
Abstract
Purpose
TAS-117 is a highly potent and selective, oral, allosteric pan-AKT inhibitor under development for advanced/metastatic solid tumors. The safety, clinical pharmacology, pharmacogenomics and efficacy were investigated.
Methods
This phase I, open-label, non-randomized, dose-escalating, first-in-human study enrolled patients with advanced/metastatic solid tumors and comprised three phases (dose escalation phase [DEP], regimen modification phase [RMP], and safety assessment phase [SAP]). The SAP dose and regimen were determined in the DEP and RMP. Once-daily and intermittent dosing (4 days on/3 days off, 21-day cycles) were investigated. The primary endpoints were dose-limiting toxicities (DLTs) in Cycle 1 of the DEP and RMP and incidences of adverse events (AEs) and adverse drug reactions (ADRs) in the SAP. Secondary endpoints included pharmacokinetics, pharmacodynamics, pharmacogenomics, and antitumor activity.
Results
Of 66 enrolled patients, 65 received TAS-117 (DEP, n = 12; RMP, n = 10; SAP, n = 43). No DLTs were reported with 24-mg/day intermittent dosing, which was selected as a recommended dose in SAP. In the SAP, 98.5% of patients experienced both AEs and ADRs (grade ≥ 3, 67.7% and 60.0%, respectively). In the dose range tested (8 to 32 mg/day), TAS-117 pharmacokinetics were dose proportional, and pharmacodynamic analysis showed a reduction of phosphorylated PRAS40, a direct substrate of AKT. Four patients in the SAP had confirmed partial response.
Conclusion
Oral doses of TAS-117 once daily up to 16 mg/day and intermittent dosing of 24 mg/day were well tolerated. TAS-117 pharmacokinetics were dose proportional at the doses evaluated. Antitumor activity may occur through AKT inhibition.
Trial registration
jRCT2080222728 (January 29, 2015).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2020, new cancer cases worldwide were estimated at 19.3 million, and there were nearly 10.0 million deaths from cancer [1]. The global burden of cancer is expected to rise over time, with a projected increase of 47% to 28.4 million cases by 2040. The phosphatidylinositol-3-kinase (PI3K)-v-akt murine thymoma viral oncogene homolog (AKT)-mammalian target of rapamycin (mTOR) signaling pathway regulates numerous cellular functions, including proliferation, differentiation, metabolism, survival, motility, and autophagy [2]. Many types of tumors have altered PI3K/AKT/mTOR signaling. In the 1980s, researchers established a clear link between this pathway and cancer. More recently, this signal transduction network has emerged as one of the most frequently dysregulated pathways in human tumors [3]. The AKT family has three highly conserved isoforms: AKT1/α and AKT2/β, which are ubiquitously expressed, and AKT3/γ, which has a more restricted tissue distribution and is abundant in nervous tissues [2, 4]. AKT activation occurs in many types of cancers, including acute myeloid leukemia, brain, breast, colon, endometrial cancer, gastric, head and neck, lung, melanoma, multiple myeloma, ovarian, pancreas, prostate, and renal cell carcinoma, and it is a key factor in the oncogenic pathway [2, 5,6,7,8]. Reportedly, activation of AKT is associated with a poor prognosis and chemotherapy resistance [9,10,11,12,13]. Thus, AKT is expected to be a promising target in PI3K/AKT/mTOR pathway-activated tumors.
Most AKT inhibitors currently in clinical development are considered pan-AKT inhibitors because they inhibit all three isoforms of AKT [14]. There are two types of pan-AKT inhibitors: adenosine triphosphate (ATP)-competitive inhibitors that bind to the active conformation of AKT, thereby exposing the ATP-binding pocket [15, 16]; and allosteric inhibitors that prevent localization of AKT, thereby blocking AKT phosphorylation and activation [17]. Allosteric AKT inhibitors are more specific than ATP-competitive inhibitors, which have a higher off-target effect [18, 19].
TAS-117 (Taiho Pharmaceutical Co., Ltd., Tokyo, Japan) is an AKT inhibitor being developed for the treatment of advanced or metastatic solid tumors. TAS-117 is a highly potent and selective, oral, allosteric pan-AKT inhibitor [20] with strong anti-proliferative activity against multiple tumor cell lines derived from human cancers, including breast, ovarian, gastric, endometrial, and myeloma (data on file, Taiho Pharmaceutical Co., Ltd.). In preclinical studies, oral dosing of TAS-117 in murine xenograft models of gastric cancer (NCI-N87; HER2 amplification) and breast cancer (KPL-4; HER2 amplification, PIK3CA mutation) has demonstrated single-agent antitumor activity and was well tolerated [20]. TAS-117 has synergistic or additive effects with chemotherapeutic agents both in vitro and in vivo [21]. The toxicity of repeated TAS-117 dosing has been evaluated in rats and monkeys, including 4-week oral toxicity studies. TAS-117-associated toxicity findings common to both species were dysregulation of carbohydrate metabolism and atrophic changes in the lymphatic/hematopoietic organs and adipocytes (data on file, Taiho Pharmaceutical Co., Ltd.). These toxicological changes tended to return to normal at the end of the recovery period.
