to ICIs [57, 58]. In NSCLC, patients with EGFR mutations or ALK rearrangements have very poor response to PD-(L)1 inhibitors. In a retrospective analysis of 58 NSCLC patients treated with PD-(L)1 inhibitors, responses were observed in only 4% of ALK-positive or EGFR-mutant tumors versus 23% of ALK-negative/unknown and EGFR wild-type tumors [59]. PD-L1 expression was observed in 47% and 16% of tumors, respectively, and the PD-L1 expression (≥ 5%) was not concurrently associated with high levels of CD8 + TILs (observed in only 1 pretreatment and 5 resistant EGFR-mutant tumor samples but not observed in any ALK-positive, pre- or post-TKI tissues). This lack of CD8 + TILs and concurrent PD-L1 expression indicates innate PD-L1 expression and underlies the limited effectiveness of PD-(L)1/PD-L1 inhibitors in a majority of EGFR-mutant and ALK-positive NSCLCs [59]. In a study of 336 treatment-naïve EGFR-mutated NSCLC cases, Liu et al. [60] reported low immunogenicity of EGFR-mutated NSCLC by analyzing the TCGA data and an independent validation cohort of patients. In mouse models of EGFRvIII-mutant GBM, CXCL2, and CXCL3 induced local and systemic recruitment of PMN-MDSCs, which correlated with resistance to PD-1 and CTLA4 checkpoint inhibition, CXCR2 antagonism resulted in systemic decrease of PMN-MDSC and enhanced the efficacy of combination immune checkpoint blockade and prolongation of survival [61]. The combination of anti-PD-L1 and EGFR-TKI has been used as a strategy to overcome immunotherapy resistance and yielded a response rate of 43%; however, further development has been halted due to an increased incidence of interstitial lung disease [62]. Amivantamab, an EGFR-MET targeting bispecific in being investigated in combination with anti-PD-1 antibody, cetrelimab in the PolyDamas trial (NCT05908734).
Interferon-gamma (IFN-γ) signaling pathway
IFN-γ plays an important role in innate and adaptive immunity and demonstrates antiviral, immune-regulatory, and antitumor activity [63, 64]. Upon tumor antigen recognition, T cells produce IFN-γ, which through the IFN-γ receptors (IFNGR1, IFNGR2), the Janus kinases (JAK1 and JAK2) and the signal transducers and activators of transcription (STATs) lead to antitumor effects, such as increased antigen presentation, transporters associated with antigen processing (TAP) and MHC, as well as increased production of chemokines [65]. IFN-γ signaling induces or enhances MHC class I antigen presentation, a process that requires coordinated expression of several genes, including TAP1, TAP2, β2M, and the immunoproteasome genes PSMB8, PSMB9, and PSMB10 [66]. Copy number alterations of the IFN-γ signaling pathway are associated with primary resistance to ICIs with the most coming being genomic loss of key IFN-γ pathway genes such as IFNGR1, IRF-1, JAK2, and IFNGR2, as well as amplification of important IFN-γ pathway inhibitors including SOCS1 and PIAS4 [67]. JAK1/2 loss-of-function mutations are a genetic mechanism of lack of reactive PD-L1 expression and response to IFN-γ, leading to primary resistance to PD-1 blockade therapy. A large analysis using TCGA melanoma dataset reported that 6% and 11% of tumors harbored alterations in JAK1 and JAK2, respectively, and were associated with worse outcomes [68]. The oncogenic activation of MYC in SCLC cells via MYC amplification downregulates JAK2 and impairs IγSGs stimulation by IFNγ [69].
