
Abstract
Despite recent progress, ischemic heart disease poses a persistent global challenge, driving significant morbidity and mortality. The pursuit of therapeutic solutions has led to the emergence of strategies such as ischemic preconditioning, postconditioning, and remote conditioning to shield the heart from myocardial ischemia/reperfusion injury (MIRI). These ischemic conditioning approaches, applied before, after, or at a distance from the affected organ, inspire future therapeutic strategies, including pharmacological conditioning. Gasotransmitters, comprising nitric oxide, hydrogen sulfide, sulfur dioxide, and carbon monoxide, play pivotal roles in physiological and pathological processes, exhibiting shared features such as smooth muscle relaxation, antiapoptotic effects, and anti-inflammatory properties. Despite potential risks at high concentrations, physiological levels of gasotransmitters induce vasorelaxation and promote cardioprotective effects. Noble gases, notably argon, helium, and xenon, exhibit organ-protective properties by reducing cell death, minimizing infarct size, and enhancing functional recovery in post-ischemic organs. The protective role of noble gases appears to hinge on their modulation of molecular pathways governing cell survival, leading to both pro- and antiapoptotic effects. Among noble gases, helium and xenon emerge as particularly promising in the field of cardioprotection. This overview synthesizes our current understanding of the roles played by gasotransmitters and noble gases in the context of MIRI and cardioprotection. In addition, we underscore potential future developments involving the utilization of noble gases and gasotransmitter donor molecules in advancing cardioprotective strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronary artery disease persists as a major contributor to worldwide mortality and morbidity, driving extensive research aimed at safeguarding the myocardium from ischemic damage. Ischemic preconditioning (PC), postconditioning (PostC) and remote conditioning (RC) are extensively studied adaptive mechanisms. These procedures involve brief exposure to ischemia/reperfusion, occurring prior (PC) to prolonged ischemia, at the immediate onset of reperfusion (PostC), or in a remote organ (RC), respectively, thereby initiating cardioprotection. Yet, pharmacological conditioning, achieved through specific drug administration, can mimic the effects of ischemic PC and PostC, providing an alternative approach to heart protection. Conditioning protection is evidenced by notable reductions in arrhythmias, decreased infarct size, and alleviated cardiac and endothelial dysfunction [e.g., see Refs. 24, 60, 61, 64, 67, 68]. While the cardioprotective benefits of ischemic or pharmacological conditioning strategies have been demonstrated across various species, including humans, the presence of cardiovascular risk factors, comorbidities, and associated medications (comedications) may disrupt cardioprotective signaling pathways [5, 44, 92, 154]. In addition, the potential cardioprotective effect of female sex, with differences possibly existing between pre- and post-menopausal women, should be considered. However, there is currently no evidence indicating sex-related disparities in infarct size and cardioprotection in pigs [93, 106].
Nevertheless, the precise cellular mechanisms underlying the cardioprotective pathways remain elusive in male and female, although several signal transduction cascades have been proposed. The initiation of cardioprotective modalities involves mainly the occupancy of specific surface receptors by various ligands, leading to intracellular signaling transduction, including redox signaling by reactive oxygen species (ROS) [7, 69, 144, 152], S-nitrosylation by nitric oxide (NO) and its derivatives, and S-sulfhydration by hydrogen sulfide (H2S) [5, 7, 144]. These modalities interact and regulate an integrated pathway, impacting one another’s function. Notably, enzymes may undergo phosphorylation and/or nitrosylation at specific or distinct sites, resulting in alterations in their activity. The cardioprotective pathways, namely the survivor activating factor enhancement (SAFE) pathway, the reperfusion injury salvage kinase (RISK) and the NO/cyclic 3′,5′-guanosine monophosphate (cGMP)/protein kinase G (PKG) pathway, were initially associated with ischemic conditioning [see Refs. 91, 93, 95, 226]. Enhanced understanding of these pathways holds promise for advancing cardioprotective strategies, including pharmacological interventions, in clinical practice. In particular, we outline the elements of RISK pathway involving intracellular mediators such as phosphatidyl-inositol-4,5-bisphosphate 3-kinase (PI3K), protein kinase C (PKC), mitogen-activated protein kinase (MAPK), glycogen synthase kinase-3β (GSK-3β), and extracellular signal-regulated kinase 1/2 (ERK1/2), as well as the SAFE pathway influenced by janus kinase-signal transducer and activator of transcription (STAT) pathways [24, 226]. Convergence between these pathways occurs at various steps but mainly on the mitochondria and, specifically, on the mitochondrial permeability transition pore (mPTP) [24, 60, 61, 226]. Indeed, these factors and organelles represent a direct or indirect target for various gases, which exhibit biological effects, and among them, oxygen (O2) and NO, along with their derivatives, ROS and reactive nitrogen species (RNS), are well known for their biological activities.
For approximately four decades, it has been recognized that our organism possesses an enzymatic apparatus capable of generating gases with distinct biological effects. Termed collectively as gasotransmitters, including NO, H2S, sulfur dioxide (SO2) and carbon monoxide (CO), these gases constitute a group of endogenous, highly reactive, and regulatory molecules. As we will see in the specific paragraphs gases play a crucial role not only as endogenous factors, but also in cardioprotection induced by pharmacological conditioning using exogenous gasotransmitters as well as noble gases. Indeed, noble gases, traditionally labeled as ‘inert gases’, have gained recognition more recently due to their manifestation of distinctive biological effects.
Here, we focus on the cardioprotective effects of gasotransmitters and noble gases, allocating only some discussion to the role of ROS/RNS. The cardioprotective effects of ROS/RNS have been comprehensively reviewed by our group and others, emphasizing the intricate signaling mechanisms involved in cardioprotection; for reviews on the role of ROS/RNS in cardioprotection, see Refs. [7, 69, 144, 152]. In addition, some volatile anesthetics are gaseous molecules known for their protective effects [12, 203, 229]. Specifically, this review aims to provide insights into the roles of gasotransmitters and noble gases in myocardial ischemia/reperfusion injury (MIRI) and cardioprotection, while also discussing potential future directions for developing agents that modulate these molecules for cardioprotective purposes. The literature search was performed using the PubMed databases, employing as keywords the distinct gases in combination with conditioning and cardioprotection. By curating recent (last 20 years) English-language publications, we present an overview of current research and anticipate innovative approaches to enhance the effectiveness and broaden the clinical applications of cardioprotection.
The vital gas, oxygen, in ischemia/reperfusion and cardioprotection: focus on supersaturated-oxygen (SSO2)
Given the vital role of O2 as a fundamental gas for life and as a component of certain gasotransmitters, it is necessary to acknowledge its significance not only for life but also for potential therapeutic applications. Therefore, before exploring the cardioprotective effects of gasotransmitters and noble gases in the context of MIRI and cardioprotection, we briefly discuss O2, considering both its essentiality and its detrimental or therapeutic paradox, such as the concept of SSO2 therapy.
The regulatory influence of O2 extends from embryonic ontogeny to pathological processes, making it a key participant in various cellular activities [112]. Oxygen is a crucial element for the survival of all mammals, playing a fundamental role in generating biological energy required by cells [112]. Fluctuating O2 levels influence cellular physiology, with evidence suggesting that variable O2 levels, rather than persistently low levels, pose the greatest harm [112].
The “oxygen paradox” arises from the dual role of O2 in both MIRI and cardioprotection [7, 144, 152, 203]. This paradox involves the harm caused by reoxygenation following ischemia, known as reperfusion injury, juxtaposed with the therapeutic potential of slow release of O2 [43, 133, 150] and gentle reperfusion [122] or super-reoxygenation [113, 190, 230]. In the context of ischemia/reperfusion, “compartment syndrome” refers to increased pressure within tissue compartments, impeding blood flow and causing potential damage [34, 110]. Although the term “compartment syndrome” is typically applied to peripheral muscle reperfusion injury rather than the heart, we employ it here to underscore the significant impact of edema on O2 delivery during reperfusion in the setting of MIRI [34, 48]. SSO2 has emerged as a potential therapeutic approach to mitigate MIRI-associated damage, particularly in addressing “compartment syndrome” [96, 230]. SSO2 is obtained through aqueous oxygen, creating a metastable saline solution with a higher concentration of dissolved O2 than the liquid carrier [191,192,193]. Catheter-delivered SSO2 serves as a pragmatic alternative to traditional hyperbaric oxygen therapy (HBOT) in critical care settings.
Research indicates that SSO2, similar to HBOT, assists in arterial vasoconstriction, addressing compartment syndrome without the risks associated with high gas-phase [O2]/tissue interfaces [191,192,193]. Preclinical studies demonstrate that intracoronary SSO2 infusion significantly reduces infarct size and the edematous area at risk in post-ST-segment elevation MI (STEMI) reperfusion scenarios [191,192,193].
The primary mechanism contributing to SSO2-induced cardioprotection is the acute improvement of microvascular flow, leading to the normalization of left ventricle ejection fraction [193]. SSO2 facilitates gradual reperfusion at relatively low pressures, reducing microvascular damage compared to abrupt reperfusion [177, 178]. The hyperosmotic effect of hyperbaric dissolved O2, coupled with reduced capillary hydrostatic pressure, aids in removing edema fluid through Starling’s principle, interrupting the cycle of MIRI and microvascular responses [177, 191,192,193].
SSO2 also addresses the inflammatory responses associated with MIRI, as evidenced by reduced myeloperoxidase levels in animal models [86, 191]. The resolution of the “oxygen paradox” is attributed to SSO2’s positive effects on microvascular flow, improvement of left ventricular function, and reduction of infarct size [10, 190]. The time course of SSO2-induced improvements, including the reduction of precapillary resistance, remains to be elucidated. In summary, SSO2 shows promise as a therapeutic intervention in MIRI scenarios by effectively addressing oxygen-related complications and inflammatory responses. Hence, here we see “the paradox of paradox”: the detriment posed by reoxygenation following ischemia (i.e., reperfusion injury), are counteracted by the therapeutic potential of SSO2. Alternative experimental strategies to mitigate the adverse effects associated with the oxygen paradox include stuttering and slow reperfusion [177, 183], or controlled O2 release by oxygen-loaded nanodevices. These nanodevices can act as biocompatible drug carriers, enabling the gradual release of O2, potentially along with therapeutic agents [43, 132, 150] (see also “Nanodevices to deliver gases”).
While preclinical and some clinical evidence suggest the potential improvement of MIRI with SSO2, it is essential to consider the implications of hyperbaric oxygen therapy, which may exacerbate heart failure [174], and it is advised to avoid hyperoxia in cases of chronic cyanosis [121]. Moreover, a meta-analysis has revealed weak and inconsistent evidence, along with modest statistical power, regarding the safety and efficacy of oxygen therapy [88]. Therefore, caution is warranted in the use of O2 in these contexts. Adequately powered studies are imperative to gain a better understanding of the role of SSO2 in patients undergoing coronary revascularization.
Biological gasotransmitters
The intricate signaling pathways in which gasotransmitters—CO, H2S, SO2 and NO—are involved represent a multifaceted network of interactions characterized by the high reactivity and diffusive capacity inherent to these gaseous molecules. The intricacy of these pathways stems from both their chemical properties and the diverse cellular responses they modulate in the dynamic cardiovascular environment.
Gasotransmitters are versatile signaling mediators. They play a pivotal role in orchestrating cellular reactions critical to the complexity of cell physiology and pathophysiology, including MIRI [9, 112, 200, 220]. Their presence is integral to the delicate equilibrium between the potentially detrimental effects of ischemia and reperfusion. For instance, as signaling molecules, CO, H2S, SO2 and NO engage in a sophisticated crosstalk with cellular components, influencing vasodilation, inflammation, apoptosis, and oxidative stress [87].
The high reactivity of gasotransmitters and the capacity to rapidly traverse cellular membranes facilitate their widespread influence on various cellular components. Their diffusive capacity enables them to permeate tissues and reach subcellular compartments. Indeed, gasotransmitters, as well as derivative, ROS and RNS, are considered among the major factors involved in signaling pathways regulating mitochondrial function involved in cardioprotection [113, 200, 220].
As researchers explore gasotransmitter-mediated cellular responses, we expect this summary to be a valuable resource for understanding the complex dynamics involved in cardioprotection. A summary of the experimental studies on gasotransmitter-induced protection can be found in Tables 1, 2, 3 and 4.
Nitric oxide and cardioprotection
The importance of NO in both normal physiological processes and pathological conditions is well established. However, for a more in-depth understanding of its various roles, readers are encouraged to refer to recent reviews that offer comprehensive insights [3, 6, 7, 85, 153].
Sources: The synthesis and primary metabolic pathways of NO are extensively discussed elsewhere [9, 26]. In brief, endogenous NO is produced enzymatically through the conversion of l-arginine by a specific group of enzymes known as NO synthases (NOSs). These NOS enzymes, which exist as homodimeric oxidoreductases, are expressed constitutively in various cell types. Neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS) are expressed by various tissues; therefore, they are also called NOS1, NOS2, and NOS3, respectively [3]. In particular, iNOS and nNOS highlight the involvement of novel cellular actors in cardioprotection. Various cell populations, such as immune cells [4] and pericytes [39], appear to play a role in the cardioprotective capacity of NO and other gasotransmitters (Fig. 1). In addition to these cells, emerging evidence highlights the significant role played by erythrocytes in mediating NO protective effects. Erythrocytes contribute to NO bioavailability and signaling pathways, thereby exerting a notable influence on cardioprotection against MIRI. Nitric oxide can also be generated in tissues through either direct disproportionation or the reduction of nitrate and nitrite to NO under acidic and highly reduced conditions that are present in disease states like ischemia [70, 94, 117, 119, 223]. Actually, during myocardial ischemia, NO synthesis increases independently of NOS pathways, impacting outcomes via concentration-dependent mechanisms [70]. Endogenous NO favors myocardial hibernation during ischemia by reducing O2 consumption and preserving function, thus preventing necrosis [117].

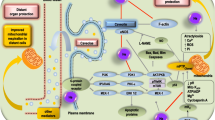
Schematic representation of endogenous gasotransmitter production and interaction on various cardiovascular cell types. In addition to the well-established role of ECs and VSMCs, immune cells and microvascular pericytes are emerging as significant contributors to cardioprotection. Green cloud: NO; yellow cloud: H2S; blue cloud: SO2; red cloud: CO. Created with BioRender.com. CO, carbon monoxide; ECs, endothelial cells; H2S, hydrogen sulfide; NO, nitric oxide; SO2, sulfur dioxide; VSMCs, vascular smooth muscular cells
Cardiac protection: Recent progress in unraveling NO’s involvement in cardiac biology has highlighted its crucial role in defending against MIRI [6, 7, 9]. The cardioprotective effects of NO are not solely induced by “conditioning” mechanisms. For instance, upon β-adrenergic stimulation, the nNOS, which is bound to ryanodine receptors in the sarcoplasmic reticulum, modulates NO levels, facilitating calcium release and controlling inotropy [49] (Figs. 2 and 3). Therefore, NO plays a pivotal role in safeguarding the β-adrenergic-mediated heart function regulation. Moreover, NO influences the calcium release through S-nitrosylation, modulating key proteins, such as calcium channels. Inhibition of nNOS by superoxide (O2(−)) as well as by peroxynitrite (ONOO(−), formed by the reaction of NO with O2(−)), hinders β-adrenergic stimulation, impacting calcium release and consequently cardiac inotropy [28, 55]. Altered NO levels and increased O2(−) result in nNOS uncoupling, and blocked phospholamban phosphorylation. The formed ONOO(−) affects cardiomyocyte action potentials, induces lipid peroxidation, and damages mitochondria. Moreover, ONOO(−) influences cardiomyocyte function through the effects on sarco-endoplasmic reticulum calcium ATPase (SERCA). Indeed, elevated ONOO(−) levels promote calcium sequestration via SERCA, affecting cardiomyocyte relaxation [15, 35]. Nitric oxide actively contributes to protection induced by various factors, such as physical exercise [42, 147] and the stimulation of β3-adrenergic receptors [126, 159]. In particular, nNOS exhibits cardioprotective effects during exercise by modulating O2 consumption. Endothelial NOS contributes to cardioprotection by inhibiting the β-adrenergic response through the regulation of the L-type calcium channel. During exercise, the increased ratio of eNOS dimer to monomer, coupled with reduced peroxynitrite levels, promotes eNOS dimerization and activation, further enhancing cardiac protection [40, 41, 124, 208]. During exercise, iNOS levels are low, while elevated levels are induced in reactive hypertrophy through various signaling mechanisms. Increased levels of NO during exercise enhance mitochondrial O2 consumption, regulated by angiotensin II in cardiomyocytes. Notably, cardiomyocytes control NO diffusion and compartmentalization, with heme-centered proteins such as cytoglobin and myoglobin scavenging locally released NO, thus regulating this gas diffusion within the heart [57, 58].

