
Abstract
Despite the advances in treatment options, cardiovascular disease (CVDs) remains the leading cause of death over the world. Chronic inflammatory response and irreversible fibrosis are the main underlying pathophysiological causes of progression of CVDs. In recent decades, cardiac macrophages have been recognized as main regulatory players in the development of these complex pathophysiological conditions. Numerous approaches aimed at macrophages have been devised, leading to novel prospects for therapeutic interventions. Our review covers the advancements in macrophage-centric treatment plans for various pathologic conditions and examines the potential consequences and obstacles of employing macrophage-targeted techniques in cardiac diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases (CVDs) encompass a spectrum of pathological conditions affecting the heart or blood vessels, including myocardial infarction (MI), arrhythmia and atherosclerosis. Despite significant advancements in research and pharmacological therapies, CVDs such as MI are still a leading cause of global health burden and major contributor to disability [1]. With an aging and expanding global population, the death from CVDs has even raised 60% over the last 30 years and is mostly linked to ischemic heart disease and ischemic stroke-related death (World Heart Federation 2021). According to the global burden of diseases, injuries and risk factors (GBD) study of CVDs related death between 1990 and 2021, metabolic risks, dietary risks and environmental risks are the primary risk factors of CVDs progression and have been found to increase the prevalence of comorbidities like chronic obstructive pulmonary disease (COPD) or diabetes [1, 2]. The most common element of all these risk factors is increased tissue inflammation, which is often negatively associated with survival in patients with CVDs. Intensive investigations in recent decades have illuminated the pivotal role of inflammation in the pathogenesis of CVDs. While acute inflammation is a host-protective response against tissue damage or external stimuli, it must be resolved in a timely manner to maintain homeostasis and facilitate optimal tissue repair [3]. In contrast, unresolved inflammation serves as a catalyst for the progression of chronic inflammation and is associated with an increased risk of heart failure (HF) [4]. Therefore, several strategies to regulate and balance inflammation have been proposed as treatment options for CVDs.
The tissue inflammation is now better appreciated by the dynamic activation status of diverse immune cells including neutrophils, macrophages and T cells in diverse disease contexts. Among them, macrophages are the most abundant immune cells in cardiac tissue, playing a crucial role in the maintenance of homeostasis and cardiac development [5, 6]. Moreover, being a key player to balance inflammation and local microenvironment upon injury, the residence and polarization of macrophages are closely related to HF progression including fibrogenesis or cardiac remodeling [7, 8]. The potential for macrophage-targeted therapies, such as modulating macrophage phenotypes, has garnered significant attention in the CVD field in recent years [9]. Advanced techniques in science and research have revealed the existence of various types of macrophages, each exhibiting distinctive characteristics under both steady-state and pathological conditions. These advancements pave the way for precise targeting of specific macrophage phenotypes and their functions in distinctive pathologies, culminating in more effective and efficient treatments. Our review provides a comprehensive overview of ongoing research and potential therapeutic strategies targeted at macrophages in specific CVDs. We also delve into the challenges of targeting macrophages specific to the heart and explore the application of macrophage-based therapies in clinical trials.
Macrophages: the shield of hearts against CVDs
The heart is a complex organ composed of a diverse array of cell types, each playing a crucial role in its function. While cardiomyocytes, the contractile cells responsible for the heart's pumping action, have traditionally been the focus of research, recent advances have highlighted the significance of non-myocytes, such as endothelial cells, fibroblasts, and immune cells, in maintaining cardiac health. These diverse cell types form a tightly interconnected network that communicates biochemically and biophysically to ensure proper heart function both under normal conditions and in response to injury [10].
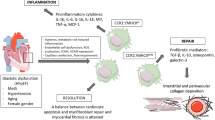
Due to the limited regenerative capacity of cardiomyocytes in adult heart, current therapeutic options are focusing in two directions: (i) increase regeneration of cardiomyocytes by stem cell therapy [11]; (ii) prevent adverse remodeling of non-myocytes including inflammation or fibrosis [12]. In this regard, macrophages have garnered significant attention in the last decades as attractive therapeutic targets for CVDs. Specifically, in the context of CVDs and HF, macrophages play active roles in all phases of the wound healing process following injury, ranging from inflammation to fibrosis and tissue remodeling [13]. Emerging studies have demonstrated the therapeutic potential of strategies aimed at modulating the activity of macrophages to prevent the development of pathological conditions associated with HF (Fig. 1).

The role of cardiac macrophages in CVDs. Cardiac macrophages play pivotal roles in the progression of cardiovascular diseases (CVDs), encompassing inflammation, fibrosis, cardiac repair, and regeneration through the secretion of mediators and interactions with other cardiac cells. TNF-α tumor necrosis factor alpha, IL interleukin, CTGF connective tissue growth factor, TIMP tissue inhibitor of metalloproteinases, MMP matrix metalloproteinases, TGF-β transforming growth factor-β, Notch 1 neurogenic locus notch homolog protein 1, VEGFaa vascular endothelial growth factor Aa
Inflammation
Macrophages are central players in the inflammatory response during progression of several types of CVDs [14, 15]. Upon cardiac damage, necrotic cells secret pathogen/damage-associated molecular patterns (PAMPs/DAMPs), which are the main triggers for the activation of immune cells including macrophages. The released PAMPs/DAMPs are then recognized by pattern recognition receptors (PRRs) on macrophages, eventually activate macrophages and initiate the inflammatory signaling cascades. Macrophages undergo polarization states during the progression of several CVDs and this status is highly associated with an inflammatory response. In the early stages of CVD, classically activated macrophages (simplified as M1 macrophages) are actively recruited into damaged myocardium [7]. They produce and release various pro-inflammatory cytokines, chemokines, and enzymes that contribute to tissue inflammation. Excessive and sustained inflammation can lead to tissue damage, impaired healing, and adverse cardiac remodeling. Over time, as the disease progresses and inflammation persists, macrophages can polarize to alternative-activated macrophages (also known as M2 macrophages). These M2 macrophages initiate a reparative healing process by releasing of anti-inflammatory cytokines. This process involves resolving inflammation and promoting tissue repair, which eventually results in decreased adverse remodeling and tissue damage [7]. However, in the context of prolonged and chronic inflammation in CVDs, the M2 polarization often becomes impaired, resulting in an imbalance between pro-inflammatory and anti-inflammatory response and contributing to HF progression [16]. Aging could also serve as a significant link between chronic inflammation and CVD risk. Notably, age-related DNA mutations that lead to a bone marrow abnormality, called clonal hematopoiesis of indeterminate potential (CHIP), can further exacerbate this imbalance by disrupting macrophage function. Specifically, mutations in TET2 and DNMT3A, two genes frequently mutated in CHIP, disrupt the delicate balance of cytokines that control macrophage behavior, leading to excessive inflammation. This further promotes the development of atherosclerotic lesions and increases CVD risk, highlighting the importance of macrophages in regulating inflammation [17].Therefore, modulation of macrophages holds promise as a therapeutic approach in CVDs. Strategies aimed at promoting the transition of macrophages from pro-inflammatory (M1) to anti-inflammatory (M2) can help mitigate chronic inflammation and hinder HF progression. Potential approaches will be discussed in further sections.
Fibrosis
Followed by resolution of inflammation, the viable part of the damaged cardiac tissue initiates extracellular matrix (ECM) remodeling to compensate for the loss of cardiomyocytes. This stage, known as fibrosis, is distinguished by an excessive accumulation of collagen and other components of the ECM, primarily attributed to the heightened activation of fibroblasts. Such aberrant deposition results in increased stiffness and irregular cardiac rhythm, ultimately leading to sudden cardiac death in individuals with CVDs. As being a key player in inflammatory response, macrophages are also indispensable effector cells during development of the cardiac fibrosis through regulating multiple pathways related to activation of fibroblasts, such as TGF-β pathway or metalloproteases (MMPs) [18]. Significant insights arising from the single-cell sequencing investigation of cardiac cells encompass the identification of potential interactions between macrophages and fibroblasts mediated by ligand-receptor signaling circuits. Cell–cell interactions have been shown to predict a co-expression pattern of fibronectin 1 (FN1), lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1), cluster of differentiation 74 (CD74), and macrophage migration inhibitory factor (MIF). This observation suggests that LYVE1 + monocytes and macrophages may interact with fibroblasts via the CD74-MIF pathway, implying a role for this interaction in the progression of fibrosis development [19, 20]. Disruptions in these interactions can result in either exaggerated fibrosis or defective fibrosis, both of which can have detrimental consequences for tissue repair and functional outcomes of the heart. The reciprocal regulatory influence of macrophages on fibroblasts and cardiac fibrosis is also highly associated with distinct subpopulations of macrophages [21]. Alternatively activated macrophages, also known as M2 phenotype, play a predominant role in wound healing and tissue remodeling. However, they also significantly contribute to cardiac fibrosis by directly promoting collagen I expression [22]. Consequently, inhibiting M2 macrophages through GATA3 depletion during fibrosis initiation has demonstrated cardioprotective effects and limited fibrotic injury in mouse MI model. However, manipulating macrophage populations demands careful consideration of their double-sword functional roles during the healing process. Depletion of M2 macrophages during resolution phase, the stage dedicated to inflammation resolution and tissue repair, results in worsened cardiac function along with exaggerated inflammation [23, 24]. This highlights that precise timing is crucial when implementing macrophage modulation strategies, ensuring alignment with their functional contributions at each stage of healing.
Additionally, specific subpopulations of M2 macrophages exhibit contrasting effects on fibrosis development in Coxsackievirus-induced myocarditis [25]. TLR4+casp-1+IL-1β+M2 macrophages promote fibrosis and inflammation, while Tim-3+M2 macrophages tend to exhibit an anti-fibrotic and anti-inflammatory phenotype [18, 25]. Furthermore, activated macrophages serve as the primary source of inflammatory cytokines such as TNF-α, IL-1, which can act as pro-fibrotic factors, promoting excessive ECM proteins production. Ultimately, this process results in poorly ordered ECM deposition and impaired tissue remodeling [26, 27]. It is important to note that macrophage phenotypes and their fibrotic effects can be influenced by various factors, including the specific pathological conditions, the stage of fibrosis, and the interplay with other cells and signaling molecules [28]. Therefore, investigating the functional properties of distinct macrophage subsets and elucidating the mechanisms governing macrophage phenotypic transitions, differentiation, and recruitment holds promise for developing strategies to attenuate and reverse pathological cardiac fibrosis.
Tissue repair
Excessive fibrosis has a further impact on the chronic remodeling of the injured heart including scar and larger vessel formation, ultimately contributing to overall long-term survival and cardiac function. These changes consequently influence on the overall microenvironment cue and the fitness of other cardiac cells such as cardiomyocytes and endothelial cells. In this context, macrophages emerge as crucial contributors to the reparative cellular responses. More importantly, macrophages play a direct role in the cardiac conduction system through their interaction with cardiomyocytes. Macrophages residing in sinoatrial (SA) node and atrioventricular (AV) node establish electrical coupling with adjacent cardiomyocytes through gap junction, which allow for direct transfer of electrical signals between cells, enabling the syncronized propagation of electrical impulses and ensuring efficient conduction throughout the heart [29]. While their specific role in cardiac repair following injury remains to be fully elucidated, macrophages undoubtedly play a critical role in preventing abnormal cardiac rhythms, which can lead to sudden cardiac death in the injured heart. By aiding in removal of debris and necrotic cells, they involve in maintenance of mitochondrial health and cardiac homeostasis. Moreover, they secret cardiomyocytes protective molecules such as FGF-1, TNF-α, and IGF-1 to maintain survival of cardiomyocytes [30]. Indeed, depletion of macrophages has been shown to result in cardiac rhythm disturbance, suggesting their essential role in maintaining cardiac health under stress conditions [31]. However, considering diversity and phenotypic plasticity, macrophages can be also served as a cellular source for promoting pathological pathways through secretion of inflammatory cytokines. Macrophages secrete cytokines such as IL-6 or IL-8, which subsequently trigger hypertrophy of cardiomyocytes. Notably, the neutralization of these cytokines has been shown to mitigate hypertrophy, highlighting their significance in this regulatory process [32].
Macrophages also play a vital role in maintaining vascular health and structural integrity. In vitro studies have demonstrated that macrophages enhanced angiogenesis, the process of new blood vessel formation, by stimulating the proliferation and vessel sprouting of ECs. This angiotrophic potential is further amplified when macrophages are polarized towards an inflammatory state by treatment with lipopolysaccharide (LPS) [33]. Different macrophage phenotypes exhibit distinct functions in vascular remodeling. Inflammatory monocytes and macrophages (Ly6C+, CCR2+) are extravasated into peripheral inflamed tissues, contributing to the vessel inflammation and formation of atherosclerotic plaque [34, 35]. Conversely, the opposing population, including Ly6Clow or Cx3cr1, Nr4a1 macrophages, are enriched within capillaries and scavenge microparticles from their luminal side and function as intravascular housekeepers for fitness of endothelial cells by promoting the safe disposal of ECs and facilitating appropriate cell turnover [36, 37]. Overall, balancing between cardioprotective and detrimental subpopulation of macrophages at the appropriate healing stages for each pathological condition is pivotal to ensure enduring therapeutic effects while minimizing secondary tissue damage.
Regeneration
The loss of cardiomyocytes following cardiac injury is irreversible. Due to limited regenerative capacity, the majority of current therapies are focused on either reducing damage on myocardium or recovering cardiomyocytes by stem cell therapy. However, these approaches face limitations due to low efficiency or high cost. Unlike in adult heart, neonatal heart has the stunning capability to reverse injury by regeneration of myocardium. Lavine et al. discovered distinct composition of macrophages between adult and neonatal cardiac tissues, both at baseline and following injury. The adult heart exhibited a coexistence of MHC-IIlow and MHC-IIhigh subsets of macrophages (CCR2−) and monocytes (CCR2+), whereas neonatal hearts exclusively contained MHC-IIlow macrophages and monocytes. These distinctive variances are associated with distinguishing outcome in cardiac remodeling post-injury [38]. In response to injury, neonatal hearts exhibit a selective expansion of resident macrophages population (MHC-IIlowCCR2−), which play critical roles in cardiac regeneration. These roles encompass the regulation of inflammation and the promotion of cardiomyocyte proliferation via Jagged-1-Notch1 axis, ultimately culminating in improved cardiac recovery post-injury [39]. Conversely, the adult heart experiences the replacement of these resident macrophages primarily by monocyte-derived macrophages that lack regenerative functions. Instead, they tend to foster a pro-inflammatory response, often leading to hypertrophy or fibrosis. Apparently, cardiac-resident macrophages (CRM) undergo a switch in their ontogeny after birth, from regenerative to adult-like phenotypes, resulting in the loss of proliferative potential [39]. Notably, preservation of resident macrophages through the inhibition of monocyte-derived macrophage recruitment has been demonstrated to yield minimal inflammation while fostering enhanced cardiomyocyte renewal in the adult heart [40]. Concordantly, studies in regenerative zebrafish also have provided compelling evidence for the indispensable role of macrophages in heart regeneration, notably by facilitating the self-renewal of cardiomyocytes. Particularly, macrophage-dependent expression of Vegfaa on epicardial cells has shown a robust association with cardiac growth signaling pathway and proliferation of cardiomyocytes [41]. The study by Rotem et al. revealed that the secretion of cytokines by macrophages plays a crucial role in determining the healing outcome of injured hearts in both neonates and adults. Among the various cytokines secreted, neonatal cardiac macrophages release high levels of osteopontin (OPN) following MI. Notably, OPN deficiency in cardiac macrophages impaired their paracrine reparative properties, highlighting the importance of macrophage-derived OPN in myocardial healing in neonatal hearts. Mechanistically, OPN secretion is essential for neonatal regeneration by stimulating cardiomyocyte cell-cycle re-entry and activating the proliferation and migration of non-CM cells. Remarkably, myocardial injection of recombinant OPN into adult hearts significantly improved infarct healing and overall cardiac function following MI, suggesting that the regenerative potential of neonatal macrophages can be harnessed to develop therapeutic strategies for acute MI in adult hearts [42].
These significant findings underscore the potential of reprogramming adult macrophages to acquire a regenerative phenotype, thereby opening up novel therapeutic avenues for treating cardiovascular diseases. The implications of these discoveries hold great promise for the field of regenerative medicine and present an exciting trajectory for future research in this domain.
Patient selection and target identification for macrophage-based therapies in pathological conditions
The benefits of macrophage-targeted therapy in CVD patients, particularly those with heart failure, can vary depending on the specific therapeutic approach and the individual patient's condition. However, individuals with chronic heart failure, especially those with evidence of persistent inflammation, excessive fibrosis, and impaired tissue repair, are more likely to benefit from such therapies. It's important to note that macrophage modulation should be carefully tailored to the specific pathological context, as macrophages have diverse functions (due to their plasticity, macrophages can exhibit both protective and pathogenic functions) and their complete abrogation or excessive suppression can have unintended consequences.
Atherosclerosis
Atherosclerosis is a chronic inflammatory condition characterized by the buildup of apolipoprotein B-containing lipoproteins and plaques within the arterial wall. It has garnered significant attention in the context of therapies targeting macrophages [43]. In the immune landscape of atherosclerotic plaques, macrophages, along with T-cells, represent the predominant cell types. Macrophages constitute approximately 50% of CD45+ cells within murine aorta upon atherosclerosis, but their prevalence decreases to 16–20% of total CD45 + cells in human carotid endarterectomies, indicating notable disparities of myeloid cell populations between humans and mice in atherosclerosis [44, 45]. Despite this difference, both pro-inflammatory and anti-inflammatory macrophage subtypes coexist within the plaque region in both species. In the pathogenesis of atherosclerosis, macrophages release a plethora of pro-inflammatory mediators, as well as anti-inflammatory factors, pro-thrombotic tissue factors, and matrix-degrading proteases. These molecular agents collectively exert significant influence over plaque growth, cellular composition, and stability. Several single-cell studies examining atherosclerotic plaques in both human and mouse models (e.g., Apoe-/- or Ldr-/- mice, under either a standard diet or a high-fat westernized diet) have unveiled the presence of three primary macrophage populations. These populations exhibit distinct gene expression signatures relevant to atherosclerotic disease, encompassing inflammatory macrophages, resident-like macrophages, and lipid-associated Trem2hi macrophages [46, 47].
Inflammatory macrophages
In response to an inflammatory environment, monocytes are recruited and differentiate into macrophages within atherosclerotic lesions [43, 48]. Inflammatory macrophages are one of the major macrophage populations within atherosclerotic aorta. Inflammatory macrophages, also known as chemokinehigh macrophages, are highly associated with atherosclerotic plaque progression. These macrophages secrete pro-inflammatory markers, digest lipoproteins, and accumulate foam cells, a hallmark of atherosclerotic lesions. This contributes to plaque growth and the release of inflammatory mediators such as IL-6 and TNF-α [43, 48]. The activated pro-inflammatory macrophages exhibit the expression of surface markers including major histocompatibility complex class II (MHCII), Fc receptor CD64, CD80, and CD86, along with the upregulation of typical pro-inflammatory transcripts such as Cxcl2, Ccl4, interleukin-1β (IL-1β), tumor necrosis factor alpha (TNF-α), NLR family pyrin domain containing 3 (NLRP3), caspase-1 (Casp1), and caspase-4 (Casp4) [49]. Inflammatory macrophages are typically enriched in non-foamy cells localized at the plaque shoulder region, which is normally mediated through inflammatory pathways, including Toll-like receptor (TLR) activation, TNF signaling, type I interferon (IFN) responses, and cytokine-chemokine interaction signaling pathways [50, 51].
Resident-like macrophages
Recent research findings reveal that the predominant myeloid cell population within the intima of the aortic arch consists of resident macrophages, which rely on CSF1 (colony-stimulating factor 1) expression and are sustained by local proliferation mechanisms. However, during the progression of atherosclerotic plaques, this resident population is gradually supplanted by recruited monocytes, primarily due to their limited capacity for self-renewal [52]. They are newly recognized population, called monocytes derived resident-like macrophages by Willemsen and Winther et al. [46]. In murine model of atherosclerosis, the resident-like macrophages primarily exhibit gene expression pattern similar to resident macrophages, including Cx3cr1, Clec4a2 (C-type lectin receptor), Lyve1, Mrc1, Forl2 and Vsig4, F13a1 [47, 53, 54]. Interestingly, atherosclerotic resident-like macrophages expressed high level of CCR2, a marker typically associated with recruited macrophages, supporting that recruited monocyte-derived macrophages replenish the resident-like macrophages upon atherosclerosis development [46, 50]. Moreover, this population express CD206, Folr2, Cbr2 and Selenoprotein-1 (Sepp1), all of which are associated with M2-like phenotype, determining the anti-inflammatory features of atherosclerotic resident-like macrophages [55]. Notable, the CD206hiCD163hi macrophages in human atherosclerotic plaques resemble to the resident-like macrophage subsets described in murine atherosclerosis [44, 51]. In contrast to a detrimental role of the inflammatory macrophages, the resident-like macrophages exhibit protective functions against atherosclerosis progression by enhancing homeostasis. During atherogenesis in mouse model, depletion of these resident-like macrophages resulted in dysregulated cholesterol metabolism, which further led to exacerbated disease progression, characterized by increased arterial stiffness and collagen deposition, thus reinforcing the significance of the resident-like macrophages in maintaining fitness of heart against atherosclerosis [53, 54]. The resident-like macrophages are mainly involved in receptor-mediated endocytosis and proliferation signaling pathways [49, 50].
