
Abstract
Introduction
In sarcoidosis granulomas, monocyte-derived macrophages are activated by pro-inflammatory cytokines including TNF and IL-6. Current drug treatment for sarcoidosis aims to suppress inflammation but disabling side effects can ensue. The macrolide azithromycin may be anti-inflammatory. We aimed to determine whether treatment with azithromycin affects blood inflammatory gene expression and monocyte functions in sarcoidosis.
Methods
Blood samples were collected from patients with chronic pulmonary sarcoidosis enrolled in a single arm, open label clinical trial who received oral azithromycin 250 mg once daily for 3 months. Whole blood inflammatory gene expression with or without LPS stimulation was measured using a 770-mRNA panel. Phenotypic analysis and cytokine production were conducted by flow cytometry and ELISA after 24h stimulation with growth factors and TLR ligands. mTOR activity was assessed by measuring phosphorylated S6RP.
Results
Differential gene expression analysis indicated a state of heightened myeloid cell activation in sarcoidosis. Compared with controls, sarcoidosis patients showed increased LPS responses for several cytokines and chemokines. Treatment with azithromycin had minimal effect on blood gene expression overall, but supervised clustering analysis identified several chemokine genes that were upregulated. At the protein level, azithromycin treatment increased LPS-stimulated TNF and unstimulated IL-8 production. No other cytokines showed significant changes following azithromycin. Blood neutrophil counts fell during azithromycin treatment whereas mononuclear cells remained stable. Azithromycin had no detectable effects on mTOR activity or activation markers.
Conclusion
Blood myeloid cells are activated in sarcoidosis, but azithromycin therapy did not suppress inflammatory gene expression or cytokine production in blood.
Trial registration: EudraCT 2019-000580-24 (17 May 2019)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sarcoidosis affects an estimated 1.2 million people worldwide [1]. Many patients with sarcoidosis exhibit a chronic progressive clinical course which is associated with significant morbidity and treatment burden. There is an unmet need for new therapies for sarcoidosis that are safe, efficacious, and cost-effective. Current treatments aim to control disease by suppressing inflammatory cytokines and dampening inflammation, but at a cost of side effects which can be distressing, disabling or dangerous [2, 3].
Granulomas, the hallmark pathology of chronic sarcoidosis, are composed primarily of macrophages derived from circulating blood monocytes, activated by pro-inflammatory cytokines including TNF [4] and IL-6 [5, 6]. Sarcoid-like granulomas can be recapitulated in mice which have been genetically modified for constitutive mTOR activation in myeloid cells [7].
Azithromycin is an attractive option for repurposing for sarcoidosis. Macrolides are widely believed to exert anti-inflammatory effects distinct from anti-microbial activity [8]. Azithromycin is rapidly and highly concentrated within human monocytes [9,10,11]. In vitro, azithromycin promotes an anti-inflammatory macrophage phenotype [12] and suppresses mTOR activity in lymphocytes [13].
In an open label, single arm study of 21 patients with chronic pulmonary sarcoidosis and self-reported cough, treatment with 250mg azithromycin daily for 3 months reduced objective and patient-reported cough metrics. [14] Here we report the results of our analysis examining whether azithromycin treatment affects i) inflammatory gene expression and cytokine production in whole blood and ii) mononuclear cell phenotype and function and iii) mTOR activation.
Methods
Patients with symptomatic chronic pulmonary sarcoidosis were enrolled in a noncontrolled, open label clinical trial of azithromycin 250 mg once daily for 3 months. Ethics committee approval was granted (19/YH/0100) and the trial was registered (EudraCT 2019–000580-24). Patients had a diagnosis of pulmonary sarcoidosis, > 6 months from diagnosis, and self-reported cough attributed to sarcoidosis. Patients with acute self-resolving disease were ineligible. Important exclusion criteria included bronchiectasis. Patients received oral azithromycin 250 mg once daily for 3 months. Assessments were performed at baseline (visit 1) and at 1 month (visit 2) and 3 months (visit 3) on azithromycin therapy. The study design including a description of subgroups and blood samples analyzed is shown in Fig. S1. The sample size was calculated based on cough counting which has been reported [14]. One patient dropped out after visit 1 for personal reasons. A further 3 patients did not have blood sampled at visit 3 due to the onset of the COVID pandemic.
Detail of the laboratory methods employed for inflammatory phenotyping are provided in the supplementary information.
Results
Inflammatory Gene Expression in Sarcoidosis Compared with Healthy Controls
Inflammatory gene expression was measured in blood samples from a subgroup of patients with sarcoidosis participating in the azithromycin trial (n = 8) and compared with healthy controls (n = 8). To minimise confounding by anti-inflammatory treatment, none of the sarcoidosis patients were taking immunosuppressant or biologic therapy. One subject was taking low dose prednisolone (5mg daily). Demographic data for the patients are shown in Table 1. All patients were white, reflecting the local population.