Schematic representation of noble gas and endogenous gasotransmitter interaction with cellular organelles. Gases can interact with various cellular organelles, including mitochondria, endoplasmic reticulum, and nucleus, influencing cellular signaling and function. Mitochondria play a major role (for more details on cardioprotective mechanisms of NO and H2S in mitochondria during ischemia/reperfusion see reference #7). Green cloud: NO; yellow cloud: H2S; blue cloud: SO2; red cloud: CO; orange cloud: He; gray cloud: Xe; light-blue cloud: Ar. Created with BioRender.com. I, II, III, IV, and V (ATP synthase) indicate respiratory chain complexes; Ar, argon; ADP, adenosine diphosphate; ATP, adenosine triphosphate; β-AR, β adrenergic receptor; BKCa, large conductance Ca2+-activated K+ channels; Ca2+, calcium; CBS, cystine beta-synthesis enzyme; CO, carbon monoxide; CSE, cystathionine gamma-lyase; Cyt c, cytochrome c; ETC, electron transport chain; H2S, hydrogen sulfide; He, helium; HO-1, heme oxygenase-1; K+, potassium; mitoKATP, mitochondrial ATP-sensitive potassium channel; mPTP, mitochondrial permeability transition pore; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; ROS, reactive oxygen species; RyR, ryanodine receptor, SERCA, sarco-endoplasmic reticulum calcium ATPase; SO2, sulfur dioxide; Xe, xenon

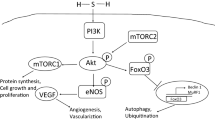
Schematic diagram illustrating the key signaling pathways of gasotransmitters and noble gases within cardiomyocytes. The diagram delineates the principal cardioprotective signaling pathways. Extracellular molecules and gases (depicted as clouds) engage with sarcolemmal receptors or function independently of receptors. This interaction triggers downstream cytosolic signaling cascades, such as the NO/PKG, RISK, and SAFE pathways, while gases also modulate or activate additional kinases not explicitly linked to the indicated pathways. Intracellularly, these pathways converge on the mitochondria, inhibiting the opening of the mPTP. Gases may also directly affect several mitochondrial components (see also Fig. 2 and reference #7). Late preconditioning involves the nucleus and transcription of enzymes such as iNOS. Gases may also modulate inotropy. Green cloud: NO; yellow cloud: H2S; blue cloud: SO2; red cloud: CO; orange cloud: He; gray cloud: Xe; light-blue cloud: Ar. Created with BioRender.com. Ade, adenosine; ADP, adenosine diphosphate; Akt/PKB, protein kinase B; Ar, argon; ATP, adenosine triphosphate; Bcl-2, B-cell leukemia/lymphoma-2; BK, bradykinin; Cav-3, caveolin-3; CBS, cystine beta-synthesis enzyme; CO, carbon monoxide; CSE, cystathionine gamma- Lyase; eNOS, endothelial nitric oxide synthase; ERK1/2, extracellular signal-regulated kinase 1/2; GFR, growth factor receptor; gp130, glycoprotein 130; GPCR, G-protein coupled receptor; GSK-3β, glycogen synthase kinase-3beta; H2S, hydrogen sulfide; He, helium; iNOS, inducible nitric oxide synthase; MAPK, mitogen-activated protein kinase; mitoKATP, mitochondrial ATP-sensitive potassium channel; MKKKs, MAPK kinase kinases; MKKs, MAPK kinases; mPTP, mitochondrial permeability transition pore; mTOR, mammalian target of rapamycin; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; OP, opioids; PI3K, phosphatidylinositol-3-kinase; PKC-ε, protein kinase C epsilon; PKG, protein kinase G; RISK, reperfusion injury salvage kinase pathway; RNS, reactive nitrogen species; ROS, reactive oxygen species; SAFE, survivor activating factor enhancement pathway; sGC, soluble guanylyl cyclase; SO2, sulfur dioxide; SOD2, superoxide dismutase; SR, sarcoplasmic reticulum; STAT3, signal transducer and activator of transcription 3; TNF-α, tumor necrosis factor α; Xe, xenon
Conditioning: In simple terms, the previously mentioned cardioprotection protocols (namely PC, PostC and RC have revealed cooperative and protective signaling pathways, including the RISK, SAFE, and NO/cGMP/PKG pathways [46, 166,167,168, 183] (Fig. 3). These pathways involve phosphorylation and dephosphorylation mechanisms with various kinases, including NOS. Furthermore, redox-dependent protective pathways are implicated, where ROS, S-nitrosylation by NO, and NO derivatives such as nitroxyl (HNO) play integral roles [113]. Therefore, NO participates in signaling cascades by activating enzymes such as guanylyl cyclase (GC) and it functions as a key element in redox signaling, engaging in reactions with O2(−) and sulfur groups.
The NO molecule plays a central role in initiating and mediating the late phase or second window of protection in ischemic PC. The cardioprotective effects of NO are particularly intriguing for the iNOS, which also displays a role in protecting the mitochondria. Actually, iNOS is primarily involved in inflammatory responses and reactive hypertrophy in the heart, but it is also involved in the second window of ischemic PC [142]. This has been demonstrated in both ischemic PC and pharmacological cardioprotection [85, 142, 195]. The widely accepted idea that increased iNOS activity in the later phase of PC enhances NO availability reinforces iNOS as a protective protein against MIRI. Yet, excessive NO production and iNOS expression and uncoupling were observed in cardiac allograft rejection, suggesting involvement of NO and O2(−) in tissue injury through ONOO(−) formation [2]. Despite limitations in current approaches, studies indicate that NO scavengers may extend acute cardiac graft survival by limiting NO’s actions and treatment with NOS inhibitors improved cardiac injury significantly [156]. However, the precise role of NO and NO/O2(−) ratio in organ rejection is still debated, necessitating further research on their effects on different cell types.
It has been suggested that NO interacts with elements of the electron transport chain (ETC) and/or the mPTP to alleviate post-ischemic myocardial damage (Fig. 2). However, the precise molecular events of this action remain elusive. Nevertheless, this interaction with mitochondria offers a fundamental molecular explanation for the mechanism behind NO-mediated cardioprotection, emphasizing NO as a common mediator of protection across various interventions against myocardial ischemia and reperfusion [6, 7, 153]. A comparable protective function is attributed to PostC, with both protection (PC and PostC) being hindered by the NOS inhibitors [81, 108, 151].
As seen above and summarized in Fig. 3 and Table 1, within the heart, NO plays a pivotal role in various signaling pathways involved in cardioprotection against MIRI. However, there is contradictory evidence that endogenous NO is involved in ischemic PC. For instance, endogenous NO did not alter infarct size development and was not implicated in the protection against infarction through classical ischemic PC in rabbits ex vivo [123] and pigs in vivo [158]. These findings underscore the complex and context-dependent nature of NO's cardioprotective mechanisms. It is also useful to differentiate between the NO that exerts cardioprotective effects within the heart and the NO that circulates in the form of cardioprotective, bioactive plasmatic nitrite. Circulating nitrite deriving from remote site contributes to cardioprotection by ischemic RC [166,167,168, 202]. These studies evidenced that circulating nitrite serves as a significant source of NO with cardioprotective properties. Furthermore, as highlighted erythrocytes contribute to the bioavailability of NO through the reduction of nitrite [51, 52, 201]. These distinct sources and mechanisms underscore the multifaceted nature of NO-mediated cardioprotection.
Pharmacological conditioning: Notably, drugs such as angiotensin-converting enzyme (ACE) inhibitors, statins, and angiotensin-receptor blockers, which boost NO levels, show benefits in mouse models of myocardial infarction (MI), supporting the potential importance of NO in conditioning cardioprotection [46, 100, 149, 150, 179]. Undoubtedly, the endothelium serves as the primary source of NO, yet its role in MIRI has often been overlooked, although the coronary circulation and endothelial dysfunction are implicated in MIRI as both causes and targets [65, 66]. In light of this, the replacement of endogenous NO with NO donors holds great promise in these conditions. Preclinical studies have unveiled various mechanisms of coronary microvascular injury that can be addressed through ischemic and pharmacological conditioning, suggesting potential clinical translation to enhance patient outcomes in MI [for reviews see 65–69]. When NO or HNO are administered before ischemia trigger protective responses similar to ischemic PC [113, 143, 198, 207]. For instance, the administration of exogenous NO, such as S-nitroso-N-acetylpenicillamine, demonstrated a beneficial effect in reducing infarct size [123]. In addition, the diethylamine-NO donor has been widely used as preconditioning mimetic [113, 143]. Moreover, the donors of HNO, a sibling of NO, are gaining recognition for their pharmacological attributes, which encompass offering functional assistance to failing hearts. In addition, HNO demonstrates the ability to precondition myocardial tissue, safeguarding it from MIRI, and exhibits actions that counteract vascular proliferation [113, 143, 200, 220].
To sum up, NO and derivatives have a plethora of targets, but the protective impact of NO and HNO operate mainly through mitochondria, interacting with different components of the ETC, mitochondrial ATP-sensitive K+ (mitoKATP) channels, and elements of the mPTP [78, 104, 150]. These interactions significantly reduce MIRI (Table 1).
Hydrogen sulfide and cardioprotection
In addition to NO, H2S is regarded as a cardioprotective gaseous mediator.
Sources: At least, three enzymes in mammalian tissues produce H2S endogenously from cysteine. Actually, endogenous H2S is generated through enzymatic or nonenzymatic routes within mammalian tissues [14, 18, 145, 221]. Key enzymes involved in this process include cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS), both utilizing homocysteine as a substrate. In addition, 3-mercaptopyruvate sulfurtransferase (3-MST) can catalyze H2S synthesis in conjunction with cysteine aminotransferase (CAT). CSE exhibits localization in the kidney, liver, vessels, and heart, CBS is found in the central nervous system, and 3-MST is expressed in the liver, heart, brain and kidney. The catabolic pathways of H2S involve: (a) oxidation to thiosulfate catalyzed by mitochondrial thioquinone oxidoreductase, S-dioxygenase, and S-transferase; (b) generation of methyl mercaptan and dimethyl sulfide through a reaction catalyzed by cytoplasmic thiol S-methyltransferase; (c) interaction with methemoglobin leading to the production of thiol hemoglobin [7, 10, 106, 168].
Cardioprotection: Apart from ischemia/reperfusion injury, heart failure, hypertrophy, fibrosis, myocardial infarction, arrhythmia, and several physiological and pathological processes have all been shown to be prevented by H2S. Its cardioprotective effect may be attributed to mechanisms such as NO interaction, ion channel regulation, antioxidative action, mitochondrial function preservation, apoptosis reduction, anti-inflammatory responses, and angiogenic actions [6, 7, 9, 20, 114, 170]. Despite identifying multiple mechanisms, additional research is necessary to determine the precise molecular mechanism of cardioprotection in various cardiac diseases to open the door to novel therapeutic targets based on H2S production and/or modulation.
Conditioning: During MIRI, the plasma level of H2S and CSE in the myocardium are reduced, while the mRNA expression level of CSE is increased following reperfusion, which contributes to a sort of positive feedback. Indeed, in KO model for CSE oxidative stress and MIRI are exacerbated [89]. In rodents, conditioning protection is mainly mediated by the RISK and SAFE pathways (Fig. 3), activated at the onset of reperfusion [87, 89, 118]. Findings indicate that naturally elevated levels of H2S act to safeguard the heart against MIRI, suggesting their potential as significant therapeutic targets.
Pharmacological conditioning: The majority of studies on this gas considered exogenous donors (Table 2). The administration of different H2S donors before reperfusion reduces infarct size and the plasma level of troponin-I significantly in MIRI mice [89]. Pretreatment with H2S, acting as a modulator of an ERK1/2-dependent pathway, also reduces endothelial cell death in vitro [235]. The protective effect of H2S is reported when the administration is either before (PC-like) or after (PostC-like) prolonged ischemia [5,6,7, 9].
In a porcine model, H2S treatment significantly improved hemodynamics, reduced infarct size, and displayed anti-inflammatory effects, suggesting potential therapeutic utility in clinical settings encountering MIRI [189].
Besides the influence on RISK and SAFE pathways, the cardioprotective effects induced by exogenous sodium hydrosulfide (NaHS) depend on mitochondrial ETC enzymes and also lead to mitoKATP channel opening (Figs. 2 and 3) [6, 7, 9]. Accumulating evidence has reported a crosstalk between H2S and NO. In fact, CSE knockout mice displayed a reduction in NO levels due to reduced eNOS expression. In this model, the treatment with H2S induces an increase in NO bioavailability and restores eNOS protein expression, which subsequently attenuated oxidative stress and MIRI. Recently, it was observed that S-sulfhydration, a post-translational modification involving the interaction of H2S with cysteine residues in proteins, alters the structure and biological activities of protein targets. In mice, application of pharmacological postconditioning using NaHS at the onset of reperfusion significantly enhanced S-nitrosylation of cardioprotective proteins, concurrently mitigating post-ischemic contractile dysfunction and reducing infarct size [198].
In the apolipoprotein E knockout model, the administration of NaHS induces increased plaque stability and blood lipid levels and reduced plaque formation [222]. Intriguingly, the Andreadou group displayed an interplay between eNOS, H2S, and CO-synthesizing enzymes in human atheroma in plaque stability and simvastatin effects [182]. Limited research has explored the interplay between CO and H2S in signal transduction, and the synergistic mechanism remains incompletely elucidated (see also “Carbon monoxide and cardioprotection”).
Sulfur dioxide and cardioprotection
Sulfur dioxide stands out as an additional gasotransmitters, showcasing diverse biological impacts, including antioxidative, anti-inflammatory, antihypertensive, and antiatherogenic effects.
Sources: Pioneering discoveries reveal that endogenous SO2 formation occurs within the cardiovascular system, exerting a prominent vasorelaxant influence [37, 74]. Subsequent findings identified SO2 products in various organs, such as the brain, stomach, lungs, liver, spleen, and heart [75, 109]. Aspartate aminotransferase is indicated as the key enzyme for SO2 synthesis. Conversely, SO2 catabolism involves the hydrogenation of bisulfite and sulfite ions, which are then oxidized to sulfate [74, 75].
Cardioprotection: Current knowledge establishes SO2’s engagement in apoptosis and oxidative stress, elevating antioxidant enzyme expression (e.g., SOD2 and GSH-Px1) and decreasing ROS production [105]. Simultaneously, in rats with isopropylarterenol-induced myocardial injury the antiapoptotic effects of SO2 are attributed to increased B-cell leukemia/lymphoma-2 (Bcl-2) expression, Bcl-2 associated protein x inhibition, mitochondrial membrane stabilization, decreased cytochrome c release, and diminished caspase activation [84]. Furthermore, SO2 plays a role in intracellular calcium homeostasis, with disruptions linked to cell death in numerous pathological conditions [74, 75]. Sulfur dioxide emerges as a pivotal regulator in various biological processes under both normal and pathological conditions associated with cardiovascular diseases. Recent investigations into the impact of SO2 on cell apoptosis have gained significant attention, revealing its regulatory influence on vascular smooth muscle cells, endothelial cells, cardiomyocytes, and other cells implicated in the pathogenesis of arterial hypertension and myocardial damage [234]. This multifaceted role positions SO2 as a crucial player in cardiovascular health, influencing cellular processes and offering potential therapeutic avenues in cardiac disorders.
Ischemic and pharmacological conditioning: Preconditioning with SO2 diminished myocardial infarct size, plasma lactate dehydrogenase, and creatine kinase activities in rats, alongside reducing myocardial caspase-3 and -9 activities. In addition, this pretreatment substantially elevated the expression levels of myocardial phosphorylated‑protein kinase B (p-PKB/p‑AKT) and phosphorylated‑PI3K-p85. Notably, the administration of the PI3K inhibitor LY294002 effectively nullified all the beneficial effects initiated by SO2 preconditioning. Furthermore, exogenous SO2 preconditioning improved cardiac function and reduced phosphorylated-ERK1/2 protein expression in isolated rat hearts with MIRI, suggesting the involvement of the ERK-MAPK pathway. Inhibition of ERK1/2 activation by the inhibitor PD98059 abolished the cardioprotective effects of SO2 [73]. Additional data on SO2 preconditioning strongly support the theory that the PI3K/Akt and ERK pathways play pivotal roles in mediating the protective effects against MIRI in rats [234] (Table 3; Fig. 3).
Carbon monoxide and cardioprotection
Sources: Carbon monoxide, being an easily diffusible gaseous molecule, originates from diverse sources within biological systems. The primary origin in mammals is attributed to heme oxygenase (HO) and HO-like activity. Carbon monoxide is generated through the degradation of the heme group via HO, which includes the constitutive forms HO-2 and HO-3, and the inducible form HO-1 [6, 19, 22, 53].
Cardioprotection: In the cardiovascular system, including the heart, CO-induced vasodilation occurs through the classical NO pathway, involving soluble GC (sGC)/cGMP, and PKG pathways. In addition, CO impedes endothelin-1 synthesis, prevents platelet aggregation, and inhibits L-type Ca2+ channels, collectively contributing to the cytoprotection of cardiomyocytes [22, 171]. Upregulation of HO-1 and its metabolites is known to activate various cell protection pathways, particularly during MI and hypoxia. Notably, during hypoxia and MI, there is a substantial increase in HO-1 expression, leading to heightened CO production [53]. Indeed, HO-1, in part, contributes to damage recovery and tissue repair by generating bioactive products such as CO. Carbon monoxide initiates a cardioprotective and antiapoptotic environment in the myocardium, akin to the effects observed during the late phase of preconditioning. This suggests that the presence of CO induces a state in the myocardium that mimics the delayed preconditioning, contributing to protection against cardiac damage and apoptosis [195].