This phase I clinical study aimed to investigate the safety, pharmacokinetics, antitumor activity, pharmacodynamics, and pharmacogenomics of TAS-117 and determine the recommended dose (RD) for further development.
Materials and methods
Patients
Patients were eligible for inclusion in this study if they were ≥ 20 years of age at enrollment, had histologically or cytologically confirmed advanced or metastatic solid tumors with no standard treatment option, had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1, were able to take medications orally, had adequate organ function, and had a life expectancy of at least 60 days. The main exclusion criteria were past or current type 1 or type 2 diabetes requiring treatment, retinopathy requiring ophthalmological therapy, past or present cardiac arrhythmia and/or conduction abnormality, major surgery or extended field radiation therapy within the 4 weeks prior to or limited field radiation therapy within the 2 weeks prior to study drug administration, anticancer treatment within 3 weeks prior to study drug administration, treatment with an investigational agent in the 3 weeks prior to study drug administration, unresolved toxicity of grade > 1 attributed to any prior therapies, current oral steroid treatment, or pregnant or lactating. A complete list of exclusion criteria is provided in Supplemental Text 1. All patients provided written informed consent for study participation.
Study design and treatments
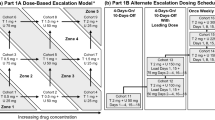
This was a phase I, open-label, non-randomized, dose-escalating first-in-human study to evaluate the safety, pharmacokinetics, antitumor activity, pharmacodynamics, and pharmacogenomics of TAS-117 in patients with advanced solid tumors. The study had three phases: a dose escalation phase (DEP), a regimen modification phase (RMP), and a safety assessment phase (SAP). The treatment cycle on each regimen lasted 21 days. The DEP used an accelerated titration and 3 + 3 design. TAS-117 was administered orally once daily (QD) on an empty stomach (1 h before or 2 h after a meal). Dosing was determined based on a 4-week repeated oral-dose toxicology study in monkeys (data on file, Taiho Pharmaceutical Co., Ltd.); the highest non-severely toxic dose was 1.2 mg/kg/day, which was converted to 3.84 mg/body/day, or 2.4 mg/m2. Therefore, the starting dose was determined to be ≤ 2.4 mg/m2. At dose level 1, TAS-117 was administered at 2 mg QD (Day − 2), and the dose was increased to 4 mg QD after 2 days (Day 1). The RMP was conducted according to a 3 + 3 design. During this phase, the dose regimen was changed from QD administration to intermittent dosing of daily TAS-117 with 4 days on/3 days off. TAS-117 was administered on an empty stomach. Dose-limiting toxicities (DLTs) were evaluated in Cycle 1 of the DEP and RMP. The maximum tolerated dose (MTD) was defined as the highest dose level at which DLTs were observed in < 33% of patients in Cycle 1. The RD and regimen for the SAP were determined based on the safety, pharmacokinetics, pharmacodynamics, and antitumor activity profile determined in the DEP and RMP. During the SAP, the safety and preliminary efficacy profiles of TAS-117 were assessed at the RD and recommended regimen. Three cohorts were included in this assessment. Cohort 1 included patients with endometrial cancer with a PIK3CA mutation; cohort 2 included patients with endometrial cancer with an AKT1 mutation, AKT1 amplification, or AKT2 amplification; and cohort 3 included patients with ovarian clear cell carcinoma.
Blood samples for pharmacokinetic analysis were collected immediately prior to dosing (0 h) and at 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h post-dose on Day 1 and Day 21 of Cycle 1 for patients in the DEP (QD dosing). For patients who received intermittent dosing regimens, blood was collected immediately prior to dosing (0 h) and at 1, 2, 3, 4, 6, 8, 10, and 24 h post-dose on Cycle 1 Day 1; immediately prior to dosing (0 h) on Cycle 1 Days 3, 4, and 8; and immediately prior to dosing (0 h) and at 1, 2, 3, 4, 6, 8, 10, 24, 48, and 96 h post-dose on Cycle 1 Day 18. Urine samples for pharmacokinetic analysis were collected immediately prior to dosing (single sample collection) and from 0 to 6 h and from 6 to 12 h and 12 to 24 h post-dose on Day 1 of Cycle 1 for both QD and intermittent dosing. Blood samples for pharmacodynamic analysis were collected at pre-dose and 4, 8, and 24 h post-dose on Cycle 1 Day 1, and at pre-dose and 4 h post-dose on Cycle 1 Day 21 (QD dosing) or Cycle 1 Day 18 (intermittent dosing).
