
Abstract
Polycythemia vera (PV) is a burdensome, chronic myeloproliferative neoplasm characterized by activating mutations in Janus kinase 2, erythrocytosis, and bone marrow hypercellularity. The goals of treatment are to achieve hematocrit and blood count control to ultimately reduce the risk of thrombohemorrhagic events and improve PV-related symptoms. Treatment is risk-stratified and typically includes cytoreduction with hydroxyurea or interferon formulations in first line for high-risk disease. However, inadequate response, resistance, or intolerance to first-line cytoreductive therapies may warrant introduction of second-line treatments, such as ruxolitinib. In this review, I detail preferred treatment and patient management approaches following inadequate response to or intolerance of first-line treatment for PV.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by erythrocytosis, bone marrow hypercellularity, and activating Janus kinase (JAK) mutations (JAK2V617F or JAK2 exon 12 mutations) that is estimated to affect more than 100,000 people in the USA [1, 2]. Patients with PV have burdensome signs and symptoms including pruritus, fatigue, night sweats, concentration problems, and splenomegaly [3, 4]. There are also increased risks of thrombosis, mortality, and disease transformation to myelofibrosis or acute myeloid leukemia [5,6,7,8,9,10].
Polycythemia vera treatment goals include controlling blood counts (hematocrit, < 45%; white blood cell [WBC] count, < 11 × 109/L; platelet count, < 400 × 109/L), resolving disease-related signs and symptoms, and reducing risk of thromboembolic events [11,12,13]. Hydroxyurea (HU) and interferon formulations are recommended first-line treatments for patients with high-risk PV (age ≥ 60 years or history of thrombosis). However, up to 25% of patients become resistant to or intolerant of HU, and historically, interferon therapy has been associated with challenging side effects [14,15,16,17]. Furthermore, although HU is effective at reducing thromboses in patients with PV [16, 18], HU treatment has not generally been shown to improve symptoms or modify the disease [19,20,21]. Ruxolitinib, a JAK1 and JAK2 inhibitor, provides clinical benefit, including hematocrit and WBC count control and reductions in splenomegaly and symptom burden, and was approved by the US Food and Drug Administration in 2014 for PV in adults who have an inadequate response to or are intolerant of HU [22,23,24,25,26,27,28,29].
Sample patient 1 — part 1
A 32-year-old male patient presented to urgent care with persistent left upper quadrant abdominal pain for the previous 4 weeks. Computed tomography scan showed a 15 × 12 × 12.5 cm spleen with multiple splenic infarcts. Laboratory work showed that hemoglobin was 19.8 g/dL, and hematocrit was 60.3%. WBC count was 10.9 × 109/L, platelets were 276 × 109/L, lactose dehydrogenase was 696 units/L, ferritin was 13.5 μg/L, and erythropoietin was 1 mIU/mL. JAK2 quantitative polymerase chain reaction showed presence of the JAK2V617F mutation with allele frequency of 36%. Bone marrow biopsy showed hypercellular marrow with erythroid hyperplasia and no increase in iron stores. Karyotype was normal. The patient was started on aspirin 81 mg daily and phlebotomy with a goal hematocrit < 45%.
Sample patient 2 — part 1
A 50-year-old female patient presented with debilitating migraines with visual auras. Blood counts were as follows: hemoglobin, 19.5 g/dL; hematocrit, 58.6%; WBC, 11.4 × 109/L; and platelets, 532 × 109/L. JAK2V617F was qualitatively positive. Bone marrow biopsy was consistent with PV without fibrosis. The patient was started on aspirin 81 mg daily and HU.
Treatment goals and first-line treatment options in PV
The primary treatment goals in my practice are to reduce the risk of thrombosis and hemorrhage and maintain blood count control [11,12,13, 30, 31]. We balance that with a comprehensive patient management approach that aims to improve symptom burden and quality of life (QoL) in patients with PV.
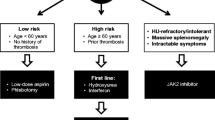
Treatment for patients with PV should be guided by the patient’s risk status (Fig. 1). Patients with low-risk disease (age < 60 years and no history of thrombosis) are treated with low-dose aspirin and therapeutic phlebotomy to maintain hematocrit < 45%. For patients with high-risk disease, the addition of cytoreductive therapy is recommended. Specifically, HU, pegylated interferon alfa-2a, or ropeginterferon alfa-2b may be considered, which have been shown to have similar clinical benefit in the treatment of PV, despite varying mechanisms of action, modes of administration, and toxicity profiles [16, 30, 32,33,34,35].

Therapeutic strategy for polycythemia vera. PV, polycythemia vera
HU, a ribonucleotide reductase inhibitor, has traditionally been the standard cytoreductive agent administered to high-risk patients with PV [36]. However, a considerable number of patients are either intolerant to HU or are HU resistant [14,15,16,17, 33]. Various interferon formulations have also been widely used in patients with PV for decades, given their antiproliferative, immunomodulating, and anticlonal effects [17]. More recently, pegylated forms of interferon, specifically pegylated interferon alfa-2a (an interferon with six monopegylated positional isomers) and ropeginterferon alfa-2b (a monopegylated interferon with a single positional isomer associated with an extended elimination half-life), have been recommended for first-line PV treatment due to their longer acting effects, less frequent administration, and tolerable side effects [16, 32, 33, 37, 38].