We measured mRNA abundance in whole blood for 770 inflammation-associated genes using a targeted Nanostring nCounter® panel in the presence or absence of ex vivo stimulation for 24h with LPS. We identified 213 differentially expressed genes (DEGs; false discovery rate (FDR) < 1%, fold change (FC) > 1.5) comparing sarcoid patients to controls (118 up, 95 down; Fig. 1 and table S1). The top upregulated genes by fold change included ALAS2 (5'-Aminolevulinate Synthase 2), GMPR (Guanosine Monophosphate Reductase), FCGR1A (Fc Gamma Receptor Ia), SERPING1 (Serpin Family G Member 1), and NUDT1 (Nudix Hydrolase 1). The top downregulated genes by fold change included AGER (Advanced Glycosylation End-Product Specific Receptor), CTPS1 (CTP Synthase 1), FAM129C (Niban Apoptosis Regulator 3), PLCG1 (Phospholipase C Gamma 1), and MTOR (Mechanistic Target of Rapamycin Kinase). We next used nSolver software to identify the pathways differentially regulated in sarcoidosis whole blood compared with healthy controls (Fig. 1B). Top positively regulated pathways in sarcoid patients included autoantigens (including histones), antigen presentation, cytotoxicity, interferon (type I and II) signaling, immunometabolism, and TLR signaling, indicating a general state of immune (especially myeloid cell) activation. Conversely, gene sets negatively regulated in sarcoidosis included Treg, Th17 and Th2 differentiation (Fig. 1C).

Expression of 770 inflammation-associated genes in whole blood. A Volcano plot comparing sarcoidosis patients (n = 8) and healthy controls (n = 8). The plot displays each gene's log2 fold change (x axis) and adjusted -log10(p-value) (y axis). Highly statistically significant genes fall at the top of the plot above the horizontal line, and highly differentially expressed genes fall to either side. The horizontal line indicates a false discovery rate (FDR) of 1%, and the vertical lines indicate fold changes of −1.5 and 1.5. B Heatmap of pathway scores. The plot compares pathway score changes across individual samples. Orange indicates high scores; blue indicates low scores. C Pathway analysis summarizing differential expression between sarcoidosis and controls. Each gene set's most differentially expressed genes are identified and the extent of differential expression in each gene set is summarized as a directed global significance score
Sarcoidosis Monocytes are Sensitive to LPS Stimulation
Monocytes are exquisitely sensitive to stimulation by LPS through binding to CD14 and TLR4, signalling through MD2 and MyD88, and activation of MAPK and NF-kB leading to production of inflammatory cytokines and chemokines including TNF and IL-6. Modulation of LPS sensitivity may thus be used to probe monocyte activation or priming. LPS stimulation led to multiple changes in gene expression compared with unstimulated blood as expected (Fig. S2). Over 95% of LPS-induced DEGs were shared between sarcoidosis patients and controls (Fig. 2A). Sarcoidosis unique upregulated DEGs in response to LPS were FOXP1 (Forkhead Box P1), DDIT4 (DNA Damage Inducible Transcript 4), and IL7R, and unique downregulated DEGs in sarcoidosis were PYCARD (Caspase Recruitment Domain-Containing Protein 5), CD4, and PLD3 (Phospholipase D Family Member 3). Unique DEGs upregulated in response to LPS in controls included IL27, TMEM176A (Transmembrane Protein 176A), DLL4 (Delta Like Canonical Notch Ligand 4), CD14, and CD74, and unique downregulated DEGs in controls were ITGA4 (Integrin Subunit Alpha 4), HIST1H3H (H3 Clustered Histone 10), and ID1 (Inhibitor Of DNA Binding 1). Further analysis focussing on inflammatory cytokines and chemokines showed that patients with sarcoidosis had greater fold change increases in abundance of many inflammatory mRNAs in response to LPS compared with controls (Fig. 2B).

Comparison of changes in blood gene expression in response to LPS stimulation. A Venn diagram comparing number of genes significantly up-regulated (red arrows), down-regulated (blue arrows), or contra-regulated (both arrows) in response to stimulation with 100 ng/ml LPS for 24h between patients with sarcoidosis (n = 8) and healthy controls (n = 8). Included genes were those with at least 1.5 × fold change with LPS stimulation compared with no LPS that was statistically significant (q < 0.01). In addition, regulated genes in only controls or only sarcoidosis had to have measurable counts in both and at least 1.5 × fold change difference between the disease states. B Heatmap illustrates fold changes in cytokine and chemokine gene expression following LPS stimulation comparing blood samples from subjects with sarcoidosis (s1–s8) and healthy controls (c1–c8). On the right, columns show averaged changes in gene expression in response to LPS for subjects with sarcoidosis and controls. Scale bar represents Log2 fold changes. Gene names in bold showed statistically significant higher upregulation in response to LPS in sarcoidosis compared with controls (* p < 0.05, unpaired t-tests)
Effect of Azithromycin Therapy on Whole Blood Gene Expression in Sarcoidosis Patients
The effect of azithromycin on whole blood gene expression was assessed in 8 sarcoidosis patients before and after 1 month of azithromycin treatment. Expression of each individual gene was analyzed in paired fashion before and after azithromycin therapy and differences presented in a volcano plot (Fig. 3). In contrast to the difference in blood transcriptome between sarcoidosis and healthy controls (Fig. 1), there was minimal effect of azithromycin therapy on whole blood gene expression, either with or without LPS stimulation (Fig. 3). No genes passed the FDR 5% and FC 1.5 threshold.