Foamy Trem2hi macrophages
The third subpopulation, Trem2hi macrophages, corresponds to lipid-laden foamy macrophages, which are recognized as principal trigger for atherosclerotic plaque formation. These macrophages are notably characterized by an elevated expression of key markers, such as Trem2, CD9, Fabp4, Apoe and Apoc1, and they are predominantly localized within the plaque intima and its necrotic core [46]. Fabp4 serves as reliable marker for identifying foamy macrophages within atherosclerotic lesions. CD9 is also known to play a pivotal role in foam cell formation through its association with CD36, a well-known stimulator of foam cell development [46]. In obese patients, CD9hi adipose tissue macrophages demonstrate a higher intracellular lipid content in comparison to CD9lo adipose tissue macrophages [56]. Particularly, Trem2hi macrophages are only found in atherosclerotic plaque, not in healthy aorta as shown by the findings from Cochain et al. [49]. A pathway analysis of these Trem2hi macrophages reveals their enrichment in processes related to lipid metabolism, regulation of cholesterol efflux, and oxidative stress, underscoring the involvement of Trem2hi macrophages in intracellular lipid accumulation and foam cell generation [49]. More interestingly, these macrophages also present pro-fibrotic characteristics, as evidenced by the expression of Galectin-3, which may indicate a potential role of Trem2hi macrophages in stabilizing plaques macrophage populations, drivers for plaque progression [57, 58]. Interestingly, these subpopulations display diminished pro-inflammatory phenotypes, hinting at an anti-inflammatory nature of Trem2hi macrophages. Similarly, Trem2 is typically expressed on anti-inflammatory macrophages and has been demonstrated to restrain macrophage activation [59].
Ischemic heart disease and myocardial infarction (MI)
Atherosclerosis can further provoke secondary complications. The progressive enlargement of atherosclerotic plaque can result in the occlusion of the vessel lumens, leading to ischemic injury or MI, one of the leading causes of death worldwide [60]. MI is a clinical condition characterized by the death of cardiomyocytes and tissue damage due to impaired oxygenation of the myocardium. The loss of cardiomyocytes concomitant changes in cell proportion, which lead to inflammatory response and cardiac remodeling in ischemic myocardium [13]. This pathological condition, commonly known as a heart attack, is another CVD that can potentially benefit from macrophage-based therapies.
Macrophages are the most abundant cell population among leukocytes infiltrated following MI, constituting 58.7% of the total CD45 + cells [61]. They are dynamically changed over the MI time continuum and play pivotal roles in balancing inflammatory response. The proportion of macrophages drastically dropped on day 1 (24.9%), but gradually recovered from day 3 (66.8%), peaking on day 7 (84.0%) post-MI [61]. More interestingly, macrophages exhibit temporal diversity in phenotypes during disease progression following MI. The polarization status of macrophages largely determines their pathological functions and impact during different phases of MI. In a murine model, within the early phase after MI (1–3 days post-MI), macrophages are actively recruited to the site of injury and initiate the clearance of necrotic tissue. During this phase, macrophages also release pro-inflammatory cytokines, which can intensify the inflammatory response. Once necrotic debris is removed, at later phase of MI (5–7 days up to months post-MI), anti-inflammatory macrophages replace the pro-inflammatory macrophages paving the way to the wound healing phase and resolution of inflammation. During this latter, macrophages release anti-inflammatory cytokines which stimulate the release of collagen via the TGF-β cascade, promoting scar formation [62, 63]. This transition of macrophages from a pro-inflammatory to a reparative phenotype should occur at the appropriate timing for the proper resolution of inflammation.
Early pro-inflammatory macrophages
Upon ischemia injury following coronary occlusion, remarkable infiltration of macrophages was observed from day 1 and persisting through day 7 post-operation [64]. Several previous scRNA-seq analyses have unveiled distinct subpopulations of cardiac macrophage during MI progression. In murine model of MI, cardiac macrophages were further subdivided into seven distinct subclusters, comprising three tissue-resident macrophages and four ischemia-associated macrophages. Among them, Olr1+ macrophages, one of ischemia associate macrophage populations, were the largest population at day 1 post-MI, accounting for 15% of total CD45 + cells. These Olr1+ macrophages exhibit pro-inflammatory characteristics and are endowed with phagocytic capabilities [64]. This finding aligns with the transcriptomic profiles of isolated cardiac macrophages post-MI, affirming that the early macrophage response following MI is marked by pro-inflammatory phenotypes [65]. In a separate study by Jin et al., the most signature genes characterizing inflammatory macrophages in the context of MI are identified as CCR2 and MHC II. CCR2hi macrophages represent infiltrating monocyte-derived macrophages, predominantly expressing canonical macrophage markers like Cd68, Fcgr1, Itgam and CCR2. This subset also peaked in abundance on day 1 and gradually decreased until day 7 post-MI [61, 64]. MHChigh subset is notably enriched with antigen processing and presentation-related genes, primarily exhibiting pro-inflammatory functions [66]. Associated signaling pathways of these post-MI inflammatory macrophages encompass NF-kB and NOD-like receptor signaling pathways, which are closely linked to activated leukocyte migration and cytokine productions, featuring by upregulated expression of Cxcl2, Ccl9, Ccl24, Il1b, and Trem1 [64].
Late reparative macrophages
Subsequent to MI, macrophages at later stages exhibit a relatively higher gene expression of Apoe, Fcrls, Rgs10, Adgre1, Trem2, Gpnmb, Fabp5, Spp1 and Timp2. Their expression gradually increases, becoming most prominent at 7 days post-MI, with potential extension over subsequent months [61]. In particular, Gpnmb+ macrophages exhibit a notable surge in infiltration at 7 days post-MI, accompanied by elevated phagocytic activity. They display enrichment of lysosome and cholesterol metabolism pathways, marked by increased expression levels of Gpnmb, Fabp5, and Trem2 [64]. Interestingly, the late macrophages demonstrate a distinctive metabolic profile compared to early macrophages post-MI. Late Gpnmb+ macrophages exhibit increased phagocytic and fatty acid oxidation scores, whereas early Olr1+ macrophages show elevated senescence‐associated secretory phenotype (SASP) and glycolysis scores [64]. As similarly observed in atherosclerosis, Tremhi macrophages are present in advanced infarct region following MI, with predominant expression in the later stage [61]. This subpopulation is marked by anti-inflammatory characteristics, evidenced by high expression of anti-inflammatory genes such as Arg1, IL-10 and Tgfb1. Of particular significance, Trem2hi macrophages also display a heightened expression of osteopontin (Spp1), which is related to pro-fibrotic potential in regulating post-MI LV remodeling [61]. In line with these findings, a recent study from Kim et al. reported the therapeutic potential of a molecule secreted by Tremhi macrophages, soluble Trem2 (sTrem2). In vivo, administration of sTrem2 significantly improved myocardial function and LV remodeling post-MI by promoting polarization of macrophage toward an anti-inflammatory phenotype, which results in effective regulation of inflammation in the infarcted myocardium [67].
Resident macrophages (steady-state macrophages)
In contrast to infiltrated macrophages, tissue-resident macrophages have been comparably less studied and were often considered of insignificance during MI progression. However, in recent years, there has been a growing awareness of their role [68, 69].
Zhuang et al. recently identified three tissue-resident macrophage subsets in the heart following MI using scRNA-seq [64]. Among those, the Timd4 + cluster represents the most conserved tissue-resident macrophage subset across multiple organs in both mice and humans [70]. This subset is enriched in lysosome and endocytosis signaling pathways and exclusively expresses Folr2, Timd4, and Lyve1 [61, 71]. Tissue-resident macrophages are primarily sustained through local proliferation, as evidenced by elevated expression of proliferation-related genes such as Jund, Tcf4, and Maf [71]. Although their relative proportion declines on the first-day post-MI, it gradually restores by day 7, yet does not return to steady-state levels. This suggests an indirect implication of their involvement during the later stages of MI. A central role of these macrophages is to attenuate the post-MI inflammatory response [71, 72]. Depletion of resident macrophages deteriorates cardiac function and impairs healing after MI, highlighting their protective role. Resident macrophages are essential for efficiently clearing necrotic and apoptotic debris from the infarcted area, which contributes to timely inflammation resolution. Additionally, pro-inflammatory signaling pathways, such as NF-κB, apoptosis, and IL-17, are significantly less pronounced in Folr2 + resident macrophages than in Folr2- monocyte-derived macrophages [71]
HFpEF (heart failure with preserved ejection fraction) and diastolic dysfunction
Unlike systolic dysfunction, HFpEF is characterized by impaired LV contractility and diastolic relaxation, while the overall function of cardiomyocytes remains preserved [73, 74]. Unfortunately, conventional pharmacological interventions employed against heart failure with reduced ejection fraction (HFrEF), such as beta blockers or ACE inhibitors, have proven ineffective in restoring cardiac function in HFpEF patients. Despite its growing prevalence, the specific treatment for HFpEF is still limited. Emerging studies reveal the significant role of inflammation in cardiac remodeling for both HFrEF and HFpEF, emphasizing the need for distinct therapeutic approaches due to distinctive pathophysiology [75]. Notably, HFpEF exhibits more pronounced systemic inflammation and fibrotic pathways. Myocardial biopsies from patients with HFpEF reveal a higher number of infiltrated inflammatory cells than those from healthy controls. These inflammatory cells play a crucial role in the intense inflammatory response and secretion of pro-fibrotic growth factors such as TGF-β [76]. This pattern is also evident in the blood of HFpEF patients, where there is a two- to fourfold increase in classical, intermediate, and non-classical monocyte subsets, indicative of a chronic state of inflammation during HFpEF [77]. Consequently, significantly elevated systemic levels of inflammatory cytokines, such as TNF-α, IL-6, and the chemokine CCL2, have been observed in HFpEF patients experiencing disease exacerbation, with increases ranging from 1.3- to 2.4-fold compared to those with stable disease [78, 79]. Interestingly, these hematopoietic activities are closely correlated with myocardial filling pressure [77]. This underscores the role of systemic inflammation as a potential trigger for the development of fibrosis by promoting the activation of myofibroblasts. The amplified fibrotic response ultimately leads to diastolic dysfunction, thereby contributing to clinical deterioration in patients with HFpEF [77].
Profibrotic macrophages
Macrophages are among the major effector cells in the inflammatory response. While the contribution of macrophages on HFpEF progression is comparably less understood than in HFrEF, emerging evidence has linked macrophages, particularly pro-fibrotic M2 macrophages, to adverse outcomes in HFpEF [77, 80]. In contrast to their beneficial effects in HFrEF by resolving inflammation and promoting wound healing, these M2 macrophages exhibit a more pathological role in HFpEF, manifesting an enhanced fibrogenic phenotype. In HFpEF, M2 macrophages play a critical role in myofibroblast activation and collagen deposition by secreting fibrosis stimulators like Galectin-3 or TGF-β, ultimately leading to myocardial stiffness and diastolic dysfunction [77, 80, 81]. Significantly, the IL-10 signaling pathway plays a central role in driving the polarization of macrophages towards a pro-fibrotic phenotype during the progression of HFpEF. Macrophage-derived IL-10 secretion creates an autocrine loop that promotes pro-fibrotic macrophage polarization and the release of fibroblast-activating molecules such as TGF-β and osteopontin [77]. Remarkably, HFpEF patient-derived serum enhances the differentiation of healthy monocytes into macrophages that express IL-10, indicating that prolonged exposure to the HFpEF patient's microenvironment directs macrophages towards a fibrogenic phenotype. These findings highlight the potential of IL-10 as a therapeutic target for regulating pro-fibrotic macrophages and mitigating HFpEF complications, including myocardial collagen deposition and diastolic dysfunction [77, 80, 81]. This contrasts with the pattern observed in HFrEF, where IL-10 is beneficial for tissue repair and inflammation resolution [82]. These findings suggest that the same macrophage pathway can have either beneficial or detrimental effects depending on the stage of disease progression and the concomitant changes in the myocardial microenvironment. Such insights have important implications for developing targeted therapeutic strategies to limit disease progression in HF of various etiologies. However, to achieve optimal and efficient therapeutic outcomes, a comprehensive understanding of macrophage behavior and function during the progression of HFpEF is needed.
Current status of macrophage-targeting strategies
In the last years, several attempts have been applied to fine-tune the biology of macrophages. In this section, we illustrate the most promising and challenging therapeutic approaches aimed to modulate the chemotaxis, inflammatory response as well as phenotype of these phagocytic cells (Fig. 1).
Atherosclerosis
Depletion of monocytes/macrophages
The negative prognostic significance of macrophage infiltration and their persistence in atherosclerosis supports the development of treatment options, which includes either blocking the recruitment of macrophages or neutralizing relative pro-inflammatory cytokines. Depletion of macrophages has emerged as an effective strategy, particularly for individuals with atherosclerosis, owing to the strong association between recruited monocytes/macrophages and the growth and destabilization of atherosclerotic plaques, which typically exhibit a large lipid core and a weakened fibrous cap [83, 84]. Pharmacological depletion of macrophages can be achieved by promoting programmed cell death, either through enhanced apoptosis or autophagy [85]. One extensively utilized method for in vivo investigation of macrophage function involves the use of clodronate liposomes (Clo-Lip). Clo-Lip depletes macrophages by instigating programmed cell death [86, 87]. Depletion of macrophages induced by Clo-Lip administration has been shown to significantly improve systolic blood velocity in atherosclerosis, underscoring the therapeutic potential of inhibiting macrophage accumulation to impede the progression of atherosclerosis [88]. However, chronic administration of Clo-Lip does present limitations. Delayed macrophage recruitment resulting from Clo-Lip intervention can lead to impaired neutrophil resolution and subsequent heart regeneration [89]. Additionally, a recent study by Culemann et al. has challenged prior notions by revealing that neutrophils are the primary effectors impacted by Clo-Lip, rather than monocytes or macrophages [90]. Clo-Lip exerts anti-inflammatory effects independently of macrophage presence, but its effectiveness is contingent on proper neutrophil function. However, the precise mechanisms through which Clo-Lip-induced impairment of neutrophil function influences monocytes or macrophages remain to be fully elucidated. This discovery prompts a re-evaluation of the intricate relationship between neutrophils and macrophages, with the goal of optimizing the use of Clo-Lip for inflammation regulation [90].
Extensive research has focused on disrupting specific chemokine-chemokine receptor pairs, such as CCL5–CCR5 and CCL2–CCR2, to impede monocyte and macrophage recruitment. Interventions involving CCL5 (RANTES) antagonists, CCL2 (MCP-1) inhibitors, or the silencing of CCR2 mRNA have shown significant reductions in macrophage infiltration within atherosclerotic plaques. These interventions have led to a reduction in lesion size and plaque stabilization [91,92,93]. However, the translation of these findings into clinical trials for agents targeting these chemokine axes has been lagging behind, primarily due to systemic inhibition and the complexity of these pathways, which can result in side effects on non-targeted tissues [94, 95]. Strategies to address this challenge include the optimization of drug delivery through tissue-specific nanoparticles or exosomes loaded with monoclonal antibodies, as well as the development of CCR2-biased or probe-dependent antagonists, which can selectively block specific chemokines or signaling pathways. These strategies hold promise for clinical implementation [94, 96]. Inhibition of macrophage migratory inhibitory factor (MIF), a cytokine-inducing adhesion molecule like ICAM1, and VCAM1, can also contribute to protective effects in limiting macrophage adhesion. Treatment with antibodies against MIF has resulted in a significant reduction in the number of infiltrated macrophages and foam cells, leading to improved cardiac function and reduced scar size [97, 98]. Statins, a class of cholesterol biosynthesis inhibitors, are widely prescribed for the treatment of atherosclerosis. Administration of statins effectively manages hyperlipidemia and results in a significant reduction in macrophage infiltration within atherosclerotic plaques within one month, supporting the clinical application of macrophage infiltration inhibition as a therapeutic strategy [99,100,101]. Similarly, interventions targeting the IL-1 pathway, such as Anakinra (IL-1R agonist; ClinicalTrials.gov, number NCT01950299, completed) [102, 103] or canakinumab (monoclonal antibody block IL-1β; ClinicalTrials.gov, number NCT01327846, completed) [104], have successfully transitioned into clinical studies. Their administration has led to a significant reduction in leukocyte production, resulting in limited adverse remodeling and attenuated systemic inflammation. This, in turn, has led to reduced mortality and hospitalization rates among patients with HF [102, 105, 106].
Induction of macrophage autophagy
As the role of macrophage-mediated inflammation at certain levels is essential for the wound healing process in various disease contexts, there is a growing interest in the modulation of macrophage behavior as an alternative to inhibition. This approach represents the next generation of macrophage-based therapeutic strategies. In the context of atherosclerosis, this approach is designed to promote plaque regression, reduce inflammation, and enhance overall plaque stability. One well-established strategy involves the induction of autophagy in macrophages [107]. Autophagy is a crucial cellular process responsible for removing and recycling damaged or long-lived intracellular materials. An increasing body of research has demonstrated the close link between autophagy and macrophage biology within the realm of innate immunity. Autophagy in macrophages plays a protective role in atherosclerosis, primarily due to its regulatory function in facilitating cholesterol efflux from macrophage foam cells [107, 108]. In the context of atherosclerotic plaque, the induction of autophagy in macrophages is largely dependent on the ATP-binding cassette transporter A1 (ABCA1)-mediated process [108]. Elevating ABCA1 levels in macrophages present a therapeutic effect by inhibiting the inflammatory response and progression of atherosclerotic lesions [109]. Selective inhibition of autophagy regulators, such as the PI3K/Akt/mTOR pathway, can effectively induce autophagy and reduce macrophage aggregation in atherosclerotic plaques, thereby promoting plaque stability by protecting cells and reducing the secretion of inflammatory factors [110]. Furthermore, autophagy serves as a regulator of efferocytosis, a process in which macrophages efficiently clear debris or necrotic cells, essential for organ repair following injury [111, 112]. Disruption of autophagy can lead to a loss of this function, resulting in prolonged inflammation, which in turn exacerbates atherosclerosis progression [113, 114]. Additionally, pro-reparative factors are secreted by macrophages after efferocytosis, further promoting wound healing [115].
Adipose tissue macrophages (ATM)
Being a lipid-driven chronic inflammatory disease, atherosclerosis is also closely linked to metabolic disorders such as diabetes or obesity [116]. Abnormal accumulation of pro-inflammatory adipose tissue macrophages (ATM) is a common feature in metabolic disorders, which can trigger the onset of other chronic diseases such as hypertension or cerebrovascular diseases [117]. The uncontrolled release of pro-inflammatory mediators like IL-6, IL-1β and TNF-α by ATMs plays a significant role in obesity-related adipose tissue inflammation and metabolic dysfunction [116, 118]. Therefore, the therapeutic options aimed at modulating the metabolic switch or shifting pro-inflammatory ATMs towards an anti-inflammatory phenotype hold promise in preventing atherosclerosis progression. These approaches attenuate inflammation, promote cholesterol clearance and improve plaque stability.
Myocardial infarction
Macrophage polarization/inflammation resolution
Incomplete resolution of inflammation, characterized by prolonged and unresolved inflammatory processes, has been linked to the pathogenesis of adverse complications, potentially contributing to detrimental cardiac remodeling that ultimately leads to impaired cardiac function and heart failure development following MI [4, 114]. Macrophages, as crucial regulators of inflammation, have emerged as promising therapeutic targets for achieving a balanced pro- and anti-inflammatory response following MI [119,120,121].
Current therapeutic strategies that target macrophage activity in the context of MI primarily aim to promote the resolution of inflammation by increasing the presence of anti-inflammatory macrophages, while concurrently reducing pro-inflammatory signals and mitigating adverse remodeling [122]. This is possible by pharmacological interventions targeting related cell-signaling molecules, such as chemokines, cytokines, or antibodies. Well-established stimulants of M2 macrophages, such as IL-4 and IL-10, drive macrophages toward reparative phenotypes. This promotes post-MI tissue repair, which leads to ECM stabilization as well as improvement of cardiac function compared to control mice [103, 123, 124]. In this regard, transplantation of M2b macrophages has been demonstrated to significantly reduce fibrosis and prevent adverse remodeling in a mouse model of ischemia/reperfusion injury, providing strong evidence for the therapeutic potential of these cells in the context of cardiac remodeling [125]. Beyond the classical stimulators IL-4, IL-13, and IL-10, various factors capable of modulating macrophage polarization have been identified. These factors include Lrg4, Activator protein-1 (AP-1), cAMP-responsive element-binding protein (CREB) activation, and ECM components [126, 127]. AP-1 is a proinflammatory transcription factor complex composed of FOS and Jun family, which plays a role in regulating the inflammatory status by enhancing macrophage polarization [128, 129]. Consequently, inhibition of the AP-1/Fos signaling pathway has been demonstrated to have a protective effect on cardiac function following MI by promoting polarization of macrophage towards a more anti-inflammatory phenotype, resulting in enhanced resolution of inflammation [128, 129]. Similarly, Lrg4 can also govern pro-inflammatory macrophage activation by exerting synergy effects on AP-1 activation and subsequent CREB-mediated Fos transactivation. Macrophages lacking Lrg4 exhibit diminished inflammatory gene signatures. In alignment with these findings, macrophage-specific Lrg4 knockout mice demonstrated reduced ischemic injury as well as improved healing process, attributed in part to the regulation of AP activity [126]. In addition to cardiac macrophages, splenic monocytes/macrophages also play a detrimental role in myocardial ischemia/reperfusion (I/R) injury. Specifically, the NLRP3 inflammasome in splenic monocytes mediates an inflammatory response shortly after reperfusion, worsening MI/R injury in a mitochondrial cell-free DNA (mt-cfDNA)/TLR9-dependent manner. Notably, depletion of NLRP3 in splenic macrophages using CY09 administration effectively reduces infarct size [130]. Macrophages are major cell source of ECM components such as fibronectin or hyaluronic acid (HA) during tissue repair and wound healing. Interestingly, these by-products of ECM can also influence on phenotype switch of macrophages. In their recent study, Wang and colleagues reported that hyaluronic acid-derived short oligosaccharides (HA-o) decreased the infarct size and apoptosis as well as improved angiogenesis in post-MI mouse model, which is attributed to augmented M2 macrophages [131]. In another study, injection of recombinant collagen type I and type III matrices increased M2 macrophages by 1.5-fold at 28 days post-MI, limiting adverse remodeling as well as promoting healing environment [132]. While less well-known, epigenetic modification can also regulate macrophage polarization. In mouse models of atherosclerosis and MI, HDAC (Histone Deacetylase 9) inhibition promoted reparative macrophage polarization and reduced inflammation, suggesting a role for HDACs in macrophage activation [133, 134].