One approach to overcome immunotherapy resistance is to induce a strong IFN response by triggering pattern recognition receptors. Synthetic CpG oligodeoxynucleotide (ODN) agonists of TLR9 are being tested in the clinic in combination with the PD-(L)1 inhibitors. The combination of intratumoral SD101 (a CpG-ODN) with pembrolizumab resulted in antitumor responses in patients with advanced melanoma who were refractory or resistant to prior anti-PD-1 therapy [70]. RNA profiling of tumor biopsies demonstrated increased CD8 + T cells, natural killer cells, cytotoxic cells, dendritic cells, and B cells. Vidutolimod, a TLR9 agonist, was investigated in a phase 1b study (CMP-001-003; NCT03438318) in combination with atezolizumab with and without radiation therapy in patients with advanced NSCLC [71]. The combination was well-tolerated and 15% to 25% of patients had stable disease as the best response. Talimogene laherparepvec (T-VEC) is an oncolytic virus that is FDA approved for the treatment of metastatic melanoma. It preferentially replicates within tumor cells and expresses the cytokine GM-CSF to promote the maturation and activation of APCs in the vicinity and therefore, does not interfere with antigen presentation in infected cells [72]. In combination with anti-PD-1 therapy, T-VEC resulted in ORR of 62% in a phase Ib study [72] in patients with metastatic melanoma. Moreover, 3 of 5 patients with low baseline IFN-γ production had CR, supporting a role for T-VEC in patients without preexisting antitumor-immune responses. SC-43, a SHP-1 agonist that inhibits STAT3, is being investigated in combination with cisplatin in a phase I/II trial for NSCLC (NCT04733521). TTI-101 is a first-in-class, orally bioavailable, selective small molecule that prevents STAT3-mediated transcriptional activity [73] and, a phase I/II study of TTI-101 in monotherapy or in combination with pembrolizumab or atezolizumab–bevacizumab is currently ongoing (NCT05440708).
Other
In an immunogenomic analysis of TCGA data and clinical trials of anti-PD-(L)1 therapy, 9p21 loss confers “cold” tumor-immune phenotypes [74], characterized by reduced abundance of TILs, particularly, T/B/NK cells, altered spatial TILs patterns, diminished immune cell trafficking/activation, decreased rate of PD-L1 positivity, along with activation of immunosuppressive signaling. Tumors with 9p21 loss exhibited significantly lower response rates to ICIs across eight trials of > 1,000 patients. Among genes mapping to the chromosomal region 9p21.3, CDKN2A was most frequently deleted (13.5%), followed by MTAP (9.3%). In addition to homozygous deletion (HD), 9p21 LOH due to hemizygous deletion of CDKN2A and MTAP was observed in 24.6% and 27.8% of cancers, respectively. In addition, although LOH of 9p21 did not lead to massive changes in CDKN2A/MTAP expression, it conferred significantly shorter OS in comparison with tumors with diploid/wild-type 9p21. The association between CDK4 gain (on 12q14.1 loci) and primary resistance to anti-PD-(L)1 therapy was validated in 85 patients with advanced melanoma (P < 0.05) [75]. RNA-Seq analysis of CDK4-normal cell lines and CDK4-normal tumors showed altered transcriptional output in TNFα signaling via NF-κB, inflammatory response, and IFNγ response gene set. In addition, CDK4/6 inhibitor (palbociclib) treatment increased PD-L1 protein levels and enhanced efficacy (P < 0.05). The phase II NEWFLAME trial investigated the combination of nivolumab, abemaciclib, and endocrine therapy (fulvestrant or letrozole) in patients with hormone receptor positive metastatic breast cancer [76]. While the ORR was 4% to 54%, there was a high incidence of severe immune-related adverse events, such as hepatotoxicity thus limiting further investigations.
The mechanism of resistance for several genomic predictors of ICI resistance remains unclear. STK11/LKB1 and KRAS co-mutated NSCLC is a distinct subgroup with primary resistance to ICIs. In this cohort of patients, the response rates to PD-(L)1 inhibitors were only 7.4% in the Stand Up To Cancer (SU2C) cohort (174 patients) with KRAS-mutant NSCLC and 0% in patients treated with nivolumab in the CheckMate-057 phase III trial [77]. In KRAS-mutant murine LUAC models, STK11/LKB1 loss promoted PD-(L)1/inhibitor resistance [77], possibly due to altered cytokine/chemokine milieu, metabolic restriction of effector T cells, or impaired antigenicity. KEAP1 loss has been identified to lead to innate ICI resistance in NSCLC [78] likely by suppressing CD103 DC-mediated CD8 T-cell immunity [78]. CB839, a glutaminase inhibitor, may be a strategy [79]. In an analysis of 155 patients with stage IV solid tumors, all six tumors with MDM2/MDM4 amplification were noted to have primary resistance as well as hyper-progression on ICIs. Two of 10 patients with EGFR alterations were also hyper-progressors [80].