Gene expression in whole blood in sarcoidosis patients before and 1 month into azithromycin therapy. A Volcano plot comparing unstimulated blood pre and 1 month post azithromycin treatment. The plot shows log2 fold change (x axis) and adjusted -log10 p-value (y axis) comparing mRNA abundance post vs pre-treatment using paired (repeated measures) analyses (n = 8). The horizontal line indicates a false discovery rate of 5%, and the vertical lines indicate FC < −1.5 and > 1.5. B Volcano plot comparing LPS-stimulated blood pre and 1 month post azithromycin treatment. The plot shows log2 fold change (x axis) and adjusted −log10 p-value (y axis) comparing mRNA abundance post vs pretreatment using paired (repeated measures) analyses (n = 8). The horizontal line indicates a false discovery rate of 5%, and the vertical lines indicate FC < −1.5 and > 1.5. C Heatmap showing selected differentially upregulated genes after azithromycin therapy (without LPS stimulation). Chemokine genes (CCL2, CCL3, CXCL2) were identified following PLS-DA and pathway analysis with variable influence scoring. CD40, FCRL1, and SRC were upregulated > twofold with unadjusted p-values < 0.05. Gene expression is illustrated in 8 subjects with sarcoidosis before (pre) and after (post) treatment with azithromycin for 1 month. Scale bar represents Log2 normalised counts
Next, we performed unsupervised and supervised clustering to give a high level exploratory view of blood gene expression before and after azithromycin therapy (Fig. S3). Unsupervised hierarchical clustering and principal component analysis did not identify clusters or variables associated with azithromycin treatment (Fig. S3 A,B). Supervised clustering with two predefined classes (pre and post azithromycin) using orthogonal partial least squares discriminant analysis (PLS-DA) and variable influence on projection (VIP) scores highlighted chemokines as the top regulated feature following azithromycin therapy (Fig. S3 C,D). Several chemokine genes showed > 1.5-fold significant upregulation following azithromycin therapy (unadjusted p values < 0.05), including CCL2 (fold change (FC) 3.32, p = 0.004), CCL3 (FC 1.6, p = 0.002), and CXCL2 (FC 2.45, p = 0.032). The normalized counts for these mRNAs in individual subjects are shown in Fig. 3C. In addition to CCL2, three further genes showed > twofold upregulation in mRNA abundance following azithromycin therapy—CD40 (p = 0.019), FCRL1 (Fc Receptor Like 1, p = 0.009), and the protein tyrosine kinase SRC (p = 0.013).
Effect of Azithromycin Therapy on Inflammatory Cytokine Production in Whole Blood
To determine whether azithromycin therapy has anti-inflammatory effects not reflected in whole blood gene expression profiles, we first analyzed TNF and IL-6 protein concentrations in the full azithromycin trial cohort (n = 21) [14]. Demographic and clinical details are shown in Table 1.
We stimulated blood samples with stimuli including TLR ligands and monocyte/macrophage growth factors. Incubation of whole blood ex vivo for 24 h under 10 individual stimulation conditions induced a range of changes in TNF and IL-6 concentrations (Fig. 4).

Effect of azithromycin on TNF and IL-6 concentrations in response to ex vivo stimulation of whole blood. A TNF and B IL-6 were measured in supernatants from whole blood stimulated ex vivo for 24h at 37°C under 10 conditions: PBS (control), M-CSF (CSF1 3ng/ml and 300ng/ml), GM-CSF (3 ng/ml and 300ng/ml), FSL1 (3 ng/ml and 300 ng/ml), lipopolysaccharide (LPS 1µg/ml and LPS 10ng/ml), or phytohemagglutinin (PHA 100 μg/ml). Individual patient data are plotted as dots and connecting lines (n = 21). Patients taking oral corticosteroid therapy are plotted in orange. Bars represent mean concentrations in plasma supernatants from samples taken at baseline (green) and following 1 month (blue) and 3 months (purple) of azithromycin therapy. Data were analyzed using a repeated measures linear mixed effects model. p values were corrected for multiple comparisons. Statistically significant results are shown on the plot
No inhibition of TNF or IL-6 production following azithromycin therapy was seen under any stimulation conditions. In contrast, treatment with azithromycin significantly increased TNF concentrations in response to 1µg/ml LPS (median 1782 pg/ml [95%CI 712–4383] at baseline to 3697 pg/ml [1969–7306] at 1 month; p = 0.0091: 5305 pg/ml [2860–6866] at 3 months; p = 0.0012). (Fig. 4).
