
Abstract
Increasing evidence implicates endo-lysosomal dysfunction in frontotemporal dementia (FTD). 18 proteins were quantified using a mass spectrometry assay panel in the cerebrospinal fluid of 36 people with the language variant of FTD, primary progressive aphasia (PPA) (including 13 with non-fluent variant (nfvPPA), 11 with semantic variant (svPPA), and 12 with logopenic variant (lvPPA)) and 19 healthy controls. The concentrations of the cathepsins (B, D, F, L1, and Z) as well as AP-2 complex subunit beta, ganglioside GM2 activator, beta-hexosaminidase subunit beta, tissue alpha l-fucosidase, and ubiquitin were decreased in nfvPPA compared with controls. In contrast, the concentrations of amyloid beta A4 protein, cathepsin Z, and dipeptidyl peptidase 2 were decreased in svPPA compared with controls. No proteins were abnormal in lvPPA. These results indicate a differential alteration of lysosomal proteins in the PPA variants, suggesting those with non-Alzheimer’s pathologies are more likely to show abnormal lysosomal function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The primary progressive aphasias (PPA) are disorders characterized by focal degeneration of the brain regions involved in language function and can be divided into three main subtypes: the non-fluent or agrammatic variant (nfvPPA), the semantic variant (svPPA), and the logopenic variant (lvPPA) [1]. These three variants are distinguished by the type of linguistic deficits with which they present, as well as their neuroanatomical signatures and underlying pathology [2]. For example, nfvPPA is most commonly a primary tauopathy as seen in progressive supranuclear palsy or corticobasal degeneration [3, 4] and svPPA is in most cases a TDP-43 proteinopathy [5], both reflecting frontotemporal lobar degeneration (FTLD) pathology, whereas lvPPA is usually caused by Alzheimer’s disease (AD) pathology [6].
Knowledge about the underlying pathophysiology of the PPA disorders, which are all usually sporadic, is limited. However, in other forms of frontotemporal dementia (FTD), one of the pathways that has been highlighted in recent years as likely to be affected, is the endo-lysosomal system [7,8,9]. There is increasing evidence that suggests alterations in this system, leading to dysfunctional proteostasis, play a key role in neurodegenerative disorders. The role of lysosomes in the cell is to break down proteins and maintain homeostasis of the cell through processes like endocytosis and autophagy. Impairment of the normal function of the lysosomal pathway leads to protein accumulation and aggregation and, therefore, neurodegeneration. Lysosomal dysfunction has been implicated particularly in the pathophysiology of progranulin-related FTD, where progranulin itself and related lysosomal proteins such as prosaposin, the cathepsins, and glucocerebrosidase are affected [9]. However, this has yet to be investigated in sporadic PPA. The ubiquitin–proteasome system (UPS) is similarly a major intracellular protein degradation system whose dysfunction has been associated with many neurological diseases including AD and amyotrophic lateral sclerosis (ALS) [10]. Disruption of the UPS leads to deficits in the clearance of misfolded proteins, in turn causing intracellular protein aggregation, cytotoxicity, and cell death [11]. Furthermore, it has been demonstrated that the UPS plays a key role degrading TDP-43 [12], and clearing hyperphosphorylated tau, as well as degrading intra-neuronal insoluble tau aggregates [13]. Overall, the endo-lysosomal network, therefore, plays an important role in the clearance of the core proteins that aggregate within FTD spectrum disorders.
One way of measuring a change in cellular function in vivo is to develop fluid biomarkers to assess a change in the concentration of proteins in different parts of the pathway [14]. In this study, we measured the cerebrospinal fluid (CSF) concentration of a panel of proteins involved in both the endo-lysosomal and ubiquitin–protease system to investigate the presence of dysfunction in these pathways within the PPA disorders.
Methods
Participants
Thirty-six people with sporadic PPA and available CSF were recruited through the Longitudinal Investigation of FTD (LIFTD) study at University College London (Table 1): 13 nfvPPA, 11 svPPA, and 12 lvPPA, diagnosed according to current consensus criteria [2]. All cases were negative for any of the genes that are causative of FTD including the C9orf72 expansion. Nineteen healthy controls were also recruited through the LIFTD study over the same time period. All patients with lvPPA had a biomarker profile consistent with underlying Alzheimer’s disease: mean (standard deviation) total tau/Aβ42 ratio of 3.2 (2.2) with a range of 1.2–8.3 where > 1 is considered abnormal. All nfvPPA and svPPA participants and all controls had a ratio of < 1.