Sample patient 1—part 2
Three years after diagnosis, the patient experienced rapidly increasing spleen size with associated abdominal discomfort and unintentional weight loss of 11 kg. Laboratory work showed that hemoglobin was 14.6 g/dL, hematocrit was 45%, and WBC count was 19 × 109/L. A repeat bone marrow biopsy was performed, which showed 100% cellularity with panmyelosis (trilineage hyperplasia), megakaryocyte clustering, and mild reticulin fibrosis. Karyotype showed 47, XY, + 9 [11] /47, XY, del(20q)(q11.2q13.2) [9], suggesting cytogenetic evolution with uniparental disomy. Next-generation sequencing showed variant allele fraction of JAK2V617F mutation to be 63%, corresponding with an additional copy of 9th chromosome.
Sample patient 2—part 2
Treatment with first-line HU at 1000 mg daily for 6 months was associated with improvements in the patient’s headaches but led to intolerable mouth ulcers and hair loss.
Optimal time to proceed to second-line treatment
In my practice, we monitor for several factors that indicate patients may benefit from a change in treatment. These include new thrombosis or disease-related major bleeding, frequent phlebotomy or phlebotomy intolerance, splenomegaly, progressive thrombocytosis and/or leukocytosis, or new or worsening disease-related symptoms (e.g., pruritus, night sweats, fatigue) [16]. For HU specifically, European LeukemiaNet established a consensus definition for resistance or intolerance that my practice follows [15]. These criteria define HU resistance or intolerance in PV as one or more of the following: (a) need for phlebotomy to maintain hematocrit < 45% after 3 months of ≥ 2 g/day HU, (b) uncontrolled myeloproliferation (i.e., platelet count > 400 × 109/L and WBC count > 10 × 109/L) after 3 months of ≥ 2 g/day HU, (c) failure to reduce massive splenomegaly (i.e., organ extending by > 10 cm from costal margin) by > 50% as measured by palpation or failure to completely relieve symptoms related to splenomegaly after 3 months of ≥ 2 g/day HU, (d) absolute neutrophil count < 1.0 × 109/L, platelet count < 100 × 109/L, or hemoglobin < 100 g/L at the lowest dose of HU required to achieve a complete or partial response, or (e) the presence of leg ulcers or other unacceptable HU-related nonhematologic toxicities (e.g., mucocutaneous manifestations, gastrointestinal symptoms, pneumonitis, or fever at any dose of HU). It is important to note that 2 g/day HU is often difficult to tolerate, in which case our practice evaluates patients using these same criteria at the patient’s maximum tolerated dose. We also recommend hematocrit monitoring at each phlebotomy or at least every 3 months to ensure levels < 45%. There are no formal guidelines that define inadequate response or intolerance for the various available interferon formulations. In my practice, emergence of new or worsening PV-related signs or symptoms, as described above, or intolerable interferon-related side effects, including flu-like symptoms, chronic fatigue and/or musculoskeletal pain, depression, thyroid dysfunction, and autoimmune diseases that cannot be managed with supportive care approaches, are indicative of a need to consider a change in therapy [17].
Sample patient 1—part 3
The patient was initiated on pegylated interferon at 90 μg weekly, which was poorly tolerated due to flu-like symptoms and arthralgias lasting 3 to 4 days after each injection for the first 4 weeks. The dose was reduced to 45 μg weekly, which was tolerated well and gradually increased to 135 μg weekly. After 9 months of interferon therapy, the patient experienced a slight decrease in phlebotomy requirements but continued splenomegaly, fatigue, and weight loss with no substantial change in WBC counts. In addition, his wife reported a withdrawn and dysphoric mood atypical for him, which is a known side effect of interferon therapy. Because of this, pegylated interferon was discontinued, and ruxolitinib was started at 10 mg twice daily (bid). This resulted in rapid improvement in mood symptoms, weight gain, and reduced splenomegaly.
Sample patient 2—part 3
After demonstrating signs of HU intolerance, the patient was switched to ruxolitinib 1 year after diagnosis, with immediate resolution of mouth sores, continued control of headaches, and improvement in energy level. This also eliminated the need for phlebotomy.
Second-line treatment
Ruxolitinib is my preferred second-line therapy for most patients who are resistant to or intolerant of first-line therapy [16, 30]. This recommendation is supported by multiple large, randomized, phase 3 clinical trials that demonstrated clinical benefit with ruxolitinib, including RESPONSE [22, 23, 29], RESPONSE-2 [39,40,41], and RELIEF [20].
The RESPONSE study consisted of patients who were unresponsive or intolerant to HU, had splenomegaly, and were phlebotomy-dependent. Patients treated with ruxolitinib were more likely to achieve hematocrit, WBC count, and platelet count control; reduction in spleen volume; reduction in symptoms; and lower phlebotomy rates compared with best available therapy (BAT) [22]. These responses were durable through 5 years of follow-up [29]. For patients originally randomized to BAT, those who crossed over to ruxolitinib experienced similar clinical benefit as those originally randomized to ruxolitinib [23]. Finally, a post hoc analysis from the RESPONSE trial that examined JAK2V617F allele burden demonstrated that patients who received ruxolitinib had greater reductions in allele burden compared with those receiving BAT [42]. Although the clinical relevance of these allele burden reductions is unclear, higher allele burden levels have been associated with more severe disease [43].
The RESPONSE-2 study evaluated ruxolitinib versus BAT in HU-resistant patients who were phlebotomy dependent but without splenomegaly. Ruxolitinib was superior to BAT at reducing phlebotomy requirement and achieving hematocrit, WBC count, and platelet count control. Patients treated with ruxolitinib also experienced better symptom control and improvements in QoL compared with BAT [40]. Similar to RESPONSE, these responses were also durable through 5 years of follow-up [41].