Pathophysiological interactions between CO and H2S have been validated in a model of myocardial ischemic PC. In a rat model of pulmonary hypertension, H2S was observed to elevate HO-1 expression in the pulmonary artery and increase CO concentration in plasma [20, 164]. Carbon monoxide demonstrates cardioprotective properties, primarily through the opening of mitoKATP channels. This process inhibits the long-lasting opening of the mPTP and regulates mitochondrial ROS production. Although information on the cardioprotective effects of CO is limited, it is suggested that at low concentrations, CO facilitates the preservation of mitochondrial function. Under these conditions, CO has been observed to maintain the stable potential of the mitochondrial membrane, particularly during the ischemia/reperfusion period [9, 131, 171]. The mitochondrial level is a key site of CO action, where it induces the opening of mitoKATP channels and inhibits mPTP opening [22, 171]. Moreover, CO modulates several signal pathways implicated in cardioprotection, including NO/GC, MAPK, and ROS action (Fig. 3). In addition, CO has the capacity to impede platelet activation through both cGMP-dependent and cGMP-independent pathways [171]. In addition to the NO and cGMP pathways, CO exerts its anti-inflammatory properties by inhibiting cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-1β (IL-1β) [135]. Apart from its anti-inflammatory effects, CO has proven its value in the field of organ transplantation, leading to improved outcomes [132]. Furthermore, in a porcine model of cardiopulmonary bypass, CO demonstrated its beneficial effects by enhancing cardiac energetics [98].
HO-1 expression and activity increase in hypoxic states, decrease during reperfusion, and correlate with heightened myocardial damage [171]. Protective action of CO is attributed to increased heme availability, a substrate for HO-1, and elevated HO-1 activity during reperfusion [22]. Indeed, the absence of HO-1 significantly increases vulnerability to ischemic injury [79].
Pharmacological conditioning: The majority of studies deal with exogenous CO, whose administration simulates HO-1 induction, resulting in heightened inotropy, reduced apoptosis, and diminished infarct size in rat hearts. Moreover, CO operates through a PKB/Akt-GSK-3β-nuclear factor erythroid 2-related factor (Nrf2) pathway, fostering mitochondrial biogenesis. Indeed, the influence of HO-1/CO on mitochondria is pivotal for the differentiation of stem cells into cardiac myocytes within the heart [155, 197]. Mitochondrial function likely plays a crucial role in CO-induced cardioprotection, impacting gene regulation, downstream signaling, and cell survival. Protective role of CO in the heart relies on HO-1 levels, triggered by tissue injury leading to the release of cellular contents, including elevated heme levels. This, subsequently, initiates HO-1 and endogenous CO generation, both highly cardioprotective and replicable by exogenous CO. Subtle concentrations of CO form transient bonds with cytochrome c oxidase, generating minimal levels of ROS. These ROS, in turn, activate signaling complexes involving Nrf2, hypoxia-inducible factor 1alpha (HIF-1α), and nuclear factor-kappa B (NF-κB)/inhibitor of NF-κB. Carbon monoxide also boosts the expression of peroxisome proliferator-activated receptor-gamma coactivator-1α, Nrf1, and Nrf2, thereby promoting CO-induced mitochondrial biogenesis for tissue preservation and regeneration [16, 21, 155].
Preconditioning triggered by CO demonstrates its ability to offer neuronal protection against apoptosis. This phenomenon highlights the capacity of CO to create a preconditioned state that safeguards cells from the process of programmed cell death [165, 206]. Two recent studies highlight the therapeutic potential of CO-releasing molecules (CORMs) in myocardial protection. Portal et al. show that CORM-3, a water-soluble CO-releasing molecule, exhibits substantial cardioprotective effects against hypoxia-reoxygenation in adult cardiomyocytes, emphasizing its potential as a therapeutic agent for MI [157]. In another study, Iqbal et al. demonstrate that CORM-A1 significantly reduce infarct size in a porcine model of acute MI [82] (Table 4).
Noble gases
Noble gases belong to a family of six naturally occurring gases: helium (He), neon (Ne), argon (Ar), krypton (Kr), xenon (Xe) and radioactive radon (Rn) [72, 209, 231].
Noble gases possess a defining trait: a fully occupied outer shell of valence electrons. This characteristic imparts inertness or, at the very least, diminished reactivity with other compounds under typical atmospheric pressure and temperature conditions. It is this distinctive feature that leads to their common designation as inert gases.
Nonetheless, a few of these noble gases exhibit potent biological effects. For instance, Xe is the only noble gas that shows anesthetic properties under normobaric conditions, while none of the other five gases has these properties [27, 72].
There is increasing evidence during the last 20 years that these so-called inert gases have strong cardioprotective and neuroprotective qualities [32, 50, 72, 116, 183, 209, 213, 214].
However, how these gases exert their cardioprotection is still a matter of debate. For Xe, it was shown that it may cause conformational changes in two proteins: annexin V, which has a hydrophilic pore inside and is meant to bind to cell membranes via a calcium-dependent mechanism, and urate oxidase, an intracellular globular protein with large hydrophobic cavities [25]. The Xe binding sites of the corresponding proteins consist of flexible gas cavities devoid of water. It has been demonstrated that Xe affects a number of cell membrane receptors. These include the nicotinic acetylcholine receptor [223], the 5-hydroxytryptamine 3 receptor [199], the plasmalemmal adenosine triphosphate-sensitive potassium channels (KATP) channel [11], the two-pore domain potassium channel TREK-1 [54], and the N-methyl-D-aspartate (NMDA) receptors [47]. In particular, it has been shown that at the glycine site of the NMDA receptor, Xe competes with the co-agonist glycine [33, 59]. However, this knowledge is primarily derived from neural cells. Thus, it is unclear if these Xe effects on neural cells also contribute to its myocardial protection. Nonetheless, it has been shown that regarding cardioprotection KATP channels—at least the mitoKATP channels—play a crucial role in the PC of the heart [134] and, therefore, might be a key mediator in noble gas-induced cardioprotection.
Here, we will briefly report and discuss the recent studies displaying cardioprotective effects of some noble gases (Ar, Xe, and He) with the main focus on the underlying molecular mechanism involved. A summary of the experimental studies on noble gas-induced cardioprotection can be found in Tables 5, 6 and 7 and Figs. 2 and 3.
Argon and cardioprotection
The name argon in Greek: αργός means inert. Argon has anesthetic properties under hyperbaric conditions and is relatively abundant in atmospheric air where it is found at a concentration of 0.93%. It is non-corrosive, non-flammable and non-toxic, with a density 38% higher than that of air and its solubility in water and plasma is 24 times less than that of carbon dioxide [125]. Despite being considered ‘biologically’ inert, recent evidence suggests that argon may have significant pharmacological effects [125, 204]. It has mainly been investigated for its neuroprotective effects but there is some evidence from both in vitro and in vivo studies that Ar exhibited cardioprotective effects and that these effects are mediated by activating ERK1/2 and Akt while regulating c-Jun N-terminal kinases (JNKs) in a biphasic manner [90, 99, 115, 141, 163] (Fig. 3 and Table 5).
Xenon and cardioprotection
Xenon is regarded as a gaseous anesthetic; despite being costly and scarce, it offers several special benefits such as cytoprotectivity and quick diffusion, along with minimal hemodynamic side effects [38, 169].
The noble gas Xe induces cardioprotection whether applied before (PC-like effect) or after (PostC-like effect) an infarcting ischemia [50, 62, 160, 175, 219]. In particular, in pathological conditions, applying 20% Xe along with 34 ºC hypothermia during early reperfusion can also reduce the area of the MI in rats [175]. Regarding the cardioprotective mechanisms of Xe, it has been suggested that the mitoKATP channel and phosphatidylinositol-dependent kinase-1 are first activated by Xe. These two in turn activate PKC-ε, which in turn activates p38 MAPK. Two downstream targets of p38 MAPK, mitogen-activated protein kinase-activated protein kinase-2 and heat-shock protein 27 (HSP27), are then phosphorylated, which causes HSP27 to translocate to the particulate fraction and increases F-actin polymerization [217,218,219]. In addition to p38 MAPK, ERK1/2 and cyclooxygenase-2 (COX-2) are critical mediators of either Xe early preconditioning [216] or Xe induced late preconditioning [211]. Xenon can also cause GSK-3β- and Akt-phosphorylation, prevent Ca2+ from causing mPTP to open, and maintain mitochondrial function [120].
The minimal side effects of Xe are in line with the minimal effects on cardiac function. Differences were observed between global or regional administration of 50% or 70% Xe in dogs. Regional administration of Xe in the left anterior descending artery only reduced local myocardial contractility when Xe was given at 70% but did not affect global hemodynamics, coronary blood flow and regional myocardial function in the circumflex coronary artery-dependent myocardium [160]. In isolated guinea pig hearts, 40% or 80% of the Xe did not significantly change the NO-dependent flow response, the electrical, mechanical, or metabolic effects. This may be because Xe did not change the major cation currents in the guinea pig cardiomyocytes [196]. Xenon (20, 50, and 65%), in addition to basic intravenous anesthesia, has been demonstrated to cause a reduction in total hepatic O2 delivery and venous hepatic O2 saturation, as well as a downregulation of heart rate and cardiac output without altering mean arterial pressure or hepatic arterial blood flow [80, 205]. However, it has not been observed to impair intestinal oxygenation in pigs. Actually, pigs with hepatic venous blood that was supplemented with pentobarbital and buprenorphine received 73–78% Xe, which increased the amount of O2 in the blood [170]. These data suggest that the effects of Xe on heart rate and hepatic O2 levels may be influenced by concomitant intravenous anesthesia.
Since Xe is scarce, expensive, and difficult to administer (only by gaseous route), it is challenging to use Xe outside of controlled respiration and hospital conditions. In addition, it is unlikely to be applied to the management of acute and chronic heart conditions. Researchers have recently solidified Xe, enabling its oral or intravenous administration, by incorporating it into cyclodextrins, starch derivative excipients commonly used as drug carriers [162, 228]. For instance, the saturation point of Xe in water is 0.22 mM only. The solubility of Xe was increased from 0.22 to 0.67 mM when 2-hydroxypropyl-β-cyclodextrin was added as a cage molecule. Supplementing these xenon-enriched solutions by gavage decreased hypertension, left ventricular hypertrophy, and cardiac dysfunction in aged ApoE-knockout mice fed a high-fat diet for six weeks [228]. The acquired information provides opportunities for the creation of medications based on the lyophilized-cyclodextrin-xenon complex that are suitable for transportation, storage, and use in medicine, including outside of hospitals (Table 6).
Helium and cardioprotection
Helium does not have any anesthetic effects, thus it may be used in awake patients in situations involving ischemia–reperfusion, such as percutaneous coronary interventions for STEMI patients. As He has a low density, it helps patients with airway diseases breath by lowering their energy requirements. Since there are ventilators that enable the application of He via both invasive and non-invasive ventilation strategies, He may also be used in patients undergoing organ transplantation or during heart or vascular surgery. Helium therefore could be a great substitute for Xe or Ar in clinical ischemia/reperfusion scenario because it is a noble gas that is significantly less expensive and there are no known adverse effects of He on regional or global hemodynamics.
Over the last 25 years, several investigations have demonstrated that He has vital cytoprotective properties on endothelial cells [185,186,187], the brain [1, 23, 31, 56, 104, 107], the gut [36], the liver [232] and the heart [1, 45, 63, 76,77,78, 127,128,129,130, 141, 214].
Helium administration prior to ischemia, known as He preconditioning, exhibits a significant reduction in the infarct size in the model of MIRI, specifically observed in young rats but not in aged rats, and in Zucker lean rats but not in Zucker obese rats [63, 76, 77] (Table 7). The elicitation of helium-induced cardioprotection is associated with the activation of PI3K, MAPK/ERK1/2, p70S6 kinase, cyclic AMP-dependent protein kinase (PKA), COX-2, opioid receptors, mPTP opening, and NO production by eNOS [48, 76,77,78, 136,137,138,139,140,141, 215]. In addition, the threshold of helium-induced preconditioning was lowered in vivo by inhibiting GSK-3β or p53 through a mPTP-dependent mechanism [140].
Furthermore, He was cardioprotective when given after ischemia (He-PostC) in the Zucker lean rat or Male Wistar rat models of MIRI. These protective effects on rats are associated with upregulating autophagy-related genes, downregulating apoptosis-related genes [128], raising calcium channel, voltage-dependent, L type (alpha-1D subunit), caveolin-1 and caveolin-3 (Cav-1 and Cav-3) protein levels, and activating ERK1/2 and Akt [45, 77, 128, 215]. According to Smit et al. [186], He preserves post-ischemic endothelial function; eNOS blocking did not reverse this effect.
However, in male adult Wistar rats, a prolonged 30- or 60-min of 70% He dose during reperfusion does not cause cardioprotection [127]. Although the burst of inflammatory cytokines did not decrease, the prolonged inhalation of He may have contributed to the proinflammatory response by raising IL-1β and cytokine-induced neutrophil chemoattractant 3 in the myocardium of the at-risk area, but not in the not-at-risk area [127]. However, when considering the clinical application of noble gases, different approaches have been undertaken for Xe and He. In a study with healthy volunteers, utilizing a forearm blood flow model to explore endothelial function, He was found to alleviate post-ischemic endothelial dysfunction. This improvement occurred without any observed impact on plasma levels of cytokines, adhesion molecules, or microparticles [186]. Furthermore, a clinical study found that neither He-PC (3 × 5 min of 70% He and 30% O2 applied before aortic cross-clamping) nor the combination (15 min of He applied before aortic cross-clamp release and continued for 5 min after start of reperfusion;) had any effect on the activation of p38 MAPK, ERK1/2, or the levels of PKC-ε and HSP27 in the hearts of patients undergoing coronary artery bypass grafting (CABG) procedures. Moreover, in these patients, postoperative troponin release was unaffected by helium-pre- or helium-postconditioning [184].
For Xe, it was shown, that unlike hypothermia alone, the addition of Xe to hypothermia reduced myocardial damage in patients following out-of-hospital cardiac arrest and return of spontaneous circulation [8]. The combination of Xe with hypothermia was linked to a more substantial recovery of left ventricular systolic function compared to hypothermia alone. This suggests that Xe possesses cardioprotective properties in this clinical context [173].
However, a multinational, randomized clinical trial examined the cardioprotective effects of Xe anesthesia in patients undergoing CABG surgery, comparing it to anesthesia based on sevoflurane or propofol. Among the 492 patients who received either propofol, sevoflurane, or Xe, a decrease in troponin I release was observed in the Xe group compared to the propofol group, as well as in the sevoflurane group compared to the propofol group. However, the difference in troponin release was minimal, and it remains uncertain whether this observed effect holds clinical significance [71]. In line with these results, a study in patients with American Society of Anesthesiologists physical status III undergoing aortic surgery under either Xe or total intravenous anesthesia did not find significant differences in global myocardial performance, myocardial contractility or laboratory values between the groups [13]. Furthermore, in a randomized trial in 30 patients who underwent elective on-pump CABG receiving balanced anesthesia of either Xe or sevoflurane, Xe rather triggers proinflammatory effects and suppresses the anti-inflammatory response [17]. The clinical relevance of these findings, however, has not been determined in this study.
Taking these results into account, even though the noble gases can be easily applied in the clinical situation, their cardioprotective effects so far have not yet been strongly supported by larger randomized trials. Therefore, additional studies are necessary to clarify these discrepancies between experimental and clinical studies. For instance, the presence of a threshold in the hypertensive heart is suggested by the fact that, in spontaneous hypertensive rats, only a triple intervention of He-PostC can reduce MIRI, in contrast to the healthy Wistar Kyoto rats [129]. According to an in vitro study He-conditioning enhanced fibroblast migration, but not the release of extracellular vesicles of protective medium or soluble factors from the cardiac fibroblasts, contributed to cardioprotection [83]. According to a recent study, a dose-dependent improvement in lipopolysaccharide-induced left ventricular dysfunction and cavity enlargement can be obtained with intraperitoneal injection of 99.999% He; the optimal dose, in this study, was found to be 1.0 ml/100 g [234]. In terms of mechanisms, in this study, He decreased the phosphorylation of NF-κB, inhibited the expression of toll-like receptor 4 and then attenuated the expression of TNF-α and IL-18 (Table 7).
Interactions between gasotransmitters and noble gases
From the preceding discussion, it can be inferred that noble gases frequently act on the same targets as gasotransmitters, either directly or indirectly, leading to the induction of endogenous gas formation and signaling pathways activation. The synergistic interactions between gasotransmitters and noble gases in cardioprotection scenario have become a focal point of scientific exploration [6, 7, 9, 139], and recent studies have delved into the experimental intricacies surrounding their collective impact. Notably, investigations suggest that these gases protect endothelial cells [209, 211, 212] and collaboratively induce vasorelaxation within the cardiovascular system, bolstering their individual and synergistic cardioprotective effects. As seen above, this phenomenon has been examined in controlled experimental settings, employing various in vitro and in vivo models of MIRI. Experiments involving perfused hearts, isolated vessels, and animal models have provided valuable insights into the specific mechanisms through which the interplay of NO, H2S, CO, and noble gases—Ar, He, and Xe in particular—manifests as a shield against MIRI (Tables 1, 2, 3, 4, 5, 6, and 7) [209, 231].