CSF samples
CSF was collected from all participants in polypropylene tubes through a lumbar puncture and centrifuged to remove insoluble material and cells. Supernatants were aliquoted and stored at − 80 °C within 2 h after withdrawal. CSF Aβ42, total tau, and phosphorylated tau concentrations were measured using commercially available enzyme-linked immunosorbent assays (INNOTEST; Fujirebio Europe, Ghent, Belgium), according to the manufacturer’s instructions.
Sample digestion and solid-phase extraction
Digestion and solid-phase extraction (SPE) were performed as described previously [15, 16]. One hundred μL of CSF from subject samples was mixed with internal standard and then reduced and alkylated with 1,4-dithiothreitol and iodoacetamide, respectively. The samples were then digested using sequencing grade modified trypsin (Promega Co., Madison, WI, USA). Trypsination was ended by the addition of trifluoroacetic acid and was followed by SPE using Oasis HLB 96-well μElution Plates (2 mg sorbent and 30 μm particle size; Waters Co., Milford, MA, USA) according to the generic protocol of the manufacturer. As the final step, the samples were eluted in methanol and dried by vacuum centrifugation. The samples were frozen and stored at − 80 °C pending analysis.
Parallel reaction monitoring-mass spectrometry
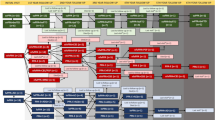
Eighteen proteins (49 peptides) were quantified by parallel reaction monitoring-mass spectrometry (PRM-MS) and are shown in Fig. 1 as well as being listed in Table 2: AP-2 complex subunit beta (AP2B1), amyloid beta A4 protein (APP), complement component C9, cathepsins B, D, F, L1, Z, dipeptidyl peptidase 2 (DPP2) ganglioside GM2 activator (GM2A), beta-hexosaminidase subunit beta (HEXB), lysosome-associated membrane glycoprotein 1 (LAMP1), lysosome-associated membrane glycoprotein 2 (LAMP2), lysozyme C, tissue alpha-l-fucosidase T-ALF, transcobalamin-2, tripeptidyl peptidase 1 (TPP1), and ubiquitin. PRM-MS analysis was performed as described previously [16] using an UltiMate 3000 standard-LC system (Thermo Fisher Scientific Inc., Waltham, MA, USA) and a Hypersil GOLD HPLC C18 column (length 200 mm; inner diameter 2.1 mm; particle size 1.9 μm; Thermo Fisher Scientific Inc.), and a Q Exactive mass spectrometer (Thermo Fisher Scientific Inc.). Electrospray ionization was performed in positive ion mode with a Heated Electrospray Ionization (HESI-II) probe (Thermo Fisher Scientific Inc.). Acquisition of single microscans was performed in PRM mode with an isolation window of m/z 3, a resolution setting 70 k, an AGC target 1 × 106, a maximum injection time 300 ms, and fragmentation with beam type collision-induced dissociation (HCD). Peak detection and area integration were performed using Skyline v3.6 [17], targeting [M + H]1+ y-ions with a data-independent acquisition method setting and a fixed isolation window of m/z 3 and an orbitrap analyzer resolution setting of 70 k at m/z 200. The concentration of the peptides is expressed as a ratio between the sum of fragment ions of the tryptic peptide against the corresponding heavy labeled internal standard peptide (L/H peptide ratio). For the proteins for which more than one peptide was quantified, the peptide with the best analytical performance (lowest coefficient of variation) was selected.

Schematic diagram showing the endo-lysosomal system and the proteins measured in the study (see main text for abbreviations)
Statistical analyses
All statistical analyses were performed in STATA (v.16) and RStudio (R version 4.0.2). The Shapiro–Wilk test was performed to determine the normality of distribution of each endo-lysosomal marker in each group. The levels of each endo-lysosomal and ubiquitin protein were compared between groups using a linear regression model adjusting for age at CSF sample collection and sex; bootstrapping with 2000 repetitions was used if the measures were not normally distributed.
Results
The concentration of ten out of the 18 proteins was lower in nfvPPA compared with both controls and lvPPA: AP2B1, cathepsins B, D, F, L1 and Z, GM2A, HEXB, T-ALF, and ubiquitin (Table 2, Fig. 2). Four further proteins were lower in nfvPPA compared with lvPPA: APP, complement 9, LAMP1, and transcobalamin-2 (Table 2, Fig. 2).