The phosphorylated proline-rich AKT substrate of 40 kDa (pPRAS40) level was found to be an indicator of AKT activation, given that PRAS40 is a substrate of AKT [22]. The pharmacodynamic activity of TAS-117 for AKT inhibition was evaluated by the change in the ratio of pPRAS40 (threonine 246) to total PRAS40 in platelet-rich plasma samples before and after TAS-117 administration. The phosphorylation status of PRAS40 in the blood was measured with an electrochemiluminescence detection assay using a Meso Scale Discovery instrument (Meso Scale Diagnostics, Rockville, MD, US). For efficacy, solid tumors were assessed using Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1). Computed tomography scans were evaluated at baseline, every 6 weeks after initiating TAS-117 administration, and at the time of study discontinuation.
Blood samples for pharmacogenomics analyses were collected at baseline (within 7 days of the first study drug administration) for the DEP and RMP and cohort 3 of the SAP. Because SAP cohorts 1 and 2 included patients positive for PIK3CA, or AKT gene abnormalities, blood samples for patients in these cohorts were collected during the biomarker screening period (i.e., after obtaining consent for pharmacogenomics pre-testing and before obtaining consent for study participation) to confirm eligibility. Thus, additional blood samples were optional for patients who had previously been reliably confirmed to be positive for PIK3CA or AKT gene abnormalities. Cell-free DNA in blood was analyzed for PIK3CA and AKT1 mutations and AKT1 and AKT2 amplifications using droplet digital polymerase chain reaction (PCR). Formalin-fixed paraffin-embedded (FFPE) tumor tissues obtained during surgery or used for definitive diagnoses were collected for pharmacogenomics analyses (required for participants in the SAP, optional for participants in the DEP and RMP). FFPE tumor tissue was evaluated for the presence of PIK3CA using PCR and for AKT1 mutation and AKT1 and AKT2 amplifications using droplet digital PCR; messenger RNA levels of AKT1, AKT2, AKT3, PIK3C2B, PHLDA1, and ACTB using real-time reverse transcriptase PCR; levels of AKT2 and pan-AKT protein using immunohistochemistry; and analysis of genomic abnormalities using next-generation sequencing.
This study was initially registered at the Japic Clinical Trials Information (JapicCTI, registered as JapicCTI-152780 on January 29, 2015). Because of the integration of JapicCTI into the Japan Registry of Clinical Trials (jRCT), the study was registered to the jRCT (jRCT2080222728) on January 29, 2015. The registration date remains the same as in the JapicCTI.
Outcomes
The primary endpoints were the proportion of patients who experienced DLTs in Cycle 1 of the DEP and RMP and the incidence of adverse events (AEs) and adverse drug reactions (ADRs) in the SAP. AEs and ADRs were coded using the Medical Dictionary for Regulatory Activities (MedDRA; version 24.1) and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE; version 4.03). Secondary endpoints were pharmacokinetic findings, changes in the ratio of phosphorylated PRAS40 (pPRAS40)/total PRAS40 from baseline, efficacy, and pharmacogenomics findings.
Statistical methods
The sample size for the DEP and RMP was not determined based on statistical considerations. Given that both phases aimed to explore different dose levels based on the occurrence of DLTs, a sample size could not be predetermined. It was assumed that the MTD and pharmacokinetics of TAS-117 could be determined and safety evaluated with a maximum of 54 patients for the DEP and a maximum of 54 patients for each regimen in the RMP. For the SAP, the sample size was determined as 20 patients for each cohort. With a sample size of 20, a null proportion of 5%, and a confidence level of 5%, the power was calculated to be > 80% when an alternative proportion was ≥ 30%.
Regarding the analysis populations, all treated patients included all patients who were enrolled and received at least one dose of TAS-117, and the full analysis set included all patients who were administered the study drug and had data for at least one efficacy endpoint.
Descriptive data were used to summarize safety data and antitumor activity. Pharmacokinetic parameters were derived using non-compartmental analysis and summarized descriptively by dose level, study day, and study phase. The dose proportionality of the maximum plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) of TAS-117 on Day 1 were assessed using linear regression and power model analyses. The accumulation of TAS-117 was assessed by comparing pharmacokinetic parameters on Day 1 and Day 21 (QD) or Day 18 (intermittent dosing). Pharmacodynamic data were presented using summary statistics of the ratio of pPRAS40/total PRAS40 to baseline values (pre-dose) on Day 1 and Day 18 or 21, by dose level and treatment regimen. Pharmacogenomics data were summarized, and the relevance between biomarkers and efficacy was analyzed using Fisher’s Exact test.
P-values < 0.05 were considered significant. Missing data were not imputed, and analyses were performed using all available data. Plasma pharmacokinetic parameters were evaluated using Phoenix® WinNonlin® software (Pharsight Corporation as part of Certara, Princeton, NJ, US, Version 8.1 or 8.3). Statistical analyses were performed using SAS version 9.4 (SAS Institute; Cary, NC, US).