The therapeutic targeting of M2 macrophages in the context of MI is an intriguing approach, but it comes with certain limitations. An overabundance of M2 macrophages might contribute to excessive fibrosis in the post-MI heart through the activation of myofibroblasts, ultimately leading to impaired cardiac function. Particularly, M2 macrophages encompass four subsets: M2a, M2b, M2c and M2d, which are often referred to collectively as M2 macrophages. While all M2 macrophages subtypes possess immunosuppressive properties, they exhibit distinct expression markers and functions. For example, M2a and M2c subtypes presented more pro-fibrotic phenotype, whereas M2b is considered regulator cells [125]. Moreover, M2 macrophages are effective in reducing pro-inflammatory signals, which may potentially hinder efficient wound healing, since a certain degree of inflammation is necessary to facilitate the clearance of necrotic or apoptotic cells in the infarct region. Therefore, ensuring the appropriate polarization of the right M2 subtype at the correct time and place is essential.
Resident macrophage survival
During the steady state of the heart, cardiac macrophages can be categorized into two major populations: embryonic monocyte-derived macrophages (CCR2+) and yolk sac-derived Cx3Cr1+CCR2− resident macrophages. These distinctions are based on their origins and physiological characteristics, as determined through various fate-mapping techniques. In the human heart, there are also distinct macrophage populations that functionally parallel the roles of mouse cardiac CCR2− and CCR2+ macrophages [135]. However, following cardiac injury, the balance between these two subtypes shifts significantly, with infiltrating circulating macrophages experiencing substantial alterations. Consequently, over the past few decades, the majority of research efforts have concentrated on understanding the dynamics of monocyte-derived macrophages. Only recently has the significance of self-renewing tissue-resident macrophages in pathological conditions gained greater recognition. A distinguishing feature of tissue-resident macrophages is their proficiency in phagocytosis, a process critical for the prompt removal of cellular debris resulting from injury. Additionally, they play a pivotal role in the maintenance of myocardial homeostasis by regulating tissue metabolic states [72]. The absence of resident macrophages, particularly those expressing Trem2, leads to impaired elimination of damaged mitochondria, resulting in heightened inflammation and myocardial dysfunction, particularly in conditions like septic heart disease [135, 136]. Similarly, their protective effects have been also presented in a murine model of MI, despite the significant loss of Cx3Cr1+ cells during maturation, with limited self-renewal capacity and regenerative potential, selective depletion of this population in adult hearts leads to impaired cardiac function and remodeling following ischemic cardiac injury, indicating that Cx3Cr1+ cells still play a role in wound healing in the adult heart [38, 71, 137]. Furthermore, cardiac-resident macrophages demonstrate robust proangiogenic and mitogenic properties, suggesting their potential in cardiac repair [138]. More interestingly, the injection of soluble triggering receptor expressed on myeloid cells 2 (Trme2), a crucial gene for the self-renewal capacity of CRM, results in improved cardiac remodeling and myocardial function following MI via polarization of macrophages toward anti-inflammatory phenotypes. This improvement is attributed to the polarization of macrophages toward anti-inflammatory phenotypes [61, 67]. Similarly, overexpression of CRM specifically expressing legumain (Lgmn) also improves cardiac function in mice after MI via efficient efferocytosis-mediated clearance of apoptotic cells [72]. These findings convey two key messages: 1. resident macrophages exhibit anti-inflammatory characteristics; 2. molecules associated with resident macrophage survival, such as Trem2, could serve as alternative therapeutic targets. Additionally, the specific role of CRM has gained attention in recent 4–5 years. The work by Hulsmans and colleagues highlights their role in facilitating electrical conduction of cardiomyocytes by interacting with connexin-43 gap junctions during steady-state conditions. While their role in disease contexts has been comparatively less explored, intriguingly, macrophage ablation has been linked to disrupted cardiac rhythm both in steady-state and after MI [139]. Moreover, the loss of resident macrophages leads to impaired ventricular remodeling and coronary angiogenesis, resulting in increased mortality in chronically failing hearts with reduced contractility [140]. These findings strongly emphasize the essential and protective role of resident macrophages in proper cardiac remodeling and maintaining cardiac function after injury. Importantly, these results underscore that not only inflammation-mediated infiltrating macrophages but also a reduced population of CRM can contribute to adverse remodeling and impaired cardiac function after MI. However, the underlying molecular basis for the distinct roles and responses to ischemic injury between circulating and tissue resident macrophages remains unclear. Future studies should aim to identify molecules or related signaling pathways to enhance the survival of CRM and their cardiac protective role while reducing inflammatory signaling in infiltrating monocyte-derived macrophages, ultimately optimizing cardiac remodeling.
HFpEF (heart failure with preserved ejection fraction) and diastolic dysfunction
Inhibition of pro-fibrotic macrophages
HFpEF often manifests with LV diastolic dysfunction, primarily stemming from systemic inflammation and resultant interstitial fibrosis [73]. Although the role of macrophages in HFpEF progression is less extensively studied compared to in HFrEF, current research efforts have revealed that the crosstalk between macrophages and fibroblasts is inevitable in HFpEF pathogenesis [75]. Myocardial biopsies from HFpEF patients have shown a twofold increase in cardiac macrophage abundance and a 59% elevation in the gene expression of pro-fibrotic TGF-β compared to control samples. This heightened TGF-β appears to contribute to fibroblast activation and excessive collagen deposition [76, 77]. Consequently, inhibiting pro-fibrotic macrophages or their associated genes has demonstrated therapeutic potential in HFpEF and diastolic dysfunction [77, 141].
C–X–C chemokine receptor 4 (Cxcr4), a critical regulator of macrophage-mediated immune responses, is prominently expressed on infiltrated macrophages in both murine HFpEF models and patients with HFpEF. Cxcr4 + macrophages not only influence the inflammatory response but also play a role in myofibroblast transition via the activation of the Cxcl3–Cxcr2 pathway [142]. These effects can be reversed by myeloid-specific Cxcr4 deficiency, suggesting that inhibiting Cxcr4 may offer a novel therapeutic option to block macrophage Cxcr4 signaling and prevent cardiac diastolic dysfunction in HFpEF patients [142]. A study conducted by Hulsmans et al. identified IL-10 as the most up-regulated fibrosis-related genes in cardiac macrophages exposed to conditions such as salty drinking water, unilateral nephrectomy, and chronic exposure to aldosterone (SAUNA) induced diastolic dysfunction. Whereas inhibition of IL-10 in cardiac macrophages results in diminished fibroblasts activation and diastolic dysfunction [77]. The correlation between macrophage abundance/function and disease progression in human HFpEF underscores the development of therapeutics that inhibit macrophage recruitment and neutralize their detrimental inflammatory functions to promote recovery of the failing heart [75, 77]. In clinical trials, administration of Anakinra, the recombinant form of the naturally occurring IL-1 receptor antagonist, successfully attenuated both systemic inflammation and disease symptoms [143]. However, the specific macrophage subtypes involved in HFpEF and their precise roles, as well as the associated signaling pathways, remain areas of ongoing research and require further understanding for effective targeting.
Non-coding RNA (ncRNA)
In recent decades, there has been a growing interest in developing innovative therapeutic strategies based on new scientific findings (Table 1). The rapid development of RNA-based approaches in basic research has inspired their application to clinical research. Within this context, the expanding knowledge surrounding various classes of non-coding RNAs (ncRNAs) and their diverse functional roles has sparked considerable enthusiasm as promising candidates for RNA interference (RNAi)-based gene regulation. The understanding of novel regulatory mechanisms involving ncRNAs and their substantial implications in the pathophysiology of numerous disorders and diseases has positioned ncRNA-based therapies as a focal point of interest. These therapies have shown great potential, particularly for addressing targets traditionally considered 'undruggable,' and for their prospective role in precision or personalized medicine across a spectrum of conditions, including CVDs [144]. By advance in transcriptomics analysis, several altered genes and ncRNAs have been determined during activation and polarization of macrophages [145].
miRNAs
MicroRNAs (miRNAs) are small (approximately 20–22 nucleotides) and among the most extensively studied ncRNAs. Recent studies have conducted a comprehensive miRNome analysis of major cardiac cell fractions, identifying several macrophage-specific enriched microRNAs (miRNAs). Among them, miR-21 is the highest expressed miRNA in cardiac macrophages both in health and disease (25% and 43% respectively of all miRNAs). MiR-21 is a key regulator of the profibrotic function of cardiac macrophages, which contributes to disease-associated fibrosis [146]. In a pressure overload-induced HF model of mice, macrophage-specific genetic deletion of miR-21 resulted in regulatory effects on fibrosis and cardiac dysfunction [146]. However, in contrast, miR-21 mimic is rather beneficial in an obstructed HF model. In vivo nanoparticle delivery of miR-21 to cardiac macrophages in the infarct area prompts the switch of macrophages towards a reparative phenotype, supporting tissue healing and curbing fibrosis [147]. These opposite effects of miRNAs describe that each pathological condition requires a proper strategy despite the common molecule. In an I/R mouse model, mesenchymal stromal cells (MSCs) generating miR-182 enriched exosomes are able to target macrophages and shift their polarization from pro- to anti-inflammatory phenotype, which results in decreased inflammation and infarct size [148]. More importantly, systemic depletion of macrophages using Clo-Lip abolished the therapeutic effects of MSC-Exo, suggesting the significant role of macrophages in mediating the effects of MSC-Exo [148]. MiR-33 is known to target genes involved in cholesterol homeostasis, including ATP binding cassette subfamily A member 1 (ABCA1) and PGC-1α in both human and mouse macrophages. This action limits cholesterol efflux from macrophages and promotes foam cell formation [149, 150]. Specific loss of miR-33 in macrophages reduces lipid accumulation and inflammation under hyperlipidemic conditions, resulting in reduced plaque burden [151]. MiRNAs such as miR-146a and miR-181b regulate macrophage polarization by their anti-inflammatory properties, thereby reducing the inflammatory response and atherosclerotic plaque development. Consequently, the targeted delivery of these miRNAs to macrophages mitigates inflammation and stabilizes atherosclerotic plaques [152, 153]. Interestingly, miRNAs are not only involved in regulating macrophage phenotypes but the enzyme responsible for generating miRNAs, Dicer, is also implicated in macrophage activation [154]. A study by Wei and colleagues demonstrated that specific ablation of Dicer in macrophages accelerated atherosclerosis in mice. This was accompanied by an enhanced inflammatory response and increased lipid accumulation in lesional macrophages. The miRNAs, generating Dicer, include miR-10a, let-7b, and miR-195a. Among them, miR-10a promotes fatty acid oxidation (FAO) in macrophages, which promotes the resolution of inflammation, limiting foam cell formation, and reducing atherosclerosis [154]. Notably, miR-10a expression was found to be negatively correlated with atherosclerosis progression in humans. Therefore, promoting Dicer/miR-10a signaling may represent a novel and promising therapeutic strategy for atherosclerosis. In human failing heart, miR-223-3p and miR-486-3p were identified as key drivers in the polarization of macrophages towards a pro-inflammatory phenotype [155], suggesting they can be potential therapeutic targets for HF via modulation of inflammatory states of macrophages.
Long non-coding RNAs (LncRNAs) and circular RNAs (CircRNAs)
Nevertheless, the modulation of macrophages can be mediated also by long non-coding RNA, which are longer than 200 nt, encompassing linear lncRNAs and circular RNAs (circRNAs).
RNA-seq profiling of atherosclerotic lesion intima has revealed the presence of a macrophage-specific lncRNA, called MAARS (Macrophage-Associated Atherosclerosis lncRNA Sequence) [156]. Targeted silencing of MAARS has been shown to significantly reduce atherosclerosis progression in a murine model by regulating macrophage apoptosis [156]. The expression of lncRNA SNHG16 is notably increased in both atherosclerosis patients and in ox-LDL-mediated atherosclerotic macrophages in vitro. Importantly, its elevated expression is associated with inflammation and foam cell formation via the NF-κB signaling pathway. Conversely, the inhibition of SNHG16 leads to opposing results, suggesting SNHG16 as a potential target for atherosclerosis treatment [157]. Another critical lncRNA involved in cholesterol metabolism in macrophages is MeXis, which plays a role in regulating ABCA1 expression. The loss of MeXis impairs macrophage Abca1 expression and accelerates atherosclerosis [158]. Like miRNAs, lncRNAs also impact macrophage polarization. For instance, NEAT1, whose levels were correlated with post-MI status, independent of statin intake and LV ejection fraction, is increased in ox-LDL-mediated atherosclerotic human THP-1 macrophages and promotes inflammation and lipid uptake [159, 160]. Whereas knockdown of NEAT1 in macrophages represses inflammation and the formation of foam cells, suggesting a role for NEAT1 in atherosclerosis development [160]. Another example is the lncRNA plasmacytoma variant translocation 1 (PVT1), which exhibits elevated expression in myocardial tissue and heart-infiltrating macrophages of sepsis mice. A major function of PVT1 is to enhance M1 macrophage polarization and promote LPS-induced myocardial injury via the miR-29a/HMGB1 axis. In murine model of lipopolysaccharide (LPS)-induced myocarditis, the knockdown of PVT1 prevents the polarization of macrophages toward a pro-inflammatory phenotype, which leads to relieved sepsis-induced myocardial injury [161].
CircRNAs represent a relatively less explored class of ncRNAs, and their role as miRNA- sponges is one of the well-established mechanisms of circRNAs in disease progression [162]. The interaction between circRNAs and miRNAs has also been observed in macrophages during the progression of CVD. For example, CircDENND1B has been identified to bind to miR-17-5p, exerting a pro-cholesterol-efflux function [162]. Increased level of circDENND1B in macrophages promotes Abca1-mediated cholesterol efflux, consequently reducing foam cell formation. This suggests that modulation of CircDENND1B/miR-17-5p axis in macrophages could be a potential therapeutic option for the regulation of foam cell formation in atherosclerosis [162]. Another circRNA, hsa_circ_0008896, was found to be significantly upregulated in both in vitro and in vivo atherosclerosis models [163]. It functionally interacts with hsa-miR-633 and enhances the proliferation, migration, and invasion of vascular smooth muscle cells (VSMCs), thereby promoting atherosclerosis progression. While previous studies have demonstrated the up-regulation of hsa_circ_0008896 in oxidized low-density lipoprotein-treated macrophages, there is limited research on its modulation in macrophages [164]. These findings suggest therapeutic potential of cell-specific ncRNAs in macrophages and emphasize the need for a deeper understanding of the interplay between macrophages and ncRNAs to expand therapeutic options for HF. Nevertheless, the translation of these therapeutic strategies to human studies has not been extensively realized. Successful clinical translation relies on a comprehensive understanding of the biology of target cells within specific organs and the specific non-coding RNAs involved, which can ensure the desired therapeutic outcomes while minimizing off-target effects.
Biomarker study
Macrophages also secrete important signaling molecules that influence neighboring cells and the tissue microenvironment. Secreted factors from pathology-associated macrophages have the potential to be valuable biomarkers for the diagnosis and management of heart failure (Table 1).
Inflammatory mediators
Cytokines/chemokines
In response to various inflammatory stimuli, such as endotoxins and various forms of chemical and physical cardiac injury, activated macrophages primarily secrete pro-inflammatory cytokines. The elevated release of pro-inflammatory cytokines such as IL-1β, IL-6, MCP-1 and TNF-α intensifies pro-inflammatory signaling, ultimately leading to chronic inflammation and cardiac dysfunction in both HFrEF and HFpEF [79, 165, 166]. Notably, chronic and systemic inflammation, coupled with pro-fibrotic signals, holds greater prominence in HFpEF [167]. Elevated serum levels of pro-inflammatory macrophage cytokines, such as TNF-a, MCP-1, IL-6 and IL-12 were observed in patients with HFpEF compared to patients with asymptomatic left ventricular diastolic dysfunction or asymptomatic hypertension [80, 168]. Particularly noteworthy, a higher serum level of IL-6 is closely linked to the onset HFpEF in the general population, while no significant association with HFrEF [169]. While these cytokines may also originate from other damaged cardiac cells such as cardiomyocytes, endothelial cells, the reduction of pro-inflammatory signals in mice on a high-fat diet was notably achieved through the depletion of pro-inflammatory macrophages, resulting in mitigated diastolic dysfunction [170]. This finding strongly suggests that activated macrophages represent a primary cell source for the secretion of pro-inflammatory cytokines and underscores their potential as a promising therapeutic target in HFpEF.
MPO
Myeloperoxidase (MPO), an enzyme that facilitates the formation of hypochlorite from chloride and hydrogen peroxide, is another promising biomarker for the early detection of CVDs. It is released by activated macrophages and neutrophils during inflammatory processes [171, 172]. In the context of coronary artery disease (CAD), MPO contributes to oxidative stress and vascular dysfunction, both of which are key factors in the progression of the disease. Studies have shown that MPO levels are elevated in the blood and plaque tissue of patients with CAD, and that these levels correlate with disease severity [173]. However, MPO's specificity to CVD is not absolute, given that macrophage and neutrophil activation may also manifest in response to infections and non-cardiovascular-related inflammatory responses [172]. Despite these limitations, MPO is a valuable tool for the early detection of CAD. Its use in conjunction with other biomarkers and clinical risk factors can help to identify patients who are at high risk for developing the disease.
Pro-fibrotic mediators
Growth factors
Macrophages are also recognized for their pro-fibrotic properties in the advancement of heart failure. In particular, pro-fibrotic macrophages secrete various factors, including transforming growth factor (TGF)-β and IL-10, which orchestrate cardiac fibroblast activity-directing migration, proliferation, and collagen expression. This cascade ultimately leads to collagen fiber deposition and the formation of mature scars within the heart [73, 168, 174]. Correspondingly, HFpEF patients exhibit elevated plasma levels of TGF-β [175]. GDF-15, previously known as macrophage-inhibitory cytokine-1, belongs to the TGF-β superfamily and is another growth factor prominently secreted by activated macrophages [176]. While GDF-15 was initially identified as a negative regulator of macrophage activation, recent studies have revealed its paradoxical role in promoting fibrosis in various contexts. In the context of acute heart failure (AHF) patients participating in the RELAX-AHF study, elevated GDF-15 levels were correlated with a higher probability of adverse outcomes [177]. These findings suggest that GDF-15, like TGF-β, may contribute to the progression of heart failure. Although TGF-β and GDF-15 are not exclusive to cardiovascular disease, as they have also been found elevated in various cancers, their strong association with disease progression in heart failure patients suggests that these growth factors could serve as valuable tools for risk assessment and guiding therapeutic decisions.
Galectin-3
Galectin-3, a β-galactoside–binding lectin primarily secreted by activated macrophages, exhibits a strong association with myocardial fibrosis and the progression of HF [178,179,180,181,182]. Its involvement in cardiac fibrosis has prompted its inclusion in the European and American HF guidelines as a biomarker for myocardial fibrosis, with a class IIb recommendation [183, 184]. Gal-3 serves as a valuable biomarker not only for diagnosing HFpEF, but also for assessing post-MI risk and LV remodeling [185,186,187,188]. Multiple clinical studies have demonstrated that plasma levels of Gal-3 can effectively predict cardiovascular mortality [189, 190]. In particular, a serum Gal-3 level exceeding 8.7 ng/mL has been identified as an independent predictor of heightened all-cause mortality risk in both MI and chronic heart failure patients [191,192,193]. The strong association between Gal-3 levels and HF severity highlights its potential as a prognostic marker and therapeutic target. Measuring Gal-3 levels can aid in risk stratification and guide treatment decisions in HF patients.