It is important to emphasize that ruxolitinib provided hematocrit control and improvements in PV-related symptoms regardless of the presence or absence of splenomegaly, indicating that splenomegaly should not be a prerequisite for initiating ruxolitinib treatment [22, 40].
Ruxolitinib has also been demonstrated to provide clinical benefit regardless of whether patients were previously treated with interferon or HU. In a post hoc analysis of patients who were treated with an interferon formulation as investigator-selected BAT in the RESPONSE and RESPONSE-2 studies, patients who crossed over to ruxolitinib experienced clinical improvements including hematologic and spleen response as well as reduced phlebotomy requirement [44].
Several studies have evaluated the effects of ruxolitinib on thrombotic events. In RESPONSE through week 32, thromboembolic events occurred in one patient receiving ruxolitinib and 6 patients receiving BAT [22]. Exposure-adjusted rates were also lower for patients receiving ruxolitinib than BAT (1.2 vs. 8.2 per 100 patient-years, respectively) through 5 years of follow-up [29]. Similarly, there was one thrombotic event in a patient receiving ruxolitinib and 3 for those receiving BAT in the primary RESPONSE-2 analysis [40]. Exposure-adjusted rates of any-grade thromboembolic events were 1.5 per 100 patient-years for patients receiving ruxolitinib and 3.7 for patients receiving BAT through 5 years of follow-up [39, 41]. Additionally, a meta-analysis covering 663 patients from RESPONSE, RESPONSE-2, RELIEF, and MAJIC reported similar rates of thrombotic events for ruxolitinib versus BAT (3.09 vs. 5.51; P = 0.98) [28]. Finally, a recent retrospective analysis evaluating real-world ruxolitinib treatment in patients with HU-resistant/intolerant PV demonstrated a significantly lower rate of arterial thrombosis with ruxolitinib versus BAT (0.4 vs. 2.3; P = 0.03) but no significant differences in venous thrombosis, major bleeding, or survival [45].
Although presence of new or worsening PV-related symptoms alone may be an indicator for treatment change, potential benefit may extend to only a subpopulation of patients. The phase 3b RELIEF study consisted of patients who had PV-related symptoms but were generally well controlled on a stable dose of HU. The study evaluated switching treatment to ruxolitinib versus remaining on HU. Among patients who switched to ruxolitinib, some experienced improvements in symptoms that contributed to a positive trend in symptom control at week 16 compared with HU; however, this did not meet statistical significance [20].
Although ruxolitinib is the preferred second-line treatment in my practice, interferon formulations (including ropeginterferon alfa-2b and pegylated interferon alfa-2a) are appropriate second-line treatment options for patients who are intolerant or have inadequate response to HU [30, 46].
In PROUD-PV and its extension study, CONTINUATION-PV, ropeginterferon alfa-2b was compared with HU in patients with early-stage PV with no history of cytoreductive treatment or less than 3 years of previous HU treatment of which they were resistant or intolerant. In PROUD-PV, noninferiority versus HU was not achieved at 1 year for the combined primary endpoint: hematologic response (defined as hematocrit < 45% with no phlebotomy in the past 3 months, platelet count < 400 × 109/L, and leukocyte count < 10 × 109/L) and normal spleen size [32]. However, response to ropeginterferon alfa-2b increased over time with improved hematologic response and improvement in disease burden compared with HU at 36 months in CONTINUATION-PV [32]. Ropeginterferon alfa-2b may also provide additional benefit in patients with low-risk PV, as assessed in the low-PV study. This trial compared ropeginterferon alfa-2b added to a standard phlebotomy regimen (including 100 mg aspirin daily) with phlebotomy alone in patients with low-risk PV. Hematocrit control was maintained in more patients receiving add-on ropeginterferon alfa-2b than phlebotomy alone [32, 47].
Additionally, a phase 2 study from the Myeloproliferative Disorders Research Consortium recently demonstrated a 60% overall response rate and 22% complete response among 50 patients with PV treated with pegylated interferon alfa-2a [33]. Statistically significant improvement in MPN-related symptoms was also observed for PV and ET patients combined; however, treatment-emergent adverse events may have offset these improvements, resulting in patient-reported measures of quality of life remaining stable [33].
Finally, rusfertide, a peptide mimetic of the master iron regulator hepcidin, is currently in development as a non-cytoreductive option in PV to reverse iron deficiency and achieve hematocrit control [48, 49]. Rusfertide demonstrated hematocrit control (< 45%) and eliminated the need for phlebotomy in two phase 2 studies in patients with PV [48, 49]. The safety and efficacy of rusfertide are also being investigated in the phase 3 randomized VERIFY study (NCT05210790).
Sample patient 1—part 4
The ruxolitinib dose was subsequently increased to 20 mg bid after 9 months of treatment due to continued need for phlebotomies. To date, the patient has been receiving ruxolitinib for 4.5 years, with the dose remaining at 20 mg bid, with nonpalpable splenomegaly, no need for phlebotomies, and good QoL.
Sample patient 2—part 4
The patient has been receiving ruxolitinib for 7 years, and her PV remains well controlled without need for phlebotomy, with good functional status and no headaches, although she struggles with weight gain that is likely attributable to ruxolitinib.