One genomic biomarker predictive of response to immunotherapy is tumor mutational burden (TMB), which is a measure of the number of mutations in a cancer [81]. Several studies have reported a relationship between TMB and immunotherapy efficacy with a higher TMB being associated with increased responsiveness to ICIs [82,83,84]. Since neoantigens are related to the number of mutations, the higher the TMB, greater is the chance that some of the neoantigens may be immunogenic [83, 85]. However, there is variability in how well TMB can predict response to ICIs for a patient due to other factors that may influence presence or absence of immunogenic neoantigens as well as the impact of histology on TMB [83, 85, 86]. There is also currently no consensus in the TMB cutoff to be used for patient stratification [81]. Another factor that influences ICI resistance is the clonality of neoantigens [87]. Genomic intratumor heterogeneity, which results in subclonal mutations and neoantigens, and high subclonal TMB is associated with an ineffective antitumor response [87, 88]. Conversely, clonal TMB is a stronger predictor of ICI responsiveness than total TMB [88, 89].
Cancer cell-intrinsic mechanism of primary resistance to immunotherapy has been outlined by the expression of a certain set of genes that are enriched in tumors from patients who did not respond to anti-PD-1 therapy [90]. This set of genes was termed innate anti-PD-1 resistance signature (IPRES) and includes regulatory genes such as epithelial–mesenchymal transition-related genes (AXL, WNT5A, ROR2, TWIST2, FAP, and TAGLN), VEGF pathway genes (IL-10, VEGFA, and VEGFC), and macrophage chemotaxis genes (CCL2, CCL7, CCL8, and CCL13) [90]. Spatial interplay between tumor and immune microenvironment influences intercellular signaling, immune recognition, and resistance to immunotherapy [91]. Spatial transcriptomics and multiplexed quantitative immunofluorescence have made it possible to annotate localization of tumor and immune cells with precision [92], which is an emerging tool to understand molecular mechanisms of primary resistance in the spatial context of tumor microenvironment. In a cohort of NSCLC patients, multiplexed tissue imaging identified enrichment of Tregs in non-responders, and these were localized to stromal and peripheral tumor margins. Spatial phenotyping of cytokine signatures in head and neck cancer identified areas of abnormal CXCL9 and CXCL10 expression associated with resistance to immunotherapy [93]. A multi-institutional study reported that spatial distribution of T cells and T-cell exhaustion marker expression predict outcomes with ICIs in NSCLC [94].
Future and conclusions
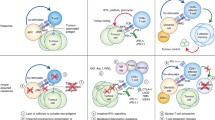
Several genomic predictors of primary response to ICIs have been identified but most are yet to be validated in prospective clinical cohorts. Further elucidation of these molecular mechanisms is key to developing risk scores as well as treatment strategies. Individual biomarkers have poor specificity and sensitivity, and the development of a validated and integrated predictive model will guide which patient will benefit from monotherapy versus combination therapy. This approach can also be applied to tumors with acquired resistance as some of the molecular alterations driving primary ICI resistance have also been noted for acquired resistance such as JAK2 or β2M mutations [95]. There are several potential approaches to target these mechanisms of primary resistance that have been tested but clinically significant benefit is yet to be seen as these trials have not been in biomarker-selected patient populations. Integration of genomic and other predictors of primary resistance in selecting patients for future trials aimed at overcoming ICI resistance is essential (Fig. 1 and Table 1).

Genomics and molecular mechanisms of primary immunotherapy resistance and current strategies under investigation to overcome resistance. Created with Biorender.com
Data availability
No datasets were generated or analyzed during the current study.