Results
Patients
Patients were enrolled between 23 February 2015 and 10 June 2019; the data cut-off date was 31 December 2021. Patient enrollment for each study phase is shown in Supplemental Fig. 1. Twelve patients were enrolled and treated in the DEP and received TAS-117 at dose levels 1 to 4 (4, 8, 16, and 24 mg QD). Ten patients were enrolled and treated in the RMP and received TAS-117 at a dose of either 24 or 32 mg daily with 4 days on/3 days off. The SAP enrolled 16 patients in cohort 1, 7 in cohort 2, and 21 in cohort 3. Patients in the SAP were treated with TAS-117 24 mg daily for 4 days on/3 days off. One patient enrolled in cohort 3 did not receive treatment.

Pharmacokinetics of TAS-117. a Mean plasma concentration–time profiles of TAS-117 in the DEP (QD dosing). Data are shown as mean (SD), blue circles represent TAS-117 concentration following Day 1 dosing, and orange circles represent TAS-117 dosing following Day 21. b Mean plasma concentration–time profiles of TAS-117 in the RMP (intermittent dosing; 4 days on/3 days off). Data are shown as mean (SD), blue circles represent TAS-117 concentration following Day 1 dosing and orange circles represent TAS-117 dosing following Day 18. c Dose proportionality of Cmax, AUClast, and AUC0–24 across TAS-117 dosing. Open orange circles represent individual values, and open blue triangles represent mean values. AUC area under the plasma concentration–time curve, AUC0–24 AUC from time 0 to 24 h, AUClast AUC up to the last observable concentration, Cmax maximum plasma concentration, DEP dose escalation phase, QD once daily, RMP regimen modification phase, SD standard deviation
Demographic and background characteristics for all treated patients are shown in Table 1. The median (min, max) age of patients was 59 years (24, 78), and 78.5% were female (100% of patients in the SAP were female). Most patients had ECOG PS of 0 (83.1%), and all were Asian. Nearly all patients had metastatic tumors (98.5%) and histories of surgery (89.2%) and chemotherapy (98.5%). The most common type of cancer among patients was endometrial (35.4%), followed by ovarian (30.8%), other (16.9%), colorectal (9.2%), and finally bladder, breast, gastrointestinal stromal tumor, pancreatic, and uterus (all, 1.5%).
Safety
In the DEP, TAS-117 was administered QD from dose levels 1 (4 mg) to 4 (24 mg). All patients at dose level 4 experienced grade 2 maculopapular rash requiring study interruption. Thus, the RD was determined to be TAS-117 16 mg QD. In the RMP, intermittent administration (4 days on/3 days off) at dose level 4 (24 mg) and dose level 5 (32 mg) were evaluated. At dose level 5, one of the three patients in whom DLT was evaluated experienced a DLT of grade 3 maculopapular rash during Cycle 1, resulting in treatment interruption. In Cycle 1 and Cycle 2 of dose level 5, two of three patients had grade 3 maculopapular rash, and treatment with TAS-117 was interrupted, and/or a dose reduction was necessary. Given that no DLTs were reported at dose level 4, 24 mg was determined to be the RD for intermittent dosing. A dose of TAS-117 24 mg intermittent administration was evaluated in the SAP.
AEs and ADRs (ADRs with a frequency of ≥ 10%) are shown in Table 2. Nearly all patients experienced any AE (98.5%) and any ADR (98.5%); 67.7% and 60.0% experienced grade ≥ 3 AEs and ADRs, respectively. ADRs with a frequency of ≥ 20% in all treated patients included all-grade nausea, stomatitis, pyrexia, neutrophil count decreased, white blood count decreased, hyperglycemia, pruritus, and maculopapular rash (both all-grade and grade ≥ 3). In total, four patients at dose level 4 (24 mg QD intermittent administration) discontinued because of AEs, which included cellulitis (n = 1; serious, not related to treatment), pneumonitis (n = 1; serious, related to treatment), rash maculopapular (n = 1; not serious, related to treatment), and γ-glutamyltransferase increased (n = 1; not serious, related to treatment). One patient (SAP, cohort 1) died because of grade 5 respiratory failure, which was related to the patient’s underlying disease and judged by the study investigator as unrelated to TAS-117 treatment.
Pharmacokinetics
Following the Cycle 1 Day 1 dose, the terminal phase elimination half-life (t1/2) of TAS-117 ranged from 22 to 32 h, and the time to maximum plasma concentration (tmax) ranged from 2 to 4 h, regardless of dose (Supplemental Table 1). The accumulation ratio of Cmax and AUC from 0 to 24 h (AUC0–24) was approximately 3- to 4-fold after repeated QD dosing and approximately 2- to 3-fold after repeated intermittent dosing (Supplemental Table 1). The t1/2 data indicate that a steady state was reached by Day 8. The urinary excretion rate ranged from 8 to 11%. The mean plasma concentration–time profiles of TAS-117 are shown in Fig. 1a and 1b. The dose proportionality analysis for the 8, 16, 24, and 32 mg doses is shown in Fig. 1c. There was a dose proportional increase in Cmax, AUC up to the last observable concentration (AUClast), and AUC0–24 in the 8 to 32 mg dose range. The 95% confidence intervals (CIs) of intercepts of linear regression lines for Cmax, AUClast, and AUC0-24 contained 0, and the 95% CI of slopes of power regression lines contained 1, thus fulfilling the criteria for dose proportionality (Supplemental Table 2).