Assessing second-line treatment safety
Patients receiving treatment with ruxolitinib should be monitored for certain adverse events that have been observed in short- and longer-term clinical trials in patients with PV. These include cytopenias, opportunistic infections, herpes zoster reactivation, nonmelanoma skin cancer, hypertension, and weight gain [20, 22, 23, 40]. One of the goals of treatment in PV is to lower elevated blood counts; as a result, cytopenias are the most common adverse events with ruxolitinib. These often present as grade 1 or 2, as observed in the RESPONSE and RESPONSE-2 trials [22, 40]. Although opportunistic infections do occur in patients treated with ruxolitinib, long-term follow-up in RESPONSE and RESPONSE-2 demonstrated that exposure-adjusted infection rates (excluding herpes zoster) were lower with ruxolitinib than with BAT [29, 41]. With respect to weight gain, one retrospective study of patients with myeloproliferative neoplasms treated with ruxolitinib found that > 50% of patients gained > 5% of their baseline body weight [50]. Physicians initiating treatment with ruxolitinib in patients with PV should consider monitoring these patients for metabolic effects (e.g., obesity, hyperglycemia, dyslipidemia, hypertension, hepatic steatosis) and providing dietary counseling or lifestyle management recommendations to help offset potential weight gain. Overall, adverse effects should be monitored closely and can be managed by dose reduction or interruption, as detailed in the next section.
For patients receiving ropeginterferon alfa-2b, results from the PROUD-PV and CONTINUATION-PV studies revealed that treatment-related toxicities overall occurred at similar rates as HU treatment. The most common adverse events with ropeginterferon alfa-2b were cytopenias (thrombocytopenia, leukopenia, and anemia; predominantly grade 1 or 2), liver laboratory abnormalities (increased γ-glutamyltransferase, alanine aminotransferase, and aspartate aminotransferase), and flu-like symptoms (fatigue, headache, dizziness).
Regarding rusfertide, although phase 3 studies are underway in PV, phase 2 studies have demonstrated a favorable safety profile, with most adverse events classified as grade 1 or 2. The most frequent adverse event was transient injection site reaction, with one study reporting its occurrence in 59% of patients [48, 49].
Ruxolitinib dose optimization
The recommended starting dose for patients with PV is 10 mg bid, and doses may be titrated based on observed safety and efficacy outcomes [24]. Guidelines for treatment interruption and restarting dosing, as well as dose modifications related to insufficient response to ruxolitinib, are included in Fig. 2.

Dosage recommendations for ruxolitinib. *Not during the first 4 weeks of therapy or more frequently than every 2 weeks. †Based on the most severe category of Hgb, platelet count, or ANC abnormality. ‡Continue treatment for ≥ 2 weeks; if dose stabilizes, it may be increased by 5 mg bid. ANC, absolute neutrophil count; bid, twice daily; BL, baseline; CBC, complete blood count; Hgb, hemoglobin; qd, once daily; ULN, upper limit of normal; WBC, white blood cell count
Ruxolitinib dosage modifications have been evaluated in a real-world setting. A medical chart review of patients with PV who switched from HU to ruxolitinib found that only half initiated treatment at the recommended dose. Dose modifications were common (27%) in the first 6 months of ruxolitinib treatment, and most patients achieved hematocrit control and had extended treatment with ruxolitinib [51]. This information reinforces the importance of selecting the proper ruxolitinib starting dose and actively titrating the dose thereafter based on safety and efficacy observations to avoid treatment interruptions and maximize clinical benefit. Nevertheless, treatment interruption may be required for some patients, especially for cytopenias (Fig. 2) [24]. Although some patients with a related myeloproliferative neoplasm, myelofibrosis, who discontinued ruxolitinib experienced ruxolitinib discontinuation syndrome, which constitutes a rebound in myelofibrosis symptoms after treatment discontinuation [52], this has not been observed in patients with PV.
Ropeginterferon alfa-2b dose optimization
The ropeginterferon alfa-2b recommended starting dose is 100 μg by subcutaneous injection every 2 weeks or 50 μg if receiving concomitant HU. Dosage should be increased by 50 μg every 2 weeks (up to a maximum of 500 μg) until target hematologic parameters are achieved. Dose interruption or discontinuation is recommended in response to certain adverse reactions such as liver enzyme elevation (with or without concomitant bilirubin elevation or other evidence of hepatic decompensation), cytopenias, or depression. Complete blood count should be monitored every 2 weeks when titrating and modifying dose. Phlebotomy may be used as needed to maintain safe hematocrit values. Following a treatment interruption and resolution of instigating factors, ropeginterferon alfa-2b should be restarted at the previously attained dose or reduced to the next lower dose level if drug-related toxicities arise. If efficacy is insufficient after a dose decrease, increase dose to the next higher dose level after recovery to grade 1 toxicity.
Conclusion
Patients with high-risk PV often experience clinical benefit with first-line cytoreductive treatments; however, treatment efficacy may diminish over time, and treatment-related toxicities may be challenging for some patients. A change in treatment is often beneficial for this patient population, with ruxolitinib the current preferred second-line treatment option. Based on evidence from 3 large phase 3 clinical trials, ruxolitinib improves PV-related signs and symptoms in the second line, and long-term treatment is feasible with dose modifications. Additional second-line options are emerging with distinct therapeutic and toxicity profiles, which reinforce the need to establish clear, patient-specific treatment goals in order to provide individualized therapy.