Others: cell-to-cell networker
Exosomes
Exosomes, extracellular vesicles released by various cell types including macrophages, play a crucial role in intercellular communication via transferring a wide range of cargos, encompassing proteins or nucleic acids. Analyzing their presence in blood and their expression profile offer valuable insights into the function of macrophages in CVDs. In atherosclerosis, macrophage foam cells release a higher quantity of exosomes compared to normal macrophages. These additional vesicles play an important role in cell-to-cell crosstalk between macrophages and vascular smooth muscle cells (VSMCs) [194]. Exosomes originating from foam cells may stimulate VSMC adhesion and migration by modulating the actin cytoskeleton and focal adhesion pathways [194]. Moreover, under hypertensive conditions, the infiltration of macrophages significantly increases, leading to the upregulation of pro-inflammatory factors within their secreted exosomes, such as intracellular adhesion molecule-1 (ICAM1) and plasminogen activator inhibitor-1 (PAI-1). These exosomes further induce inflammation in endothelial cells, contributing to the progression of the pathological state [195].
Micro RNAs
MicroRNAs constitute a significant portion of the cargo within exosomes, and macrophage-derived miRNAs serve as potential biomarkers in CVDs. In the context of atherosclerosis, Zhang YG et al. demonstrated that macrophages involved in atherosclerotic processes release exosomal miR-146a. This miRNA exacerbates atherosclerosis progression by inducing neutrophil extracellular traps (NETs) and increasing oxidative stress in neutrophils. They also showed higher serum levels of miR-146a in atherosclerosis patients compared to healthy counterparts [196]. Interestingly, intracellular miR-146a and secreted miR-146a exhibited opposite effects on atherosclerosis progression. Intracellular miR-146a displayed anti-inflammatory properties in macrophages, leading to reduced atherosclerotic plaque formation [152, 153]. These contrasting effects may be attributed to differences in effector cells and cellular environments. In the context of MI, pro-inflammatory macrophages generate exosomal miR-155, which suppress angiogenesis in endothelial cells, consequently accelerating MI-associated injury [197]. Moreover, macrophage-derived miR-155 functions as a paracrine regulator, influencing fibroblast proliferation and inflammation. Mimicking miR-155 increases the risk of cardiac rupture by suppressing fibroblast proliferation and amplifying inflammation. Inhibiting miR-155 has demonstrated therapeutic potential, with improved cardiac function in a mouse model of MI [198]. Consistent with these findings, serum levels of miR-155 were significantly elevated in post-MI heart failure patients compared to both healthy individuals and MI patients without HF [199]. These findings highlight the potential of macrophage-derived miRNAs as biomarkers and therapeutic targets in CVDs.
Gene expression
Analyzing gene expression patterns offers valuable insights into macrophage function and their impact on HF progression, particularly following MI. Chen et al. conducted a transcriptomic analysis of peripheral blood mononuclear cells (PBMCs) from AMI patients (post-MI HF vs. post-MI non-HF) as well as performed single-cell RNA-seq (scRNA-seq) of recruited macrophages from a mouse model of MI (AMI vs. control mice). They further identified 25 common genes from both gene profiling datasets. These 25 genes were enriched in myeloid leukocyte activation, collagen metabolic process, and response to hypoxia, suggesting a close relationship with cardiac remodeling. Among the 25 identified genes, three genes emerged as promising biomarkers for early heart failure detection following acute MI: CUX1, CTSD, and ADD3. CUX1 and CTSD expression in cardiac macrophages exhibited a steady increase during the first week post-MI in a mouse model. Similarly, these two genes were upregulated in the peripheral blood of MI patients with heart failure compared to those without HF development. Conversely, ADD3 expression was significantly decreased in the HF context. Collectively, macrophage-associated genes hold promise as reliable biomarkers for cardiac remodeling and heart failure manifestation following MI. However, further extensive clinical investigations are warranted [200].
Challenges and opportunity of macrophage-targeting strategies
Heterogeneity: monocyte-derived macrophages vs resident macrophages
One of the challenges in macrophage-targeting therapies lies in the inherent heterogeneity and plasticity. Cardiac macrophages can be sub-divided into two populations by their origins: monocyte-derived macrophages from hematopoietic stem cells (HSCs) and embryonic yolk sac-oriented resident macrophages [69]. Current advances in transcriptomic analysis have enabled the clear differentiation of these two distinct subsets of macrophages in the heart. CCR2− resident macrophages specifically express Lyve1, Cd163, Ccl24, while CCR2+ infiltrating macrophages are characterized by the expression of antigen-presenting genes (MHC II clusters), monocytes expressed gene (Ly6c2) genes [201]. Despite sharing common macrophage characteristics such as phagocytosis, immune response, and cytokine secretion, these two subsets exhibit divergent responses to injuries. Reiterating the points made earlier, monocyte-derived macrophages are primarily linked to inflammatory responses and pathogenic characteristics, while resident macrophages exhibit cardioprotective functions and play specific roles in cardiac physiology [69, 71, 104, 139, 165]. Consequently, depleting the entire macrophage population in the heart can lead to compromised LV remodeling, partially due to the loss of resident macrophages, which is crucial for the maintenance of myocardial homeostasis and cardiac function [202, 203]. Therefore, it is crucial to selectively target between bone marrow-derived macrophages and tissue-resident macrophages, while further discerning subsets and functions within the mixed population. Moreover, the inflammatory environment may shift the characteristics of cardiac macrophages from a reparative to a detrimental phenotype in the context of a diseased heart [201]. Therefore, the same intervention can have opposite outcomes depending on the timing and subset of macrophages targeted. To optimize therapeutic efficacy, it is imperative to comprehend the diverse macrophage types and their specific functions at the right timing to develop appropriate therapeutic approaches aimed at maintaining a balanced transition between protective and pathogenic responses in cardiac tissue.
Targeted drug delivery to tissue macrophages
Despite of promising therapeutic potential of the strategies to target pathology-associated cardiac macrophages, to translate this promise to clinical application, challenges for the specific target delivery to tissue macrophages still remain to be overcome. Therefore, the development of advanced drug delivery strategies that precisely target macrophages is expected to bring the vision of successful clinical translation to reality. In this section, we will highlight the strengths and limitations of these strategies, with the ultimate goal of facilitating successful clinical translation (Fig. 2).

Targeted drug delivery to tissue macrophages. Effectively targeting cardiac macrophages remains a significant challenge in the field of cardiovascular medicine. With the development of advanced drug delivery strategies that precisely home in on macrophages, the promise of successful clinical translation can be realized. Several promising strategies are emerging, including tissue or cell-specific promoters, cell surface modification, exosome-based delivery CD68 Cluster of Differentiation 68, PolyA polyadenylic acid, PEG polyethylene glycol
Utilize tissue or cell specific promoters
In the field of gene therapy, the utilization of tissue- or cell-specific promoters is a well-established method to ensure that the therapeutic effect is confined to the desired tissue cells. This technique has been widely applied in virus-mediated delivery systems, such as lentiviruses and adeno-associated viruses (AAV). Among these, the CD68 promoter stands out as one of the most commonly used promoters for directing transgene expression specifically to macrophages in in vivo settings via viral vectors. A study led by Levin et al. aimed to compare three variants of the CD68 promoter to determine the most efficient variant for macrophage-specific targeting using lentivirus. They observed that the 150 bp proximal region of the CD68 promoter outperformed the other variants and resulted in the highest macrophage specificity and significant protein expression, which could lead to maximal target efficiency [204]. Similarly, macrophage-specific promoters (F4/80 or CD68) also were applied to AAV vectors. Moreover, the cell specific transduction efficiency of recombinant AAV can be also enhanced by modification of capsids. Rosario et al. developed a recombinant AAV6 with triply mutated capsid Y731F/Y705F/T492V, under the control of macrophage-specific promoters (F4/80 or CD68), which allowed for selective gene modification of tissue macrophages while minimizing off-target effects [205]. This study was the first to use unique capsid/promoter combinations for tissue-macrophage-specific gene targeting, significantly enhancing safety and efficiency. AAV-mediated systems have been actively developed to deliver target molecules to the heart, and combining these systems with macrophage-specific promoters could boost target efficiency without altering other organ macrophages. However, since CD68 targets general macrophages regardless of whether they are resident or circulating, additional information for targeting pathological macrophages would also be beneficial for efficiency. Additionally, since viral vectors have a size limitation for cell uptake, combining them with nanoparticles is another emerging option to achieve specific targeting of cardiac macrophages with minimal side effects.
Surface modification and ligand targeting
As a result of challenges in delivering exogenous nucleic acids to macrophages via viral methods, several non-viral strategies have been explored. Nanoparticles and liposomes offer promising alternatives for delivery of small molecule such as siRNAs or miRNAs. These delivery vehicles can enhance cell binding and uptake specificity by incorporating ligands that specifically recognize cell surface receptors or markers expressed in target tissue cells. Recent advances in RNAi technology have enabled the selective targeting of desired cells by combining cell-specific targeting siRNAs with organ-specific biocompatible nanoparticles. For example, Tao et al. constructed PLGA-based siRNA nanoparticle conjugated with the peptide S2P, which binds to the scavenger receptor stabilin-2 on plaque macrophages. This nanoparticle was used to deliver siRNA targeting Ca2 + /calmodulin-dependent protein kinase γ (CamKIIγ), a plaque-destabilizing molecule, to atherosclerotic macrophages. In a mouse model of atherosclerosis, western diet-fed Ldlr−/− mouse, intravenous injection of this nanoparticle significantly improved efferocytosis of macrophages and reduced atherosclerotic lesions [206]. Another example is the development of macrophage-targeting/polarizing graphene oxide (GO) complex (MGC) by Han et al. MGC was constructed by conjugating GO with polyethyleneimine (PEI) and folic acid-polyethylene glycol (FA-PEG), which has shown specific affinity for folate receptors (FR) on macrophages. In a mouse model of MI, intramyocardial injection of the MGC has delivered IL-4 pDNA selectively to cardiac macrophages, with limited uptake in cardiomyocytes and fibroblasts. From a therapeutic perspective, this delivery of IL-4 pDNA via MGC led to attenuated inflammation, thanks to the early shift of macrophages to a reparative phenotype and improved cardiac function [207]. An additional illustrative instance involves the high affinity between dextran sulfate (DXS) and the scavenger receptor class AI (SR-AI), which has been harnessed for drug delivery to macrophages. SR-AI plays a pivotal role in the recognition of modified lipoproteins and the recruitment of macrophages. Its specific ligand, DXS, has been shown to inhibit the cellular internalization of ox-LDL by blocking SR-AI. Based on this knowledge, Zhao and colleagues devised nanoparticles that target atherosclerotic macrophages by decorating their surfaces with DXS. Interestingly, when these nanoparticles were incubated with Apo-I, not only decrease the formation of macrophage-derived foam cells, but cholesterol efflux was also promoted [208]. Nevertheless, their therapeutic efficacy in an in vivo model of atherosclerosis remains to be demonstrated. Additionally, membrane surface glycoproteins, such as mannose, which are predominantly expressed on M2 (CD206+) macrophages, have also been investigated as targeting motifs for efficient drug delivery. Notably, mannosylated nanoparticles increase siRNA delivery to primary macrophages by fourfold compared to the same carrier without targeting moieties [209]. Despite the advancements in non-viral nanocarriers for gene delivery, certain challenges remain. Further research is needed to address issues such as suboptimal intracellular trafficking, low gene transfer efficiency, and the sustainable maintenance of stable gene expression.
Exosome-based delivery
Exosomes are extracellular vesicles (EVs), produced in the endosomal compartment, which mediate cell-to-cell communication by transferring biological material to neighboring cells. Taking advantage of this property, exosomes are coming to the light as a surrogate for drug delivery strategies. Exosomes carry diverse constituents including proteins, lipids, DNA, mRNA, miRNA, lncRNA, and circular RNA. Among these molecules, exosomal miRNAs are the most abundant type of exosomal cargo. Nguyen et al. reported the enrichment of miRNAs in atherogenic macrophage-secreted EVs from mouse and human. Among them, miR-146a is the most enriched miRNAs and EV-mediated transfer of miR-146a plays a pivotal role in the progression of atherosclerosis [210]. Likely, exosome contents from other cells can be also transferred to macrophages. This is the case of miR-182 containing exosomes released from mesenchymal stem cells (MSC) to macrophages, which are able to modulate the polarization of macrophages. In a mouse model of I/R injury, the intramyocardial injection of miR-182 enriched MSC-derived exosomes elicited the polarization of macrophages towards a reparative phenotype as well as improved the cardiac functional parameters. While these therapeutic effects have been significantly hindered when macrophages are depleted or miR-182 in MSC-exosomes is reduced, suggesting the exosomes can be applied for miRNA delivery to macrophages [148]. MSC-derived miR-101a also presented protective effects on cardiac remodeling post-MI via macrophage polarization [211]. Additionally, cardiosphere-derived cells (CDCs) derived exosomes or EVs also have been used as macrophage-targeting gene delivery cargos. In mouse and pig models of ischemic injury, the intramyocardial injection of miR-181b enriched CDC-exosomes favored the polarization of macrophages to M2 phenotype, which in turn alleviated the inflammatory and scar formation process [212]. Interestingly, exosomes can be structurally modified to enhance their delivery efficiency to cardiac tissue. For example, in the study conducted by Vandergriff et al., exosomes derived from cardiac stem cells were conjugated with a cardiac homing peptide (CHP) [213], suggesting macrophage-targeting conjugates may enhance the delivery specificity of exosomes to cardiac macrophages. While exosomes are emerging as delivery platform to target desired cells with their ability to be administered via various routes, limited toxicity, and structural stability, they still face critical challenges, including immunogenicity, a short half-life in circulation, limited target specificity, and a lack of standardized protocols for isolation, purification, and production. More importantly, for efficient macrophage targeting, nonspecific uptake and degradation of target molecules should be further considered.
Ex vivo cell therapy (engineered cell therapy)
However, selective targeting of organ-specific macrophages, especially cardiac macrophages without influencing other cell types or organs is still challenging, due to the lack of studies of the unique marker for distinctive organ macrophages. Recent evidence has highlighted ex vivo reprogrammed macrophages could be an alternative option to avoid the massive in vivo delivery obstacles [214]. They can be re-educated to promote regeneration and reduce inflammation ex vivo. One of the key advantages of ex vivo reprogrammed macrophages is that they can be personalized to the individual patient. This is because they are derived from the patient's own cells, which reduces the risk of rejection. Additionally, ex vivo reprogramming allows for the creation of large numbers of macrophages with specific desired phenotypes, such as a reparative phenotype.
Ex vivo reprogrammed macrophages have been shown to be effective in a variety of preclinical models of cardiac repair. Bone marrow-derived macrophages (BMDMs) are a major cell source and are widely employed in macrophage-based cell therapeutic strategies for in vivo applications. While the multilineage potential of CD34+ cells may induce adverse remodeling by promoting inflammatory characteristics, hindering long-term improvements in cardiac function, direct transplantation of fully differentiated macrophages has emerged as an alternative cell therapy strategy. Notably, macrophage modification has substantially reduced the rejection risk of transplanted cells [215,216,217]. Transplantation of reparative macrophages, generated by re-educating BMDMs with the combination of M2 stimulators like IL-4, IL-10, and TGF-β1 [215, 216], has shown a superior therapeutic efficacy with improved cell engraftment in donor tissue compared to naïve BMDMs. Hypoxia priming is one of multiple strategies to educate macrophages toward reparative phenotype ex vivo. Transplantation of hypoxic macrophages primed with 1% oxygen immediately after the induction of MI in mice at 3 sites along the infarct border zone results in enhanced resolution of inflammation and subsequent better cardiac repair [218]. In line with this, Ben-Mordechai et al. have confirmed the crucial role of macrophages in the in vivo treatment of MI. The administration of mesenchymal stem cells (MSC) to mice following MI resulted in a greater polarization of macrophages towards an M2 phenotype within 3–4 days. While, temporary elimination of macrophages, with or without MSC treatment, was associated with a higher mortality rate due to left ventricular dysfunction and the development of excessive scarring. This effect was reverted by macrophage restoration and treatment with MSC, supporting the crucial role of these phagocytic cells in cardiac repair [219]. Moreover, the re-educated macrophages can function as adjuvant and positively influence regenerative stem cell therapies. In a rat model of MI, the therapeutic effects of the bone marrow-derived mesenchymal cells (BMMSCs) were strongly enhanced when they were co-injected with BMDMs compared to the only BMMSCs injected group. Injection of the mixture of BMMSCs and BMDMs notably improved cardiac function in terms of increased micro-vessel density as well as decreased fibrosis, and more importantly, facilitated resolution of inflammation with a higher presence of anti-inflammatory macrophages [220]. To date, the attempts to induce cardiac regeneration by stem cells have still not successfully translated to clinical studies, mainly due to the limited survival of the transplanted cells within a prolonged phase of tissue inflammation. These results suggest that macrophages can be an alternative adjuvant to overcome the challenges of current stem cell therapies including improved engraft survival and regulatory effects on inflammation.
Currently, several clinical trials employing ex vivo macrophage polarization and subsequent adoptive transfer for the treatment of cardiovascular diseases, such as ischemia, cardiomyopathy, or arterial diseases, have advanced to phase 2 or 3 [221]. In clinical trials, cell therapy mainly uses two sources of cells: autologous and allogeneic. Autologous transplants use the patient's own cells as the cell donor, while allogeneic transplants use cells from healthy donors. Autologous transplantation is a more traditional method and does not require the identification of an HLA-matched donor. Moreover, graft failure of autologous transplants is rare due to rapid immune reconstitution and a lower risk of life-threatening complications. Autologous re-educated macrophages have been an alternative source for cell transplantation in clinical studies. In a clinical pilot study of 21 children with severe cerebral palsy, autologous M2 macrophages significantly boost motor and cognitive activities of patients without adverse effects and comorbidities during 5-year follow-up [222]. Clinical efficacy of M2 macrophages also has been proved in patients with stroke (ClinicalTrials.gov, NCT01845350, completed). Intrathecal injection of autologous M2 macrophages improved neurological recovery with no serious adverse events or cell rejection for 6-month follow-up via immunomodulatory activity of M2 macrophages [223]. In contrast, a separate study by Perin et al. found that patients with chronic ischemic heart disease who were administered non-expanded autologous bone marrow macrophages via transendocardial injection did not experience any improvement in cardiac function compared to the placebo group (ClinicalTrials.gov, number NCT00824005, completed) [224], highlighting the ex vivo expansion of macrophages is crucial for a positive outcome in cardiac repair of patients with HF. In the cardiac field, an innovative patient-specific multicellular therapeutic approach called Ixmyelocel-T was established to treat patients with ischemic HF (ClinicalTrials.gov, NCT01670981, completed) [225, 226]. Ixmyelocel-T, an expanded multicellular therapy, is generated by expanding mixture of patient-derived autologous BMMCs enriched for CD45+CD14+M2 BMDMs and CD90+BMMSCs [225, 226]. In phase 2b, patients with dilated cardiomyopathy treated with transendocardial injection of Ixmyelocel-T showed an improvement in cardiac functions compared to patients treated with placebo during the 12 months following treatment administration [225, 226]. More importantly, the Ixmyelocel-T cells containing M2-reparative macrophages showed efficient clearance of apoptotic cells, which limit tissue injury and facilitate wound healing [227], indicating the therapeutic potential of reparative macrophages. Despite of their therapeutic potential, the development of Ixmyelocel-T as drug has been halted due to no further plan to initiate a phase III trial (Vericel Corporation, formerly Aastrom Biosciences). While the largest trial of Ixmyelocel-T, RESTORE-CLI clinical trial for limb ischemia has been downgraded in recent reports, due to the concerns about risk of bias, imprecision, and inconsistency (ClinicalTrials.gov, number NCT00468000, completed) [228]. Moreover, adverse events including amputations, heart attacks and strokes have been detected in patients with Ixmyelocel-T [228]. These clinical outcomes serve as a reminder that while cell therapy has the potential to revolutionize the treatment of CVDs, the safety and efficacy of new cell therapies must be carefully assessed before they are made widely available.
Induced pluripotent stem cell (iPSC)-derived macrophages
However, standard autologous immune cell therapies have yet several limitations: (1) the transplanted cells are not able to effectively integrate into ischemic tissue; (2) autologous approaches using macrophages derived from patients who are often heavily pre-treated, which have hindrances in terms of manufacturing and efficacy (Table 2). Given these considerations, reprogramming of allogeneic macrophages, generated from healthy donors would strengthen the efficiency of macrophage-based cell therapy, iPSC-derived macrophages, therefore, have continued to improve and have endured as an effective treatment option against cardiac diseases. The therapeutic potential of human(h)iPSC-derived macrophages has been studied in different diseases. This is the case of pulmonary alveolar proteinosis, a pathological condition in which a mutation in the granulocyte–macrophage colony-stimulating factor (GM-CSF) receptor impedes the differentiation and function of alveolar macrophages. Happle et al. demonstrated that, in a murine model of pulmonary alveolar proteinosis, the intratracheal administration of hiPSC-derived macrophages results in efficient pulmonary engraftment, differentiation to tissue-specific alveolar macrophages and improvement of pulmonary functions [229]. Differently, in an immunodeficient mouse model of liver injury, Pouyanfard et al. demonstrated the beneficial effects of applying hiPSC-derived macrophages with an anti-inflammatory phenotype. In this case, hiPSC-derived macrophages were polarized towards an anti-inflammatory phenotype and injected intraperitoneally in mice with liver injury resulting in reduced fibrosis and inflammation [230].