Pharmacodynamics
The percentage change in pPRAS40/total PRAS40 values from before to after TAS-117 administration is shown in Fig. 2. After TAS-117 administration, a reduction in the level of pPRAS40 (> 50% at any time point) was observed in the DEP, RMP, and SAP for the range of TAS-117 dose levels 2 to 5 (8 to 32 mg/body/day).

Mean changes in the ratio of pPRAS40/total PRAS40 after TAS-117 administration on Cycle 1 Day 1 compared with pre-dose. The phosphorylation status of PRAS40 in the blood was measured with an electrochemiluminescence detection assay and a Meso Scale Discovery instrument (Meso Scale Diagnostics, Rockville, MD, US). aRatio of pPRAS40/total PRAS40 (percentage compared with Day 1 pre-dose). C cohort, DEP dose escalation phase, p phosphorylated, QD once daily, RMP regimen modification phase, SAP safety assessment phase
Efficacy
No patients in the DEP or RMP had a complete response (CR) or partial response (PR); in the SAP, no patients had CR, and 1 of 16 patients (6.3%) in cohort 1 and 3 of 20 patients (15.0%) in cohort 3 had confirmed PR (Table 3). However, the DCR was low for patients in the DEP or RMP (0 to 33.3%) except for dose level 2 in the DEP (100%). The DCR was 68.8%, 85.7%, and 40.0% for patients in cohort 1, cohort 2, and cohort 3, respectively, in the SAP.
Pharmacogenomics
Pharmacogenomics results of AKT and PIK3CA gene abnormalities tested by PCR or droplet digital PCR in tumor tissue samples for patients in the SAP with endometrial cancer (cohorts 1 and 2; n = 23) and those with ovarian clear cell carcinoma (cohort 3; n = 20) are shown in Fig. 3. Given the lack of adequate clinical response in patients in the DEP, RMP, and cohorts 1 and 2 in the SAP, those groups were not considered suitable for examining predictive biomarkers. Among patients with ovarian clear cell carcinoma (cohort 3, SAP), 3 of 20 patients had PR; thus, candidate predictive biomarkers were analyzed in this population. However, no factors predictive of clinical response were identified.

Maximum tumor change from baseline and pharmacogenomics (SAP). The status of PIK3CA mutation, AKT1 mutation, and AKT1 and AKT2 amplification is displayed. Formalin-fixed paraffin-embedded tumor tissue specimens were tested at a central laboratory for the following: PIK3CA:PIK3CA mutation, AKT1M:AKT1 mutation, AKT1A:AKT1 amplification, and AKT2A:AKT2 amplification. PIK3CA data were obtained using PCR, and AKT1M, AKT1A, and AKT2A data were obtained using droplet digital PCR. Endometrial cancer patients with unlabeled PIK3CA or AKT gene abnormality were tested by next-generation sequencing for tumor tissues, droplet digital PCR for blood samples, or local tests. PCR polymerase chain reaction, PD progressive disease, PR partial response, SAP safety assessment phase, SD stable disease
Discussion
The RD dosing for TAS-117 was determined as 16 mg per day for QD dosing and 24 mg per day for intermittent dosing in patients with advanced or metastatic solid tumors. TAS-117 demonstrated acceptable safety and pharmacokinetic profiles as well as inhibitory activity against AKT; anti-tumor activity was suggested in some patients.
Skin-related and gastrointestinal AEs, myelosuppression, and hyperglycemia were frequently observed with TAS-117 treatment. These AEs are known class side effects associated with PI3K/AKT/mTOR inhibitors [23]. Most patients (93.8%) developed AEs related to skin disorders; maculopapular rash was the most common (89.2%), followed by pruritis, dry skin, and rash. Maculopapular rash was grade ≥ 3 for 35.4% of patients with this AE. Skin-related AEs resolved on their own or with TAS-117 discontinuation or dose reduction, or with the administration of topical steroids, antihistamines, or oral steroids. The safety profile of TAS-117 was acceptable at doses up to 16 mg per day with QD dosing and up to 24 mg per day with intermittent dosing.