Multiplex Cytokine Assays
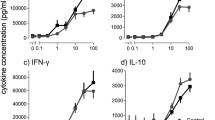
To look for an effect of azithromycin therapy on a wider range of inflammatory mediators, 13 cytokines were measured in plasma from ex vivo stimulated blood samples from a subgroup of six patients in the azithromycin trial who were not taking oral corticosteroid, immunomodulator, or biologic therapy. Compared with pre-treatment, azithromycin therapy for 3 months led to an increase in TNF production in response to M-CSF (p = 0.0278). Azithromycin did not influence concentrations of any of the other 13 cytokines in response to any stimulant (Fig. S4).
In addition, 13 cytokines were measured before and after 1 month of azithromycin therapy in sarcoidosis patients who underwent sampling using the TruCulture® (Myriad-RBM) system. Supernatants were analyzed following ex vivo stimulation with LPS or left unstimulated. IL-8 concentrations increased significantly in unstimulated TruCulture® samples following 1 month of azithromycin (median 128 pg/ml at 1 month vs 36.5 pg/ml at baseline, p < 0.0098, paired samples Wilcoxon test) (Fig. S5). No other changes in cytokine production were seen following azithromycin therapy.
Effect of Azithromycin Therapy on Blood Counts and Lymphocyte and Monocyte Subsets
Blood neutrophil counts fell on azithromycin therapy (Fig. 5). The median blood neutrophil count was 3.98 × 109/L (95%CI 3.41–4.68) at baseline, 3.2 (2.73–4.02) at 1 month (median difference −0.36 (−1.14 to −0.15), p = 0.134), and 2.98 (2.6–3.94) at 3 months (median difference −0.32 (95%CI −1.41 to -0.07), p = 0.0036).

Blood cell counts and subsets in sarcoidosis patients taking azithromycin. Blood lymphocyte and monocyte subsets were assessed by expression of cell surface markers using flow cytometry. Individual patient data are plotted as dots and connecting lines (n = 21). Patients taking oral corticosteroid therapy are plotted in orange. Bars represent mean results in blood samples taken at baseline (green) and following 1 month (blue) and 3 months (purple) of azithromycin therapy. Data were analyzed using a linear mixed effects model. p values were corrected for multiple comparisons. Statistically significant results are shown on the plot
There were no significant changes in lymphocyte, monocyte, eosinophil, or basophil counts. Total white blood cell counts mirrored neutrophil counts, but the fall was not statistically significant (mean difference at 3 months −0.54 × 109/L (−1.21–0.13), p = 0.118).
No effects of azithromycin therapy on lymphocytes or monocyte subsets were seen (Fig. 5).
Effect of Azithromycin Therapy on Cell Activation and Regulatory Markers and mTOR
Monocytes and lymphocytes were analyzed for surface activation markers CD25 and CD11b, regulatory molecules CD200L and CD200R, and intracellular mTOR activity. Azithromycin had no effect on expression of activation markers or regulatory molecules, or intracellular mTOR (mTORC1) activity as assessed by phosphorylation of S6RP (Fig.S6).
Discussion
The blood transcriptome of patients with sarcoidosis is generally indicative of a state of heightened myeloid cell activation [15,16,17,18,19,20]. Apparent contrasts in differential expression of genes and pathways between individual studies probably reflect the different clinical and demographic features of the populations studied. Treatment with corticosteroids or non-steroid immunosuppressants is particularly likely to influence transcriptomic signatures. In our cohort patients were treatment naïve (except one who was taking low dose prednisolone), similar to cohorts described by Ascoli et al. [20] and Yoshioka et al. [19]. Other published cohorts comprised both treated and untreated patients [15, 16, 18], or treatment was not stated [17]. We focused on sarcoidosis patients with a chronic progressive clinical course since this phenotype is associated with most morbidity and treatment burden. Differences in blood transcriptomes between patients with sarcoidosis and controls reflected biologically plausible sarcoidosis-related pathways. Our finding that genes encoding several histones were upregulated in sarcoidosis is of interest since histones may be autoantibody targets in sarcoidosis [21].
Anti-inflammatory and immunosuppressive effects of azithromycin [8] have long been proposed to explain how long-term low dose therapy reduces exacerbations in patients with chronic respiratory diseases [22]. Sarcoidosis is characterised by hyper-responsiveness of monocytes in peripheral blood [23] and macrolides are concentrated within monocytes [11]. We hypothesized that treatment with azithromycin may be beneficial in chronic pulmonary sarcoidosis, and that analysis of inflammatory responses in peripheral blood would provide insights into the mechanism of azithromycin’s immunomodulatory effect.