Change history
24 April 2023
A Correction to this paper has been published: https://doi.org/10.1007/s00277-023-05235-0
References
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20):2391–2405. https://doi.org/10.1182/blood-2016-03-643544
Mehta J, Wang H, Iqbal SU, Mesa R (2014) Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma 55(3):595–600. https://doi.org/10.3109/10428194.2013.813500
Mesa R, Miller CB, Thyne M, Mangan J, Goldberger S, Fazal S, Ma X, Wilson W, Paranagama DC, Dubinski DG, Boyle J, Mascarenhas JO (2016) Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer 16:167. https://doi.org/10.1186/s12885-016-2208-2
Emanuel RM, Dueck AC, Geyer HL, Kiladjian JJ, Slot S, Zweegman S, te Boekhorst PA, Commandeur S, Schouten HC, Sackmann F, Kerguelen Fuentes A, Hernandez-Maraver D, Pahl HL, Griesshammer M, Stegelmann F, Doehner K, Lehmann T, Bonatz K, Reiter A, Boyer F, Etienne G, Ianotto JC, Ranta D, Roy L, Cahn JY, Harrison CN, Radia D, Muxi P, Maldonado N, Besses C, Cervantes F, Johansson PL, Barbui T, Barosi G, Vannucchi AM, Passamonti F, Andreasson B, Ferrari ML, Rambaldi A, Samuelsson J, Birgegard G, Tefferi A, Mesa RA (2012) Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 30(33):4098–4103. https://doi.org/10.1200/jco.2012.42.3863
Price GL, Davis KL, Karve S, Pohl G, Walgren RA (2014) Survival patterns in United States (US) Medicare enrollees with non-CML myeloproliferative neoplasms (MPN). PLoS One 9(3):e90299. https://doi.org/10.1371/journal.pone.0090299
Hultcrantz M, Bjorkholm M, Dickman PW, Landgren O, Derolf AR, Kristinsson SY, Andersson TML (2018) Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: a population-based cohort study. Ann Intern Med 168(5):317–325. https://doi.org/10.7326/M17-0028
Hultcrantz M, Kristinsson SY, Andersson TM, Landgren O, Eloranta S, Derolf AR, Dickman PW, Bjorkholm M (2012) Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol 30(24):2995–3001. https://doi.org/10.1200/JCO.2012.42.1925
Grunwald MR, Stein BL, Boccia RV, Oh ST, Paranagama D, Parasuraman S, Colucci P, Mesa R (2018) Clinical and disease characteristics from REVEAL at time of enrollment (baseline): prospective observational study of patients with polycythemia vera in the United States. Clin Lymphoma Myeloma Leuk 18(12):788–795. https://doi.org/10.1016/j.clml.2018.08.009. (e782)
Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F, Randi ML, Vaidya R, Cazzola M, Rambaldi A, Gisslinger B, Pieri L, Ruggeri M, Bertozzi I, Sulai NH, Casetti I, Carobbio A, Jeryczynski G, Larson DR, Müllauer L, Pardanani A, Thiele J, Passamonti F, Barbui T (2013) Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 27(9):1874–1881. https://doi.org/10.1038/leu.2013.163
Nicol C, Ajzenberg N, Lacut K, Couturaud F, Pan-Petesch B, Lippert E, Ianotto JC (2022) Hemorrhages in polycythemia vera and essential thrombocythemia: epidemiology, description, and risk factors–learnings from a large cohort. Thromb Haemost 122(10):1712–1722. https://doi.org/10.1055/a-1849-8477
Barosi G, Mesa R, Finazzi G, Harrison C, Kiladjian JJ, Lengfelder E, McMullin MF, Passamonti F, Vannucchi AM, Besses C, Gisslinger H, Samuelsson J, Verstovsek S, Hoffman R, Pardanani A, Cervantes F, Tefferi A, Barbui T (2013) Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 121(23):4778–4781. https://doi.org/10.1182/blood-2013-01-478891
Marchioli R, Vannucchi AM, Barbui T (2013) Treatment target in polycythemia vera. N Engl J Med 368(16):1556
Barbui T, Masciulli A, Marfisi MR, Tognoni G, Finazzi G, Rambaldi A, Vannucchi A (2015) White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood 126(4):560–561. https://doi.org/10.1182/blood-2015-04-638593
Alvarez-Lárran A, Pereira A, Cervantes F, Arellano-Rodrigo E, Hernández-Boluda JC, Ferrer-Marín F, Angona A, Gómez M, Muiña B, Guillén H, Teruel A, Bellosillo B, Burgaleta C, Vicente V, Besses C (2012) Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 119(6):1363–1369. https://doi.org/10.1182/blood-2011-10-387787
Barosi G, Birgegard G, Finazzi G, Griesshammer M, Harrison C, Hasselbalch H, Kiladijan JJ, Lengfelder E, Mesa R, Mc Mullin MF, Passamonti F, Reilly JT, Vannucchi AM, Barbui T (2010) A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol 148(6):961–963. https://doi.org/10.1111/j.1365-2141.2009.08019.x
National Comprehensive Cancer Network NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for myeloproliferative neoplasms, version 1.2022. https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1477. Accessed November 5 2022
Hasselbalch HC, Holmström MO (2019) Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: minimal residual disease and cure? Semin Immunopathol 41(1):5–19. https://doi.org/10.1007/s00281-018-0700-2