The pharmacokinetics of TAS-117 were dose proportional in the dosing range evaluated (8 to 32 mg). In the SAP, four patients (cohort 1 PIK3CA mutation-positive endometrial cancer, n = 1; cohort 3 ovarian clear cell carcinoma, n = 3) had confirmed PR, suggesting that TAS-117 has antitumor activity in patients with selected advanced solid cancers. Given the limited availability of effective treatment in ovarian clear cell carcinoma overall, this finding in three patients with ovarian clear cell carcinoma is worth further investigation. Ovarian clear cell carcinoma is relatively rare in the US and Europe (< 10% of ovarian cancers) [24, 25], but in Asian countries, the incidence rates are higher (13 to 25% of ovarian cancers) and have been increasing over time [26,27,28]. With later-stage disease, the response rate to conventional platinum-based therapy is lower compared with other major histological subtypes [29], and few clinical trials have specifically targeted ovarian clear cell carcinoma [30]. Thus, there is a great unmet need for effective treatment strategies for these patients.
Although the efficacy findings of this study are encouraging, this first-in-human study was limited by the small number of participants and the absence of a control group. Further studies investigating TAS-117 as monotherapy, and in combination with other approved oncology drugs, in larger patient populations are needed.
In conclusion, this study demonstrated that TAS-117 was well tolerated at doses up to 16 mg/day with QD dosing and at intermittent doses of 24 mg/day (4 days on/3 days off). All AEs were manageable by dose interruption, dose reduction, or targeted therapy. TAS-117 pharmacokinetics were dose proportional in the doses evaluated and its pharmacodynamics suggest that TAS-117 exhibits inhibitory activity against AKT in humans. Altogether with the antitumor activity findings, these findings suggest that TAS-117 exhibits antitumor activity by inhibiting AKT.
Data and/or code availability
Data will not be shared according to the Sponsor’s policy on data sharing, which can be found at: https://www.taiho.co.jp/en/science/policy/clinical_trial_information_disclosure_policy/.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71:209–249. https://doi.org/10.3322/caac.21660
Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, McCubrey JA (2010) The emerging role of the phosphatidylinositol 3-kinase/ akt/mammalian target of rapamycin signaling network in cancer stem cell biology. Cancers (Basel) 2:1576–1596. https://doi.org/10.3390/cancers2031576
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27:5497–5510. https://doi.org/10.1038/onc.2008.245
Brazil DP, Yang ZZ, Hemmings BA (2004) Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 29:233–242. https://doi.org/10.1016/j.tibs.2004.03.006
Staal SP (1987) Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A 84:5034–5037. https://doi.org/10.1073/pnas.84.14.5034
Stanciu S, Ionita-Radu F, Stefani C, Miricescu D, Stanescu-Spinu II, Greabu M, Ripszky Totan A, Jinga M (2022) Targeting PI3K/AKT/mTOR signaling pathway in pancreatic cancer: from molecular to clinical aspects. Int J Mol Sci 23:10132. https://doi.org/10.3390/ijms231710132
Vander Broek R, Mohan S, Eytan DF, Chen Z, Van Waes C (2015) The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross-talk, and therapies. Oral Dis 21:815–825. https://doi.org/10.1111/odi.12206
Isa R, Horinaka M, Tsukamoto T, Mizuhara K, Fujibayashi Y, Taminishi-Katsuragawa Y, Okamoto H, Yasuda S, Kawaji-Kanayama Y, Matsumura-Kimoto Y, Mizutani S, Shimura Y, Taniwaki M, Sakai T, Kuroda J (2022) The rationale for the dual-targeting therapy for RSK2 and AKT in multiple myeloma. Int J Mol Sci 23:2919. https://doi.org/10.3390/ijms23062919
Huang WC, Hung MC (2009) Induction of Akt activity by chemotherapy confers acquired resistance. J Formos Med Assoc 108:180–194. https://doi.org/10.1016/S0929-6646(09)60051-6
VanderWeele DJ, Zhou R, Rudin CM (2004) Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther 3:1605–1613
Fraser M, Bai T, Tsang BK (2008) Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int J Cancer 122:534–546. https://doi.org/10.1002/ijc.23086
Yoshioka A, Miyata H, Doki Y, Yasuda T, Yamasaki M, Motoori M, Okada K, Matsuyama J, Makari Y, Sohma I, Takiguchi S, Fujiwara Y, Monden M (2008) The activation of Akt during preoperative chemotherapy for esophageal cancer correlates with poor prognosis. Oncol Rep 19:1099–1107
Terakawa N, Kanamori Y, Yoshida S (2003) Loss of PTEN expression followed by Akt phosphorylation is a poor prognostic factor for patients with endometrial cancer. Endocr Relat Cancer 10:203–208. https://doi.org/10.1677/erc.0.0100203