There were minimal changes in inflammatory gene expression following treatment with azithromycin. Using a permissive FDR threshold to minimise false negatives, four genes were upregulated more than twofold following azithromycin therapy, including CCL2. Published data also show CCL2 upregulation by macrolides [24, 25]. CCL2 can prime immune cells for release of granules and secretion of pro-inflammatory cytokines [26], potentially explaining the increased cytokine responses observed in response to azithromycin without commensurate changes in cytokine gene expression.
We did not find evidence that azithromycin suppressed production of TNF, IL-6 or other cytokines in response to multiple stimuli. We showed increases in TNF and IL-8 under some conditions, suggesting that azithromycin can be pro-inflammatory. This is not the first study to report pro-inflammatory effects of macrolides. “Reversal of immune paralysis” was hypothesized to explain enhanced peripheral blood TNF and IL-6 responses in two clinical trials of macrolide therapy in patients with sepsis [27, 28]. Pro-inflammatory effects have also been reported on blood neutrophils in healthy subjects given 500mg azithromycin for 3 days [29], on IL-8 following ex vivo stimulation of whole blood with azithromycin or clarithromycin [30], on lymphocyte proliferation ex vivo [31], and on IL-6 on IL-8 production by human fibroblasts via phosphorylation of NF-κB in vitro [32]. It remains to be determined whether augmentation of cytokine responses in response to macrolide therapy is clinically meaningful, for better or worse.
Small but statistically significant falls in blood neutrophil counts were seen in patients taking azithromycin. Variable effects of macrolide therapy on blood neutrophil counts have been reported [33]. A drop in neutrophil count could be a valuable marker of adherence to therapy.
Vallet et al. described reduced T cells and altered T cell subsets in patients taking azithromycin [34]. We did not find an effect of azithromycin on lymphocyte counts or subsets. T cell lymphopenia was present in many sarcoidosis patients at baseline but was not impacted during the 3 months of the trial. Blood monocyte subsets are altered in sarcoidosis, with increased proportions of intermediate or non-classical monocytes, and fewer classical monocytes. Despite being highly concentrated within monocytes, azithromycin did not impact monocyte subsets, nor did it affect monocyte expression of the activation marker CD11b or regulatory receptor CD200R.
Our work has limitations. The relatively small sample size in the present study may have been insufficient to confirm small biological effects. Furthermore, it is possible that whole blood samples do not recapitulate local tissue inflammation in the lung. Our study was not an exhaustive analysis of immunomodulatory mechanisms. We did not study neutrophil functions directly [29, 35,36,37]. Azithromycin could also impact cell types not included in the blood assay, such as epithelial cells [38, 39]. Basal levels of cytokines may be derived from a variety tissues, whereas our stimulation assay will only detect changes in blood cell-derived mediators. Some cytokines may be present in granules rather than generated de novo through RNA transcription, which may explain mismatches between transcriptomic and protein data. CCL2 for example, is released from vascular endothelial cells, and moderate basal blood levels may have masked a change in CCL2 production by blood immune cells. Likewise, our assay may have missed changes in mediators with long plasma half-lives. We attempted to mitigate plateau effects, but the highest concentration of LPS and PHA used in stimulation assays may be too strong to be able to detect an inhibitory effect (9). In the absence of a placebo-treated group, increases in cytokines over time could reflect random variation or a consequence of intercurrent illness. The lack of any conditions showing a reduction in cytokine production on azithromycin therapy argues against random variation. Whilst the single arm azithromycin trial can be criticized because there was no placebo group, the repeated measures design means we expect that a meaningful immunomodulatory effect of azithromycin, if present, would have been detected with the multiple metrics that were applied to the samples.
In vitro, azithromycin blocks intracellular killing of mycobacteria [40]. It is intriguing to speculate that if, as various lines of evidence suggest, sarcoidosis is driven by microbial antigens, the increased cytokine production that we observed could reflect alterations in mycobacterial processing. However, a double-blind, placebo-controlled trial showed that 16 weeks’ treatment with azithromycin in conjunction with levofloxacin, ethambutol, and rifabutin had no effect on forced vital capacity in patients with pulmonary sarcoidosis [41].