The beneficial effects of allogeneic macrophages have been also shown in cardiac disease model, rats subjected to MI. Here, human macrophages were isolated from the blood of donors and activated ex vivo by hypo-osmotic shock, which is a method to stimulate macrophages by transferring them into distilled water for a short time. This triggers cytokine production of macrophages and increases their phagocytosis efficiency [231]. Administration of the activated human macrophages into infarcted myocardium of rats leads to a significant improvement in heart function, tissue vascularization, and tissue repair within 5 weeks following treatment compared to the control group [232]. These favorable effects might be due to the replenishment of immune activity via the exchange of impaired old macrophages with healthy macrophages.
Chimeric antigen receptor (CAR) has become a promising approach to enhance the target recognition of immune cells. In a recent study on cancer research conducted by Klichinsky et al., macrophages were transduced by AAV to express CAR. The CAR-macrophages demonstrated a specific affinity and efficient antigen-specific phagocytosis for cancer cell targeting [233]. Emerging studies have addressed the importance of macrophage-mediated removal of apoptosis in cardiac remodeling post-injury. CAR-macrophage-targeting necrotic cardiomyocytes or activated fibroblasts could extend their therapeutic possibilities to cardiac conditions, in addition to cancers.
Future outlook
Advance macrophage study model in context of CVDs
To facilitate the successful translation of therapeutic strategies targeting macrophages, it is imperative to establish appropriate frameworks for evaluating these modalities. In this regard, we provide a concise review of existing macrophage study models and propose a novel model to bridge the gap between in vitro and in vivo findings, ultimately advancing their clinical applicability in the context of CVDs (Fig. 3).

Advanced cardiac macrophage study model. For successful translation of research findings into clinical applications, more clinically relevant study models for cardiac macrophages, particularly in the context of cardiovascular diseases (CVDs), should be considered. We have proposed possible models ranging from in vitro to ex vivo settings using human induced pluripotent stem cell (iPSC)-derived macrophages or novel patient-derived heart slice models, living myocardial slices (LMS). hiPSC human pluripotent stem cells, LPS lipopolysaccharide, IFN-γ Interferon gamma, TNF-α tumour necrosis factor alpha, GM-CSF granulocyte–macrophage colony-stimulating factor, IL interleukin, TGF-β transforming growth factor-β, KO knock out
In vitro study
First and foremost, the current in vitro studies employing the classic M1/M2 macrophage paradigm are overly simplistic and fail to account for the heterogeneity and plasticity of macrophage physiology in response to pathological conditions. The traditional in vitro models involve inducing M1 macrophages through cytokines like LPS, IFN-γ, TNF-α, and GM-CSF, while M2 macrophages are activated using IL-4, IL-10, IL-13, and TGF-β [234]. This oversimplified model does not adequately represent the complexity of in vivo macrophage behavior. Recent advancements in high-throughput single-cell RNA sequencing have revealed diverse macrophage subsets associated with the progression of cardiovascular diseases, each with a distinct transcriptome [235]. Depending on the specific subsets of macrophages involved in different disease contexts, it is essential to use more precise stimulants and signaling pathways to generate the desired macrophage phenotype and function. Secondly, the choice of an appropriate cell model is crucial. For studying macrophage function, readily available immortalized cell lines such as murine Raw264.7 or human THP-1 cells are commonly used [236,237,238]. Despite their convenience and reproducibility, the findings from these cells often do not translate well into in vivo or clinical studies. This disconnect arises from the differences between these cell lines and tissue-resident macrophages [237,238,239].
While cardiac macrophages can be isolated from mouse or rat hearts and cultured ex vivo, there are challenges related to chronic culturing, limited cell yield, and disparities in genomic profiles when compared to humans. Consequently, human-induced pluripotent stem cell (hiPSC)-derived macrophages have emerged as an alternative in vitro model that better represents patient-derived pathology. Navarro-Guerrero and her colleagues have developed an efficient CRISPR/Cas9-knockout hiPSC-derived macrophage protocol through lentiviral transduction [240]. This approach has paved the way for the generation of clinically relevant human pathologic macrophages, which can be employed in drug screening. More importantly, exposure to organ-specific cues enabled human or murine iPSC-derived macrophages to differentiate into their respective tissue-resident macrophages. For example, murine iMacs, when injected into the brain, differentiated into microglia in vivo, while those engrafted in the lung developed into functional alveolar macrophages. These observations suggest that cardiac-specific cues can induce iMacs to differentiate into cardiac-resident macrophages, potentially providing a clinically relevant in vitro model of cardiac macrophages [241].
In vivo study
Animal models of CVDs have been comparably well-established and closely resemble clinical scenarios, encompassing conditions like MI, transverse aortic constriction (TAC) pressure overload, and high-fat diet-induced models. Consequently, these models have been instrumental in exploring the dynamic changes and heterogeneity of macrophage populations in various HF etiologies and disease progressions. Many studies involving gene functions during macrophage polarization or phagocytosis have employed animal models with gene knockouts. Several gene functions during macrophage polarization or phagocytosis have often been studied in animal model of knockout. For a successful knockout model, a deep understanding of the lineage-specific factors and markers within the target organ or cell is crucial. Variations in macrophage phenotypes and the corresponding markers are identified by various research groups. For example, CSF1 and its receptor CSF1R are well-known primary factors involved in general macrophage differentiation. Genetic manipulations targeting these factors have led to the generation of macrophage depletion models. The selective restriction of CSF1R through the genetic deletion of the fms-intronic regulatory element (Fire), a super-enhancer of CSF1R, has effectively suppressed the differentiation of tissue macrophages [242]. Additionally, mice with the CSF1R gene floxed (CSF1Rfl/fl) have been widely used to ablate specific macrophage genes. Common phenotypic markers for macrophages include CD11b, F4/80, or CD68. Recent advances in imaging techniques, such as spatio-temporal single-cell RNA sequencing, have enabled the visualization and analysis of cardiac macrophage behavior and interactions within various in vivo CVD settings. While it is important to note that the immune milieu can be significantly influenced by the induction of specific pathologies. For example, different in vivo models of atherosclerosis, such as APOE-/- and LDLR-/- mice or high-fat diet-fed animals, may yield slightly different outcomes in terms of macrophage behavior and immune responses [243, 244]. Therefore, it's crucial to carefully select the appropriate model to ensure the accurate interpretation of research findings. Furthermore, many CVDs silently progress over extended periods, involving multiple etiologies and comorbidities [1]. To gain a more comprehensive understanding of macrophage roles and phenotypic shifts over time, long-term in vivo studies are necessary. Additionally, addressing the translation gap between small rodents and humans is vital [245, 246]. To bridge this gap, the use of large animal models or humanized mice is strongly recommended, as they better recapitulate the individual variations seen in clinical cases.
Ex vivo study
While in vivo studies provide valuable insights into diverse human CVDs, they still present several challenges, including cost, time, and ethical considerations. An emerging alternative experimental model to study cardiac macrophages is the ex vivo cell culture model, which serves as a compromise between in vitro and in vivo models. A protocol developed by Aktories et al. has enabled the establishment of long-term monocultures of tissue macrophages derived from distinct organs, offering a more clinically relevant ex vivo macrophage model [247]. Co-culturing ex vivo-isolated cardiac macrophages with other cardiac cells provides an indirect approach to understand their network dynamics from the acute to chronic phases of disease [248].
Furthermore, there has been significant progress in developing patient-derived heart or cell-based ex vivo culture platforms that mimic macrophage dynamics more closely to the clinical scenario. These platforms include precision heart cut slices, Engineered Heart Tissue (EHT), and hiPSC-derived organoids. Waleczek et al. employed the ex vivo culture model of precision heart slices, referred to as living myocardial slices (LMS), as a tool to investigate CRM. LMS are ultrathin multicellular cardiac preparations with the circulatory network interrupted, enabling the study of tissue-macrophage responses to immunomodulatory and mechanical stimulations within the preserved intricate myocardial architecture [248]. Importantly, LMS can also be generated from heart failure patients, allowing researchers to understand and visualize CVDs-associated behavior of cardiac macrophages, including their number, phenotype, and related signaling. Similarly, EHT has been introduced as another powerful ex vivo model for both healthy and diseased hearts. While EHTs are typically fabricated using cardiomyocytes, they better recapitulate the contractile function and complexity of the native myocardium with the addition of non-myocytes, such as fibroblasts and endothelial cells [249]. Given the emerging importance of resident macrophages in cardiac function, including electrical conduction and regeneration, the addition of macrophages in EHT is being strongly considered to create more physiologically relevant models [250]. In addition to LMS and EHT, a 3D culture model of mixed cell populations, iPSC-derived organoids, have emerged as promising tools for studying human cardiac cells [251,252,253,254]. Co-culture platforms containing human iPSC–derived cardiomyocytes and macrophages have demonstrated the intimate interactions between these two cell types [255, 256]. While these models are still in the early stages of development, the incorporation of damaged cardiomyocytes or fibrosis-associated fibroblasts into these systems holds promise as an experimental tool to study cardiac macrophages in the context of CVDs. Subsequent high-throughput transcriptomic or proteomic analyses of these settings could identify novel therapeutic targets for cardiac macrophages. However, to enhance clinical translation, further exploration of accurate toxicity assessment and experiment reproducibility is needed.
Conclusion
In this review, we have outlined the latest advances in macrophage-based therapies for CVDs, from preclinical animal models to clinical trials, highlighting their potential for developing therapeutic interventions in the future. Over the past few decades, there has been tremendous growth in the development of macrophage-based therapeutic strategies, as these cells play a crucial role in diverse pathological conditions. In particular, the successful outcomes of immune cell therapies such as CAR-T cells have drawn attention to macrophages, the largest immune cell population. While macrophages are excellent phagocytes with homing properties and have shown great efficacy against cancers, their proportion and network are more complex in the heart, where they continuously change phenotype in response to environmental stimuli and closely interact with both diseased and healthy cells. Moreover, delivering drugs to cardiac macrophages remains challenging. These limitations make the use of therapeutic macrophages for treating CVDs more difficult. Therefore, novel interventions are required to improve selective targeting and durability without causing unspecific off-target effects for safe and long-lasting therapeutic effects. Precise and targeted modulation of macrophages holds potential for therapeutic interventions to prevent adverse cardiac remodeling, enhance tissue repair, and improve cardiac function in various CVDs. Our review has summarized the progress of macrophage-based therapeutic strategies to date and discussed additional ways to improve their efficacy, such as through the use of nanoparticles or cell transplantation. It is important to note that macrophage-targeted therapies are still in the early stages of development, and further research is needed to fully understand their efficacy and safety profiles in clinical settings. Although further research is needed to translate these basic findings into clinical treatments, these promising therapeutic strategies are paving the way for a new era of personalized medicine based on cellular treatments that could extend to a broader range of inflammatory pathologies.
References
Vaduganathan M, Mensah GA, Turco JV, Fuster V, Roth GA (2022) The global burden of cardiovascular diseases and risk: a compass for future health. J Am Coll Cardiol 80:2361–2371. https://doi.org/10.1016/j.jacc.2022.11.005
Buddeke J, Bots ML, Van Dis I, Visseren FLJ, Hollander M, Schellevis FG, Vaartjes I (2019) Comorbidity in patients with cardiovascular disease in primary care: a cohort study with routine healthcare data. Br J Gen Pract 69:e398–e406. https://doi.org/10.3399/bjgp19X702725
Sorriento D, Iaccarino G (2019) Inflammation and cardiovascular diseases: the most recent findings. Int J Mol Sci 20:3879. https://doi.org/10.3390/ijms20163879
Halade GV, Lee DH (2022) Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine 79:103992. https://doi.org/10.1016/j.ebiom.2022.103992
Bajpai G, Lavine KJ (2019) Isolation of macrophage subsets and stromal cells from human and mouse myocardial specimens. J Vis Exp. https://doi.org/10.3791/60015
Zaman R, Epelman S (2022) Resident cardiac macrophages: Heterogeneity and function in health and disease. Immunity 55:1549–1563. https://doi.org/10.1016/j.immuni.2022.08.009
Lafuse WP, Wozniak DJ, Rajaram MVS (2021) Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells 10:51. https://doi.org/10.3390/cells10010051
Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J, Rosenthal N, Kovacic JC (2018) The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (Part 4). J Am Coll Cardiol 72:2213–2230. https://doi.org/10.1016/j.jacc.2018.08.2149
Rhee AJ, Lavine KJ (2020) New approaches to target inflammation in heart failure: harnessing insights from studies of immune cell diversity. Annu Rev Physiol 82:1–20. https://doi.org/10.1146/annurev-physiol-021119-034412
Lother A, Kohl P (2023) The heterocellular heart: identities, interactions, and implications for cardiology. Basic Res Cardiol 118:30. https://doi.org/10.1007/s00395-023-01000-6
He L, Nguyen NB, Ardehali R, Zhou B (2020) Heart regeneration by endogenous stem cells and cardiomyocyte proliferation: controversy, fallacy, and progress. Circulation 142:275–291. https://doi.org/10.1161/CIRCULATIONAHA.119.045566
Yang D, Liu HQ, Liu FY, Tang N, Guo Z, Ma SQ, An P, Wang MY, Wu HM, Yang Z, Fan D, Tang QZ (2020) The roles of noncardiomyocytes in cardiac remodeling. Int J Biol Sci 16:2414–2429. https://doi.org/10.7150/ijbs.47180
Frangogiannis NG (2014) The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11:255–265. https://doi.org/10.1038/nrcardio.2014.28
Koenig AL, Shchukina I, Amrute J, Andhey PS, Zaitsev K, Lai L, Bajpai G, Bredemeyer A, Smith G, Jones C, Terrebonne E, Rentschler SL, Artyomov MN, Lavine KJ (2022) Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat Cardiovasc Res 1:263–280. https://doi.org/10.1038/s44161-022-00028-6
Molenaar B, Timmer LT, Droog M, Perini I, Versteeg D, Kooijman L, Monshouwer-Kloots J, de Ruiter H, Gladka MM, van Rooij E (2021) Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun Biol 4:146. https://doi.org/10.1038/s42003-020-01636-3
Dick SA, Epelman S (2016) Chronic heart failure and inflammation: what do we really know? Circ Res 119:159–176. https://doi.org/10.1161/CIRCRESAHA.116.308030
Stein A, Metzeler K, Kubasch AS, Rommel KP, Desch S, Buettner P, Rosolowski M, Cross M, Platzbecker U, Thiele H (2022) Clonal hematopoiesis and cardiovascular disease: deciphering interconnections. Basic Res Cardiol 117:55. https://doi.org/10.1007/s00395-022-00969-w
Van Linthout S, Miteva K, Tschöpe C (2014) Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res 102:258–269. https://doi.org/10.1093/cvr/cvu062
Chen CJ, Kajita H, Takaya K, Aramaki-Hattori N, Sakai S, Asou T, Kishi K (2022) Single-Cell RNA-seq analysis reveals cellular functional heterogeneity in dermis between fibrotic and regenerative wound healing fates. Front Immunol 13:875407. https://doi.org/10.3389/fimmu.2022.875407
Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N, Teichmann SA (2020) Cells of the adult human heart. Nature 588:466–472. https://doi.org/10.1038/s41586-020-2797-4
Butoi E, Gan AM, Tucureanu MM, Stan D, Macarie RD, Constantinescu C, Calin M, Simionescu M, Manduteanu I (2016) Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochim Biophys Acta 1863:1568–1578. https://doi.org/10.1016/j.bbamcr.2016.04.001
Meznarich J, Malchodi L, Helterline D, Ramsey SA, Bertko K, Plummer T, Plawman A, Gold E, Stempien-Otero A (2013) Urokinase plasminogen activator induces pro-fibrotic/M2 phenotype in murine cardiac macrophages. PLoS ONE 8:e57837. https://doi.org/10.1371/journal.pone.0057837
Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP (2005) Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115:56–65. https://doi.org/10.1172/JCI200522675
Leblond AL, Klinkert K, Martin K, Turner EC, Kumar AH, Browne T, Caplice NM (2015) Systemic and cardiac depletion of M2 macrophage through CSF-1R signaling inhibition alters cardiac function post myocardial infarction. PLoS ONE 10:e0137515. https://doi.org/10.1371/journal.pone.0137515
Fairweather DL, Cihakova D (2009) Alternatively activated macrophages in infection and autoimmunity. J Autoimmun 33:222–230. https://doi.org/10.1016/j.jaut.2009.09.012
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: In vivo veritas. J Clin Invest 122:787–795. https://doi.org/10.1172/JCI59643
Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 117:524–529. https://doi.org/10.1172/JCI31487
Hu S, Yang M, Huang S, Zhong S, Zhang Q, Ding H, Xiong X, Hu Z, Yang Y (2022) Different roles of resident and non-resident macrophages in cardiac fibrosis. Front Cardiovasc Med 9:818188. https://doi.org/10.3389/fcvm.2022.818188
Hitscherich PG, Xie LH, Del Re D, Lee EJ (2019) The effects of macrophages on cardiomyocyte calcium-handling function using in vitro culture models. Physiol Rep 7:e14137. https://doi.org/10.14814/phy2.14137
Trial JA, Rossen RD, Rubio J, Knowlton AA (2004) Inflammation and ischemia: macrophages activated by fibronectin fragments enhance the survival of injured cardiac myocytes. Exp Biol Med 229:538–545. https://doi.org/10.1177/153537020422900612