Popova NV, Jücker M (2021) The role of mTOR signaling as a therapeutic target in cancer. Int J Mol Sci 22:1743. https://doi.org/10.3390/ijms22041743
Brown JS, Banerji U (2017) Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol Ther 172:101–115. https://doi.org/10.1016/j.pharmthera.2016.12.001
Huck BR, Mochalkin I (2017) Recent progress towards clinically relevant ATP-competitive Akt inhibitors. Bioorg Med Chem Lett 27:2838–2848. https://doi.org/10.1016/j.bmcl.2017.04.090
Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP, Brandhuber BJ (2010) Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 5:e12913. https://doi.org/10.1371/journal.pone.0012913
Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE, Kahana JA, Kral AM, Leander K, Lee LL, Malinowski J, McAvoy EM, Nahas DD, Robinson RG, Huber HE (2005) Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J 385:399–408. https://doi.org/10.1042/BJ20041140
DeFeo-Jones D, Barnett SF, Fu S, Hancock PJ, Haskell KM, Leander KR, McAvoy E, Robinson RG, Duggan ME, Lindsley CW, Zhao Z, Huber HE, Jones RE (2005) Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol Cancer Ther 4:271–279
Abe T, Ichikawa K, Fujita R, Okada M, Tanaka K, Fujino N, Ohkubo M, Yonekura K, Shimomura T, Utsugi T (2012) Characterization of TAS-117, a novel, highly potent and selective inhibitor of AKT. Eur J Cancer 48:108–109. https://doi.org/10.1016/S0959-8049(12)72154-8
Ichikawa K, Abe T, Fukushima H, Fujita R, Okada M, Yonekura K, Shimomura T, Utsugi T (2012) A novel highly potent AKT inhibitor TAS-117 demonstrated synergistic antitumor activity in combination with paclitaxel through enhancement of apoptosis induction. Eur J Cancer 48:108. https://doi.org/10.1016/S0959-8049(12)72153-6
Wang H, Zhang Q, Wen Q, Zheng Y, Lazarovici P, Jiang H, Lin J, Zheng W (2012) Proline-rich Akt substrate of 40kDa (PRAS40): a novel downstream target of PI3k/Akt signaling pathway. Cell Signal 24:17–24. https://doi.org/10.1016/j.cellsig.2011.08.010. (Erratum in: Cell Signal. 2012;24:2226. Philip, Lazarovici [corrected to Lazarovici, Philip])
Zhang Y, Yan H, Xu Z, Yang B, Luo P, He Q (2019) Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin Drug Metab Toxicol 15:767–774. https://doi.org/10.1080/17425255.2019.1663169
Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, Gaudet MM, Jemal A, Siegel RL (2018) Ovarian cancer statistics, 2018. CA Cancer J Clin 68:284–296. https://doi.org/10.3322/caac.21456
Coburn SB, Bray F, Sherman ME, Trabert B (2017) International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Int J Cancer 140:2451–2460. https://doi.org/10.1002/ijc.30676
Yamagami W, Nagase S, Takahashi F, Ino K, Hachisuga T, Aoki D, Katabuchi H (2017) Clinical statistics of gynecologic cancers in Japan. J Gynecol Oncol 28:e32. https://doi.org/10.3802/jgo.2017.28.e32
Kim SI, Ha HI, Eoh KJ, Lim J, Won YJ, Lim MC (2022) Trends in the incidence and survival rates of primary ovarian clear cell carcinoma compared to ovarian serous carcinoma in Korea. Front Oncol 12:874037. https://doi.org/10.3389/fonc.2022.874037
Chiang YC, Chen CA, Chiang CJ, Hsu TH, Lin MC, You SL, Cheng WF, Lai MS (2013) Trends in incidence and survival outcome of epithelial ovarian cancer: 30-year national population-based registry in Taiwan. J Gynecol Oncol 24:342–351. https://doi.org/10.3802/jgo.2013.24.4.342
Oliver KE, Brady WE, Birrer M, Gershenson DM, Fleming G, Copeland LJ, Tewari K, Argenta PA, Mannel RS, Secord AA, Stephan JM, Mutch DG, Stehman FB, Muggia FM, Rose PG, Armstrong DK, Bookman MA, Burger RA, Farley JH (2017) An evaluation of progression free survival and overall survival of ovarian cancer patients with clear cell carcinoma versus serous carcinoma treated with platinum therapy: An NRG Oncology/Gynecologic Oncology Group experience. Gynecol Oncol 147:243–249. https://doi.org/10.1016/j.ygyno.2017.08.004
Takahashi K, Takenaka M, Kawabata A, Yanaihara N, Okamoto A (2020) Rethinking of treatment strategies and clinical management in ovarian clear cell carcinoma. Int J Clin Oncol 25:425–431. https://doi.org/10.1007/s10147-020-01625-w
Acknowledgements
The authors wish to thank the patients and their families, who made this trial possible through their participation. We also thank the clinical study teams that were involved. This study was sponsored by Taiho Pharmaceutical, Co., Ltd. Finally, the authors thank Sarah Bubeck, PhD, of Edanz (www.edanz.com) for providing medical writing support, which was funded by Taiho Pharmaceutical Co., Ltd., in accordance with Good Publication Practice (GPP 2022) guidelines (https://www.ismpp.org/gpp-2022).