Our findings should raise further questions on the mechanism of action of azithromycin. Indirect effects may explain anti-inflammatory endpoints in clinical trials. Reductions in inflammatory markers in response to azithromycin therapy in patients with bacterial airways colonisation (bronchiectasis, chronic obstructive pulmonary disease, cystic fibrosis) are confounded by the antibiotic action of macrolides. A strength of our study is that airways colonisation with bacteria is not a recognised feature in sarcoidosis (patients with bronchiectasis were excluded in the protocol). In lung transplant recipients, reduction in chronic rejection with macrolide therapy occurs in the context of concurrent potent immunosuppression [42, 43], with a likely explanation being an impact of azithromycin on gastrointestinal motility, hence reducing micro-aspiration events. This may also explain the benefit of azithromycin in airways disease and in some patients with chronic cough, including sarcoidosis cough [14]. Uptake of long-term azithromycin therapy is tempered by concerns about potential effects on the microbiome and antimicrobial drug resistance [44]. Non-antibiotic alternatives would be valuable. Further research is needed to understand the direct and indirect effects of macrolide therapy.
Availability of Data and Materials
Data are openly available through the Gene Expression Omnibus, accession GSE274707, at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE274707.
Abbreviations
- FSL:
-
Fibroblast-stimulating lipopeptide
- GM-CSF:
-
Granulocyte–macrophage colony stimulating factor
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- M-CSF:
-
Macrophage colony stimulating factor
- mTOR:
-
Mechanistic target of rapamycin
- PHA:
-
Phytohemagglutinin
- S6RP:
-
Phosphorylated S6 ribosomal protein
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
References
Baughman RP, Field S, Costabel U, Crystal RG, Culver DA, Drent M et al (2016) Sarcoidosis in America. analysis based on health care use. Ann Am Thorac Soc 13(8):1244–1252
Thillai M, Atkins CP, Crawshaw A, Hart SP, Ho LP, Kouranos V et al (2021) BTS clinical statement on pulmonary sarcoidosis. Thorax 76(1):4–20. https://doi.org/10.1136/thoraxjnl-2019-214348
Robert PB, Dominique V, Peter K, Alexander GM, Wim AW, Athol W et al (2021) ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J. https://doi.org/10.1183/13993003.04079-2020
Baughman RP, Iannuzzi M (2003) Tumour necrosis factor in sarcoidosis and its potential for targeted therapy. BioDrugs 17(6):425–431. https://doi.org/10.2165/00063030-200317060-00005
Sahashi K, Ina Y, Takada K, Sato T, Yamamoto M, Morishita M (1994) Significance of interleukin 6 in patients with sarcoidosis. Chest 106(1):156–160. https://doi.org/10.1378/chest.106.1.156
Grutters JC, Sato H, Pantelidis P, Ruven HJ, McGrath DS, Wells AU et al (2003) Analysis of IL6 and IL1A gene polymorphisms in UK and dutch patients with sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 20(1):20–27
Linke M, Pham HT, Katholnig K, Schnoller T, Miller A, Demel F et al (2017) Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol 18(3):293–302. https://doi.org/10.1038/ni.3655
Pollock J, Chalmers JD (2021) The immunomodulatory effects of macrolide antibiotics in respiratory disease. Pulm Pharmacol Ther 71:102095. https://doi.org/10.1016/j.pupt.2021.102095
Bosnar M, Kelnerić Z, Munić V, Eraković V, Parnham MJ (2005) Cellular uptake and efflux of azithromycin, erythromycin, clarithromycin, telithromycin, and cethromycin. Antimicrob Agents Chemother 49(6):2372–2377. https://doi.org/10.1128/aac.49.6.2372-2377.2005
Fietta A, Merlini C, Gialdroni GG (1997) Requirements for intracellular accumulation and release of clarithromycin and azithromycin by human phagocytes. J Chemother 9(1):23–31. https://doi.org/10.1179/joc.1997.9.1.23
Wildfeuer A, Laufen H, Zimmermann T (1996) Uptake of azithromycin by various cells and its intracellular activity under in vivo conditions. Antimicrob Agents Chemother 40(1):75–79. https://doi.org/10.1128/aac.40.1.75
Haydar D, Cory TJ, Birket SE, Murphy BS, Pennypacker KR, Sinai AP et al (2019) Azithromycin polarizes macrophages to an M2 phenotype via Inhibition of the STAT1 and NF-κB signaling pathways. J Immunol 203(4):1021–1030. https://doi.org/10.4049/jimmunol.1801228
Ratzinger F, Haslacher H, Poeppl W, Hoermann G, Kovarik JJ, Jutz S et al (2014) Azithromycin suppresses CD4(+) T-cell activation by direct modulation of mTOR activity. Sci Rep 4:7438. https://doi.org/10.1038/srep07438