As seen above, the metabolic alterations observed in cardiovascular system cells during chronic illnesses (comorbidities) and their response to hypoxia are intricately linked with the involvement of endogenous gasotransmitters as well as with exogenous gases, including noble gases. As concern gasotransmitters, we have mentioned that they are generated as part of metabolic processes. For instance, NO is produced during the breakdown of arginine to citrulline, H2S is formed during cysteine metabolism, and CO is a byproduct of heme metabolism. The reader is redirect to recent reviews for the discussion on how these metabolic processes contribute to cardioprotection, specifically regarding gasotransmitters in the context of comorbidities [5,6,7, 9, 148, 180, 239]. These gaseous molecules play pivotal roles in modulating vascular tone, promoting angiogenesis, and ensuring cellular survival against hypoxic conditions and oxidative stress induced by IR. As seen, their effects are mediated via the modulation of signaling pathways that influence mitochondrial function and metabolism, and the activity of key regulators of intracellular processes, including but not limited to PKG, PI3K, MAPK, JNK1/2, sGC, cGMP, NF-κB, HIF-1α, as well as ion channels such as mitoKATP channels and large conductance calcium-activated potassium (BKCa) channels (Figs. 2 and 3). Indeed, BKCa channels are paradigmatic in elucidating the interaction and cardiovascular protective effects of gasotransmitters. Gases activating BKCa channels may confer protective effects and reduce vascular resistance during episodes of hypoxia and MIRI [180]. BKCa channels are target of H2S on cardiomyocytes under hypoxic conditions as well as upon endogenous CO stimulation of cardiomyocytes. Carbon monoxide modulates the function of these channels by interacting with reduced heme. Nitric oxide triggers the activation of BKCa channels indirectly through pathways associated with PKG and PKA signaling. Cellular functions related to Ca2+ storage and energy synthesis heavily rely on the capacity of these channels across cell and organelle membranes. Indeed, effective strategies to mitigate MIRI include activating energetic metabolic pathways such as glycolysis, glucose and ketone oxidation, as well as hexosamine biosynthesis, and deacetylation. Yet, protection can be achieved by reducing the activity of the malate-aspartate shuttle, mitochondrial O2 consumption, fatty acid oxidation, and mitochondrial succinate metabolism, requiring novel reversible and specific inhibitors. Currently, the most promising metabolic therapy for MIRI seems to involve targeting glycolysis, O-GlcNAcylation, and the metabolism of ketones, fatty acids, and succinate, either individually or in combination [239]. We have reported above the impact of gases on mitochondrial function and O2 consumption. Moreover, evidence suggests that their involvement in cellular metabolism extends to regulating energetic substrate utilization [131, 146, 169]. For instance, tyrosine residue nitration in mitochondrial key proteins contributes to regulate mitochondrial and cellular function after MIRI [239]. Consequently, the combination of gasotransmitters holds the potential to modulate metabolism, mitigate mitochondrial damage, and ultimately reduce MIRI. The crosstalk between gasotransmitters and noble gases has been probed at the molecular level shedding light on potential molecular targets for advanced cardioprotective strategies [181, 188, 213]. Controlled studies utilizing gasotransmitter-releasing compounds and precise administration protocols have demonstrated the therapeutic promise of these molecules in mitigating MIRI [82, 133]. Similarly, experiments employing various delivery methods for noble gases, including inhalation and controlled release, have showcased their organ-protective properties with a focus on detailed dose–response relationships and pharmacokinetics [227].
In conclusion, the ongoing research in the intricate interplay between gasotransmitters (NO, H2S, SO2, CO) and noble gases (Ar, He, Xe) paves the way for novel therapeutic strategies, underlining the potential of these molecular shields against the global health challenge of ischemic heart disease.
Nanodevices to deliver gases
Nanoparticle-based drug delivery has gained significant popularity as a strategy to optimize the therapeutic potential of drugs. Despite notable improvements, formulating gases presents unique challenges not encountered with liquid and solid active ingredients. The challenges and failures of gases-oriented therapies in the field of cardioprotection have been recently and extensively reviewed [6, 7, 9, 146]. In brief, drug formulation and tolerance as well as comedications and comorbidities may affect the outcomes [44, 92]. Specifically, in comorbidities, elevated production of ROS/RNS can influence cardiac gases-dependent signaling and induce adaptive changes in the expression and activity of enzymes producing gases as well as antioxidant enzymes. Initially, these changes may serve as protective mechanisms in metabolic syndrome and diabetes [148]. However, prolonged exposure to oxidative, nitrosative, and nitrative stress can deplete these protective mechanisms, leading to increased ROS/RNS production and reduced gases bioavailability (especially for NO and H2S) in the myocardium. Furthermore, heightened oxidative and nitrosative stress can impair the NO-sGC signaling pathway, limiting the ability of NO to perform its essential signaling functions in the heart. The upregulation of ROS/RNS production in the presence of risk factors also promotes the activation of redox-dependent transcription factors like NF-κB, which stimulates the expression of various proinflammatory mediators, contributing to the development of cardiac dysfunction and remodeling. The dysregulation of gases-dependent signaling may affect the therapeutic effectiveness of conventional drugs used in managing metabolic syndrome, hypertension and diabetes. Conversely, modulation of gases-dependent signaling could underlie the therapeutic benefits of established and newly developed treatment approaches, such as ACE inhibitors, specific β-blockers, and sGC activators as well as NO or H2S donors conjugated with other pharmacological agents such as non-steroidal anti-inflammatory drugs [6, 7, 9, 114, 146] A deeper understanding of these pathological processes and pharmacology of gas-enhancers holds promise for developing more effective therapeutic strategies to combat comorbidities and its cardiac consequences related to gases-dependent signaling. Gas molecules or noble gases, released from formulations for therapeutic purposes, have been studied, although insufficiently. Gasses can be modified into prodrugs called gas-releasing molecules (GRMs) and subsequently released from these GRMs. For instance, the development of CORMs stemmed from the need for compounds capable of transporting and releasing controlled amounts of toxic CO within cellular systems. This approach offered a promising avenue for studying the pharmacological effects of the gas and elucidating its mechanisms of action [82, 133, 157]. Extensive coverage is given to various nanosystems and their roles in efficient shuttling, targeting, and release of therapeutic gases. Diverse ways have been proposed in which GRM prodrugs, in delivery nanosystems, respond to intrinsic and extrinsic stimuli for sustained release, offering a comprehensive overview of their design and application in nanomedicine [133]. Considerable investigation has been conducted into the utilization of inorganic materials for the loading and delivery of drugs. These include, photothermal agents [100, 102, 233], photon‐to‐photon conversion agents such as lanthanide [237], catalytic metal ionic species [111], photocatalytic nanomaterials [210, 236] and polymer‐based nanomaterials [162, 176, 224, 228], to name only a few used for gas-loading in different pathological conditions (Table 8). In a related context, recent developments introduce two types of O2-loaded nanodevices (ND) based on alpha-cyclodextrin and cyclic nigerosyl-nigerose. These NDs demonstrate protection against hypoxia/reperfusion-induced cell death in vitro, serving as biocompatible and biodegradable drug carriers for simultaneous O2 and therapeutic delivery [43, 150]. Advances in solidifying Xe within cyclodextrins make oral or intravenous administration feasible [162, 228]. Hence, O2- NO- and/or Xe-loaded NDs, conjugated with an ischemic myocardium-targeting peptide (that is protein/peptide‐modified materials), could efficiently deliver to the infarcted heart. This methodology involves synthesizing ND formulations that integrate O2, gasotransmitters, and/or specific noble gases, achieved through thoughtful modifications around the glucose ring to enhance efficiency. In addition, lipid nanoparticles have proven effective in drug delivery and play a significant role in controlling the release and diffusion of gases within biological tissues in gasotransmitter applications [133]. Several stimuli-responsive GRMs have been tested for therapeutic gas release, emphasizing the importance of efficient and targeted delivery at diseased sites. Intrinsic stimuli, such as pH, offer high biocompatibility but demonstrate weaker driving forces for gas release compared to external stimuli such as magnetic, ultrasound, and near-infrared light (NIR)-triggers. Controllable external stimuli generally exhibit high release efficiency and specificity. Combining intrinsic and external stimuli can enhance responsiveness. Critical challenges for intrinsic stimuli include the need for improved sensitivity. External stimuli, such magnetic, ultrasound, and NIR triggers due to their superior characteristics might be preferred (Table 8). However, external stimuli may cause tissue irritation at high powers [133].
While there have been a few clinical trials conducted on the gasotransmitter H2S (refer to Table 1 in [9]), a multitude of studies have explored NO donors, nitrates, and nitrites, which are beyond the scope of this review. In particular, evidence from clinical practice indicates that H2S may produce inconclusive outcomes in patients with cardiovascular diseases. A clinical trial investigating the impact of Na2S on patients with acute STEMI was withdrawn by the researchers (NCT01007461), while another study, known as the Groningen Intervention study for the Preservation of cardiac function with sodium thiosulfate after STEMI (GIPS-IV; NCT02899364), demonstrated neutral findings [30]. To our knowledge, CO is often studied as toxic agent and only one trial has been conducted thus far involving CO in patients with stable chronic obstructive pulmonary disease (NCT00122694). Overall, further experimentation is crucial to advance the GRMs and/or stimuli for potential clinical applications in gasotransmitter and noble gases research. Nevertheless, the synthesis and testing of these formulations are deemed essential, particularly in the cardiovascular field and relevant myocardial infarct models, providing a promising avenue for future clinical applications, recognizing the imperative of adopting a multitarget approach in addressing MIRI [29]. This strategy underscores the potential efficacy of exploiting gaseous diffusibility as a readily attainable means of achieving multifaceted therapeutic goals.
Conclusions
In summary, we highlight the diverse yet interconnected roles of gasotransmitters and noble gases in modulating molecular pathways to promote cell survival and overall tissue health. While gasotransmitters such as NO and H2S, produced by various cell types, contribute significantly to various physiological processes, noble gases such as He and Xe emerge as promising candidates for organ protection. Their ability to modulate molecular pathways and promote cell survival underscores their cardioprotective potential. However, collaborative efforts between experimental and clinical researchers are essential to bridge the gap between laboratory findings and real-world applications. Prospective studies may focus on establishing optimal dosages, timings, and combination therapies involving both noble gases and gasotransmitter donor molecules. Integrating advanced imaging techniques, molecular profiling, and systems biology approaches will contribute to a comprehensive understanding of the intricate molecular pathways involved. Of course, it is crucial to acknowledge the influence of cardiovascular disease, confounders and risk factors on endogenous cardioprotective mechanisms. Here, we highlight the need to address the impact of cardiovascular comorbidities on the regulation of gasotransmitters and noble gases, as well as the challenges associated with the clinical application of their cardioprotective potential [5, 44, 154, 184, 187]. Moving forward, incorporating information on the effects of cardiovascular risk factors on individual gasotransmitters and exploring new avenues for therapeutic strategies will be essential for advancing the field and optimizing patient outcomes. Research in various areas has emphasized the significance of interactions among gasses, suggesting the need for integrated investigation into their role in MIRI and cardioprotection. Gasses influenced pathways intersect in common downstream effectors during processes such as cardioprotection, vasodilation and angiogenesis. Therefore, effective therapeutic approaches may require the utilization of multiple gases or their pharmacological modulators, considering also their potential in affecting cell metabolism [9, 131, 146, 169, 239]. This multidimensional approach is crucial for developing more effective and targeted cardioprotective strategies.
Data availability
This is a review article, and therefore, there are no original data to be made available.
References
Aehling C, Weber NC, Zuurbier CJ, Preckel B, Galmbacher R, Stefan K, Hollmann MW, Popp E, Knapp J (2018) Effects of combined helium pre/post-conditioning on the brain and heart in a rat resuscitation model. Acta Anaesthesiol Scand 62:63–74. https://doi.org/10.1111/aas.13041
Akizuki E, Akaike T, Okamoto S, Fujii S, Yamaguchi Y, Ogawa M, Maeda H (2000) Role of nitric oxide and superoxide in acute cardiac allograft rejection in rats. Proc Soc Exp Biol Med 225:151–159. https://doi.org/10.1046/j.1525-1373.2000.22519.x
Andrabi SM, Sharma NS, Karan A, Shahriar SMS, Cordon B, Ma B, Xie J (2023) Nitric oxide: physiological functions, delivery, and biomedical applications. Adv Sci 10:e2303259. https://doi.org/10.1002/advs.202303259
Andreadou I, Cabrera-Fuentes HA, Devaux Y, Frangogiannis NG, Frantz S, Guzik T, Liehn EA, Gomes CPC, Schulz R, Hausenloy DJ (2019) Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res 115:1117–1130. https://doi.org/10.1093/cvr/cvz050
Andreadou I, Daiber A, Baxter GF, Brizzi MF, Di Lisa F, Kaludercic N, Lazou A, Varga ZV, Zuurbier CJ, Schulz R, Ferdinandy P (2021) Influence of cardiometabolic comorbidities on myocardial function, infarction, and cardioprotection: role of cardiac redox signaling. Free Radic Biol Med 166:33–52. https://doi.org/10.1016/j.freeradbiomed.2021.02.012