Grune J, Lewis AJM, Yamazoe M, Hulsmans M, Rohde D, Xiao L, Zhang S, Ott C, Calcagno DM, Zhou Y, Timm K, Shanmuganathan M, Pulous FE, Schloss MJ, Foy BH, Capen D, Vinegoni C, Wojtkiewicz GR, Iwamoto Y, Grune T, Brown D, Higgins J, Ferreira VM, Herring N, Channon KM, Neubauer S, Shanmuganathan M, Ferreira VM, Channon KM, Sosnovik DE, Milan DJ, Swirski FK, King KR, Aguirre AD, Ellinor PT, Nahrendorf M (2022) Neutrophils incite and macrophages avert electrical storm after myocardial infarction. Nat Cardiovasc Res 1:649–664. https://doi.org/10.1038/s44161-022-00094-w
Braune J, Weyer U, Hobusch C, Mauer J, Brüning JC, Bechmann I, Gericke M (2017) IL-6 regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. J Immunol 198:2927–2934. https://doi.org/10.4049/jimmunol.1600476
Tattersall IW, Du J, Cong Z, Cho BS, Klein AM, Dieck CL, Chaudhri RA, Cuervo H, Herts JH, Kitajewski J (2016) In vitro modeling of endothelial interaction with macrophages and pericytes demonstrates Notch signaling function in the vascular microenvironment. Angiogenesis 19:201–215. https://doi.org/10.1007/s10456-016-9501-1
Woollard KJ, Geissmann F (2010) Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol 7:77–86. https://doi.org/10.1038/nrcardio.2009.228
Yang J, Zhang L, Yu C, Yang XF, Wang H (2014) Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2:1. https://doi.org/10.1186/2050-7771-2-1
Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, Hedrick CC, Cook HT, Diebold S, Geissmann F (2013) Nr4a1-dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell 153:362–375. https://doi.org/10.1016/j.cell.2013.03.010
Kumar AG, Ballantyne CM, Michael LH, Kukielka GL, Youker KA, Lindsey ML, Hawkins HK, Birdsall HH, MacKay CR, LaRosa GJ, Rossen RD, Smith CW, Entman ML (1997) Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation 95:693–700. https://doi.org/10.1161/01.CIR.95.3.693
Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL (2014) Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A 111:16029–16034. https://doi.org/10.1073/pnas.1406508111
Chen R, Zhang S, Liu F, Xia L, Wang C, Sandoghchian Shotorbani S, Xu H, Chakrabarti S, Peng T, Su Z (2023) Renewal of embryonic and neonatal-derived cardiac-resident macrophages in response to environmental cues abrogated their potential to promote cardiomyocyte proliferation via Jagged-1–Notch1. Acta Pharm Sin B 13:128–141. https://doi.org/10.1016/j.apsb.2022.08.016
Gao Y, Qian N, Xu J, Wang Y (2021) The roles of macrophages in heart regeneration and repair after injury. Front Cardiovasc Med 8:744615. https://doi.org/10.3389/fcvm.2021.744615
Bruton FA, Kaveh A, Ross-Stewart KM, Matrone G, Oremek MEM, Solomonidis EG, Tucker CS, Mullins JJ, Lucas CD, Brittan M, Taylor JM, Rossi AG, Denvir MA (2022) Macrophages trigger cardiomyocyte proliferation by increasing epicardial vegfaa expression during larval zebrafish heart regeneration. Dev Cell 57:1512-1528.e5. https://doi.org/10.1016/j.devcel.2022.05.014
Rotem I, Konfino T, Caller T, Schary Y, Shaihov-Teper O, Palevski D, Lewis N, Lendengolts D, Naftali-Shani N, Leor J (2022) Osteopontin promotes infarct repair. Basic Res Cardiol 117:51. https://doi.org/10.1007/s00395-022-00957-0
Fazio S, Shapiro MD (2017) Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. F1000Res 6:134. https://doi.org/10.12688/f1000research.9845.1
Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir EA, Amadori L, Khan NS, Wong CK, Shamailova R, Hill CA, Wang Z, Remark R, Li JR, Pina C, Faries C, Awad AJ, Moss N, Bjorkegren JLM, Kim-Schulze S, Gnjatic S, Ma’ayan A, Mocco J, Faries P, Merad M, Giannarelli C (2019) Single-cell immune landscape of human atherosclerotic plaques. Nat Med 25:1576–1588. https://doi.org/10.1038/s41591-019-0590-4
Da LJ, Nishi H, Poles J, Niu X, Mccauley C, Rahman K, Brown EJ, Yeung ST, Vozhilla N, Weinstock A, Ramsey SA, Fisher EA, Loke P (2019) Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 4:e124574. https://doi.org/10.1172/jci.insight.124574
Willemsen L, de Winther MPJ (2020) Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol 250:705–714. https://doi.org/10.1002/path.5392
Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, Robbins CS, Monaco C, Park I, McNamara CA, Binder CJ, Cybulsky MI, Scipione CA, Hedrick CC, Galkina EV, Kyaw T, Ghosheh Y, Dinh HQ, Ley K (2020) Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res 127:402–426. https://doi.org/10.1161/CIRCRESAHA.120.316903
Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H (2014) Atherosclerosis: process, indicators, risk factors and new hopes. Int J Prev Med 5:927–946
Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A (2018) Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res 122:1661–1674. https://doi.org/10.1161/CIRCRESAHA.117.312509
Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, Jang MY, Jang HS, Yun TJ, Lee SH, Yoon WK, Prat A, Seidah NG, Choi J, Lee SP, Yoon SH, Nam JW, Seong JK, Oh GT, Randolph GJ, Artyomov MN, Cheong C, Choi JH (2018) Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res 123:1127–1142. https://doi.org/10.1161/CIRCRESAHA.118.312804
Stöger JL, Gijbels MJJ, van der Velden S, Manca M, van der Loos CM, Biessen EAL, Daemen MJAP, Lutgens E, de Winther MPJ (2012) Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 225:461–468. https://doi.org/10.1016/j.atherosclerosis.2012.09.013
Williams JW, Zaitsev K, Kim KW, Ivanov S, Saunders BT, Schrank PR, Kim K, Elvington A, Kim SH, Tucker CG, Wohltmann M, Fife BT, Epelman S, Artyomov MN, Lavine KJ, Zinselmeyer BH, Choi JH, Randolph GJ (2020) Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol 21:1194–1204. https://doi.org/10.1038/s41590-020-0768-4
Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, Chew SHS, See P, Chakarov S, Wang XN, Lim LH, Johnson LA, Lum J, Fong CY, Bongso A, Biswas A, Goh C, Evrard M, Yeo KP, Basu R, Wang JK, Tan Y, Jain R, Tikoo S, Choong C, Weninger W, Poidinger M, Stanley RE, Collin M, Tan NS, Ng LG, Jackson DG, Ginhoux F, Angeli V (2018) Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49:1191. https://doi.org/10.1016/j.immuni.2018.06.008
Park I, Goddard ME, Cole JE, Zanin N, Lyytikäinen LP, Lehtimäki T, Andreakos E, Feldmann M, Udalova I, Drozdov I, Monaco C (2022) C-type lectin receptor CLEC4A2 promotes tissue adaptation of macrophages and protects against atherosclerosis. Nat Commun 13:215. https://doi.org/10.1038/s41467-021-27862-9
Colin S, Chinetti-Gbaguidi G, Staels B (2014) Macrophage phenotypes in atherosclerosis. Immunol Rev 262:153–166. https://doi.org/10.1111/imr.12218
Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, Nguyen HCB, Chegireddy K, Kim J, Habertheuer A, Vallabhajosyula P, Kambayashi T, Won KJ, Lazar MA (2018) Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A 115:E5096–E5105. https://doi.org/10.1073/pnas.1802611115
Depuydt MAC, Prange KHM, Slenders L, Örd T, Elbersen D, Boltjes A, De Jager SCA, Asselbergs FW, De Borst GJ, Aavik E, Lönnberg T, Lutgens E, Glass CK, Den Ruijter HM, Kaikkonen MU, Bot I, Slütter B, Van Der Laan SW, Yla-Herttuala S, Mokry M, Kuiper J, De Winther MPJ, Pasterkamp G (2020) Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res 127:1437–1455. https://doi.org/10.1161/CIRCRESAHA.120.316770
De Winther MPJ, Bäck M, Evans P, Gomez D, Goncalves I, Jørgensen HF, Koenen RR, Lutgens E, Norata GD, Osto E, Dib L, Simons M, Stellos K, Ylä-Herttuala S, Winkels H, Bochaton-Piallat ML, Monaco C (2023) Translational opportunities of single-cell biology in atherosclerosis. Eur Heart J 44:1216–1230. https://doi.org/10.1093/eurheartj/ehac686
Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M (2006) Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 177:3520–3524. https://doi.org/10.4049/jimmunol.177.6.3520
Palasubramaniam J, Wang X, Peter K (2019) Myocardial infarction—from atherosclerosis to thrombosis. Arterioscler Thromb Vasc Biol 39:e176–e185. https://doi.org/10.1161/atvbaha.119.312578
Jung SH, Hwang BH, Shin S, Park EH, Park SH, Kim CW, Kim E, Choo E, Choi IJ, Swirski FK, Chang K, Chung YJ (2022) Spatiotemporal dynamics of macrophage heterogeneity and a potential function of Trem2hi macrophages in infarcted hearts. Nat Commun 13:4580. https://doi.org/10.1038/s41467-022-32284-2
Duncan SE, Gao S, Sarhene M, Coffie JW, Linhua D, Bao X, Jing Z, Li S, Guo R, Su J, Fan G (2020) Macrophage activities in myocardial infarction and heart failure. Cardiol Res Pract 22(2020):4375127. https://doi.org/10.1155/2020/4375127
Molinaro C, Scalise M, Leo I, Salerno L, Sabatino J, Salerno N, De Rosa S, Torella D, Cianflone E, Marino F (2023) Polarizing macrophage functional phenotype to foster cardiac regeneration. Int J Mol Sci 24:10747. https://doi.org/10.3390/ijms241310747
Zhuang L, Wang Y, Chen Z, Li Z, Wang Z, Jia K, Zhao J, Zhang H, Xie H, Lu L, Chen K, Chen L, Fukuda K, Sano M, Zhang R, Liu J, Yan X (2022) Global characteristics and dynamics of single immune cells after myocardial infarction. J Am Heart Assoc 11:e027228. https://doi.org/10.1161/JAHA.122.027228
Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML (2018) Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol 113:26. https://doi.org/10.1007/s00395-018-0686-x
Jin K, Gao S, Yang P, Guo R, Li D, Zhang Y, Lu X, Fan G, Fan X (2022) Single-cell RNA sequencing reveals the temporal diversity and dynamics of cardiac immunity after myocardial infarction. Small Methods 6:e2100752. https://doi.org/10.1002/smtd.202100752
Kim SH, Lee KY, Chang K (2023) The protective role of TREM2 in the heterogenous population of macrophages during post-myocardial infarction inflammation. Int J Mol Sci 24:5556. https://doi.org/10.3390/ijms24065556
Chen C, Wang J, Liu C, Hu J (2023) Cardiac resident macrophages: key regulatory mediators in the aftermath of myocardial infarction. Front Immunol 14:1207100. https://doi.org/10.3389/fimmu.2023.1207100
Sansonetti M, Waleczek FJG, Jung M, Thum T, Perbellini F (2020) Resident cardiac macrophages: crucial modulators of cardiac (patho)physiology. Basic Res Cardiol 115:77. https://doi.org/10.1007/s00395-020-00836-6
Dick SA, Wong A, Hamidzada H, Nejat S, Nechanitzky R, Vohra S, Mueller B, Zaman R, Kantores C, Aronoff L, Momen A, Nechanitzky D, Li WY, Ramachandran P, Crome SQ, Becher B, Cybulsky MI, Billia F, Keshavjee S, Mital S, Robbins CS, Mak TW, Epelman S (2022) Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol 7:eabf7777. https://doi.org/10.1126/sciimmunol.abf7777
Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, Robbins CS, Epelman S (2019) Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20:29–39. https://doi.org/10.1038/s41590-018-0272-2
Jia D, Chen S, Bai P, Luo C, Liu J, Sun A, Ge J (2022) Cardiac resident macrophage-derived legumain improves cardiac repair by promoting clearance and degradation of apoptotic cardiomyocytes after myocardial infarction. Circulation 145:1542–1556. https://doi.org/10.1161/CIRCULATIONAHA.121.057549
Paulus WJ, Tschöpe C (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62:263–271. https://doi.org/10.1016/j.jacc.2013.02.092
Sharma K, Kass DA (2014) Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ Res 115:79–96. https://doi.org/10.1161/CIRCRESAHA.115.302922
DeBerge M, Shah SJ, Wilsbacher L, Thorp EB (2019) Macrophages in heart failure with reduced versus preserved ejection fraction. Trends Mol Med 25:328–340. https://doi.org/10.1016/j.molmed.2019.01.002
Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, Von SJ, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschöpe C (2011) Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4:44–52. https://doi.org/10.1161/CIRCHEARTFAILURE.109.931451
Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F, Nahrendorf M (2018) Cardiac macrophages promote diastolic dysfunction. J Exp Med 215:423–440. https://doi.org/10.1084/jem.20171274
Abernethy A, Raza S, Sun JL, Anstrom KJ, Tracy R, Steiner J, VanBuren P, LeWinter MM (2018) Pro-inflammatory biomarkers in stable versus acutely decompensated heart failure with preserved ejection fraction. J Am Heart Assoc 7:e007385. https://doi.org/10.1161/JAHA.117.007385
Collier P, Watson CJ, Voon V, Phelan D, Jan A, Mak G, Martos R, Baugh JA, Ledwidge MT, McDonald KM (2011) Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur J Heart Fail 13:1087–1095. https://doi.org/10.1093/eurjhf/hfr079
Glezeva N, Voon V, Watson C, Horgan S, McDonald K, Ledwidge M, Baugh J (2015) Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J Card Fail 21:167–177. https://doi.org/10.1016/j.cardfail.2014.11.004
Edelmann F, Holzendorf V, Wachter R, Nolte K, Schmidt AG, Kraigher-Krainer E, Duvinage A, Unkelbach I, Düngen HD, Tschöpe C, Herrmann-Lingen C, Halle M, Hasenfuss G, Gelbrich G, Stough WG, Pieske BM (2015) Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail 17:214–223. https://doi.org/10.1002/ejhf.203
Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R (2009) IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res 104:e9-18. https://doi.org/10.1161/CIRCRESAHA.108.188243
Kockx MM, De Meyer GRY, Muhring J, Jacob W, Bult H, Herman AG (1998) Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation 97:2307–2315. https://doi.org/10.1161/01.CIR.97.23.2307
Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT (1996) Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol 7:330–335. https://doi.org/10.1097/00041433-199610000-00012
De Meyer I, Martinet W, De Meyer GRY (2012) Therapeutic strategies to deplete macrophages in atherosclerotic plaques. Br J Clin Pharmacol 74:246–263. https://doi.org/10.1111/j.1365-2125.2012.04211.x
Han X, Li Q, Lan X, EL-Mufti L, Ren H, Wang J (2019) Microglial depletion with clodronate liposomes increases proinflammatory cytokine levels, induces astrocyte activation, and damages blood vessel integrity. Mol Neurobiol 56:6184–6196. https://doi.org/10.1007/s12035-019-1502-9
Yang L, Rojas AM, Shiau CE (2021) Liposomal clodronate-mediated macrophage depletion in the zebrafish model. Bio Protoc 11:e3951. https://doi.org/10.21769/BioProtoc.3951
Shoulders H, Garner KH, Singla DK (2019) Macrophage depletion by clodronate attenuates bone morphogenetic protein-7 induced M2 macrophage differentiation and improved systolic blood velocity in atherosclerosis. Transl Res 203:1–14. https://doi.org/10.1016/j.trsl.2018.07.006
Lai SL, Marín-Juez R, Moura PL, Kuenne C, Lai JKH, Tsedeke AT, Guenther S, Looso M, Stainier DYR (2017) Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. Elife 6:e25605. https://doi.org/10.7554/eLife.25605
Culemann S, Knab K, Euler M, Wegner A, Garibagaoglu H, Ackermann J, Fischer K, Kienhöfer D, Crainiciuc G, Hahn J, Grüneboom A, Nimmerjahn F, Uderhardt S, Hidalgo A, Schett G, Hoffmann MH, Krönke G (2023) Stunning of neutrophils accounts for the anti-inflammatory effects of clodronate liposomes. J Exp Med 220:e20220525. https://doi.org/10.1084/jem.20220525
Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z (2008) Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117:1649–1657. https://doi.org/10.1161/CIRCULATIONAHA.107.745091
Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein-Barash H, Cantley W, Wong J, Cortez-Retamozo V, Newton A, Love K, Libby P, Pittet MJ, Swirski FK, Koteliansky V, Langer R, Weissleder R, Anderson DG, Nahrendorf M (2011) Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol 29:1005–1010. https://doi.org/10.1038/nbt.1989
Veillard NR, Kwak B, Pelli G, Mulhaupt F, James RW, Proudfoot AEI, Mach F (2004) Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res 94:253–261. https://doi.org/10.1161/01.RES.0000109793.17591.4E
Georgakis MK, Bernhagen J, Heitman LH, Weber C, Dichgans M (2022) Targeting the CCL2–CCR2 axis for atheroprotection. Eur Heart J 43:1799–1808. https://doi.org/10.1093/eurheartj/ehac094
Lim GB (2018) Distinct macrophage subsets in the human heart. Nat Rev Cardiol 15:502. https://doi.org/10.1038/s41569-018-0053-5
Wang J, Seo MJ, Deci MB, Weil BR, Canty JM, Nguyen J (2018) Effect of CCR2 inhibitor-loaded lipid micelles on inflammatory cell migration and cardiac function after myocardial infarction. Int J Nanomed 13:6441–6451. https://doi.org/10.2147/IJN.S178650
Burger-Kentischer A, Göbel H, Kleemann R, Zernecke A, Bucala R, Leng L, Finkelmeier D, Geiger G, Schaefer HE, Schober A, Weber C, Brunner H, Rütten H, Ihling C, Bernhagen J (2006) Reduction of the aortic inflammatory response in spontaneous atherosclerosis by blockade of macrophage migration inhibitory factor (MIF). Atherosclerosis 184:28–38. https://doi.org/10.1016/j.atherosclerosis.2005.03.028
Pan JH, Sukhova GK, Yang JT, Wang B, Xie T, Fu H, Zhang Y, Satoskar AR, David JR, Metz CN, Bucala R, Fang K, Simon DI, Chapman HA, Libby P, Shi GP (2004) Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 109:3149–3153. https://doi.org/10.1161/01.CIR.0000134704.84454.D2
Chen Y, Zhang H, Hu L, Shi H, Liu X, Jia J, Sun S, Ou Y, Luo X, Zhou G, Shen W (2020) Pravastatin attenuates atherosclerosis after myocardial infarction by inhibiting inflammatory Ly6Chigh monocytosis in apolipoprotein E knockout mice. J Int Med Res 48:300060520932816. https://doi.org/10.1177/0300060520932816
Kwak B, Mulhaupt F, Myit S, Mach F (2000) Statins as a newly recognized type of immunomodulator. Nat Med 6:1399–1402. https://doi.org/10.1038/82219
Martín-Ventura JL, Blanco-Colio LM, Gómez-Hernández A, Muñoz-García B, Vega M, Serrano J, Ortega L, Hernández G, Tuñón J, Egido J (2005) Intensive treatment with atorvastatin reduces inflammation in mononuclear cells and human atherosclerotic lesions in one month. Stroke 36:1796–1800. https://doi.org/10.1161/01.STR.0000174289.34110.b0
Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GGL, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, Gelwix CC, Salloum FN, Hastillo A, Dinarello CA, Vetrovec GW (2010) Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra remodeling trial [VCU-ART] pilot study). Am J Cardiol 105:1371-1377.e1. https://doi.org/10.1016/j.amjcard.2009.12.059
Abdelaziz MH, Abdelwahab SF, Wan J, Cai W, Huixuan W, Jianjun C, Kumar KD, Vasudevan A, Sadek A, Su Z, Wang S, Xu H (2020) Alternatively activated macrophages; a double-edged sword in allergic asthma. J Transl Med 18:58. https://doi.org/10.1186/s12967-020-02251-w
Rizzo G, Gropper J, Piollet M, Vafadarnejad E, Rizakou A, Bandi SR, Arampatzi P, Krammer T, DiFabion N, Dietrich O, Arias-Loza AP, Prinz M, Mack M, Schlepckow K, Haass C, Silvestre JS, Zernecke A, Saliba AE, Cochain C (2022) Dynamics of monocyte-derived macrophage diversity in experimental myocardial infarction. Cardiovasc Res 119:772–785. https://doi.org/10.1093/cvr/cvac113
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377:1119–1131. https://doi.org/10.1056/nejmoa1707914
Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, Nahrendorf M (2015) Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation 132:1880–1890. https://doi.org/10.1161/CIRCULATIONAHA.115.016160
Shao BZ, Han BZ, Zeng YX, Su DF, Liu C (2016) The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol Sin 37:150–156. https://doi.org/10.1038/aps.2015.87
Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL (2011) Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 13:655–667. https://doi.org/10.1016/j.cmet.2011.03.023
Kim JD, Zhu L, Sun Q, Fang L (2021) Systemic metabolite profiling reveals sexual dimorphism of AIBP control of metabolism in mice. PLoS ONE 16:e0248964. https://doi.org/10.1371/journal.pone.0248964
Zhai C, Cheng J, Mujahid H, Wang H, Kong J, Yin Y, Li J, Zhang Y, Ji X, Chen W (2014) Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PLoS ONE 9(3):e90563. https://doi.org/10.1371/journal.pone.0090563