Fraser SD, Thackray-Nocera S, Shepherd M, Flockton R, Wright C, Sheedy W et al (2020) Azithromycin for sarcoidosis cough: an open-label exploratory clinical trial. ERJ open research 6(4):00534–02020. https://doi.org/10.1183/23120541.00534-2020
Koth LL, Solberg OD, Peng JC, Bhakta NR, Nguyen CP, Woodruff PG (2011) Sarcoidosis blood transcriptome reflects lung inflammation and overlaps with tuberculosis. Am J Respir Crit Care Med 184(10):1153–1163
Bloom CI, Graham CM, Berry MP, Rozakeas F, Redford PS, Wang Y et al (2013) Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PLoS ONE 8(8):e70630. https://doi.org/10.1371/journal.pone.0070630
Maertzdorf J, Weiner J 3rd, Mollenkopf HJ, Bauer T, Prasse A, Muller-Quernheim J et al (2012) Common patterns and disease-related signatures in tuberculosis and sarcoidosis. Proc Natl Acad Sci U S A 109(20):7853–7858
Garman L, Pelikan RC, Rasmussen A, Lareau CA, Savoy KA, Deshmukh US et al (2020) Single cell transcriptomics implicate novel monocyte and T cell immune dysregulation in sarcoidosis. Front Immunol 11:567342. https://doi.org/10.3389/fimmu.2020.567342
Yoshioka K, Sato H, Kawasaki T, Ishii D, Imamoto T, Abe M et al (2022) Transcriptome analysis of peripheral blood mononuclear cells in pulmonary sarcoidosis. Front Med (Lausanne) 9:822094. https://doi.org/10.3389/fmed.2022.822094
Ascoli C, Schott CA, Huang Y, Turturice BA, Wang W, Ecanow N et al (2022) Altered transcription factor targeting is associated with differential peripheral blood mononuclear cell proportions in sarcoidosis. Front Immunol 13:848759. https://doi.org/10.3389/fimmu.2022.848759
Khassawneh B, Zhu C, Barkes B, Vestal B, Shrock S, Gillespie M et al (2022) Autoantibody profile in sarcoidosis, analysis from the GRADS sarcoidosis cohort. PLoS ONE 17(10):e0274381. https://doi.org/10.1371/journal.pone.0274381
Smith D, Du Rand I, Addy CL, Collyns T, Hart SP, Mitchelmore PJ et al (2020) British thoracic society guideline for the use of long-term macrolides in adults with respiratory disease. Thorax 75(5):370–404. https://doi.org/10.1136/thoraxjnl-2019-213929
Fraser SD, Sadofsky LR, Kaye PM, Hart SP (2016) Reduced expression of monocyte CD200R is associated with enhanced proinflammatory cytokine production in sarcoidosis. Sci Rep 6:38689. https://doi.org/10.1038/srep38689
Vrančić M, Banjanac M, Nujić K, Bosnar M, Murati T, Munić V et al (2012) Azithromycin distinctively modulates classical activation of human monocytes in vitro. Br J Pharmacol 165(5):1348–1360. https://doi.org/10.1111/j.1476-5381.2011.01576.x
Iwanaga N, Nakamura S, Oshima K, Kajihara T, Takazono T, Miyazaki T et al (2015) Macrolides promote CCL2-mediated macrophage recruitment and clearance of nasopharyngeal pneumococcal colonization in mice. J Infect Dis 212(7):1150–1159. https://doi.org/10.1093/infdis/jiv157
Gschwandtner M, Derler R, Midwood KS (2019) More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol 10:2759. https://doi.org/10.3389/fimmu.2019.02759
Spyridaki A, Raftogiannis M, Antonopoulou A, Tsaganos T, Routsi C, Baziaka F et al (2012) Effect of clarithromycin in inflammatory markers of patients with ventilator-associated pneumonia and sepsis caused by Gram-negative bacteria: results from a randomized clinical study. Antimicrob Agents Chemother 56(7):3819–3825. https://doi.org/10.1128/aac.05798-11
Giamarellos-Bourboulis EJ, Siampanos A, Bolanou A, Doulou S, Kakavoulis N, Tsiakos K et al (2024) Clarithromycin for early anti-inflammatory responses in community-acquired pneumonia in Greece (ACCESS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. https://doi.org/10.1016/s2213-2600(23)00412-5
Culić O, Eraković V, Cepelak I, Barisić K, Brajsa K, Ferencić Z et al (2002) Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol 450(3):277–289. https://doi.org/10.1016/s0014-2999(02)02042-3
Kurdowska A, Noble JM, Griffith DE (2001) The effect of azithromycin and clarithromycin on ex vivo interleukin-8 (IL-8) release from whole blood and IL-8 production by human alveolar macrophages. J Antimicrob Chemother 47(6):867–870. https://doi.org/10.1093/jac/47.6.867
Tomazic J, Kotnik V, Wraber B (1993) In vivo administration of azithromycin affects lymphocyte activity in vitro. Antimicrob Agents Chemother 37(9):1786–1789. https://doi.org/10.1128/aac.37.9.1786