Kaplan ME, Mack K, Goldberg JD, Donovan PB, Berk PD, Wasserman LR (1986) Long-term management of polycythemia vera with hydroxyurea: a progress report. Semin Hematol 23(3):167–171
Antonioli E, Carobbio A, Pieri L, Pancrazzi A, Guglielmelli P, Delaini F, Ponziani V, Bartalucci N, Tozzi L, Bosi A, Rambaldi A, Barbui T, Vannucchi AM (2010) Hydroxyurea does not appreciably reduce JAK2 V617F allele burden in patients with polycythemia vera or essential thrombocythemia. Haematologica 95(8):1435–1438. https://doi.org/10.3324/haematol.2009.021444
Mesa R, Vannucchi AM, Yacoub A, Zachee P, Garg M, Lyons R, Koschmieder S, Rinaldi C, Byrne J, Hasan Y, Passamonti F, Verstovsek S, Hunter D, Jones MM, Zhen H, Habr D, Martino B (2017) The efficacy and safety of continued hydroxycarbamide therapy versus switching to ruxolitinib in patients with polycythaemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Br J Haematol 176(1):76–85
Geyer H, Scherber R, Kosiorek H, Dueck AC, Kiladjian JJ, Xiao Z, Slot S, Zweegman S, Sackmann F, Fuentes AK, Hernández-Maraver D, Döhner K, Harrison CN, Radia D, Muxi P, Besses C, Cervantes F, Johansson PL, Andreasson B, Rambaldi A, Barbui T, Bonatz K, Reiter A, Boyer F, Etienne G, Ianotto JC, Ranta D, Roy L, Cahn JY, Maldonado N, Barosi G, Ferrari ML, Gale RP, Birgegard G, Xu Z, Zhang Y, Sun X, Xu J, Zhang P, Te Boekhorst PA, Commandeur S, Schouten H, Pahl HL, Griesshammer M, Stegelmann F, Lehmann T, Senyak Z, Vannucchi AM, Passamonti F, Samuelsson J, Mesa RA (2016) Symptomatic profiles of patients with polycythemia vera: implications of inadequately controlled disease. J Clin Oncol 34(2):151–159. https://doi.org/10.1200/JCO.2015.62.9337
Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, Harrison CN, Pane F, Zachee P, Mesa R, He S, Jones MM, Garrett W, Li J, Pirron U, Habr D, Verstovsek S (2015) Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 372(5):426–435. https://doi.org/10.1056/NEJMoa1409002
Verstovsek S, Vannucchi AM, Griesshammer M, Masszi T, Durrant S, Passamonti F, Harrison CN, Pane F, Zachee P, Kirito K, Besses C, Hino M, Moiraghi B, Miller CB, Cazzola M, Rosti V, Blau I, Mesa R, Jones MM, Zhen H, Li J, Francillard N, Habr D, Kiladjian J-J (2016) Ruxolitinib versus best available therapy in patients with polycythemia vera: 80 week follow-up from the RESPONSE trial. Haematologica 101(7):821–829. https://doi.org/10.3324/haematol.2016.143644
JAKAFI® (ruxolitinib). Full prescribing information, Incyte Corporation, Wilmington, DE, USA, 2020
Coltoff A, Mesa R, Gotlib J, Shulman J, Rampal RK, Siwoski O, Yacoub A, Moliterno A, Yang A, Braunstein E, Gerds AT, Hobbs GS, Winton EF, Goel S, Wadleigh M, Tremblay D, Moshier E, Mascarenhas J (2020) Real-world outcomes of ruxolitinib treatment for polycythemia vera. Clin Lymphoma Myeloma Leuk 20(10):697–703. https://doi.org/10.1016/j.clml.2020.05.019
Kirito K, Suzuki K, Miyamura K, Takeuchi M, Handa H, Okamoto S, Gadbaw B, Yamauchi K, Amagasaki T, Ito K, Hino M (2018) Ruxolitinib is effective and safe in Japanese patients with hydroxyurea-resistant or hydroxyurea-intolerant polycythemia vera with splenomegaly. Int J Hematol 107(2):173–184. https://doi.org/10.1007/s12185-017-2333-y
Foltz L, Pica GM, Zerazhi H, Van Droogenbroeck J, Visanica S, Baez de la Fuente E, Leber B, de Almeida AM, Ranta D, Kiladjian JJ, Chrit L, Kandra A, Morando J, Devos T (2019) Safety and efficacy findings from the open-label, multicenter, phase 3b, expanded treatment protocol study of ruxolitinib for treatment of patients with polycythemia vera who are resistant/intolerant to hydroxyurea and for whom no alternative treatments are available. Leuk Lymphoma 60(14):3493–3502. https://doi.org/10.1080/10428194.2019.1636985
Masciulli A, Ferrari A, Carobbio A, Ghirardi A, Barbui T (2020) Ruxolitinib for the prevention of thrombosis in polycythemia vera: a systematic review and meta-analysis. Blood Adv 4(2):380–386. https://doi.org/10.1182/bloodadvances.2019001158
Kiladjian JJ, Zachee P, Hino M, Pane F, Masszi T, Harrison CN, Mesa R, Miller CB, Passamonti F, Durrant S, Griesshammer M, Kirito K, Besses C, Moiraghi B, Rumi E, Rosti V, Blau IW, Francillard N, Dong T, Wroclawska M, Vannucchi AM, Verstovsek S (2020) Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol 7(3):e226–e237. https://doi.org/10.1016/S2352-3026(19)30207-8
Barbui T, Tefferi A, Vannucchi AM, Passamonti F, Silver RT, Hoffman R, Verstovsek S, Mesa R, Kiladjian JJ, Hehlmann R, Reiter A, Cervantes F, Harrison C, Mc Mullin MF, Hasselbalch HC, Koschmieder S, Marchetti M, Bacigalupo A, Finazzi G, Kroeger N, Griesshammer M, Birgegard G, Barosi G (2018) Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia 32(5):1057–1069. https://doi.org/10.1038/s41375-018-0077-1
Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, Harrison C, Hasselbalch HC, Hehlmann R, Hoffman R, Kiladjian JJ, Kröger N, Mesa R, McMullin MF, Pardanani A, Passamonti F, Vannucchi AM, Reiter A, Silver RT, Verstovsek S, Tefferi A, LeukemiaNet E (2011) Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 29(6):761–770. https://doi.org/10.1200/JCO.2010.31.8436
Gisslinger H, Klade C, Georgiev P, Krochmalczyk D, Gercheva-Kyuchukova L, Egyed M, Rossiev V, Dulicek P, Illes A, Pylypenko H, Sivcheva L, Mayer J, Yablokova V, Krejcy K, Grohmann-Izay B, Hasselbalch HC, Kralovics R, Kiladjian JJ, PROUD-PV Study Group (2020) Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol 7(3):e196–e208. https://doi.org/10.1016/S2352-3026(19)30236-4
Yacoub A, Mascarenhas J, Kosiorek H, Prchal JT, Berenzon D, Baer MR, Ritchie E, Silver RT, Kessler C, Winton E, Finazzi MC, Rambaldi A, Vannucchi AM, Leibowitz D, Rondelli D, Arcasoy MO, Catchatourian R, Vadakara J, Rosti V, Hexner E, Kremyanskaya M, Sandy L, Tripodi J, Najfeld V, Farnoud N, Papaemmanuil E, Salama M, Singer-Weinberg R, Rampal R, Goldberg JD, Barbui T, Mesa R, Dueck AC, Hoffman R (2019) Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood 134(18):1498–1509. https://doi.org/10.1182/blood.2019000428
Masarova L, Patel KP, Newberry KJ, Cortes J, Borthakur G, Konopleva M, Estrov Z, Kantarjian H, Verstovsek S (2017) Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: a post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol 4(4):e165–e175. https://doi.org/10.1016/S2352-3026(17)30030-3
Mascarenhas J, Kosiorek HE, Prchal JT, Rambaldi A, Berenzon D, Yacoub A, Harrison CN, McMullin MF, Vannucchi AM, Ewing J, O’Connell CL, Kiladjian J-J, Mead AJ, Winton EF, Leibowitz DS, De Stefano V, Arcasoy MO, Kessler CM, Catchatourian R, Rondelli D, Silver RT, Bacigalupo A, Nagler A, Kremyanskaya M, Levine MF, Arango Ossa JE, McGovern E, Sandy L, Salama ME, Najfeld V, Tripodi J, Farnoud N, Penson AV, Weinberg RS, Price L, Goldberg JD, Barbui T, Marchioli R, Tognoni G, Rampal RK, Mesa RA, Dueck AC, Hoffman R (2022) A randomized phase 3 trial of interferon-α vs hydroxyurea in polycythemia vera and essential thrombocythemia. Blood 139(19):2931–2941. https://doi.org/10.1182/blood.2021012743
Ferrari A, Carobbio A, Masciulli A, Ghirardi A, Finazzi G, De Stefano V, Vannucchi AM, Barbui T (2019) Clinical outcomes under hydroxyurea treatment in polycythemia vera: a systematic review and meta-analysis. Haematologica 104(12):2391–2399. https://doi.org/10.3324/haematol.2019.221234
Foser S, Schacher A, Weyer KA, Brugger D, Dietel E, Marti S, Schreitmüller T (2003) Isolation, structural characterization, and antiviral activity of positional isomers of monopegylated interferon alpha-2a (PEGASYS). Protein Expr Purif 30(1):78–87. https://doi.org/10.1016/s1046-5928(03)00055-x
Verger E, Soret-Dulphy J, Maslah N, Roy L, Rey J, Ghrieb Z, Kralovics R, Gisslinger H, Grohmann-Izay B, Klade C, Chomienne C, Giraudier S, Cassinat B, Kiladjian J-J (2018) Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J 8(10):94. https://doi.org/10.1038/s41408-018-0133-0
Griesshammer M, Saydam G, Palandri F, Benevolo G, Egyed M, Callum J, Devos T, Sivgin S, Guglielmelli P, Bensasson C, Khan M, Ronco JP, Passamonti F (2018) Ruxolitinib for the treatment of inadequately controlled polycythemia vera without splenomegaly: 80-week follow-up from the RESPONSE-2 trial. Ann Hematol 97(9):1591–1600. https://doi.org/10.1007/s00277-018-3365-y
Passamonti F, Griesshammer M, Palandri F, Egyed M, Benevolo G, Devos T, Callum J, Vannucchi AM, Sivgin S, Bensasson C, Khan M, Mounedji N, Saydam G (2017) Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 18(1):88–99. https://doi.org/10.1016/S1470-2045(16)30558-7
Passamonti F, Palandri F, Saydam G, Callum J, Devos T, Guglielmelli P, Vannucchi AM, Zor E, Zuurman M, Gilotti G, Zhang Y, Griesshammer M (2022) Ruxolitinib versus best available therapy in inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): 5-year follow up of a randomised, phase 3b study. Lancet Haematol 9(7):e480–e492. https://doi.org/10.1016/S2352-3026(22)00102-8
Vannucchi AM, Verstovsek S, Guglielmelli P, Griesshammer M, Burn TC, Naim A, Paranagama D, Marker M, Gadbaw B, Kiladjian JJ (2017) Ruxolitinib reduces JAK2 p V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol 96(7):1113–1120. https://doi.org/10.1007/s00277-017-2994-x
Vannucchi AM, Antonioli E, Guglielmelli P, Longo G, Pancrazzi A, Ponziani V, Bogani C, Ferrini PR, Rambaldi A, Guerini V, Bosi A, Barbui T, MPD Research Consortium (2007) Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 21(9):1952–1959. https://doi.org/10.1038/sj.leu.2404854