Li R, Ji Z, Qin H, Kang X, Sun B, Wang M, Chang CH, Wang X, Zhang H, Zou H, Nel AE, Xia T (2014) Interference in autophagosome fusion by rare earth nanoparticles disrupts autophagic flux and regulation of an interleukin-1β producing inflammasome. ACS Nano 8:10280–10292. https://doi.org/10.1021/nn505002w
Viaud M, Ivanov S, Vujic N, Duta-Mare M, Aira LE, Barouillet T, Garcia E, Orange F, Dugail I, Hainault I, Stehlik C, Marchetti S, Boyer L, Guinamard R, Foufelle F, Bochem A, Hovingh KG, Thorp EB, Gautier EL, Kratky D, Dasilva-Jardine P, Yvan-Charvet L (2018) Lysosomal cholesterol hydrolysis couples efferocytosis to anti-inflammatory oxysterol production. Circ Res 122:1369–1384. https://doi.org/10.1161/CIRCRESAHA.117.312333
Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I (2012) Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15:545–553. https://doi.org/10.1016/j.cmet.2012.01.022
Liu B, Zhang B, Guo R, Li S, Xu Y (2014) Enhancement in efferocytosis of oxidized low-density lipoprotein-induced apoptotic RAW264.7 cells through Sirt1-mediated autophagy. Int J Mol Med 33:523–533. https://doi.org/10.3892/ijmm.2013.1609
Martin-Rodriguez O, Gauthier T, Bonnefoy F, Couturier M, Daoui A, Chagué C, Valmary-Degano S, Gay C, Saas P, Perruche S (2021) Pro-resolving factors released by macrophages after efferocytosis promote mucosal wound healing in inflammatory bowel disease. Front Immunol 12:754475. https://doi.org/10.3389/fimmu.2021.754475
Lackey DE, Olefsky JM (2016) Regulation of metabolism by the innate immune system. Nat Rev Endocrinol 12:15–28. https://doi.org/10.1038/nrendo.2015.189
Liang W, Qi Y, Yi H, Mao C, Meng Q, Wang H, Zheng C (2022) The roles of adipose tissue macrophages in human disease. Front Immunol 13:908749. https://doi.org/10.3389/fimmu.2022.908749
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830. https://doi.org/10.1172/JCI200319451
Kim Y, Nurakhayev S, Nurkesh A, Zharkinbekov Z, Saparov A (2021) Macrophage polarization in cardiac tissue repair following myocardial infarction. Int J Mol Sci 22:2715. https://doi.org/10.3390/ijms22052715
Krzyszczyk P, Schloss R, Palmer A, Berthiaume F (2018) The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front Physiol 9:419. https://doi.org/10.3389/fphys.2018.00419
Lu W, Wang Q, Sun X, He H, Wang Q, Wu Y, Liu Y, Wang Y, Li C (2019) Qishen granule improved cardiac remodeling via balancing M1 and M2 macrophages. Front Pharmacol 10:1399. https://doi.org/10.3389/fphar.2019.01399
Zhang Z, Tang J, Cui X, Qin B, Zhang J, Zhang L, Zhang H, Liu G, Wang W, Zhang J (2021) New insights and novel therapeutic potentials for macrophages in myocardial infarction. Inflammation 44:1696–1712. https://doi.org/10.1007/s10753-021-01467-2
Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML (2017) IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol 112:33. https://doi.org/10.1007/s00395-017-0622-5
Shintani Y, Ito T, Fields L, Shiraishi M, Ichihara Y, Sato N, Podaru M, Kainuma S, Tanaka H, Suzuki K (2017) IL-4 as a repurposed biological drug for myocardial infarction through augmentation of reparative cardiac macrophages: proof-of-concept data in mice. Sci Rep 7:6877. https://doi.org/10.1038/s41598-017-07328-z
Yue Y, Huang S, Wang L, Wu Z, Liang M, Li H, Lv L, Li W, Wu Z (2020) M2b macrophages regulate cardiac fibroblast activation and alleviate cardiac fibrosis after reperfusion injury. Circ J 84:626–635. https://doi.org/10.1253/circj.CJ-19-0959
Huang CK, Dai D, Xie H, Zhu Z, Hu J, Su M, Liu M, Lu L, Shen W, Ning G, Wang J, Zhang R, Yan X (2020) Lgr4 governs a pro-inflammatory program in macrophages to antagonize post-infarction cardiac repair. Circ Res 127:953–973. https://doi.org/10.1161/CIRCRESAHA.119.315807
Jian Y, Zhou X, Shan W, Chen C, Ge W, Cui J, Yi W, Sun Y (2023) Crosstalk between macrophages and cardiac cells after myocardial infarction. Cell Commun Signal 21:109. https://doi.org/10.1186/s12964-023-01105-4
Srivastava M, Saqib U, Naim A, Roy A, Liu D, Bhatnagar D, Ravinder R, Baig MS (2017) The TLR4–NOS1–AP1 signaling axis regulates macrophage polarization. Inflamma Res 66:323–334. https://doi.org/10.1007/s00011-016-1017-z
Ye N, Ding Y, Wild C, Shen Q, Zhou J (2014) Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem 57:6930–6948. https://doi.org/10.1021/jm5004733
Xie D, Guo H, Li M, Jia L, Zhang H, Liang D, Wu N, Yang Z, Tian Y (2023) Splenic monocytes mediate inflammatory response and exacerbate myocardial ischemia/reperfusion injury in a mitochondrial cell-free DNA-TLR9-NLRP3-dependent fashion. Basic Res Cardiol 118:44. https://doi.org/10.1007/s00395-023-01014-0
Wang N, Liu C, Wang X, He T, Li L, Liang X, Wang L, Song L, Wei Y, Wu Q, Gong C (2019) Hyaluronic acid oligosaccharides improve myocardial function reconstruction and angiogenesis against myocardial infarction by regulation of macrophages. Theranostics 9:1980–1992. https://doi.org/10.7150/thno.31073
McLaughlin S, McNeill B, Podrebarac J, Hosoyama K, Sedlakova V, Cron G, Smyth D, Seymour R, Goel K, Liang W, Rayner KJ, Ruel M, Suuronen EJ, Alarcon EI (2019) Injectable human recombinant collagen matrices limit adverse remodeling and improve cardiac function after myocardial infarction. Nat Commun 10:4866. https://doi.org/10.1038/s41467-019-12748-8
Cao Q, Rong S, Repa JJ, Clair RS, Parks JS, Mishra N (2014) Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol 34:1871–1879. https://doi.org/10.1161/ATVBAHA.114.303393
Kimbrough D, Wang SH, Wright LH, Mani SK, Kasiganesan H, LaRue AC, Cheng Q, Nadig SN, Atkinson C, Menick DR (2018) HDAC inhibition helps post-MI healing by modulating macrophage polarization. J Mol Cell Cardiol 119:51–63. https://doi.org/10.1016/j.yjmcc.2018.04.011
Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, Shankar TS, Selzman CH, Drakos SG, Lavine KJ (2018) The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 24:1234–1245. https://doi.org/10.1038/s41591-018-0059-x
Zhang K, Wang Y, Chen S, Mao J, Jin Y, Ye H, Zhang Y, Liu X, Gong C, Cheng X, Huang X, Hoeft A, Chen Q, Li X, Fang X (2023) TREM2hi resident macrophages protect the septic heart by maintaining cardiomyocyte homeostasis. Nat Metab 5:129–146. https://doi.org/10.1038/s42255-022-00715-5
Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, Pinto AR, Klapproth K, Henri S, Malissen B, Rodewald HR, Rosenthal NA, Bajenoff M, Prinz M, Jung S, Sieweke MH (2014) Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med 211:2151–2158. https://doi.org/10.1084/jem.20140639
Gomez I, Duval V, Silvestre JS (2018) Cardiomyocytes and macrophages discourse on the method to govern cardiac repair. Front Cardiovasc Med 5:134. https://doi.org/10.3389/fcvm.2018.00134
Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wülfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M (2017) Macrophages facilitate electrical conduction in the heart. Cell 169:510-522.e20. https://doi.org/10.1016/j.cell.2017.03.050
Wong NR, Mohan J, Kopecky BJ, Guo S, Du L, Leid J, Feng G, Lokshina I, Dmytrenko O, Luehmann H, Bajpai G, Ewald L, Bell L, Patel N, Bredemeyer A, Weinheimer CJ, Nigro JM, Kovacs A, Morimoto S, Bayguinov PO, Fisher MR, Stump WT, Greenberg M, Fitzpatrick JAJ, Epelman S, Kreisel D, Sah R, Liu Y, Hu H, Lavine KJ (2021) Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity 54:2072-2088.e7. https://doi.org/10.1016/j.immuni.2021.07.003
Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial JA, Taffet GE, Entman ML (2010) Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol 49:499–507. https://doi.org/10.1016/j.yjmcc.2010.05.005
Zhang N, Ma Q, You Y, Xia X, Xie C, Huang Y, Wang Z, Ye F, Yu Z, Xie X (2022) CXCR4-dependent macrophage-to-fibroblast signaling contributes to cardiac diastolic dysfunction in heart failure with preserved ejection fraction. Int J Biol Sci 18:1271–1287. https://doi.org/10.7150/ijbs.65802
Van Tassell BW, Arena R, Biondi-Zoccai G, McNair Canada J, Oddi C, Abouzaki NA, Jahangiri A, Falcao RA, Kontos MC, Shah KB, Voelkel NF, Dinarello CA, Abbate A (2014) Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol 113:321–327. https://doi.org/10.1016/j.amjcard.2013.08.047
Kaikkonen MU, Lam MTY, Glass CK (2011) Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res 90:430–440. https://doi.org/10.1093/cvr/cvr097
Sansonetti M, De Windt LJ (2021) Non-coding RNAs in cardiac inflammation: key drivers in the pathophysiology of heart failure. Cardiovasc Res 118:2058–2073. https://doi.org/10.1093/cvr/cvab192
Ramanujam D, Schön AP, Beck C, Vaccarello P, Felician G, Dueck A, Esfandyari D, Meister G, Meitinger T, Schulz C, Engelhardt S (2021) MicroRNA-21–dependent macrophage-to-fibroblast signaling determines the cardiac response to pressure overload. Circulation 143:1513–1525. https://doi.org/10.1161/CIRCULATIONAHA.120.050682
Bejerano T, Etzion S, Elyagon S, Etzion Y, Cohen S (2018) Nanoparticle delivery of miRNA-21 mimic to cardiac macrophages improves myocardial remodeling after myocardial infarction. Nano Lett 18:5885–5891. https://doi.org/10.1021/acs.nanolett.8b02578
Zhao J, Li X, Hu J, Chen F, Qiao S, Sun X, Gao L, Xie J, Xu B (2019) Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc Res 115:1205–1216. https://doi.org/10.1093/cvr/cvz040
Karunakaran D, Thrush AB, Nguyen MA, Richards L, Geoffrion M, Singaravelu R, Ramphos E, Shangari P, Ouimet M, Pezacki JP, Moore KJ, Perisic L, Maegdefessel L, Hedin U, Harper ME, Rayner KJ (2015) Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by Anti-miR33 in atherosclerosis. Circ Res 117:266–278. https://doi.org/10.1161/CIRCRESAHA.117.305624
Rayner KJ, Suárez Y, Dávalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernández-Hernando C (2010) MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328:1570–1573. https://doi.org/10.1126/science.1189862
Price NL, Rotllan N, Canfrán-Duque A, Zhang X, Pati P, Arias N, Moen J, Mayr M, Ford DA, Baldán Á, Suárez Y, Fernández-Hernando C (2017) Genetic dissection of the impact of miR-33a and miR-33b during the progression of atherosclerosis. Cell Rep 21:1317–1330. https://doi.org/10.1016/j.celrep.2017.10.023
An TH, He QW, Xia YP, Chen SC, Baral S, Mao L, Jin HJ, Li YN, Wang MD, Chen JG, Zhu LQ, Hu B (2017) MiR-181b antagonizes atherosclerotic plaque vulnerability through modulating macrophage polarization by directly targeting Notch1. Mol Neurobiol 54:6329–6341. https://doi.org/10.1007/s12035-016-0163-1
Bai Q, Xiao Y, Hong H, Cao X, Zhang L, Han R, Lee LKC, Xue EY, Tian XY, Choi CHJ (2022) Scavenger receptor-targeted plaque delivery of microRNA-coated nanoparticles for alleviating atherosclerosis. Proc Natl Acad Sci U S A 119:e2201443119. https://doi.org/10.1073/pnas.2201443119
Wei Y, Corbalán-Campos J, Gurung R, Natarelli L, Zhu M, Exner N, Erhard F, Greulich F, Geißler C, Uhlenhaut NH, Zimmer R, Schober A (2018) Dicer in macrophages prevents atherosclerosis by promoting mitochondrial oxidative metabolism. Circulation 138:2007–2020. https://doi.org/10.1161/CIRCULATIONAHA.117.031589
Verjans R, Derks WJA, Korn K, Sönnichsen B, van Leeuwen REW, Schroen B, van Bilsen M, Heymans S (2019) Functional screening identifies MicroRNAs as multi-cellular regulators of heart failure. Sci Rep 9:6055. https://doi.org/10.1038/s41598-019-41491-9
Simion V, Zhou H, Haemmig S, Pierce JB, Mendes S, Tesmenitsky Y, Pérez-Cremades D, Lee JF, Chen AF, Ronda N, Papotti B, Marto JA, Feinberg MW (2020) A macrophage-specific lncRNA regulates apoptosis and atherosclerosis by tethering HuR in the nucleus. Nat Commun 11:6135. https://doi.org/10.1038/s41467-020-19664-2
An JH, Chen ZY, Ma QL, Wang HJ, Zhang JQ, Shi FW (2019) LncRNA SNHG16 promoted proliferation and inflammatory response of macrophages through MIR-17-5p/NF-κB signaling pathway in patients with atherosclerosis. Eur Rev Med Pharmacol Sci 23:8665–8677. https://doi.org/10.26355/eurrev_201910_19184
Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K, Eskin A, Casero D, Zhang Z, Sandhu J, Salisbury D, Rajbhandari P, Civelek M, Hong C, Ito A, Liu X, Daniel B, Lusis AJ, Whitelegge J, Nagy L, Castrillo A, Smale S, Tontonoz P (2018) Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat Med 24:304–312. https://doi.org/10.1038/nm.4479
Gast M, Rauch BH, Haghikia A, Nakagawa S, Haas J, Stroux A, Schmidt D, Schumann P, Weiss S, Jensen L, Kratzer A, Kraenkel N, Müller C, Börnigen D, Hirose T, Blankenberg S, Escher F, Kühl AA, Kuss AW, Meder B, Landmesser U, Zeller T, Poller W (2019) Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc Res 115:1886–1906. https://doi.org/10.1093/cvr/cvz085
Wang L, Xia JW, Ke ZP, Zhang BH (2019) Blockade of NEAT1 represses inflammation response and lipid uptake via modulating miR-342-3p in human macrophages THP-1 cells. J Cell Physiol 234:5319–5326. https://doi.org/10.1002/jcp.27340
Luo YY, Yang ZQ, Lin XF, Zhao FL, Tu HT, Wang LJ, Wen MY, Xian SX (2021) Knockdown of lncRNA PVT1 attenuated macrophage M1 polarization and relieved sepsis induced myocardial injury via miR-29a/HMGB1 axis. Cytokine 143:155509. https://doi.org/10.1016/j.cyto.2021.155509
Panda AC (2018) Circular RNAs act as miRNA sponges. Adv Exp Med Biol 1087:67–79. https://doi.org/10.1007/978-981-13-1426-1_6
Hou X, Dai H, Zheng Y (2022) Circular RNA hsa_circ_0008896 accelerates atherosclerosis by promoting the proliferation, migration and invasion of vascular smooth muscle cells via hsa-miR-633/CDC20B (cell division cycle 20B) axis. Bioengineered 13:5987–5998. https://doi.org/10.1080/21655979.2022.2039467
Wang L, Zheng Z, Feng X, Zang X, Ding W, Wu F, Zhao Q (2019) CircRNA/lncRNA-miRNA-mRNA network in oxidized, low-density, lipoprotein-induced foam cells. DNA Cell Biol 38:1499–1511. https://doi.org/10.1089/dna.2019.4865
Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL (2014) Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40:91–104. https://doi.org/10.1016/j.immuni.2013.11.019
Moskalik A, Niderla-Bielińska J, Ratajska A (2022) Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail Rev 27:1413–1430. https://doi.org/10.1007/s10741-021-10156-z
Sanders-Van Wijk S, Van Empel V, Davarzani N, Maeder MT, Handschin R, Pfisterer ME, Brunner-La Rocca HP (2015) Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur J Heart Fail 17:1006–1014. https://doi.org/10.1002/ejhf.414
Shen JL, Xie XJ (2020) Insight into the pro-inflammatory and profibrotic role of macrophage in heart failure with preserved ejection fraction. J Cardiovasc Pharmacol 76:276–285. https://doi.org/10.1097/FJC.0000000000000858
Chia YC, Kieneker LM, van Hassel G, Binnenmars SH, Nolte IM, van Zanden JJ, van der Meer P, Navis G, Voors AA, Bakker SJL, De Borst MH, Eisenga MF (2021) Interleukin 6 and development of heart failure with preserved ejection fraction in the general population. J Am Heart Assoc 10:e018549. https://doi.org/10.1161/JAHA.120.018549
Liu H, Huang Y, Zhao Y, Kang GJ, Feng F, Wang X, Liu M, Shi G, Revelo X, Bernlohr D, Dudley SC (2023) Inflammatory macrophage interleukin-1β mediates high-fat diet-induced heart failure with preserved ejection fraction. JACC Basic Transl Sci 8:174–185. https://doi.org/10.1016/j.jacbts.2022.08.003
Heslop CL, Frohlich JJ, Hill JS (2010) Myeloperoxidase and C-reactive protein have combined utility for long-term prediction of cardiovascular mortality after coronary angiography. J Am Coll Cardiol 55:1102–1109. https://doi.org/10.1016/j.jacc.2009.11.050
Hochholzer W, Morrow DA, Giugliano RP (2010) Novel biomarkers in cardiovascular disease: update 2010. Am Heart J 160:583–594. https://doi.org/10.1016/j.ahj.2010.06.010
Calmarza P, Lapresta C, Martínez M, Lahoz R, Povar J (2018) Utility of myeloperoxidase in the differential diagnosis of acute coronary syndrome. Arch Cardiol Mex 88:391–396. https://doi.org/10.1016/j.acmx.2017.11.003
Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K (2016) Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest 126:2151–2166. https://doi.org/10.1172/JCI85782
Bielecka-Dabrowa A, Sakowicz A, Misztal M, von Haehling S, Ahmed A, Pietrucha T, Rysz J, Banach M (2016) Differences in biochemical and genetic biomarkers in patients with heart failure of various etiologies. Int J Cardiol 221:1073–1080. https://doi.org/10.1016/j.ijcard.2016.07.150
Wiklund FE, Bennet AM, Magnusson PKE, Eriksson UK, Lindmark F, Wu L, Yaghoutyfam N, Marquis CP, Stattin P, Pedersen NL, Adami HO, Grönberg H, Breit SN, Brown DA (2010) Macrophage inhibitory cytokine-1 (MIC-1/GDF15): a new marker of all-cause mortality. Aging Cell 9:1057–1064. https://doi.org/10.1111/j.1474-9726.2010.00629.x
Cotter G, Voors AA, Prescott MF, Felker GM, Filippatos G, Greenberg BH, Pang PS, Ponikowski P, Milo O, Hua TA, Qian M, Severin TM, Teerlink JR, Metra M, Davison BA (2015) Growth differentiation factor 15 (GDF-15) in patients admitted for acute heart failure: results from the RELAX-AHF study. Eur J Heart Fail 17:1133–1143. https://doi.org/10.1002/ejhf.331
De Boer RA, Voors AA, Muntendam P, Van Gilst WH, Van Veldhuisen DJ (2009) Galectin-3: a novel mediator of heart failure development and progression. Eur J Heart Fail 11:811–817. https://doi.org/10.1093/eurjhf/hfp097
De Boer RA, Yu L, Van Veldhuisen DJ (2010) Galectin-3 in cardiac remodeling and heart failure. Curr Heart Fail Rep 7:1–8. https://doi.org/10.1007/s11897-010-0004-x
Ho JE, Liu C, Lyass A, Courchesne P, Pencina MJ, Vasan RS, Larson MG, Levy D (2012) Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol 60:1249–1256. https://doi.org/10.1016/j.jacc.2012.04.053
Sharma UC, Pokharel S, Van Brakel TJ, Van Berlo JH, Cleutjens JPM, Schroen B, André S, Crijns HJGM, Gabius HJ, Maessen J, Pinto YM (2004) Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 110:3121–3128. https://doi.org/10.1161/01.CIR.0000147181.65298.4D
Zaborska B, Sikora-Frąc M, Smarż K, Pilichowska-Paszkiet E, Budaj A, Sitkiewicz D, Sygitowicz G (2023) The role of galectin-3 in heart failure—the diagnostic, prognostic and therapeutic potential—where do we stand? Int J Mol Sci 24:13111. https://doi.org/10.3390/ijms241713111
Ponikowski P, Voors A (2016) 2016 Esc guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European society of cardiology (ESC)—developed with the special contribution of the heart failure association (HFA) of the ESC. Eur J Heart Fail 18:891–975. https://doi.org/10.1002/ejhf.592
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld JA, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C (2017) 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the american college of cardiology/American heart association task force on clinical practice guidelines and the heart failure society of America. J Am Coll Cardiol 70:776–803. https://doi.org/10.1016/j.jacc.2017.04.025
Di Tano G, Caretta G, De Maria R, Parolini M, Bassi L, Testa S, Pirelli S (2017) Galectin-3 predicts left ventricular remodelling after anterior-wall myocardial infarction treated by primary percutaneous coronary intervention. Heart 103:71–77. https://doi.org/10.1136/heartjnl-2016-309673
Tymińska A, Kapłon-Cieślicka A, Ozierański K, Budnik M, Wancerz A, Sypień P, Peller M, Maksym J, Balsam P, Opolski G, Filipiak KJ (2019) Association of galectin-3 and soluble ST2 with in-hospital and 1-year outcomes in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Pol Arch Intern Med 129:770–780. https://doi.org/10.20452/pamw.15030
Watson CJ, Gallagher J, Wilkinson M, Russell-Hallinan A, Tea I, James S, O’Reilly J, O’Connell E, Zhou S, Ledwidge M, McDonald K (2021) Biomarker profiling for risk of future heart failure (HFpEF) development. J Transl Med 19:61. https://doi.org/10.1186/s12967-021-02735-3