Funding
This study was funded by Taiho Pharmaceutical Co., Ltd.
Author information
Authors and Affiliations
Contributions
TD: contributed to the conception or design of the study, acquisition of data, analysis of data, and interpretation of data. ST, DA, KY, HH, KHasegawa, KT, KHarano, MY, HN, EN, KHorie, AO, and SO: contributed to acquisition of data, analysis of data, and interpretation of data. All authors were involved in reviewing and editing, and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
TD has received institutional grants or contracts from PRA Health Sciences, MSD, Daiichi Sankyo, Amgen, Taiho, GSK, ONO Pharma, Janssen Pharma, Boehringer Ingelheim, Pfizer, BMS, Abbvie, Eisai, RIN Institute, Chugai Pharma, and SHIONOGI; consulting fees from Sumitomo Pharma, Oncolys BioPharma, Takeda, Chugai Pharma, Boehringer Ingelheim, Nano Carrier, Rakuten Medical, Otsuka Pharma, KAKEN Pharma, KYOWA KIRIN, SHIONOGI, PRA Health Science, A2 Health Care, and Noil-Immune Biotech; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Daiichi Sankyo; and has participated on a Data Safety Monitoring Board or Advisory Board for Gilead, Pfizer, Amgen, and ZYMEWORKS BIOPHARMA. ST has received institutional grants from Astellas, Bayer, Chugai, Daiichi Sankyo, Eisai, Eli Lilly, MSD, Novartis, Ono, Sanofi, and Taiho; and honoraria from Astellas, Bayer, Chugai, Daiichi Sankyo, Eisai, MSD, Ono, Sanofi, and Taiho. DA has received honoraria from AstraZeneca, Chugai, Eisai, Genmab, MSD, Myriad Genetics, and Takeda. KY has received honoraria from Pfizer, Eisai, AstraZeneca, Eli Lilly, Takeda, Chugai, Fuji Film Pharma, PDR pharma, MSD, Boehringer Ingelheim, Ono, Daiichi Sankyo, Bayer, Janssen, and Sanofi; served on the advisory boards of Eisai, AstraZeneca, Sanofi, Genmab, Gilead, OncXerna, Takeda, Novartis, and MSD; and has received institutional research grants and been a Principal Investigator for MSD, Daiichi Sankyo, AstraZeneca, Taiho, Pfizer, Novartis, Takeda, Chugai, Ono, Sanofi, Seattle Genetics, Eisai, Eli Lilly, Genmab, Boehringer Ingelheim, Kyowa Hakko Kirin, Nihon Kayaku, and Haihe. HH has received honoraria from Bayer, Bristol-Myers Squibb, Chugai, Daiichi Sankyo, Kyowa Hakko Kirin, Lilly, Merck Biopharma, MSD, Ono, Taiho, Takeda, and Yakult; held consulting or advisory roles for Bristol-Myers Squibb, Boehringer Ingelheim, and Daiichi Sankyo; and received research grants from ALX Oncology, Amgen, Astellas, AstraZeneca, Bayer, BeiGene, Boehringer Ingelheim, Bristol-Myers Squibb, Chugai, Daiichi Sankyo, Eisai, Janssen, Merck Biopharma, MSD, Ono, and Taiho. KHasegawa has received consultancy fees from Takeda, MSD, Sanofi, and Eisai; institutional grants from Takeda, MSD, and Ono; honoraria from AstraZeneca, Takeda, MSD, Sanofi, Chugai, Eisai, and Genmab; and travel/accommodation expenses from Regeneron. KT has received honoraria from Taiho Pharmaceutical Co. Ltd. KHarano has received consultancy fees from Taiho and Takeda; institutional grants from Merck and Daiichi-Sankyo; and payment for development of educational presentations including service on speakers' bureaus from AstraZeneca, Chugai, MSD, Taiho, and Takeda. MY has received honoraria from Taiho Pharmaceutical Co., Ltd, AstraZeneca, Takeda Pharmaceutical Co., MSD, Merck, Chugai Pharmaceutical Co., Mochida Pharmaceutical Co., and Eisai Pharmaceutical Co. HN, TS, KHorie, AO, and SO have no conflicts of interest to declare.
Ethical approval
This study was conducted in accordance with the study protocol, Good Clinical Practice guidelines, International Council for Harmonisation Guidelines, the ethical principles originating in the Declaration of Helsinki, and all applicable regulatory requirements.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Consent to publish
Patients signed informed consent regarding publishing their data.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Doi, T., Takahashi, S., Aoki, D. et al. A first-in-human phase I study of TAS-117, an allosteric AKT inhibitor, in patients with advanced solid tumors. Cancer Chemother Pharmacol 93, 605–616 (2024). https://doi.org/10.1007/s00280-023-04631-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-023-04631-7