References
Shergold AL, Millar R, Nibbs RJB (2019) Understanding and overcoming the resistance of cancer to PD-1/PD-L1 blockade. Pharmacol Res 145:104258
Malhotra J, Jabbour SK, Aisner J (2017) Current state of immunotherapy for non-small cell lung cancer. Transl Lung Cancer Res 6(2):196–211
Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14(10):1014–1022
Neurath MF, Finotto S (2012) The emerging role of T cell cytokines in non-small cell lung cancer. Cytokine Growth Factor Rev 23(6):315–322
Eiz-Vesper B, Schmetzer HM (2020) Antigen-presenting cells: potential of proven und new players in immune therapies. Transfus Med Hemother 47(6):429–431
Greenwald RJ, Freeman GJ, Sharpe AH (2005) The B7 family revisited. Annu Rev Immunol 23:515–548
Arasanz H et al (2017) PD1 signal transduction pathways in T cells. Oncotarget 8(31):51936–51945
Kluger HM et al (2020) Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC immunotherapy resistance taskforce. J Immunother Cancer 8(1):e000398
Fares CM et al (2019) Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book 39:147–164
Shui IM et al (2022) Resistance to anti-PD1 therapies in patients with advanced melanoma: systematic literature review and application of the society for immunotherapy of cancer immunotherapy resistance taskforce anti-PD1 resistance definitions. Melanoma Res 32(6):393–404
Robert C et al (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372(4):320–330
Shah S et al (2018) Clinical and molecular features of innate and acquired resistance to anti-PD-1/PD-L1 therapy in lung cancer. Oncotarget 9(4):4375–4384
Diem S et al (2016) Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br J Cancer 114(3):256–261
Nishino M et al (2017) Immune-related tumor response dynamics in melanoma patients treated with pembrolizumab: identifying markers for clinical outcome and treatment decisions. Clin Cancer Res 23(16):4671–4679
Taube JM et al (2014) Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 20(19):5064–5074
Gide TN et al (2018) Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin Cancer Res 24(6):1260–1270
Uryvaev A et al (2018) The role of tumor-infiltrating lymphocytes (TILs) as a predictive biomarker of response to anti-PD1 therapy in patients with metastatic non-small cell lung cancer or metastatic melanoma. Med Oncol 35(3):25
Tumeh PC et al (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515(7528):568–571
Snyder A et al (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371(23):2189–2199
Ayers M et al (2017) IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127(8):2930–2940
Sharma P et al (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168(4):707–723
Sade-Feldman M et al (2017) Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8(1):1136
Torrejon DY et al (2020) Overcoming genetically based resistance mechanisms to PD-1 blockade. Cancer Discov 10(8):1140–1157
Germano G et al (2021) CD4 T cell-dependent rejection of Beta-2 microglobulin null mismatch repair-deficient tumors. Cancer Discov 11(7):1844–1859
de Vries NL et al (2023) gammadelta T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature 613(7945):743–750
Charych DH et al (2016) NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res 22(3):680–690
Sharma M et al (2020) Bempegaldesleukin selectively depletes intratumoral Tregs and potentiates T cell-mediated cancer therapy. Nat Commun 11(1):661
Wang C et al (2022) The immune-related role of beta-2-microglobulin in melanoma. Front Oncol 12:944722
Karre K et al (1986) Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319(6055):675–678
del Campo AB et al (2009) Efficient recovery of HLA class I expression in human tumor cells after beta2-microglobulin gene transfer using adenoviral vector: implications for cancer immunotherapy. Scand J Immunol 70(2):125–135
Bedikian AY, Del Vecchio M (2008) Allovectin-7 therapy in metastatic melanoma. Expert Opin Biol Ther 8(6):839–844
Nabel GJ et al (1996) Immune response in human melanoma after transfer of an allogeneic class I major histocompatibility complex gene with DNA-liposome complexes. Proc Natl Acad Sci USA 93(26):15388–15393
Stopeck AT et al (1997) Phase I study of direct gene transfer of an allogeneic histocompatibility antigen, HLA-B7, in patients with metastatic melanoma. J Clin Oncol 15(1):341–349