Andreadou I, Iliodromitis EK, Rassaf T, Schulz R, Papapetropoulos A, Ferdinandy P (2015) The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br J Pharmacol 172:1587–1606. https://doi.org/10.1111/bph.12811
Andreadou I, Schulz R, Papapetropoulos A, Turan B, Ytrehus K, Ferdinandy P, Daiber A, Di Lisa F (2020) The role of mitochondrial reactive oxygen species, NO and H2S in ischaemia/reperfusion injury and cardioprotection. J Cell Mol Med 24:6510–6522. https://doi.org/10.1111/jcmm.15279
Arola O, Saraste A, Laitio R, Airaksinen J, Hynninen M, Bäcklund M, Ylikoski E, Wennervirta J, Pietilä M, Roine RO, Harjola VP, Niiranen J, Korpi K, Varpula M, Scheinin H, Maze M, Vahlberg T, Laitio T, Xe-HYPOTHECA Study Group (2017) Inhaled xenon attenuates myocardial damage in comatose survivors of out-of-hospital cardiac arrest: the xe-hypotheca trial. J Am Coll Cardiol 70:2652–2660. https://doi.org/10.1016/j.jacc.2017.09.1088
Arrigo E, Comità S, Pagliaro P, Penna C, Mancardi D (2023) Clinical applications for gasotransmitters in the cardiovascular system: are we there yet? Int J Mol Sci 24:12480. https://doi.org/10.3390/ijms241512480
Babchin A, Levich E, Melamed MDY, Sivashinsky G (2011) Osmotic phenomena in application for hyperbaric oxygen treatment. Colloids Surf B Biointerfaces 83:128–132. https://doi.org/10.1016/j.colsurfb.2010.11.019
Bantel C, Maze M, Trapp S (2009) Neuronal preconditioning by inhalational anesthetics: evidence for the role of plasmalemmal adenosine triphosphate-sensitive potassium channels. Anesthesiology 110:986–995. https://doi.org/10.1097/ALN.0b013e31819dadc7
Behmenburg F, van Caster P, Bunte S, Brandenburger T, Heinen A, Hollmann MW, Huhn R (2018) Impact of anesthetic regimen on remote ischemic preconditioning in the rat heart in vivo. Anesth Analg 126:1377–1380. https://doi.org/10.1213/ANE.0000000000002563
Bein B, Turowski P, Renner J, Hanss R, Steinfath M, Scholz J, Tonner PH (2005) Comparison of xenon-based anaesthesia compared with total intravenous anaesthesia in high risk surgical patients. Anaesthesia 60:960–967. https://doi.org/10.1111/j.1365-2044.2005.04326.x.Erratum.In:Anaesthesia.201469:653
Bełtowski J, Jamroz-Wiśniewska A (2014) Hydrogen sulfide and endothelium-dependent vasorelaxation. Molecules 19:21183–21199. https://doi.org/10.3390/molecules191221183
Bencsik P, Kupai K, Giricz Z, Görbe A, Huliák I, Fürst S, Dux L, Csont T, Jancsó G, Ferdinandy P (2008) Cardiac capsaicin-sensitive sensory nerves regulate myocardial relaxation via S-nitrosylation of SERCA: role of peroxynitrite. Br J Pharmacol 153:488–496. https://doi.org/10.1038/sj.bjp.0707599
Bonello S, Zähringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Görlach A (2007) Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol 27:755–761. https://doi.org/10.1161/01.ATV.0000258979.92828.bc
Breuer T, Emontzpohl C, Coburn M, Benstoem C, Rossaint R, Marx G, Schälte G, Bernhagen J, Bruells CS, Goetzenich A, Stoppe C (2015) Xenon triggers pro-inflammatory effects and suppresses the anti-inflammatory response compared to sevoflurane in patients undergoing cardiac surgery. Crit Care 19:365. https://doi.org/10.1186/s13054-015-1082-7
Cao X, Ding L, Xie ZZ, Yang Y, Whiteman M, Moore PK, Bian JS (2019) A review of hydrogen sulfide synthesis, metabolism, and measurement: is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid Redox Signal 31:1–38. https://doi.org/10.1089/ars.2017.7058
Chen B, Guo L, Fan C, Bolisetty S, Joseph R, Wright MM, Agarwal A, George JF (2009) Carbon monoxide rescues heme oxygenase-1-deficient mice from arterial thrombosis in allogeneic aortic transplantation. Am J Pathol 175:422–429. https://doi.org/10.2353/ajpath.2009.081033
Chen Y, Zhang F, Yin J, Wu S, Zhou X (2020) Protective mechanisms of hydrogen sulfide in myocardial ischemia. J Cell Physiol 235:9059–9070. https://doi.org/10.1002/jcp.29761
Choi YK, Park JH, Baek YY, Won MH, Jeoung D, Lee H, Ha KS, Kwon YG, Kim YM (2016) Carbon monoxide stimulates astrocytic mitochondrial biogenesis via L-type Ca(2+) channel-mediated PGC-1alpha/ERRalpha activation. Biochem biophysical Res Commun 479:297–304. https://doi.org/10.1016/j.bbrc.2016.09.063
Chu LM, Shaefi S, Byrne JD, Alves de Souza RW, Otterbein LE (2021) Carbon monoxide and a change of heart. Redox Biol 48:102183. https://doi.org/10.1016/j.redox.2021.102183
Coburn M, Maze M, Franks NP (2008) The neuroprotective effects of xenon and helium in an in vitro model of traumatic brain injury. Crit Care Med 36:588–595. https://doi.org/10.1097/01.CCM.0B013E3181611F8A6
Cohen MV, Downey JM (2015) Signaling pathways and mechanisms of protection in pre- and postconditioning: historical perspective and lessons for the future. Br J Pharmacol 172:1913–1932. https://doi.org/10.1111/bph.12903
Colloc’h N, Sopkova-de Oliveira Santos J, Retailleau P, Vivarès D, Bonneté F, Langlois d’Estainto B, Gallois B, Brisson A, Risso JJ, Lemaire M, Prangé T, Abraini JH (2007) Protein crystallography under xenon and nitrous oxide pressure: comparison with in vivo pharmacology studies and implications for the mechanism of inhaled anesthetic action. Biophys J 92:217–224. https://doi.org/10.1529/biophysj.106.093807
Csonka C, Páli T, Bencsik P, Görbe A, Ferdinandy P, Csont T (2015) Measurement of NO in biological samples. Br J Pharmacol 172:1620–1632. https://doi.org/10.1111/bph.12832
Cullen SC, Gross EG (1951) The anesthetic properties of xenon in animals and human beings, with additional observations on krypton. Science 113:580–582. https://doi.org/10.1126/science.113.2942.580
Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, Bers DM, Mohler PJ, Ziolo MT, Shannon TR (2014) Nitric oxide-dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS ONE 9:e87495. https://doi.org/10.1371/journal.pone.0087495
Davidson SM, Ferdinandy P, Andreadou I, Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, Garcia-Dorado D, CARDIOPROTECTION COST Action (CA16225) (2019) Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol 73:89–99. https://doi.org/10.1016/j.jacc.2018.09.086
de Koning MLY, van Dorp P, Assa S, Pundziute-Do Prado G, Voskuil M, Anthonio RL, Veen D, Leiner T, Sibeijn-Kuiper AJ, van Goor H, van Veldhuisen DJ, van der Meer P, Nijveldt R, Lipšic E, van der Harst P (2023) Sodium thiosulfate in acute myocardial infarction: a randomized clinical trial. JACC Basic Transl Sci 8:1285–1294. https://doi.org/10.1016/j.jacbts.2023.06.001
Deng RM, Li HY, Li X, Shen HT, Wu DG, Wang Z, Chen G (2021) Neuroprotective effect of helium after neonatal hypoxic ischemia: a narrative review. Med Gas Res 11:121–123. https://doi.org/10.4103/2045-9912.314332
Dickinson R, Franks NP (2010) Bench-to-bedside review: Molecular pharmacology and clinical use of inert gases in anesthesia and neuroprotection. Crit Care 14:229. https://doi.org/10.1186/cc9051
Dickinson R, Peterson BK, Banks P, Simillis C, Martin JC, Valenzuela CA, Maze M, Franks NP (2007) Competitive inhibition at the glycine site of the n-methyl-d-aspartate receptor by the anesthetics xenon and isoflurane: evidence from molecular modeling and electrophysiology. Anesthesiology 107:756–767. https://doi.org/10.1097/01.anes.0000287061.77674.71
Dongaonkar RM, Stewart RH, Geissler HJ, Laine GA (2010) Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc Res 87:331–339. https://doi.org/10.1093/cvr/cvq145
Dries E, Santiago DJ, Johnson DM, Gilbert G, Holemans P, Korte SM, Roderick HL, Sipido KR (2016) Calcium/calmodulin-dependent kinase II and nitric oxide synthase 1-dependent modulation of ryanodine receptors during β-adrenergic stimulation is restricted to the dyadic cleft. J Physiol 594:5923–5939. https://doi.org/10.1113/JP271965
Du L, Zhang R, Luo T, Nie M, Bi J (2015) Effects of helium preconditioning on intestinal ischemia and reperfusion injury in rats. Shock 44:365–370. https://doi.org/10.1097/SHK.0000000000000418
Du SX, Jin HF, Bu DF, Zhao X, Geng B, Tang CS, Du JB (2008) Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta Pharmacol Sin 29:923–930. https://doi.org/10.1111/j.1745-7254.2008.00845.x
Dworschak M (2008) Pharmacologic neuroprotection–is xenon the light at the end of the tunnel? Crit Care Med 36:2477–2479. https://doi.org/10.1097/CCM.0b013e31818113d2
Fang J, Wei Z, Zheng D, Ying T, Hong H, Hu D, Lin Y, Jiang X, Wu L, Lan T, Yang Z, Zhou X, Chen L (2020) Recombinant extracellular domain (p75ECD) of the neurotrophin receptor p75 attenuates myocardial ischemia-reperfusion injury by inhibiting the p-JNK/caspase-3 signaling pathway in rat microvascular pericytes. J Am Heart Assoc 9:e016047. https://doi.org/10.1161/JAHA.119.016047
Farah C, Kleindienst A, Bolea G, Meyer G, Gayrard S, Geny B, Obert P, Cazorla O, Tanguy S, Reboul C (2013) Exercise-induced cardioprotection: a role for eNOS uncoupling and NO metabolites. Basic Res Cardiol 108:389. https://doi.org/10.1007/s00395-013-0389-2
Farah C, Nascimento A, Bolea G, Meyer G, Gayrard S, Lacampagne A, Cazorla O, Reboul C (2017) Key role of endothelium in the eNOS-dependent cardioprotection with exercise training. J Mol Cell Cardiol 102:26–30. https://doi.org/10.1016/j.yjmcc.2016.11.008
Fasipe B, Li S, Laher I (2023) Exercise and vascular function in sedentary lifestyles in humans. Pflugers Arch 475:845–856. https://doi.org/10.1007/s00424-023-02828-6
Femminò S, Penna C, Bessone F, Caldera F, Dhakar N, Cau D, Pagliaro P, Cavalli R, Trotta F (2018) α-Cyclodextrin and α-cyclodextrin polymers as oxygen nanocarriers to limit hypoxia/reoxygenation injury: implications from an in vitro model. Polymers 10:211. https://doi.org/10.3390/polym10020211
Ferdinandy P, Andreadou I, Baxter GF, Bøtker HE, Davidson SM, Dobrev D, Gersh BJ, Heusch G, Lecour S, Ruiz-Meana M, Zuurbier CJ, Hausenloy DJ, Schulz R (2023) Interaction of cardiovascular nonmodifiable risk factors, comorbidities and comedications with ischemia/reperfusion injury and cardioprotection by pharmacological treatments and ischemic conditioning. Pharmacol Rev 75:159–216. https://doi.org/10.1124/pharmrev.121.000348
Flick M, Albrecht M, Oei GTML, Steenstra R, Kerindongo RP, Zuurbier CJ, Patel HH, Hollmann MW, Preckel B, Weber NC (2016) Helium postconditioning regulates expression of caveolin-1 and -3 and induces RISK pathway activation after ischaemia/reperfusion in cardiac tissue of rats. Eur J Pharmacol 791:718–725. https://doi.org/10.1016/j.ejphar.2016.10.012
Frankenreiter S, Groneberg D, Kuret A, Krieg T, Ruth P, Friebe A, Lukowski R (2018) Cardioprotection by ischemic postconditioning and cyclic guanosine monophosphate-elevating agents involves cardiomyocyte nitric oxide-sensitive guanylyl cyclase. Cardiovasc Res 114:822–829. https://doi.org/10.1093/cvr/cvy039
Franks NP, Dickinson R, de Sousa SL, Hall AC, Lieb WR (1998) How does xenon produce anaesthesia? Nature 396:324. https://doi.org/10.1038/24525
Garcia-Dorado D, Andres-Villarreal M, Ruiz-Meana M, Inserte J, Barba I (2012) Myocardial edema: a translational view. J Mol Cell Cardiol 52:931–939. https://doi.org/10.1016/j.yjmcc.2012.01.010
Ghimire K, Altmann HM, Straub AC, Isenberg JS (2017) Nitric oxide: what’s new to NO? Am J Physiol Cell Physiol 312:C254–C262. https://doi.org/10.1152/ajpcell.00315.2016
Goetzenich A, Hatam N, Preuss S, Moza A, Bleilevens C, Roehl AB, Autschbach R, Bernhagen J, Stoppe C (2014) The role of hypoxia-inducible factor-1α and vascular endothelial growth factor in late-phase preconditioning with xenon, isoflurane and levosimendan in rat cardiomyocytes. Interact Cardiovasc Thorac Surg 18:321–328. https://doi.org/10.1093/icvts/ivt450
Grau M, Kollikowski A, Bloch W (2016) Remote ischemia preconditioning increases red blood cell deformability through red blood cell-nitric oxide synthase activation. Clin Hemorheol Microcirc 63:185–197. https://doi.org/10.3233/CH-152039
Grau M, Seeger B, Mozigemba L, Roth R, Baumgartner L, Predel HG, Bloch W, Tomschi F (2022) Effects of recurring IPC vs. rIPC maneuvers on exercise performance, pulse wave velocity, and red blood cell deformability: special consideration of reflow varieties. Biology 11:163. https://doi.org/10.3390/biology11020163
Grilli A, De Lutiis MA, Patruno A, Speranza L, Gizzi F, Taccardi AA, Di Napoli P, De Caterina R, Conti P, Felaco M (2003) Inducible nitric oxide synthase and heme oxygenase-1 in rat heart: direct effect of chronic exposure to hypoxia. Ann Clin Lab Sci 33:208–215
Gruss M, Bushell TJ, Bright DP, Lieb WR, Mathie A, Franks NP (2004) Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol Pharmacol 65:443–452. https://doi.org/10.1124/mol.65.2.443
Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J, Niggli E (2013) NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc Res 100:392–401. https://doi.org/10.1093/cvr/cvt201
Haelewyn B, David HN, Blatteau JE, Vallée N, Meckler C, Risso JJ, Abraini JH (2016) Modulation by the noble gas helium of tissue plasminogen activator: effects in a rat model of thromboembolic stroke. Crit Care Med 44:e383-389. https://doi.org/10.1097/CCM.0000000000001424
Halligan KE, Jourd’heuil FL, Jourd’heuil D (2009) Cytoglobin is expressed in the vasculature and regulates cell respiration and proliferation via nitric oxide dioxygenation. J Biol Chem 284:8539–8547. https://doi.org/10.1074/jbc.M808231200
Hamilton DJ, Zhang A, Li S, Cao TN, Smith JA, Vedula I, Cordero-Reyes AM, Youker KA, Torre-Amione G, Gupte AA (2016) Combination of angiotensin II and l-NG-nitroarginine methyl ester exacerbates mitochondrial dysfunction and oxidative stress to cause heart failure. Am J Physiol Heart Circ Physiol 310:H667–H680. https://doi.org/10.1152/ajpheart.00746.2015
Harris K, Armstrong SP, Campos-Pires R, Kiru L, Franks NP, Dickinson R (2013) Neuroprotection against traumatic brain injury by xenon, but not argon, is mediated by inhibition at the n-methyl-d-aspartate receptor glycine site. Anesthesiology 119:1137–1148. https://doi.org/10.1097/ALN.0b013e3182a2a265
Hausenloy DJ, Lecour S, Yellon DM (2011) Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid Redox Signal 14:893–907. https://doi.org/10.1089/ars.2010.3360
Hausenloy DJ, Ong SB, Yellon DM (2009) The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res Cardiol 104:189–202. https://doi.org/10.1007/s00395-009-0010-x
Hein M, Roehl AB, Baumert JH, Bantes B, Bleilevens C, Bernstein N, Steendijk P, Rossaint R (2008) Establishment of a porcine right ventricular infarction model for cardioprotective actions of xenon and isoflurane. Acta Anaesthesiol Scand 52:1194–1203. https://doi.org/10.1111/j.1399-6576.2008.01757.x
Heinen A, Huhn R, Smeele KM, Zuurbier CJ, Schlack W, Preckel B, Weber NC, Hollmann MW (2008) Helium-induced preconditioning in young and old rat heart: impact of mitochondrial Ca(2+) -sensitive potassium channel activation. Anesthesiology 109:830–836. https://doi.org/10.1097/ALN.0b013e3181895aa0
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. https://doi.org/10.1161/CIRCRESAHA.116.305348
Heusch G (2016) The coronary circulation as a target of cardioprotection. Circ Res 118:1643–1658. https://doi.org/10.1161/CIRCRESAHA.116.308640
Heusch G (2019) Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res Cardiol 114:45. https://doi.org/10.1007/s00395-019-0756-8
Heusch G (2020) Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 17:773–789. https://doi.org/10.1038/s41569-020-0403-y