Kiladjian JJ, Guglielmelli P, Griesshammer M, Saydam G, Masszi T, Durrant S, Passamonti F, Jones M, Zhen H, Li J, Gadbaw B, Perez Ronco J, Khan M, Verstovsek S (2018) Efficacy and safety of ruxolitinib after and versus interferon use in the RESPONSE studies. Ann Hematol 97(4):617–627. https://doi.org/10.1007/s00277-017-3225-1
Alvarez-Larran A, Garrote M, Ferrer-Marin F, Perez-Encinas M, Mata-Vazquez MI, Bellosillo B, Arellano-Rodrigo E, Gomez M, Garcia R, Garcia-Gutierrez V, Gasior M, Cuevas B, Angona A, Gomez-Casares MT, Martinez CM, Magro E, Ayala R, Del Orbe-Barreto R, Perez-Lopez R, Fox ML, Raya JM, Guerrero L, Garcia-Hernandez C, Caballero G, Murillo I, Xicoy B, Ramirez MJ, Carreno-Tarragona G, Hernandez-Boluda JC, Pereira A, MPN Spanish Group (2022) Real-world analysis of main clinical outcomes in patients with polycythemia vera treated with ruxolitinib or best available therapy after developing resistance/intolerance to hydroxyurea. Cancer 128(13):2441–2448. https://doi.org/10.1002/cncr.34195
Tefferi A, Vannucchi AM, Barbui T (2021) Polycythemia vera: historical oversights, diagnostic details, and therapeutic views. Leukemia 35(12):3339–3351. https://doi.org/10.1038/s41375-021-01401-3
Barbui T, Vannucchi AM, De Stefano V, Masciulli A, Carobbio A, Ferrari A, Ghirardi A, Rossi E, Ciceri F, Bonifacio M, Iurlo A, Palandri F, Benevolo G, Pane F, Ricco A, Carli G, Caramella M, Rapezzi D, Musolino C, Siragusa S, Rumi E, Patriarca A, Cascavilla N, Mora B, Cacciola E, Mannarelli C, Loscocco GG, Guglielmelli P, Betti S, Lunghi F, Scaffidi L, Bucelli C, Vianelli N, Bellini M, Finazzi MC, Tognoni G, Rambaldi A (2021) Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (low-PV study): a multicentre, randomised phase 2 trial. Lancet Haematol 8(3):e175–e184. https://doi.org/10.1016/S2352-3026(20)30373-2
Hoffman R, Ginzburg Y, Kremyanskaya M, Khanna S, Modi N, Valone FH, O’Connor PG, Gupta S, Saks SR (2022) Rusfertide (PTG-300) treatment in phlebotomy-dependent polycythemia vera patients. J Clin Oncol 40(16 suppl):7003–7003. https://doi.org/10.1200/JCO.2022.40.16_suppl.7003
Kuykendall A, Kremyanskaya M, Ginsburg Y, Pemmaraju N, Ritchie E, Gotlib J, Valone F, Khanna S, Gupta S, Hoffman R, Verstovsek S (2022) PTG-300 (Rusfertide) treatment interruption reverses hematological gains and restores therapeutic benefit on reinitiation in subjects with polycythemia vera. Presented at: European Hematology Association June 9–12, Vienna, Austria
Sapre M, Tremblay D, Wilck E, James A, Leiter A, Coltoff A, Koshy AG, Kremyanskaya M, Hoffman R, Mascarenhas JO, Gallagher EJ (2019) Metabolic effects of JAK1/2 inhibition in patients with myeloproliferative neoplasms. Sci Rep 9(1):16609. https://doi.org/10.1038/s41598-019-53056-x
Altomare I, Parasuraman S, Paranagama D, Kish J, Lord K, Yu J, Colucci P (2021) Real-world dosing patterns of ruxolitinib in patients with polycythemia vera who are resistant to or intolerant of hydroxyurea. Clin Lymphoma Myeloma Leuk 21(11):e915–e921. https://doi.org/10.1016/j.clml.2021.06.023
Palandri F, Palumbo GA, Elli EM, Polverelli N, Benevolo G, Martino B, Abruzzese E, Tiribelli M, Tieghi A, Latagliata R, Cavazzini F, Bergamaschi M, Binotto G, Crugnola M, Isidori A, Caocci G, Heidel F, Pugliese N, Bosi C, Bartoletti D, Auteri G, Cattaneo D, Scaffidi L, Trawinska MM, Stella R, Ciantia F, Pane F, Cuneo A, Krampera M, Semenzato G, Lemoli RM, Iurlo A, Vianelli N, Cavo M, Breccia M, Bonifacio M (2021) Ruxolitinib discontinuation syndrome: incidence, risk factors, and management in 251 patients with myelofibrosis. Blood Cancer J 11(1):4. https://doi.org/10.1038/s41408-020-00392-1
Acknowledgements
This study was funded by the Incyte Corporation (Wilmington, DE, USA). Writing assistance was provided by Nicole Farra, PhD, an employee of ICON (Blue Bell, PA, USA), and was funded by the Incyte Corporation.
Author information
Authors and Affiliations
Contributions
ATK contributed substantially to the conception, content development, writing, and review, and approved submission.
Corresponding author
Ethics declarations
Competing interests
AK received consulting and honoraria fees from the Incyte, Pharmaessentia, Protagonist, and Imago Biosciences; and research fees from Protagonist.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: This article was originally published without an Open Access but due to the authors final decision to opt for Open Choice this correction was created.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kuykendall, A.T. Treatment of hydroxyurea-resistant/intolerant polycythemia vera: a discussion of best practices. Ann Hematol 102, 985–993 (2023). https://doi.org/10.1007/s00277-023-05172-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-023-05172-y