Bedikian AY et al (2010) A phase 2 study of high-dose Allovectin-7 in patients with advanced metastatic melanoma. Melanoma Res 20(3):218–226
Agarwala SS (2015) Intralesional therapy for advanced melanoma: promise and limitation. Curr Opin Oncol 27(2):151–156
Wang X et al (2017) Suppression of Type I IFN signaling in tumors mediates resistance to Anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res 77(4):839–850
Kumar S et al (2020) Mitogen-activated protein kinase inhibitors and T-Cell-dependent immunotherapy in cancer. Pharmaceuticals (Basel) 13(1):9
Bedognetti D et al (2017) The MAPK hypothesis: immune-regulatory effects of MAPK-pathway genetic dysregulations and implications for breast cancer immunotherapy. Emerg Top Life Sci 1(5):429–445
Loi S et al (2016) RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res 22(6):1499–1509
Ribas A et al (2020) PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma. Nat Commun 11(1):6262
Atkins MB et al (2023) Combination dabrafenib and trametinib versus combination nivolumab and ipilimumab for patients with advanced BRAF-mutant melanoma: the DREAMseq trial-ECOG-ACRIN EA6134. J Clin Oncol 41(2):186–197
Dong Y et al (2014) PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene 33(38):4632–4642
Peng W et al (2016) Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 6(2):202–216
George S et al (2017) Loss of PTEN is associated with resistance to Anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 46(2):197–204
Parsa AT et al (2007) Loss of tumor suppressor PTEN function increases B7–H1 expression and immunoresistance in glioma. Nat Med 13(1):84–88
O’Donnell JS et al (2018) PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol 48:91–103
Kaneda MM et al (2016) PI3Kgamma is a molecular switch that controls immune suppression. Nature 539(7629):437–442
Spranger S et al (2017) Tumor-Residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31(5):711–723
Chehrazi-Raffle A et al (2021) Wnt/beta-catenin signaling and immunotherapy resistance: lessons for the treatment of urothelial carcinoma. Cancers (Basel) 13(4):711–723
Holtzhausen A et al (2015) Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol Res 3(9):1082–1095
Zhao F et al (2018) Paracrine Wnt5a-beta-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity 48(1):147–160
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523(7559):231–235
Li J et al (2022) Disruption of Wnt/beta-catenin pathway elevates the sensitivity of gastric cancer cells to PD-1 antibody. Curr Mol Pharmacol 15(3):557–569
Grasso CS et al (2018) Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov 8(6):730–749
Abril-Rodriguez G et al (2020) PAK4 inhibition improves PD-1 blockade immunotherapy. Nat Cancer 1(1):46–58
Klempner SJ et al (2022) DKN-01 and tislelizumab ± chemotherapy as a first-line (1L) and second-line (2L) investigational therapy in advanced gastroesophageal adenocarcinoma (GEA): DisTinGuish Trial. J Clin Oncol 40(4_suppl):292–292
Ribas A, Hu-Lieskovan S (2016) What does PD-L1 positive or negative mean? J Exp Med 213(13):2835–2840
Ma Y et al (2022) Tumor-intrinsic PD-L1 exerts an oncogenic function through the activation of the Wnt/beta-catenin pathway in human non-small cell lung cancer. Int J Mol Sci 23(19):11031
Gainor JF et al (2016) EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res 22(18):4585–4593
Liu SY et al (2018) Clinical relevance of PD-L1 expression and CD8+ T cells infiltration in patients with EGFR-mutated and ALK-rearranged lung cancer. Lung Cancer 125:86–92
Yeo AT et al (2023) Driver mutations dictate the immunologic landscape and response to checkpoint immunotherapy of glioblastoma. Cancer Immunol Res 11(5):629–645
Oxnard GR et al (2020) TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol 31(4):507–516
Hu X, Ivashkiv LB (2009) Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 31(4):539–550
Platanias LC (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5(5):375–386
Bach EA, Aguet M, Schreiber RD (1997) The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol 15:563–591
Kalbasi A, Ribas A (2020) Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 20(1):25–39