Heusch G (2024) Myocardial ischemia/reperfusion: Translational pathophysiology of ischemic heart disease. Med 5:10–31. https://doi.org/10.1016/j.medj.2023.12.007
Heusch G, Andreadou I, Bell R, Bertero E, Botker HE, Davidson SM, Downey J, Eaton P, Ferdinandy P, Gersh BJ, Giacca M, Hausenloy DJ, Ibanez B, Krieg T, Maack C, Schulz R, Sellke F, Shah AM, Thiele H, Yellon DM, Di Lisa F (2023) Health position paper and redox perspectives on reactive oxygen species as signals and targets of cardioprotection. Redox Biol 67:102894. https://doi.org/10.1016/j.redox.2023.102894
Heusch G, Post H, Michel MC, Kelm M, Schulz R (2000) Endogenous nitric oxide and myocardial adaptation to ischemia. Circ Res 87:146–152. https://doi.org/10.1161/01.res.87.2.146
Hofland J, Ouattara A, Fellahi JL, Gruenewald M, Hazebroucq J, Ecoffey C, Joseph P, Heringlake M, Steib A, Coburn M, Amour J, Rozec B, Liefde I, Meybohm P, Preckel B, Hanouz JL, Tritapepe L, Tonner P, Benhaoua H, Roesner JP, Bein B, Hanouz L, Tenbrinck R, Bogers AJJC, Mik BG, Coiffic A, Renner J, Steinfath M, Francksen H, Broch O, Haneya A, Schaller M, Guinet P, Daviet L, Brianchon C, Rosier S, Lehot JJ, Paarmann H, Schön J, Hanke T, Ettel J, Olsson S, Klotz S, Samet A, Laurinenas G, Thibaud A, Cristinar M, Collanges O, Levy F, Rossaint R, Stevanovic A, Schaelte G, Stoppe C, Hamou NA, Hariri S, Quessard A, Carillion A, Morin H, Silleran J, Robert D, Crouzet AS, Zacharowski K, Reyher C, Iken S, Weber NC, Hollmann M, Eberl S, Carriero G, Collacchi D, Di Persio A, Fourcade O, Bergt S, Alms A, Xenon-CABG Study Group (2017) Effect of xenon anesthesia compared to sevoflurane and total intravenous anesthesia for coronary artery bypass graft surgery on postoperative cardiac troponin release: an international, multicenter, phase 3, single-blinded, randomized noninferiority trial. Anesthesiology 127:918–933. https://doi.org/10.1097/ALN.0000000000001873
Hollig A, Coburn M (2021) Noble gases and neuroprotection: summary of current evidence. Curr Opin Anaesthesiol 34:603–606. https://doi.org/10.1097/ACO.0000000000001033
Huang P, Sun Y, Yang J, Chen S, Liu AD, Holmberg L, Huang X, Tang C, Du J, Jin H (2013) The ERK1/2 signaling pathway is involved in sulfur dioxide preconditioning-induced protection against cardiac dysfunction in isolated perfused rat heart subjected to myocardial ischemia/reperfusion. Int J Mol Sci 14:22190–22201. https://doi.org/10.3390/ijms141122190
Huang Y, Tang C, Du J, Jin H (2016) Endogenous sulfur dioxide: a new member of gasotransmitter family in the cardiovascular system. Oxid Med Cell Longev 2016:8961951. https://doi.org/10.1155/2016/8961951
Huang Y, Zhang H, Lv B, Tang C, Du J, Jin H (2022) Sulfur dioxide: endogenous generation, biological effects, detection, and therapeutic potential. Antioxid Redox Signal 36:256–274. https://doi.org/10.1089/ars.2021.0213
Huhn R, Heinen A, Weber NC, Hieber S, Hollmann MW, Schlack W, Preckel B (2009) Helium-induced late preconditioning in the rat heart in vivo. Br J Anaesth 102:614–619. https://doi.org/10.1093/bja/aep042
Huhn R, Heinen A, Weber NC, Kerindongo RP, Oei GT, Hollmann MW, Schlack W, Preckel B (2009) Helium-induced early preconditioning and postconditioning are abolished in obese Zucker rats in vivo. J Pharmacol Exp Ther 329:600–607. https://doi.org/10.1124/jpet.108.149971
Huhn R, Weber NC, Preckel B, Schlack W, Bauer I, Hollmann MW, Heinen A (2012) Age-related loss of cardiac preconditioning: impact of protein kinase A. Exp Gerontol 47:116–121. https://doi.org/10.1016/j.exger.2011.11.003
Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B, Joseph R, Prabhu SD, Suliman HB, Piantadosi CA, Agarwal A, George JF (2016) Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 1:e85817. https://doi.org/10.1172/jci.insight.85817
Iber T, Hecker K, Vagts DA, Roesner JP, Otto B, Steinicke A, Nöldge-Schomburg GF, Rossaint R (2008) Xenon anesthesia impairs hepatic oxygenation and perfusion in healthy pigs. Minerva Anestesiol 74:511–519
Inserte J, Hernando V, Vilardosa Ú, Abad E, Poncelas-Nozal M, Garcia-Dorado D (2013) Activation of cGMP/protein kinase G pathway in postconditioned myocardium depends on reduced oxidative stress and preserved endothelial nitric oxide synthase coupling. J Am Heart Assoc 2:e005975. https://doi.org/10.1161/JAHA.112.005975
Iqbal J, Chamberlain J, Alfaidi M, Hughes M, Alizadeh T, Casbolt H, Evans P, Mann B, Motterlini R, Francis S, Gunn J (2021) Carbon monoxide releasing molecule A1 reduces myocardial damage after acute myocardial infarction in a porcine model. J Cardiovasc Pharmacol 78:e656–e661. https://doi.org/10.1097/FJC.0000000000001067
Jelemenský M, Kovácsházi C, Ferenczyová K, Hofbauerová M, Kiss B, Pállinger É, Kittel Á, Sayour VN, Görbe A, Pelyhe C, Hambalkó S, Kindernay L, Barančík M, Ferdinandy P, Barteková M, Giricz Z (2021) Helium conditioning increases cardiac fibroblast migration which effect is not propagated via soluble factors or extracellular vesicles. Int J Mol Sci 22:10504. https://doi.org/10.3390/ijms221910504
Jin H, Liu AD, Holmberg L, Zhao M, Chen S, Yang J, Sun Y, Chen S, Tang C, Du J (2013) The role of sulfur dioxide in the regulation of mitochondrion-related cardiomyocyte apoptosis in rats with isopropylarterenol-induced myocardial injury. Int J Mol Sci 14:10465–10482. https://doi.org/10.3390/ijms140510465
Jones SP, Bolli R (2006) The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol 40:16–23. https://doi.org/10.1016/j.yjmcc.2005.09.011
Kaluza GL, Creech JL, Furer A, Afari ME, Milewski K, Yi GH, Cheng Y, Conditt GB, McGregor JC, Blum D, Rousselle SD, Granada JF, Burkhoff D (2022) Chronic myocardial and coronary arterial effects of intracoronary supersaturated oxygen therapy in swine with normal and ischemic-reperfused myocardium. Sci Rep 12:5785. https://doi.org/10.1038/s41598-022-09776-8
Karwi QG, Bice JS, Baxter GF (2017) Pre- and postconditioning the heart with hydrogen sulfide (H2S) against ischemia/reperfusion injury in vivo: a systematic review and meta-analysis. Basic Res Cardiol 113:6. https://doi.org/10.1007/s00395-017-0664-8
Khan AR, Abdulhak AB, Luni FK, Farid TA, Riaz H, Ruzieh M, Pham L, Hirsch G, Bolli R (2019) Oxygen administration does not influence the prognosis of acute myocardial infarction: a meta-analysis. Am J Ther 26:e151–e160. https://doi.org/10.1097/MJT.0000000000000475
King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ (2014) Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci USA 111:3182–3187. https://doi.org/10.1073/pnas.1321871111
Kiss A, Shu H, Hamza O, Santer D, Tretter EV, Yao S, Markstaller K, Hallström S, Podesser BK, Klein KU (2018) Argon preconditioning enhances postischaemic cardiac functional recovery following cardioplegic arrest and global cold ischaemia. Eur J Cardiothorac Surg 54:539–546. https://doi.org/10.1093/ejcts/ezy104
Kleinbongard P (2023) Perspective: mitochondrial STAT3 in cardioprotection. Basic Res Cardiol 118:32. https://doi.org/10.1007/s00395-023-01003-3
Kleinbongard P, Bøtker HE, Ovize M, Hausenloy DJ, Heusch G (2020) Co-morbidities and co-medications as confounders of cardioprotection-does it matter in the clinical setting? Br J Pharmacol 177:5252–5269. https://doi.org/10.1111/bph.14839
Kleinbongard P, Lieder H, Skyschally A, Heusch G (2023) No sex-related differences in infarct size, no-reflow, and protection by ischaemic pre-conditioning in Göttingen minipigs. Cardiovasc Res 119:561–570. https://doi.org/10.1093/cvr/cvac062
Kleinbongard P, Schulz R, Rassaf T, Lauer T, Dejam A, Jax T, Kumara I, Gharini P, Kabanova S, Ozüyaman B, Schnürch HG, Gödecke A, Weber AA, Robenek M, Robenek H, Bloch W, Rösen P, Kelm M (2006) Red blood cells express a functional endothelial nitric oxide synthase. Blood 107:2943–2951. https://doi.org/10.1182/blood-2005-10-3992
Kleinbongard P, Skyschally A, Gent S, Pesch M, Heusch G (2017) STAT3 as a common signal of ischemic conditioning: a lesson on “rigor and reproducibility” in preclinical studies on cardioprotection. Basic Res Cardiol 113:3. https://doi.org/10.1007/s00395-017-0660-z
Kloner RA, Creech JL, Stone GW, O’Neill WW, Burkhoff D, Spears JR (2021) Update on cardioprotective strategies for STEMI: focus on supersaturated oxygen delivery. JACC Basic Transl Sci 6:1021–1033. https://doi.org/10.1016/j.jacbts.2021.07.011
Kottenberg E, Thielmann M, Bergmann L, Heine T, Jakob H, Heusch G, Peters J (2012) Protection by remote ischemic preconditioning during coronary artery bypass graft surgery with isoflurane but not propofol—a clinical trial. Acta Anaesthesiol Scand 56:30–38. https://doi.org/10.1111/j.1399-6576.2011.02585.x
Lavitrano M, Smolenski RT, Musumeci A, Maccherini M, Slominska E, Di Florio E, Bracco A, Mancini A, Stassi G, Patti M, Giovannoni R, Froio A, Simeone F, Forni M, Bacci ML, D’Alise G, Cozzi E, Otterbein LE, Yacoub MH, Bach FH, Calise F (2004) Carbon monoxide improves cardiac energetics and safeguards the heart during reperfusion after cardiopulmonary bypass in pigs. FASEB J 18:1093–1095. https://doi.org/10.1096/fj.03-0996fje
Lemoine S, Blanchart K, Souplis M, Lemaitre A, Legallois D, Coulbault L, Simard C, Allouche S, Abraini JH, Hanouz JL, Rouet R, Sallé L, Guinamard R, Manrique A (2017) Argon exposure induces postconditioning in myocardial ischemia-reperfusion. J Cardiovasc Pharmacol Ther 22:564–573. https://doi.org/10.1177/1074248417702891
Li H, Yao Y, Shi H, Lei Y, Huang Y, Wang K, He X, Liu J (2019) A near-infrared light-responsive nanocomposite for photothermal release of H2S and suppression of cell viability. J Mater Chem B 7:5992–5997. https://doi.org/10.1039/c9tb01611b
Li Q, Lian C, Zhou R, Li T, Xiang X, Liu B (2013) Pretreatment with xenon protected immature rabbit heart from ischaemia/reperfusion injury by opening of the mitoKATP channel. Heart Lung Circ 22:276–283. https://doi.org/10.1016/j.hlc.2012.10.016
Li Y, Dang J, Liang Q, Yin L (2019) Thermal-responsive carbon monoxide (CO) delivery expedites metabolic exhaustion of cancer cells toward reversal of chemotherapy resistance. ACS Cent Sci 5:1044–1058. https://doi.org/10.1021/acscentsci.9b00216
Li Y, Zhang P, Liu Y, Liu W, Yin N (2016) Helium preconditioning protects the brain against hypoxia/ischemia injury via improving the neurovascular niche in a neonatal rat model. Behav Brain Res 314:165–172. https://doi.org/10.1016/j.bbr.2016.08.015
Li YW, Li YM, Hon Y, Wan QL, He RL, Wang ZZ, Zhao CH (2017) AT1 receptor modulator attenuates the hypercholesterolemia-induced impairment of the myocardial ischemic post-conditioning benefits. Korean Circ J 47:182–192. https://doi.org/10.4070/kcj.2015.0295
Liang Y, Liu D, Ochs T, Tang C, Chen S, Zhang S, Geng B, Jin H, Du J (2011) Endogenous sulfur dioxide protects against isoproterenol-induced myocardial injury and increases myocardial antioxidant capacity in rats. Lab Invest 91:12–23. https://doi.org/10.1038/labinvest.2010.156
Lieder HR, Irmert A, Kamler M, Heusch G, Kleinbongard P (2019) Sex is no determinant of cardioprotection by ischemic preconditioning in rats, but ischemic/reperfused tissue mass is for remote ischemic preconditioning. Physiol Rep 7:e14146. https://doi.org/10.14814/phy2.14146
Liu K, Kang ZM, Sun XJ, Liu WW, Mao YF (2016) Helium preconditioning protects against neonatal hypoxia-ischemia via nitric oxide mediated upregulation of antioxidases in a rat model. Behav Brain Res 300:31–37. https://doi.org/10.1016/j.bbr.2015.12.001
Lochner A, Marais E, Du Toit E, Moolman J (2002) Nitric oxide triggers classic ischemic preconditioning. Ann N Y Acad Sci 962:402–414. https://doi.org/10.1111/j.1749-6632.2002.tb04084.x
Luo L, Chen S, Jin H, Tang C, Du J (2011) Endogenous generation of sulfur dioxide in rat tissues. Biochem Biophys Res Commun 415:61–67. https://doi.org/10.1016/j.bbrc.2011.10.012
Lyden SP, Shortell CK, Illig KA (2001) Reperfusion and compartment syndromes: strategies for prevention and treatment. Semin Vasc Surg 14:107–113. https://doi.org/10.1053/svas.2001.23166
Major TC, Brant DO, Burney CP, Amoako KA, Annich GM, Meyerhoff ME, Handa H, Bartlett RH (2011) The hemocompatibility of a nitric oxide generating polymer that catalyzes S-nitrosothiol decomposition in an extracorporeal circulation model. Biomaterials 32:5957–5969. https://doi.org/10.1016/j.biomaterials.2011.03.036
Mancardi D, Ottolenghi S, Attanasio U, Tocchetti CG, Paroni R, Pagliaro P, Samaja M (2022) Janus, or the inevitable battle between too much and too little oxygen. Antioxid Redox Signal 37:972–989. https://doi.org/10.1089/ars.2021.0232
Mancardi D, Pagliaro P, Ridnour LA, Tocchetti CG, Miranda K, Juhaszova M, Sollott SJ, Wink DA, Paolocci N (2022) HNO protects the myocardium against reperfusion injury, inhibiting the mPTP opening via PKCε activation. Antioxidants 11:382. https://doi.org/10.3390/antiox11020382
Mancardi D, Penna C, Merlino A, Del Soldato P, Wink DA, Pagliaro P (2009) Physiological and pharmacological features of the novel gasotransmitter: hydrogen sulfide. Biochim Biophys Acta 1787:864–872. https://doi.org/10.1016/j.bbabio.2009.03.005
Mayer B, Soppert J, Kraemer S, Schemmel S, Beckers C, Bleilevens C, Rossaint R, Coburn M, Goetzenich A, Stoppe C (2016) Argon induces protective effects in cardiomyocytes during the second window of preconditioning. Int J Mol Sci 17:1159. https://doi.org/10.3390/ijms17071159
Maze M, Laitio T (2020) Neuroprotective properties of xenon. Mol Neurobiol 57:118–124. https://doi.org/10.1007/s12035-019-01761-z
Martin C, Schulz R, Post H, Boengler K, Kelm M, Kleinbongard P, Gres P, Skyschally A, Konietzka I, Heusch G (2007) Microdialysis-based analysis of interstitial NO in situ: NO synthase-independent NO formation during myocardial ischemia. Cardiovasc Res 74:46–55. https://doi.org/10.1016/j.cardiores.2006.12.020
Meng G, Wang J, Xiao Y, Bai W, Xie L, Shan L, Moore PK, Ji Y (2015) GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J Biomed Res 29:203–213. https://doi.org/10.7555/JBR.28.20140037
Merx MW, Gorressen S, van de Sandt AM, Cortese-Krott MM, Ohlig J, Stern M, Rassaf T, Gödecke A, Gladwin MT, Kelm M (2014) Depletion of circulating blood NOS3 increases severity of myocardial infarction and left ventricular dysfunction. Basic Res Cardiol 109:398. https://doi.org/10.1007/s00395-013-0398-1
Mio Y, Shim YH, Richards E, Bosnjak ZJ, Pagel PS, Bienengraeber M (2009) Xenon preconditioning: the role of prosurvival signaling, mitochondrial permeability transition and bioenergetics in rats. Anesth Analg 108:858–866. https://doi.org/10.1213/ane.0b013e318192a520
Morita K (2012) Surgical reoxygenation injury of the myocardium in cyanotic patients: clinical relevance and therapeutic strategies by normoxic management during cardiopulmonary bypass. Gen Thorac Cardiovasc Surg 60:549–556. https://doi.org/10.1007/s11748-012-0115-2
Musiolik J, van Caster P, Skyschally A, Boengler K, Gres P, Schulz R, Heusch G (2010) Reduction of infarct size by gentle reperfusion without activation of reperfusion injury salvage kinases in pigs. Cardiovasc Res 85:110–117. https://doi.org/10.1093/cvr/cvp271
Nakano A, Liu GS, Heusch G, Downey JM, Cohen MV (2000) Exogenous nitric oxide can trigger a preconditioned state through a free radical mechanism, but endogenous nitric oxide is not a trigger of classical ischemic preconditioning. J Mol Cell Cardiol 32:1159–1167. https://doi.org/10.1006/jmcc.2000.1152