Yin QS, Shi B, Dong L, Bi L (2014) Comparative study of galectin-3 and B-type natriuretic peptide as biomarkers for the diagnosis of heart failure. J Geriatr Cardiol 11:79–82. https://doi.org/10.3969/j.issn.1671-5411.2014.01.014
Hara A, Niwa M, Kanayama T, Noguchi K, Niwa A, Matsuo M, Kuroda T, Hatano Y, Okada H, Tomita H (2020) Galectin-3: a potential prognostic and diagnostic marker for heart disease and detection of early stage pathology. Biomolecules 10:1277. https://doi.org/10.3390/biom10091277
Shirazi LF, Bissett J, Romeo F, Mehta JL (2017) Role of inflammation in heart failure. Curr Atheroscler Rep 19:27. https://doi.org/10.1007/s11883-017-0660-3
Cheng Z, Cai K, Xu C, Zhan Q, Xu X, Xu D, Zeng Q (2022) Prognostic value of serum galectin-3 in chronic heart failure: a meta-analysis. Front Cardiovasc Med 9:783707. https://doi.org/10.3389/fcvm.2022.783707
Lisowska A, Knapp M, Tycińska A, Motybel E, Kamiński K, Świecki P, Musiał WJ, Dymicka-Piekarska V (2016) Predictive value of Galectin-3 for the occurrence of coronary artery disease and prognosis after myocardial infarction and its association with carotid IMT values in these patients: a mid-term prospective cohort study. Atherosclerosis 246:309–317. https://doi.org/10.1016/j.atherosclerosis.2016.01.022
Maiolino G, Rossitto G, Pedon L, Cesari M, Frigo AC, Azzolini M, Plebani M, Rossi GP (2015) Galectin-3 predicts long-term cardiovascular death in high-risk patients with coronary artery disease. Arterioscler Thromb Vasc Biol 35:725–732. https://doi.org/10.1161/ATVBAHA.114.304964
Niu C, Wang X, Zhao M, Cai T, Liu P, Li J, Willard B, Zu L, Zhou E, Li Y, Pan B, Yang F, Zheng L (2016) Macrophage foam cell-derived extracellular vesicles promote vascular smooth muscle cell migration and adhesion. J Am Heart Assoc 5:e004099. https://doi.org/10.1161/JAHA.116.004099
Osada-Oka M, Shiota M, Izumi Y, Nishiyama M, Tanaka M, Yamaguchi T, Sakurai E, Miura K, Iwao H (2017) Macrophage-derived exosomes induce inflammatory factors in endothelial cells under hypertensive conditions. Hypertens Res 40:353–360. https://doi.org/10.1038/hr.2016.163
Zhang YG, Song Y, Guo XL, Miao RY, Fu YQ, Miao CF, Zhang C (2019) Exosomes derived from oxLDL-stimulated macrophages induce neutrophil extracellular traps to drive atherosclerosis. Cell Cycle 18:2674–2684. https://doi.org/10.1080/15384101.2019.1654797
Liu S, Chen J, Shi J, Zhou W, Wang L, Fang W, Zhong Y, Chen X, Chen Y, Sabri A, Liu S (2020) M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res Cardiol 115:22. https://doi.org/10.1007/s00395-020-0781-7
Wang C, Zhang C, Liu L, Xi A, Chen B, Li Y, Du J (2017) Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther 25:192–204. https://doi.org/10.1016/j.ymthe.2016.09.001
Zhang B, Li B, Qin F, Bai F, Sun C, Liu Q (2019) Expression of serum microRNA-155 and its clinical importance in patients with heart failure after myocardial infarction. J Int Med Res 47:6294–6302. https://doi.org/10.1177/0300060519882583
Chen Q, Yin Q, Song J, Liu C, Chen H, Li S (2021) Identification of monocyte-associated genes as predictive biomarkers of heart failure after acute myocardial infarction. BMC Med Genom 14:44. https://doi.org/10.1186/s12920-021-00890-6
Revelo XS, Parthiban P, Chen C, Barrow F, Fredrickson G, Wang H, Yücel D, Herman A, Van Berlo JH (2021) Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circ Res. https://doi.org/10.1161/CIRCRESAHA.121.319737
Van Amerongen MJ, Harmsen MC, Van Rooijen N, Petersen AH, Van Luyn MJA (2007) Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol 170:818–829. https://doi.org/10.2353/ajpath.2007.060547
Frantz S, Hofmann U, Fraccarollo D, Schäfer A, Kranepuhl S, Hagedorn I, Nieswandt B, Nahrendorf M, Wagner H, Bayer B, Pachel C, Schön MP, Kneitz S, Bobinger T, Weidemann F, Ertl G, Bauersachs J (2013) Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J 27:871–881. https://doi.org/10.1096/fj.12-214049
Levin MC, Lidberg U, Jirholt P, Adiels M, Wramstedt A, Gustafsson K, Greaves DR, Li S, Fazio S, Linton MF, Olofsson SO, Borén J, Gjertsson I (2012) Evaluation of macrophage-specific promoters using lentiviral delivery in mice. Gene Ther 19:1041–1047. https://doi.org/10.1038/gt.2011.195
Rosario AM, Cruz PE, Ceballos-Diaz C, Strickland MR, Siemienski Z, Pardo M, Schob KL, Li A, Aslanidi GV, Srivastava A, Golde TE, Chakrabarty P (2016) Microglia-specific targeting by novel capsid-modified AAV6 vectors. Mol Ther Methods Clin Dev 3:16026. https://doi.org/10.1038/mtm.2016.26
Tao W, Yurdagul A, Kong N, Li W, Wang X, Doran AC, Feng C, Wang J, Islam MA, Farokhzad OC, Tabas I, Shi J (2020) SiRNA nanoparticles targeting CaMKIIγ in lesional macrophages improve atherosclerotic plaque stability in mice. Sci Transl Med 12:eaay1063. https://doi.org/10.1126/SCITRANSLMED.AAY1063
Han J, Kim YS, Lim MY, Kim HY, Kong S, Kang M, Choo YW, Jun JH, Ryu S, Jeong HY, Park J, Jeong GJ, Lee JC, Eom GH, Ahn Y, Kim BS (2018) Dual roles of graphene oxide to attenuate inflammation and elicit timely polarization of macrophage phenotypes for cardiac repair. ACS Nano 12:1959–1977. https://doi.org/10.1021/acsnano.7b09107
Zhao Y, Jiang C, He J, Guo Q, Lu J, Yang Y, Zhang W, Liu J (2017) Multifunctional dextran sulfate-coated reconstituted high density lipoproteins target macrophages and promote beneficial antiatherosclerotic mechanisms. Bioconjug Chem 28:438–448. https://doi.org/10.1021/acs.bioconjchem.6b00600
Yu SS, Lau CM, Barham WJ, Onishko HM, Nelson CE, Li H, Smith CA, Yull FE, Duvall CL, Giorgio TD (2013) Macrophage-specific RNA interference targeting via “click”, mannosylated polymeric micelles. Mol Pharm 10:975–987. https://doi.org/10.1021/mp300434e
Nguyen MA, Karunakaran D, Geoffrion M, Cheng HS, Tandoc K, Perisic Matic L, Hedin U, Maegdefessel L, Fish JE, Rayner KJ (2018) Extracellular vesicles secreted by atherogenic macrophages transfer MicroRNA to inhibit cell migration. Arterioscler Thromb Vasc Biol 38:49–63. https://doi.org/10.1161/ATVBAHA.117.309795
Wang J, Lee CJ, Deci MB, Jasiewicz N, Verma A, Canty JM, Nguyen J (2020) MiR-101a loaded extracellular nanovesicles as bioactive carriers for cardiac repair. Nanomedicine 27:102201. https://doi.org/10.1016/j.nano.2020.102201
De Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, Berman BP, Marbán E (2017) Exosomal MicroRNA transfer into macrophages mediates cellular postconditioning. Circulation 136:200–214. https://doi.org/10.1161/CIRCULATIONAHA.116.024590
Vandergriff A, Huang K, Shen D, Hu S, Hensley MT, Caranasos TG, Qian L, Cheng K (2018) Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics 8:1869–1878. https://doi.org/10.7150/thno.20524
Poltavets AS, Vishnyakova PA, Elchaninov AV, Sukhikh GT, Fatkhudinov TK (2020) Macrophage modification strategies for efficient cell therapy. Cells 9:1535. https://doi.org/10.3390/cells9061535
Liu Y, Wu M, Zhong C, Xu B, Kang L (2022) M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer. Biomater Res 26:14. https://doi.org/10.1186/s40824-022-00260-y
Podaru MN, Fields L, Kainuma S, Ichihara Y, Hussain M, Ito T, Kobayashi K, Mathur A, D’Acquisto F, Lewis-McDougall F, Suzuki K (2019) Reparative macrophage transplantation for myocardial repair: a refinement of bone marrow mononuclear cell-based therapy. Basic Res Cardiol 114:34. https://doi.org/10.1007/s00395-019-0742-1
Xie J, Jiang L, Wang J, Yin Y, Wang R, Du L, Chen T, Ni Z, Qiao S, Gong H, Xu B, Xu Q (2023) Multilineage contribution of CD34+ cells in cardiac remodeling after ischemia/reperfusion injury. Basic Res Cardiol 118:17. https://doi.org/10.1007/s00395-023-00981-8
Zhu Y, Yang W, Wang H, Tang F, Zhu Y, Zhu Q, Ma R, Jian Z, Xiao Y (2022) Hypoxia-primed monocytes/macrophages enhance postinfarction myocardial repair. Theranostics 12:307–323. https://doi.org/10.7150/THNO.63642
Ben-Mordechai T, Holbova R, Landa-Rouben N, Harel-Adar T, Feinberg MS, Abd Elrahman I, Blum G, Epstein FH, Silman Z, Cohen S, Leor J (2013) Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol 62:1890–1901. https://doi.org/10.1016/j.jacc.2013.07.057
Lim SY, Cho DI, Jeong HY, Kang HJ, Kim MR, Cho M, Kim YS, Ahn Y (2018) Adjuvant role of macrophages in stem cell-induced cardiac repair in rats. Exp Mol Med 50:1–10. https://doi.org/10.1038/s12276-018-0171-5
Na YR, Kim SW, Seok SH (2023) A new era of macrophage-based cell therapy. Exp Mol Med 55:1945–1954. https://doi.org/10.1038/s12276-023-01068-z
Chernykh ER, Kafanova MY, Shevela EY, Sirota SI, Adonina EI, Sakhno LV, Ostanin AA, Kozlov VV (2014) Clinical experience with autologous M2 macrophages in children with severe cerebral palsy. Cell Transplant 23(Suppl 1):S97-104. https://doi.org/10.3727/096368914x684925
Chernykh ER, Shevela EY, Starostina NM, Morozov SA, Davydova MN, Menyaeva EV, Ostanin AA (2016) Safety and therapeutic potential of m2 macrophages in stroke treatment. Cell Transplant 25:1461–1471. https://doi.org/10.3727/096368915X690279
Perin EC, Willerson JT, Pepine CJ, Henry TD, Ellis SG, Zhao DXM, Silva GV, Lai D, Thomas JD, Kronenberg MW, Martin AD, Anderson RD, Traverse JH, Penn MS, Anwaruddin S, Hatzopoulos AK, Gee AP, Taylor DA, Cogle CR, Smith D, Westbrook L, Chen J, Handberg E, Olson RE, Geither C, Bowman S, Francescon J, Baraniuk S, Piller LB, Simpson LM, Loghin C, Aguilar D, Richman S, Zierold C, Bettencourt J, Sayre SL, Vojvodic RW, Skarlatos SI, Gordon DJ, Ebert RF, Kwak M, Moyé LA, Simari RD (2012) Effect of transendocardial delivery of autologous bone marrow mononuclear cells on functional capacity, left ventricular function, and perfusion in chronic heart failure: the FOCUS-CCTRN trial. JAMA 307:1717–1726. https://doi.org/10.1001/jama.2012.418
Bartel RL, Cramer C, Ledford K, Longcore A, Parrish C, Stern T, Watling S, Zeigler F (2012) The Aastrom experience. Stem Cell Res Ther 3:26. https://doi.org/10.1186/scrt117
Patel AN, Henry TD, Quyyumi AA, Schaer GL, Anderson RD, Toma C, East C, Remmers AE, Goodrich J, Desai AS, Recker D, DeMaria A (2016) Ixmyelocel-T for patients with ischaemic heart failure: a prospective randomised double-blind trial. Lancet 387:2412–2421. https://doi.org/10.1016/S0140-6736(16)30137-4
Ledford KJ, Zeigler F, Bartel RL (2013) Ixmyelocel-T, an expanded multicellular therapy, contains a unique population of M2-like macrophages. Stem Cell Res Ther 4:134. https://doi.org/10.1186/scrt345
Moazzami B, Mohammadpour Z, Zabala ZE, Farokhi E, Roohi A, Dolmatova E, Moazzami K (2022) Local intramuscular transplantation of autologous bone marrow mononuclear cells for critical lower limb ischaemia. Cochrane Database of Syst Rev 7:CD008347. https://doi.org/10.1002/14651858.CD008347.pub4
Happle C, Lachmann N, Ackermann M, Mirenska A, Göhring G, Thomay K, Mucci A, Hetzel M, Glomb T, Suzuki T, Chalk C, Glage S, Dittrich-Breiholz O, Trapnell B, Moritz T, Hansen G (2018) Pulmonary transplantation of human induced pluripotent stem cell-derived macrophages ameliorates pulmonary alveolar proteinosis. Am J Respir Crit Care Med 198:350–360. https://doi.org/10.1164/rccm.201708-1562OC
Pouyanfard S, Meshgin N, Cruz LS, Diggle K, Hashemi H, Pham TV, Fierro M, Tamayo P, Fanjul A, Kisseleva T, Kaufman DS (2021) Human induced pluripotent stem cell-derived macrophages ameliorate liver fibrosis. Stem Cells 39:1701–1717. https://doi.org/10.1002/stem.3449
Frenkel O, Shani E, Ben-Bassat I, Brok-Simoni F, Shinar E, Danon D (2001) Activation of human monocytes/macrophages by hypo-osmotic shock. Clin Exp Immunol 124:103–109. https://doi.org/10.1046/j.1365-2249.2001.01496.x
Leor J, Rozen L, Zuloff-Shani A, Feinberg MS, Amsalem Y, Barbash IM, Kachel E, Holbova R, Mardor Y, Daniels D, Ocherashvilli A, Orenstein A, Danon D (2006) Ex vivo activated human macrophages improve healing, remodeling, and function of the infarcted heart. Circulation 114:I94-100. https://doi.org/10.1161/CIRCULATIONAHA.105.000331
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, Schmierer M, Gabrusiewicz K, Anderson NR, Petty NE, Cummins KD, Shen F, Shan X, Veliz K, Blouch K, Yashiro-Ohtani Y, Kenderian SS, Kim MY, O’Connor RS, Wallace SR, Kozlowski MS, Marchione DM, Shestov M, Garcia BA, June CH, Gill S (2020) Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol 38:947–953. https://doi.org/10.1038/s41587-020-0462-y
Huang X, Li Y, Fu M, Xin HB (2018) Polarizing macrophages in vitro. Methods Mol Biol 1784:119–126. https://doi.org/10.1007/978-1-4939-7837-3_12
Fu M, Song J (2021) Single-cell transcriptomics reveals the cellular heterogeneity of cardiovascular diseases. Front Cardiovasc Med 8:643519. https://doi.org/10.3389/fcvm.2021.643519
Forrester MA, Wassall HJ, Hall LS, Cao H, Wilson HM, Barker RN, Vickers MA (2018) Similarities and differences in surface receptor expression by THP-1 monocytes and differentiated macrophages polarized using seven different conditioning regimens. Cell Immunol 332:58–76. https://doi.org/10.1016/j.cellimm.2018.07.008
Li P, Hao Z, Wu J, Ma C, Xu Y, Li J, Lan R, Zhu B, Ren P, Fan D, Sun S (2021) Comparative proteomic analysis of polarized human THP-1 and mouse RAW264.7 macrophages. Front Immunol 12:700009. https://doi.org/10.3389/fimmu.2021.700009
Mendoza-Coronel E, Castañón-Arreola M (2016) Comparative evaluation of in vitro human macrophage models for mycobacterial infection study. Pathog Dis 74:ftw052. https://doi.org/10.1093/femspd/ftw052
Hoppenbrouwers T, Bastiaan-Net S, Garssen J, Pellegrini N, Willemsen LEM, Wichers HJ (2022) Functional differences between primary monocyte-derived and THP-1 macrophages and their response to LCPUFAs. PharmaNutrition 22:100322. https://doi.org/10.1016/j.phanu.2022.100322
Navarro-Guerrero E, Tay C, Whalley JP, Cowley SA, Davies B, Knight JC, Ebner D (2021) Genome-wide CRISPR/Cas9-knockout in human induced pluripotent stem cell (iPSC)-derived macrophages. Sci Rep 11:4245. https://doi.org/10.1038/s41598-021-82137-z
Takata K, Kozaki T, Lee CZW, Thion MS, Otsuka M, Lim S, Utami KH, Fidan K, Park DS, Malleret B, Chakarov S, See P, Low D, Low G, Garcia-Miralles M, Zeng R, Zhang J, Goh CC, Gul A, Hubert S, Lee B, Chen J, Low I, Shadan NB, Lum J, Wei TS, Mok E, Kawanishi S, Kitamura Y, Larbi A, Poidinger M, Renia L, Ng LG, Wolf Y, Jung S, Önder T, Newell E, Huber T, Ashihara E, Garel S, Pouladi MA, Ginhoux F (2017) Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity 47:183-198.e6. https://doi.org/10.1016/j.immuni.2017.06.017
Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, Bradford B, Caruso M, Gazova I, Sánchez A, Lisowski ZM, Alves J, Molina-Gonzalez I, Davtyan H, Lodge RJ, Glover JD, Wallace R, Munro DAD, David E, Amit I, Miron VE, Priller J, Jenkins SJ, Hardingham GE, Blurton-Jones M, Mabbott NA, Summers KM, Hohenstein P, Hume DA, Pridans C (2019) Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun 10:3215. https://doi.org/10.1038/s41467-019-11053-8
Emini Veseli B, Perrotta P, De Meyer GRA, Roth L, Van der Donckt C, Martinet W, De Meyer GRY (2017) Animal models of atherosclerosis. Eur J Pharmacol 816:3–13. https://doi.org/10.1016/j.ejphar.2017.05.010
Getz GS, Reardon CA (2016) Do the Apoe-/- and Ldlr-/- mice yield the same insight on atherogenesis? Arterioscler Thromb Vasc Biol 36:1734–1741. https://doi.org/10.1161/ATVBAHA.116.306874
Schneemann M, Schoeden G (2007) Macrophage biology and immunology: man is not a mouse. J Leukoc Biol 81:579. https://doi.org/10.1189/jlb.1106702
Schneemann M, Schoedon G (2002) Species differences in macrophage NO production are important (multiple letters). Nat Immunol 3:102. https://doi.org/10.1038/ni0202-102a
Aktories P, Petry P, Glatz P, Andrieux G, Oschwald A, Botterer H, Gorka O, Erny D, Boerries M, Henneke P, Groß O, Prinz M, Kierdorf K (2022) An improved organotypic cell culture system to study tissue-resident macrophages ex vivo. Cell Rep Methods 2:100260. https://doi.org/10.1016/j.crmeth.2022.100260
Waleczek FJG, Sansonetti M, Xiao K, Jung M, Mitzka S, Dendorfer A, Weber N, Perbellini F, Thum T (2022) Chemical and mechanical activation of resident cardiac macrophages in the living myocardial slice ex vivo model. Basic Res Cardiol 117:63. https://doi.org/10.1007/s00395-022-00971-2
Caspi O, Lesman A, Basevitch Y, Gepstein A, Arbel G, Huber I, Habib M, Gepstein L, Levenberg S (2007) Tissue engineering of vascularized cardiac muscle from human embryonic stem cells. Circ Res 100:263–272. https://doi.org/10.1161/01.RES.0000257776.05673.ff
Suku M, Forrester L, Biggs M, Monaghan MG (2022) Resident macrophages and their potential in cardiac tissue engineering. Tissue Eng Part B Rev 28:579–591. https://doi.org/10.1089/ten.TEB.2021.0036
Ergir E, Oliver-De La Cruz J, Fernandes S, Cassani M, Niro F, Pereira-Sousa D, Vrbský J, Vinarský V, Perestrelo AR, Debellis D, Vadovičová N, Uldrijan S, Cavalieri F, Pagliari S, Redl H, Ertl P, Forte G (2022) Generation and maturation of human iPSC-derived 3D organotypic cardiac microtissues in long-term culture. Sci Rep 12:17409. https://doi.org/10.1038/s41598-022-22225-w
Goldfracht I, Protze S, Shiti A, Setter N, Gruber A, Shaheen N, Nartiss Y, Keller G, Gepstein L (2020) Generating ring-shaped engineered heart tissues from ventricular and atrial human pluripotent stem cell-derived cardiomyocytes. Nat Commun 11:75. https://doi.org/10.1038/s41467-019-13868-x
Lee SG, Kim YJ, Son MY, Oh MS, Kim J, Ryu B, Kang KR, Baek J, Chung G, Woo DH, Kim CY, Chung HM (2022) Generation of human iPSCs derived heart organoids structurally and functionally similar to heart. Biomaterials 290:121860. https://doi.org/10.1016/j.biomaterials.2022.121860
Querdel E, Reinsch M, Castro L, Köse D, Bähr A, Reich S, Geertz B, Ulmer B, Schulze M, Lemoine MD, Krause T, Lemme M, Sani J, Shibamiya A, Stüdemann T, Köhne M, Von BC, Hornaschewitz N, Pecha S, Nejahsie Y, Mannhardt I, Christ T, Reichenspurner H, Hansen A, Klymiuk N, Krane M, Kupatt C, Eschenhagen T, Weinberger F (2021) Human engineered heart tissue patches remuscularize the injured heart in a dose-dependent manner. Circulation 143:1991–2006. https://doi.org/10.1161/CIRCULATIONAHA.120.047904
Long C, Guo R, Han R, Li K, Wan Y, Xu J, Gong X, Zhao Y, Yao X, Liu J (2022) Effects of macrophages on the proliferation and cardiac differentiation of human induced pluripotent stem cells. Cell Commun Signal 20:108. https://doi.org/10.1186/s12964-022-00916-1
Yang L, Han Y, Jaffré F, Nilsson-Payant BE, Bram Y, Wang P, Zhu J, Zhang T, Redmond D, Houghton S, Uhl S, Borczuk A, Huang Y, Richardson C, Chandar V, Acklin JA, Lim JK, Chen Z, Xiang J, Ho DD, Tenoever BR, Schwartz RE, Evans T, Chen S (2021) An immuno-cardiac model for macrophage-mediated inflammation in COVID-19 hearts. Circ Res 129:33–46. https://doi.org/10.1161/CIRCRESAHA.121.319060
Acknowledgements
This review was supported by funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 813716 as well as from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 465131031.
Funding
Open Access funding enabled and organized by Projekt DEAL. Deutsche Forschungsgemeinschaft, 465131031, Mira Jung, HORIZON EUROPE Marie Sklodowska-Curie Actions, 813716, Marida Sansonetti.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests. TT filed and licensed patents in the field of noncoding RNAs. TT is founder and shareholder of Cardior Pharmaceuticals GmbH.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sansonetti, M., Al Soodi, B., Thum, T. et al. Macrophage-based therapeutic approaches for cardiovascular diseases. Basic Res Cardiol 119, 1–33 (2024). https://doi.org/10.1007/s00395-023-01027-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-023-01027-9