Gao J et al (2016) Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to Anti-CTLA-4 therapy. Cell 167(2):397–404
Shin DS et al (2017) Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 7(2):188–201
Alburquerque-Bejar JJ et al (2023) MYC activation impairs cell-intrinsic IFNgamma signaling and confers resistance to anti-PD1/PD-L1 therapy in lung cancer. Cell Rep Med 4(4):101006
Ribas A et al (2018) SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib. Multicenter Study Cancer Discov 8(10):1250–1257
Negrao MV et al (2023) Vidutolimod in combination with atezolizumab with and without radiation therapy in patients with programmed cell death protein 1 or programmed death-ligand 1 blockade-resistant advanced NSCLC. JTO Clin Res Rep 4(3):100423
Ribas A et al (2017) Oncolytic virotherapy promotes intratumoral T Cell infiltration and improves Anti-PD-1 immunotherapy. Cell 170(6):1109–1119
Jung KH et al (2017) Multifunctional effects of a small-molecule STAT3 inhibitor on NASH and hepatocellular carcinoma in mice. Clin Cancer Res 23(18):5537–5546
Han G et al (2021) 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat Commun 12(1):5606
Yu J et al (2019) Genetic aberrations in the CDK4 pathway are associated with innate resistance to PD-1 blockade in Chinese patients with non-cutaneous melanoma. Clin Cancer Res 25(21):6511–6523
Masuda J et al (2023) Efficacy, safety, and biomarker analysis of nivolumab in combination with abemaciclib plus endocrine therapy in patients with HR-positive HER2-negative metastatic breast cancer: a phase II study (WJOG11418B NEWFLAME trial). J Immunother Cancer 11(9):e007126
Skoulidis F et al (2018) STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 8(7):822–835
Zavitsanou AM et al (2023) KEAP1 mutation in lung adenocarcinoma promotes immune evasion and immunotherapy resistance. Cell Rep 42(11):113295
Romero R et al (2017) Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23(11):1362–1368
Kato S et al (2017) Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 23(15):4242–4250
Jardim DL et al (2021) The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell 39(2):154–173
Carbone DP et al (2017) First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med 376(25):2415–2426
Chabanon RM et al (2016) Mutational landscape and sensitivity to immune checkpoint blockers. Clin Cancer Res 22(17):4309–4321
Campesato LF et al (2015) Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 6(33):34221–34227
Chalmers ZR et al (2017) Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9(1):34
Goodman AM et al (2020) MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med 12(1):45
Wolf Y, Sameuls Y (2023) Neoantigens in cancer immunotherapy: quantity vs. quality. Mol Oncol 17(8):1457–1459
Wolf Y et al (2019) UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell 179(1):219–235
Litchfield K et al (2021) Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 184(3):596–614
Hugo W et al (2017) Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell 168(3):542
Kim SJ, Khadka D, Seo JH (2022) Interplay between solid tumors and tumor microenvironment. Front Immunol 13:882718
Wang WJ et al (2023) Spatial transcriptomics: recent developments and insights in respiratory research. Mil Med Res 10(1):38
Ji H et al (2023) Spatial phenotyping of cytokine signatures reflecting the immunotherapy responses in head and neck cancer. J Immunol 210(1_Supplement):171.09-171.09
Lopez de Rodas M et al (2022) Role of tumor infiltrating lymphocytes and spatial immune heterogeneity in sensitivity to PD-1 axis blockers in non-small cell lung cancer. J Immunother Cancer 10(6):e004440
Zaretsky JM et al (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375(9):819–829
Funding
Open access funding provided by SCELC, Statewide California Electronic Library Consortium.
Author information
Authors and Affiliations
Contributions
J.M., S.D., K.N., V.V., and P.L. wrote the main manuscript text, and J.M. prepared the figure. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Malhotra, J., De, S., Nguyen, K. et al. Genomic and molecular alterations associated with primary resistance to immune checkpoint inhibitors. Cancer Immunol Immunother 73, 234 (2024). https://doi.org/10.1007/s00262-024-03825-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-024-03825-z