Ndongson-Dongmo B, Heller R, Hoyer D, Brodhun M, Bauer M, Winning J, Hirsch E, Wetzker R, Schlattmann P, Bauer R (2015) Phosphoinositide 3-kinase gamma controls inflammation-induced myocardial depression via sequential cAMP and iNOS signalling. Cardiovasc Res 108:243–253. https://doi.org/10.1093/cvr/cvv217
Nespoli F, Redaelli S, Ruggeri L, Fumagalli F, Olivari D, Ristagno G (2019) A complete review of preclinical and clinical uses of the noble gas argon: evidence of safety and protection. Ann Card Anaesth 22:122–135. https://doi.org/10.4103/aca.ACA_111_18
Niu X, Watts VL, Cingolani OH, Sivakumaran V, Leyton-Mange JS, Ellis CL, Miller KL, Vandegaer K, Bedja D, Gabrielson KL, Paolocci N, Kass DA, Barouch LA (2012) Cardioprotective effect of beta-3 adrenergic receptor agonism: role of neuronal nitric oxide synthase. J Am Coll Cardiol 59:1979–1987. https://doi.org/10.1016/j.jacc.2011.12.046.Erratum.In:JAmCollCardiol60:481
Oei GT, Aslami H, Kerindongo RP, Steenstra RJ, Beurskens CJ, Tuip-de Boer AM, Juffermans NP, Hollmann MW, Preckel B, Weber NC (2015) Prolonged helium postconditioning protocols during early reperfusion do not induce cardioprotection in the rat heart in vivo: role of inflammatory cytokines. J Immunol Res 2015:216798. https://doi.org/10.1155/2015/216798
Oei GT, Heger M, van Golen RF, Alles LK, Flick M, van der Wal AC, van Gulik TM, Hollmann MW, Preckel B, Weber NC (2015) Reduction of cardiac cell death after helium postconditioning in rats: transcriptional analysis of cell death and survival pathways. Mol Med 20:516–526. https://doi.org/10.2119/molmed.2014.00057
Oei GT, Huhn R, Heinen A, Hollmann MW, Schlack WS, Preckel B, Weber NC (2012) Helium-induced cardioprotection of healthy and hypertensive rat myocardium in vivo. Eur J Pharmacol 684:125–131. https://doi.org/10.1016/j.ejphar.2012.03.045
Oei GT, Weber NC, Hollmann MW, Preckel B (2010) Cellular effects of helium in different organs. Anesthesiology 112:1503–1510. https://doi.org/10.1097/ALN.0b013e3181d9cb5e
Oliveira S, Queiroga CS, Vieira HL (2016) Mitochondria and carbon monoxide: cytoprotection and control of cell metabolism—a role for Ca(2+)? J Physiol 594:4131–4138. https://doi.org/10.1113/JP270955
Ollinger R, Wang H, Yamashita K, Wegiel B, Thomas M, Margreiter R, Bach FH (2007) Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid Redox Signal 9:2175–2185. https://doi.org/10.1089/ars.2007.1807
Opoku-Damoah Y, Zhang R, Ta HT, Xu ZP (2022) Therapeutic gas-releasing nanomedicines with controlled release: advances and perspectives. Exploration (Beijing) 2:20210181. https://doi.org/10.1002/EXP.20210181
O’Rourke B (2004) Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res 94:420–432. https://doi.org/10.1161/01.RES.0000117583.66950.43
Otterbein LE, Bach FH, Alam J, Soares M, Tao LuH, Wysk M, Davis RJ, Flavell RA, Choi AM (2000) Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med 6:422–428. https://doi.org/10.1038/74680
Pagel PS, Krolikowski JG (2009) Transient metabolic alkalosis during early reperfusion abolishes helium preconditioning against myocardial infarction: restoration of cardioprotection by cyclosporin A in rabbits. Anesth Analg 108:1076–1082. https://doi.org/10.1213/ane.0b013e318193e934
Pagel PS, Krolikowski JG, Amour J, Warltier DC, Weihrauch D (2009) Morphine reduces the threshold of helium preconditioning against myocardial infarction: the role of opioid receptors in rabbits. J CardiothoracVasc Anesth 23:619–624. https://doi.org/10.1053/j.jvca.2008.12.020
Pagel PS, Krolikowski JG, Pratt PF Jr, Shim YH, Amour J, Warltier DC, Weihrauch D (2008) Reactive oxygen species and mitochondrial adenosine triphosphate-regulated potassium channels mediate helium-induced preconditioning against myocardial infarction in vivo. J Cardiothorac Vasc Anesth 22:554–559. https://doi.org/10.1053/j.jvca.2008.04.005
Pagel PS, Krolikowski JG, Pratt PF Jr, Shim YH, Amour J, Warltier DC, Weihrauch D (2008) The mechanism of helium-induced preconditioning: a direct role for nitric oxide in rabbits. Anesth Analg 107:762–768. https://doi.org/10.1213/ane.0b013e3181815995
Pagel PS, Krolikowski JG, Pratt PF Jr, Shim YH, Amour J, Warltier DC, Weihrauch D (2008) Inhibition of glycogen synthase kinase or the apoptotic protein p53 lowers the threshold of helium cardioprotection in vivo: the role of mitochondrial permeability transition. Anesth Analg 107:769–775. https://doi.org/10.1213/ane.0b013e3181815b84
Pagel PS, Krolikowski JG, Shim YH, Venkatapuram S, Kersten JR, Weihrauch D, Warltier DC, Pratt PF Jr (2007) Noble gases without anesthetic properties protect myocardium against infarction by activating prosurvival signaling kinases and inhibiting mitochondrial permeability transition in vivo. Anesth Analg 105:562–569. https://doi.org/10.1213/01.ane.0000278083.31991.36
Pagliaro P, Gattullo D, Rastaldo R, Losano G (2001) Ischemic preconditioning: from the first to the second window of protection. Life Sci 69:1–15. https://doi.org/10.1016/s0024-3205(01)01113-4
Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, Feelisch M, Wink DA, Kass DA, Paolocci N (2003) Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic Biol Med 34:33–43. https://doi.org/10.1016/s0891-5849(02)01179-6
Pagliaro P, Penna C (2015) Redox signalling and cardioprotection: translatability and mechanism. Br J Pharmacol 172:1974–1995. https://doi.org/10.1111/bph.12975
Panthi S, Chung HJ, Jung J, Jeong NY (2016) Physiological importance of hydrogen sulfide: emerging potent neuroprotector and neuromodulator. Oxid Med Cell Longev 2016:9049782. https://doi.org/10.1155/2016/9049782
Pechánová O, Varga ZV, Cebová M, Giricz Z, Pacher P, Ferdinandy P (2015) Cardiac NO signalling in the metabolic syndrome. Br J Pharmacol 172:1415–1433. https://doi.org/10.1111/bph.12960
Penna C, Alloatti G, Crisafulli A (2020) Mechanisms involved in cardioprotection induced by physical exercise. Antioxid Redox Signal 32:1115–1134. https://doi.org/10.1089/ars.2019.8009
Penna C, Andreadou I, Aragno M, Beauloye C, Bertrand L, Lazou A, Falcão-Pires I, Bell R, Zuurbier CJ, Pagliaro P, Hausenloy DJ (2020) Effect of hyperglycaemia and diabetes on acute myocardial ischaemia-reperfusion injury and cardioprotection by ischaemic conditioning protocols. Br J Pharmacol 177:5312–5335. https://doi.org/10.1111/bph.14993
Penna C, Cappello S, Mancardi D, Raimondo S, Rastaldo R, Gattullo D, Losano G, Pagliaro P (2006) Post-conditioning reduces infarct size in the isolated rat heart: role of coronary flow and pressure and the nitric oxide/cGMP pathway. Basic Res Cardiol 101:168–179. https://doi.org/10.1007/s00395-005-0543-6
Penna C, Femminò S, Caldera F, Rubin Pedrazzo A, Cecone C, Alfì E, Comità S, Higashiyama T, Trotta F, Pagliaro P, Cavalli R (2021) Cyclic nigerosyl-nigerose as oxygen nanocarrier to protect cellular models from hypoxia/reoxygenation injury: implications from an in vitro model. Int J Mol Sci 22:4208. https://doi.org/10.3390/ijms22084208
Penna C, Mancardi D, Rastaldo R, Losano G, Pagliaro P (2007) Intermittent activation of bradykinin B2 receptors and mitochondrial KATP channels trigger cardiac postconditioning through redox signaling. Cardiovasc Res 75:168–177. https://doi.org/10.1016/j.cardiores.2007.03.001
Penna C, Perrelli MG, Pagliaro P (2013) Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxid Redox Signal 18:556–599. https://doi.org/10.1089/ars.2011.4459
Perrelli MG, Pagliaro P, Penna C (2011) Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World J Cardiol 3:186–200. https://doi.org/10.4330/wjc.v3.i6.186
Perrino C, Ferdinandy P, Bøtker HE, Brundel BJJM, Collins P, Davidson SM, den Ruijter HM, Engel FB, Gerdts E, Girao H, Gyöngyösi M, Hausenloy DJ, Lecour S, Madonna R, Marber M, Murphy E, Pesce M, Regitz-Zagrosek V, Sluijter JPG, Steffens S, Gollmann-Tepeköylü C, Van Laake LW, Van Linthout S, Schulz R, Ytrehus K (2021) Improving translational research in sex-specific effects of comorbidities and risk factors in ischaemic heart disease and cardioprotection: position paper and recommendations of the ESC working group on cellular biology of the heart. Cardiovasc Res 117:367–385. https://doi.org/10.1093/cvr/cvaa155
Piantadosi CA, Carraway MS, Babiker A, Suliman HB (2008) Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 103:1232–1240. https://doi.org/10.1161/01.RES.0000338597.71702.ad
Pieper GM, Khanna AK, Roza AM (2002) Prolonging organ allograft survival: potential role of nitric oxide scavengers. BioDrugs 16:37–45. https://doi.org/10.2165/00063030-200216010-00004
Portal L, Morin D, Motterlini R, Ghaleh B, Pons S (2019) The CO-releasing molecule CORM-3 protects adult cardiomyocytes against hypoxia-reoxygenation by modulating pH restoration. Eur J Pharmacol 862:172636. https://doi.org/10.1016/j.ejphar.2019.172636
Post H, Schulz R, Behrends M, Gres P, Umschlag C, Heusch G (2000) No involvement of endogenous nitric oxide in classical ischemic preconditioning in swine. J Mol Cell Cardiol 32:725–733. https://doi.org/10.1006/jmcc.2000.1117
Pott C, Brixius K, Bloch W, Ziskoven C, Napp A, Schwinger RH (2006) Beta3-adrenergic stimulation in the human heart: signal transduction, functional implications and therapeutic perspectives. Pharmazie 61:255–260
Preckel B, Ebel D, Müllenheim J, Frässdorf J, Thämer V, Schlack W (2002) The direct myocardial effects of xenon in the dog heart in vivo. Anesth Analg 94:545–551. https://doi.org/10.1097/00000539-200203000-00012
Preckel B, Müllenheim J, Moloschavij A, Thämer V, Schlack W (2000) Xenon administration during early reperfusion reduces infarct size after regional ischemia in the rabbit heart in vivo. Anesth Analg 91:1327–1332. https://doi.org/10.1097/00000539-200012000-00003
Prokhorov DA, Kutyshenko VP, Tarahovsky YS, Kukushkin NI, Khrenov MO, Kovtun AL, Zakharova NM (2023) Solid xenon carrier based on α-cyclodextrin: properties, preparation, and application. J Pharm Sci 112:344–349. https://doi.org/10.1016/j.xphs.2022.08.014
Qi H, Soto-Gonzalez L, Krychtiuk KA, Ruhittel S, Kaun C, Speidl WS, Kiss A, Podesser BK, Yao S, Markstaller K, Klein KU, Tretter V (2018) Pretreatment with argon protects human cardiac myocyte-like progenitor cells from oxygen glucose deprivation-induced cell death by activation of AKT and differential regulation of mapkinases. Shock 49:556–563. https://doi.org/10.1097/SHK.0000000000000998
Qingyou Z, Junbao D, Weijin Z, Hui Y, Chaoshu T, Chunyu Z (2004) Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem Biophys Res Commun 317:30–37. https://doi.org/10.1016/j.bbrc.2004.02.176
Queiroga CS, Tomasi S, Widerøe M, Alves PM, Vercelli A, Vieira HL (2012) Preconditioning triggered by carbon monoxide (CO) provides neuronal protection following perinatal hypoxia-ischemia. PLoS ONE 7:e42632. https://doi.org/10.1371/journal.pone.0042632
Rassaf T, Kleinbongard P, Preik M, Dejam A, Gharini P, Lauer T, Erckenbrecht J, Duschin A, Schulz R, Heusch G, Feelisch M, Kelm M (2002) Plasma nitrosothiols contribute to the systemic vasodilator effects of intravenously applied NO: experimental and clinical Study on the fate of NO in human blood. Circ Res 91(6):470–477. https://doi.org/10.1161/01.res.0000035038.41739.cb. (PMID: 12242264)
Rassaf T, Preik M, Kleinbongard P, Lauer T, Heiss C, Strauer BE, Feelisch M, Kelm M (2002) Evidence for in vivo transport of bioactive nitric oxide in human plasma. J Clin Invest 109:1241–1248. https://doi.org/10.1172/JCI14995
Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M (2014) Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res 114:1601–16010. https://doi.org/10.1161/CIRCRESAHA.114.303822
Recchia FA (2002) Role of nitric oxide in the regulation of substrate metabolism in heart failure. Heart Fail Rev 7:141–148. https://doi.org/10.1023/a:1015324508556
Reinelt H, Marx T, Kotzerke J, Topalidis P, Luederwald S, Armbruster S, Schirmer U, Schmidt M (2002) Hepatic function during xenon anesthesia in pigs. Acta Anaesthesiol Scand 46:713–716. https://doi.org/10.1034/j.1399-6576.2002.460614.x
Russo I, Barale C, Melchionda E, Penna C, Pagliaro P (2023) Platelets and cardioprotection: the role of nitric oxide and carbon oxide. Int J Mol Sci 24:6107. https://doi.org/10.3390/ijms24076107
Sanders RD, Ma D, Maze M (2005) Anaesthesia induced neuroprotection. Best Pract Res Clin Anaesthesiol 19:461–474. https://doi.org/10.1016/j.bpa.2005.01.005
Saraste A, Ballo H, Arola O, Laitio R, Airaksinen J, Hynninen M, Bäcklund M, Ylikoski E, Wennervirta J, Pietilä M, Roine RO, Harjola VP, Niiranen J, Korpi K, Varpula M, Scheinin H, Maze M, Vahlberg T, Laitio T (2021) Effect of inhaled xenon on cardiac function in comatose survivors of out-of-hospital cardiac arrest-a substudy of the xenon in combination with hypothermia after cardiac arrest trial. Crit Care Explor 3:e0502. https://doi.org/10.1097/CCE.0000000000000502
Schiavo S, Brenna CTA, Albertini L, Djaiani G, Marinov A, Katznelson R (2024) Safety of hyperbaric oxygen therapy in patients with heart failure: a retrospective cohort study. PLoS ONE 19:e0293484. https://doi.org/10.1371/journal.pone.0293484
Schwiebert C, Huhn R, Heinen A, Weber NC, Hollmann MW, Schlack W, Preckel B (2010) Postconditioning by xenon and hypothermia in the rat heart in vivo. Eur J Anaesthesiol 27:734–739. https://doi.org/10.1097/EJA.0b013e328335fc4c
Senapati S, Mahanta AK, Kumar S, Maiti P (2018) Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct Target Ther 3:7. https://doi.org/10.1038/s41392-017-0004-3
Sezer M, Escaned J, Broyd CJ, Umman B, Bugra Z, Ozcan I, Sonsoz MR, Ozcan A, Atici A, Aslanger E, Sezer ZI, Davies JE, van Royen N, Umman S (2022) Gradual versus abrupt reperfusion during primary percutaneous coronary interventions in ST-segment-elevation myocardial infarction (GUARD). J Am Heart Assoc 11:e024172. https://doi.org/10.1161/JAHA.121.024172
Sezer M, van Royen N, Umman B, Bugra Z, Bulluck H, Hausenloy DJ, Umman S (2018) Coronary microvascular injury in reperfused acute myocardial infarction: a view from an integrative perspective. J Am Heart Assoc 7:e009949. https://doi.org/10.1161/JAHA.118.009949
Shao Q, Casin KM, Mackowski N, Murphy E, Steenbergen C, Kohr MJ (2017) Adenosine A1 receptor activation increases myocardial protein S-nitrosothiols and elicits protection from ischemia-reperfusion injury in male and female hearts. PLoS ONE 12:e0177315. https://doi.org/10.1371/journal.pone.0177315
Shemarova I, Nesterov V, Emelyanova L, Korotkov S (2021) Mitochondrial mechanisms by which gasotransmitters (H2S, NO and CO) protect cardiovascular system against hypoxia. Front Biosci 13:105–130. https://doi.org/10.52586/S556
Shen Y, Shen Z, Luo S, Guo W, Zhu YZ (2015) The cardioprotective effects of hydrogen sulfide in heart diseases: from molecular mechanisms to therapeutic potential. Oxid Med Cell Longev 2015:925167. https://doi.org/10.1155/2015/925167
Sigala F, Efentakis P, Karageorgiadi D, Filis K, Zampas P, Iliodromitis EK, Zografos G, Papapetropoulos A, Andreadou I (2017) Reciprocal regulation of eNOS, H2S and CO-synthesizing enzymes in human atheroma: correlation with plaque stability and effects of simvastatin. Redox Biol 12:70–81. https://doi.org/10.1016/j.redox.2017.02.006
Skyschally A, Walter B, Heusch G (2013) Coronary microembolization during early reperfusion: infarct extension, but protection by ischaemic postconditioning. Eur Heart J 34:3314–3321. https://doi.org/10.1093/eurheartj/ehs434