Nagano T, Yamaguchi T, Kajiyama S, Suzuki T, Matsushima Y, Yashima A et al (2021) Effect of azithromycin on proinflammatory cytokine production in gingival fibroblasts and the remodeling of periodontal tissue. J Clin Med. https://doi.org/10.3390/jcm10010099
Zimmermann P, Ziesenitz VC, Curtis N, Ritz N (2018) The immunomodulatory effects of macrolides-a systematic review of the underlying mechanisms. Front Immunol 9:302. https://doi.org/10.3389/fimmu.2018.00302
Vallet N, Le Grand S, Bondeelle L, Hoareau B, Corneau A, Bouteiller D et al (2022) Azithromycin promotes relapse by disrupting immune and metabolic networks after allogeneic stem cell transplantation. Blood 140(23):2500–2513. https://doi.org/10.1182/blood.2022016926
Tsai WC, Rodriguez ML, Young KS, Deng JC, Thannickal VJ, Tateda K et al (2004) Azithromycin blocks neutrophil recruitment in Pseudomonas endobronchial infection. Am J Respir Crit Care Med 170(12):1331–1339. https://doi.org/10.1164/rccm.200402-200OC
Wenisch C, Parschalk B, Zedtwitz-Liebenstein K, Weihs A, el Menyawi I, Graninger W (1996) Effect of single oral dose of azithromycin, clarithromycin, and roxithromycin on polymorphonuclear leukocyte function assessed ex vivo by flow cytometry. Antimicrob Agents Chemother 40(9):2039–2042
Parnham MJ, Culić O, Eraković V, Munić V, Popović-Grle S, Barisić K et al (2005) Modulation of neutrophil and inflammation markers in chronic obstructive pulmonary disease by short-term azithromycin treatment. Eur J Pharmacol 517(1–2):132–143. https://doi.org/10.1016/j.ejphar.2005.05.023
Ribeiro CM, Hurd H, Wu Y, Martino ME, Jones L, Brighton B et al (2009) Azithromycin treatment alters gene expression in inflammatory, lipid metabolism, and cell cycle pathways in well-differentiated human airway epithelia. PLoS ONE 4(6):e5806. https://doi.org/10.1371/journal.pone.0005806
Varenyiova Z, Rojas-Hernandez LS, Spano J, Capek V, Rosenberg-Hasson Y, Holmes T et al (2023) Azithromycin promotes proliferation, and inhibits inflammation in nasal epithelial cells in primary ciliary dyskinesia. Sci Rep 13(1):14453. https://doi.org/10.1038/s41598-023-41577-5
Renna M, Schaffner C, Brown K, Shang S, Tamayo MH, Hegyi K et al (2011) Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Investig 121(9):3554–3563. https://doi.org/10.1172/JCI46095
Drake WP, Culver DA, Baughman RP, Judson MA, Crouser ED, James WE et al (2021) Phase II investigation of the efficacy of antimycobacterial therapy in chronic pulmonary sarcoidosis. Chest 159(5):1902–1912. https://doi.org/10.1016/j.chest.2020.12.027
Vos R, Vanaudenaerde BM, Verleden SE, Vleeschauwer SID, Willems-Widyastuti A, Raemdonck DEV et al (2011) A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 37(1):164–172. https://doi.org/10.1183/09031936.00068310
Corris PA, Ryan VA, Small T, Lordan J, Fisher AJ, Meachery G et al (2015) A randomised controlled trial of azithromycin therapy in bronchiolitis obliterans syndrome (BOS) post lung transplantation. Thorax 70(5):442–450. https://doi.org/10.1136/thoraxjnl-2014-205998
Li H, Liu DH, Chen LL, Zhao Q, Yu YZ, Ding JJ et al (2014) Meta-analysis of the adverse effects of long-term azithromycin use in patients with chronic lung diseases. Antimicrob Agents Chemother 58(1):511–517. https://doi.org/10.1128/aac.02067-13
Funding
This work was funded by a research grant from SarcoidosisUK and consumables grant from NanoString® and Myriad-RBM®. PMK is supported by a Wellcome Trust Senior Investigator Award (WT104726).
Author information
Authors and Affiliations
Contributions
SDF, MGC, PMK, and SPH designed the study. STN, CW, RF, MGC, and SPH performed the clinical assessments. SDF conducted the cytokine assays and flow cytometry. SRJ performed the transcriptomic analyses. SDF and SPH analyzed the data. SPH has full access to all the data and takes responsibility for the integrity of the data and the accuracy of the data analysis. SDF, PMK, and SPH drafted the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics Approval
Ethics approval was granted by the Yorkshire & The Humber—Sheffield Research Ethics Committee (19/YH/0100).
Consent to Participate
Informed consent was obtained from all study participants.
Consent to Publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fraser, S.D., Thackray-Nocera, S., Wright, C. et al. Effects of Azithromycin on Blood Inflammatory Gene Expression and Cytokine Production in Sarcoidosis. Lung (2024). https://doi.org/10.1007/s00408-024-00743-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00408-024-00743-w