Smit KF, Brevoord D, De Hert S, de Mol BA, Kerindongo RP, van Dieren S, Schlack WS, Hollmann MW, Weber NC, Preckel B (2016) Effect of helium pre- or postconditioning on signal transduction kinases in patients undergoing coronary artery bypass graft surgery. J Transl Med 14:294. https://doi.org/10.1186/s12967-016-1045-z
Smit KF, Konkel M, Kerindongo R, Landau MA, Zuurbier CJ, Hollmann MW, Preckel B, Nieuwland R, Albrecht M, Weber NC (2018) Helium alters the cytoskeleton and decreases permeability in endothelial cells cultured in vitro through a pathway involving caveolin-1. Sci Rep 8:4768. https://doi.org/10.1038/s41598-018-23030-0
Smit KF, Oei GT, Brevoord D, Stroes ES, Nieuwland R, Schlack WS, Hollmann MW, Weber NC, Preckel B (2013) Helium induces preconditioning in human endothelium in vivo. Anesthesiology 118:95–104. https://doi.org/10.1097/ALN.0b013e3182751300
Smit KF, Oei GTML, Konkel M, Augustijn QJJ, Hollmann MW, Preckel B, Patel HH, Weber NC (2019) Plasma from volunteers breathing helium reduces hypoxia-induced cell damage in human endothelial cells-mechanisms of remote protection against hypoxia by helium. Cardiovasc Drugs Ther 33:297–306. https://doi.org/10.1007/s10557-019-06880-2
Smit KF, Weber NC, Hollmann MW, Preckel B (2015) Noble gases as cardioprotectants—translatability and mechanism. Br J Pharmacol 172:2062–2073. https://doi.org/10.1111/bph.12994
Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, Szabo C, Stahl GL, Sellke FW (2009) Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J Thorac Cardiovasc Surg 138:977–984. https://doi.org/10.1016/j.jtcvs.2008.08.074
Spears JR (2019) Reperfusion microvascular ischemia after prolonged coronary occlusion: implications and treatment with local supersaturated oxygen delivery. Hypoxia 7:65–79. https://doi.org/10.2147/HP.S217955
Spears JR, Henney C, Prcevski P, Xu R, Li L, Brereton GJ, DiCarli M, Spanta A, Crilly R, Sulaiman AM, Hadeed S, Lavine S, Patterson WR, Creech J, Vander Heide R (2002) Aqueous oxygen hyperbaric reperfusion in a porcine model of myocardial infarction. J Invasive Cardiol 14:160–166
Spears JR, Prcevski P, Jiang A, Brereton GJ, Vander Heide R (2006) Intracoronary aqueous oxygen perfusion, performed 24 h after the onset of postinfarction reperfusion, experimentally reduces infarct size and improves left ventricular function. Int J Cardiol 113:371–375. https://doi.org/10.1016/j.ijcard.2005.11.099
Spears JR, Prcevski P, Xu R, Li L, Brereton G, DiCarli M, Spanta A, Crilly R, Lavine S, van der Heide R (2003) Aqueous oxygen attenuation of reperfusion microvascular ischemia in a canine model of myocardial infarction. ASAIO J 49:716–720. https://doi.org/10.1097/01.mat.0000094665.72503.3c
Spears JR, Wang B, Wu X, Prcevski P, Jiang AJ, Spanta AD, Crilly RJ, Brereton GJ (1997) Aqueous oxygen: a highly O2-supersaturated infusate for regional correction of hypoxemia and production of hyperoxemia. Circulation 96:4385–4391. https://doi.org/10.1161/01.cir.96.12.4385
Stein AB, Bolli R, Dawn B, Sanganalmath SK, Zhu Y, Wang OL, Guo Y, Motterlini R, Xuan YT (2012) Carbon monoxide induces a late preconditioning-mimetic cardioprotective and antiapoptotic milieu in the myocardium. J Mol Cell Cardiol 52:228–236. https://doi.org/10.1016/j.yjmcc.2011.11.005
Stowe DF, Rehmert GC, Kwok WM, Weigt HU, Georgieff M, Bosnjak ZJ (2000) Xenon does not alter cardiac function or major cation currents in isolated guinea pig hearts or myocytes. Anesthesiology 92:516–522. https://doi.org/10.1097/00000542-200002000-00035
Suliman HB, Zobi F, Piantadosi CA (2016) Heme oxygenase-1/carbon monoxide system and embryonic stem cell differentiation and maturation into cardiomyocytes. Antioxid Redox Signal 24:345–360. https://doi.org/10.1089/ars.2015.6342
Sun J, Aponte AM, Menazza S, Gucek M, Steenbergen C, Murphy E (2016) Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation Cardiovasc Res 110:96–106. https://doi.org/10.1093/cvr/cvw037
Suzuki T, Koyama H, Sugimoto M, Uchida I, Mashimo T (2002) The diverse actions of volatile and gaseous anesthetics on human-cloned 5-hydroxytryptamine 3 receptors expressed in xenopus oocytes. Anesthesiology 96:699–704. https://doi.org/10.1097/00000542-200203000-00028
Tocchetti CG, Stanley BA, Murray CI, Sivakumaran V, Donzelli S, Mancardi D, Pagliaro P, Gao WD, van Eyk J, Kass DA, Wink DA, Paolocci N (2011) Playing with cardiac “redox switches”: the “HNO way” to modulate cardiac function. Antioxid Redox Signal 14:1687–1698. https://doi.org/10.1089/ars.2010.3859
Tomschi F, Niemann D, Bloch W, Predel HG, Grau M (2018) Ischemic preconditioning enhances performance and erythrocyte deformability of responders. Int J Sports Med 39:596–603. https://doi.org/10.1055/a-0631-2887
Totzeck M, Hendgen-Cotta UB, Rassaf T (2017) Nitrite-nitric oxide signaling and cardioprotection. Adv Exp Med Biol 982:335–346. https://doi.org/10.1007/978-3-319-55330-6_18
Tullio F, Angotti C, Perrelli MG, Penna C, Pagliaro P (2013) Redox balance and cardioprotection. Basic Res Cardiol 108:392. https://doi.org/10.1007/s00395-013-0392-7
Ulbrich F, Goebel U (2016) The molecular pathway of argon-mediated neuroprotection. Int J Mol Sci 17:1816. https://doi.org/10.3390/ijms17111816
Vagts DA, Hecker K, Iber T, Roesner JP, Spee A, Otto B, Rossaint R, Nöldge-Schomburg GF (2004) Effects of xenon anaesthesia on intestinal oxygenation in acutely instrumented pigs. Br J Anaesth 93:833–841. https://doi.org/10.1093/bja/aeh271
Vieira HL, Queiroga CS, Alves PM (2008) Pre-conditioning induced by carbon monoxide provides neuronal protection against apoptosis. J Neurochem 107:375–384. https://doi.org/10.1111/j.1471-4159.2008.05610.x
Wakeno-Takahashi M, Otani H, Nakao S, Uchiyama Y, Imamura H, Shingu K (2004) Adenosine and a nitric oxide donor enhances cardioprotection by preconditioning with isoflurane through mitochondrial adenosine triphosphate-sensitive K+ channel-dependent and -independent mechanisms. Anesthesiology 100:515–524. https://doi.org/10.1097/00000542-200403000-00009
Wang H, Kohr MJ, Wheeler DG, Ziolo MT (2008) Endothelial nitric oxide synthase decreases beta-adrenergic responsiveness via inhibition of the L-type Ca2+ current. Am J Physiol Heart Circ Physiol 294:H1473–H1480. https://doi.org/10.1152/ajpheart.01249.2007
Wang Q, Zuurbier CJ, Huhn R, Torregroza C, Hollmann MW, Preckel B, van den Brom CE, Weber NC (2023) Pharmacological cardioprotection against ischemia reperfusion injury-the search for a clinical effective therapy. Cells 12:1432. https://doi.org/10.3390/cells12101432
Wang SB, Zhang C, Chen ZX, Ye JJ, Peng SY, Rong L, Liu CJ, Zhang XZ (2019) A versatile carbon monoxide nanogenerator for enhanced tumor therapy and anti-inflammation. ACS Nano 13:5523–5532. https://doi.org/10.1021/acsnano.9b00345
Weber NC, Frässdorf J, Ratajczak C, Grueber Y, Schlack W, Hollmann MW, Preckel B (2008) Xenon induces late cardiac preconditioning in vivo: a role for cyclooxygenase 2? Anesth Analg 107:1807–1813. https://doi.org/10.1213/ane.Ob013e31818874bf
Weber NC, Kandler J, Schlack W, Grueber Y, Fradorf J, Preckel B (2008) Intermitted pharmacologic pretreatment by xenon, isoflurane, nitrous oxide, and the opioid morphine prevents tumor necrosis factor alpha-induced adhesion molecule expression in human umbilical vein endothelial cells. Anesthesiology 108:199–207. https://doi.org/10.1097/01.anes.0000299441.32091.ed
Weber NC, Preckel B (2019) Gaseous mediators: an updated review on the effects of helium beyond blowing up balloons. Intensive Care Med Exp 7:73. https://doi.org/10.1186/s40635-019-0288-4
Weber NC, Schilling JM, Warmbrunn MV, Dhanani M, Kerindongo R, Siamwala J, Song Y, Zemljic-Harpf AE, Fannon MJ, Hollmann MW, Preckel B, Roth DM, Patel HH (2019) Helium-induced changes in circulating caveolin in mice suggest a novel mechanism of cardiac protection. Int J Mol Sci 20:2640. https://doi.org/10.3390/ijms20112640
Weber NC, Smit KF, Hollmann MW, Preckel B (2015) Targets involved in cardioprotection by the non-anesthetic noble gas helium. Curr Drug Targets 16:786–792. https://doi.org/10.2174/1389450116666150120104459
Weber NC, Stursberg J, Wirthle NM, Toma O, Schlack W, Preckel B (2006) Xenon preconditioning differently regulates p44/42 MAPK (ERK 1/2) and p46/54 MAPK (JNK 1/2 and 3) in vivo. Br J Anaesth 97:298–306. https://doi.org/10.1093/bja/ael153
Weber NC, Toma O, Damla H, Wolter JI, Schlack W, Preckel B (2006) Upstream signaling of protein kinase C-epsilon in xenon-induced pharmacological preconditioning. Implication of mitochondrial adenosine triphosphate dependent potassium channels and phosphatidylinositol-dependent kinase-1. Eur J Pharmacol 539:1–9. https://doi.org/10.1016/j.ejphar.2006.03.054
Weber NC, Toma O, Wolter JI, Obal D, Müllenheim J, Preckel B, Schlack W (2005) The noble gas xenon induces pharmacological preconditioning in the rat heart in vivo via induction of PKC-epsilon and p38 MAPK. Br J Pharmacol 144:123–132. https://doi.org/10.1038/sj.bjp.0706063
Weber NC, Toma O, Wolter JI, Wirthle NM, Schlack W, Preckel B (2005) Mechanisms of xenon- and isoflurane-induced preconditioning—a potential link to the cytoskeleton via the MAPKAPK-2/HSP27 pathway. Br J Pharmacol 146:445–455. https://doi.org/10.1038/sj.bjp.0706324
Wink DA, Miranda KM, Katori T, Mancardi D, Thomas DD, Ridnour L, Espey MG, Feelisch M, Colton CA, Fukuto JM, Pagliaro P, Kass DA, Paolocci N (2003) Orthogonal properties of the redox siblings nitroxyl and nitric oxide in the cardiovascular system: a novel redox paradigm. Am J Physiol Heart Circ Physiol 285:H2264–H2276. https://doi.org/10.1152/ajpheart.00531.2003
Wu D, Gu Y, Zhu D (2021) Cardioprotective effects of hydrogen sulfide in attenuating myocardial ischemia-reperfusion injury. Mol Med Rep 24:875. https://doi.org/10.3892/mmr.2021.12515
Xiong Q, Wang Z, Yu Y, Wen Y, Suguro R, Mao Y, Zhu YZ (2019) Hydrogen sulfide stabilizes atherosclerotic plaques in apolipoprotein E knockout mice. Pharmacol Res 144:90–98. https://doi.org/10.1016/j.phrs.2019.04.006
Yamakura T, Harris RA (2000) Effects of gaseous anesthetics nitrous oxide and xenon on ligand-gated ion channels. Comparison with isoflurane and ethanol. Anesthesiology 93:1095–1101. https://doi.org/10.1097/00000542-200010000-00034
Yang C, Hwang HH, Jeong S, Seo D, Jeong Y, Lee DY, Lee K (2018) Inducing angiogenesis with the controlled release of nitric oxide from biodegradable and biocompatible copolymeric nanoparticles. Int J Nanomed 13:6517–6530. https://doi.org/10.2147/IJN.S174989
Yang J, Gonon AT, Sjöquist PO, Lundberg JO, Pernow J (2013) Arginase regulates red blood cell nitric oxide synthase and export of cardioprotective nitric oxide bioactivity. Proc Natl Acad Sci USA 110:15049–15054. https://doi.org/10.1073/pnas.1307058110
Yellon DM, Beikoghli Kalkhoran S, Davidson SM (2023) The RISK pathway leading to mitochondria and cardioprotection: how everything started. Basic Res Cardiol 118:22. https://doi.org/10.1007/s00395-023-00992-5
Yin H, Chen Z, Zhao H, Huang H, Liu W (2022) Noble gas and neuroprotection: from bench to bedside. Front Pharmacol 13:1028688. https://doi.org/10.3389/fphar.2022.1028688
Yin X, Moody MR, Hebert V, Klegerman ME, Geng YJ, Dugas TR, McPherson DD, Kim H, Huang SL (2019) Oral delivery of xenon for cardiovascular protection. Sci Rep 9:14035. https://doi.org/10.1038/s41598-019-50515-3
Zangrillo A, Musu M, Greco T, Di Prima AL, Matteazzi A, Testa V, Nardelli P, Febres D, Monaco F, Calabrò MG, Ma J, Finco G, Landoni G (2015) Additive effect on survival of anaesthetic cardiac protection and remote ischemic preconditioning in cardiac surgery: a bayesian network meta-analysis of randomized trials. PLoS ONE 10:e0134264. https://doi.org/10.1371/journal.pone.0134264
Zhang H, Hu H, Zhai C, Jing L, Tian H (2024) Cardioprotective strategies after ischemia-reperfusion injury. Am J Cardiovasc Drugs 24:5–18. https://doi.org/10.1007/s40256-023-00614-4
Zhang J, Liu W, Bi M, Xu J, Yang H, Zhang Y (2022) Noble gases therapy in cardiocerebrovascular diseases: the novel stars? Front Cardiovasc Med 9:802783. https://doi.org/10.3389/fcvm.2022.802783
Zhang R, Zhang L, Manaenko A, Ye Z, Liu W, Sun X (2014) Helium preconditioning protects mouse liver against ischemia and reperfusion injury through the PI3K/Akt pathway. J Hepatol 61:1048–1055. https://doi.org/10.1016/j.jhep.2014.06.020
Zhang X, Du J, Guo Z, Yu J, Gao Q, Yin W, Zhu S, Gu Z, Zhao Y (2018) Efficient near infrared light triggered nitric oxide release nanocomposites for sensitizing mild photothermal therapy. Adv Sci 6:1801122. https://doi.org/10.1002/advs.201801122
Zhang Y, Zhang J, Xu K, Chen Z, Xu X, Xu J, Zheng S, Dai M, Yang H (2020) Helium protects against lipopolysaccharide-induced cardiac dysfunction in mice via suppressing toll-like receptor 4-nuclear factor κB-tumor necrosis factor-alpha/ interleukin-18 signaling. Chin J Physiol 63:276–285. https://doi.org/10.4103/CJP.CJP_66_20
Zhao MM, Yang JY, Wang XB, Tang CS, Du JB, Jin HF (2013) The PI3K/Akt pathway mediates the protection of SO(2) preconditioning against myocardial ischemia/reperfusion injury in rats. Acta Pharmacol Sin 34:501–506. https://doi.org/10.1038/aps.2012.204
Zheng DW, Li B, Li CX, Xu L, Fan JX, Lei Q, Zhang XZ (2017) Photocatalyzing CO2 to CO for enhanced cancer therapy. Adv Mater. https://doi.org/10.1002/adma.201703822
Zhou X, Meng Z, She J, Zhang Y, Yi X, Zhou H, Zhong J, Dong Z, Han X, Chen M, Fan Q, Yang K, Wang C (2020) Near-infrared light-responsive nitric oxide delivery platform for enhanced radioimmunotherapy. Nanomicro Lett 12:100. https://doi.org/10.1007/s40820-020-00431-3
Zicola E, Arrigo E, Mancardi D (2021) H2S pretreatment is promigratory and decreases ischemia/reperfusion injury in human microvascular endothelial cells. Oxid Med Cell Longev 2021:8886666. https://doi.org/10.1155/2021/8886666
Zuurbier CJ, Bertrand L, Beauloye CR, Andreadou I, Ruiz-Meana M, Jespersen NR, Kula-Alwar D, Prag HA, Eric Botker H, Dambrova M, Montessuit C, Kaambre T, Liepinsh E, Brookes PS, Krieg T (2020) Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J Cell Mol Med 24:5937–5954. https://doi.org/10.1111/jcmm.15180
Acknowledgements
C.P. and P.P. are supported by the Progetto di Ricerca di Rilevante Interesse Nazionale (PRIN) with identification numbers 2022S74XWB and 2022AA37N3, respectively.
Funding
Open access funding provided by Università degli Studi di Torino within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
P.P. and C.P. wrote the first draft of the manuscript. G.A. and N.W. revised the manuscript critically for important intellectual content. S.F. contributed significantly to the manuscript revisions, including ensuring accuracy in figures, tables, and references. All the authors revised the manuscript. All the authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
On behalf of all the authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pagliaro, P., Weber, N.C., Femminò, S. et al. Gasotransmitters and noble gases in cardioprotection: unraveling molecular pathways for future therapeutic strategies. Basic Res Cardiol 119, 509–544 (2024). https://doi.org/10.1007/s00395-024-01061-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-024-01061-1