Altered CSF endo-lysosomal protein concentration in nfvPPA and svPPA groups compared to controls and lvPPA. Concentrations are expressed as a ratio between measured area of the tryptic peptide against the corresponding internal standard heavy label peptide (L/H peptide ratio). p values: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001. The bars indicate the median and interquartile range. AP2B1 AP-2 complex subunit beta, APP amyloid beta A4 protein, DPP2 dipeptidyl peptidase 2, GM2A ganglioside GM2 activator, HEXB beta-hexosaminidase subunit beta, LAMP1 lysosome-associated membrane glycoprotein 1, T-ALF tissue alpha-l-fucosidase
Three proteins were at lower levels in svPPA compared with both controls and lvPPA: APP, cathepsin Z, and DPP2 (Table 2, Fig. 2). Three additional proteins were lower in svPPA compared with lvPPA: cathepsin B, LAMP1, and lysozyme C (Table 2, Fig. 2). There were no significant differences between the lvPPA and controls, although for many of the peptides, there was a trend to higher levels in the lvPPA group.
Group differences are summarized in Table 3.
Discussion
In this study, we show that there are abnormalities in the CSF concentrations of proteins associated with endocytosis, lysosomal function, and the ubiquitin–proteasome system in PPA. Interestingly, there was a decrease in the concentrations of proteins in those with a form of FTLD pathology (i.e., svPPA and nfvPPA) with an opposite trend in those with underlying AD pathology (lvPPA). The nfvPPA (usually due to a tauopathy) showed changes across multiple proteins, suggesting dysfunction in endocytosis and chaperone-mediated autophagy in this disorder. A more limited set of proteins were decreased in svPPA (usually a TDP-43 proteinopathy), but nonetheless, indicating abnormalities in the endo-lysosomal pathway in this condition.
The cathepsins are proteases whose key function in the lysosome is the degradation of proteins [18, 19]. To our knowledge, there are no previous reports on the levels of cathepsins in the biofluids of people with PPA, with the only previous study in an unspecified FTD cohort showing an increase in cathepsin D in plasma exosomes [7]. CSF cathepsins have been poorly studied in general in neurodegenerative diseases with few studies investigating their concentrations, e.g., a previous study reported a decrease in cathepsin B and cathepsin F in Parkinson’s disease (PD) when compared to controls and prodromal AD [16]; while in another study, cathepsin D was increased in AD [20]. In our study, while multiple cathepsins are decreased in nfvPPA (B, D, F, L1, and Z), only cathepsins B and Z are decreased in both nfvPPA and svPPA, particularly in relation to lvPPA. Interestingly, cathepsin D has been particularly implicated in progranulin-related FTD pathophysiology before [21] and clearly, further work is needed to understand how these other cathepsins are involved in these other pathological forms of FTD.
We further report a significant decrease in HEXB, a protein involved in chaperone-mediated autophagy, in nfvPPA in this study. To our knowledge, this has not been previously reported to be altered in the CSF of neurodegenerative disorders [16]. Lysosomal β-hexosaminidase A is a heterodimeric complex composed of HEXB and subunit alpha [22]. β-hexosaminidase A hydrolyses ganglioside GM2 with the aid of ganglioside GM2 activator [23], which of particular interest, is also decreased in the nfvPPA.
The LAMP proteins are also implicated in lysosomal autophagy, with functions in vesicle fusion [24], preserving lysosomal integrity and lysosomal exocytosis [25]. LAMP1 has been recently studied as a potential candidate marker of lysosomal alteration in neurodegenerative diseases with decreases shown in CSF in PD [8, 16, 26], although it has been previously reported to be unaltered in FTD [7]. Both proteins have been investigated in AD, with increases shown in previous studies [27]. However, in this study, there is a decrease in LAMP1 but not LAMP2, in the FTLD-related disorders, a key difference from lvPPA, a disorder with AD pathology.
Endocytosis and clathrin-mediated formation of the early endosome are the starting points of the endo-lysosomal pathway. AP2B1 and APP are implicated in this early stage of the lysosomal system [28,29,30]. AP2B is specifically altered in the nfvPPA group and has been reported to be significantly increased in AD and decreased in PD [16]. Our results regarding AP2B1 and APP suggest that the early stages of the lysosomal pathway are specifically altered in the FTLD-linked disorders (and potentially particularly the primary tauopathies) when compared to those disorders with underlying AD pathology.
Finally, we also studied the levels of ubiquitin in CSF. A key role of ubiquitin involves labeling proteins for degradation by the proteasome [31], but it also has a multitude of functions as a post-translational modification [32]. It has been reported to be increased in AD [16, 33]; however, our results in the FTLD-associated disorders parallel what has been found in other neurodegenerative diseases such as PD in which ubiquitin levels are decreased when compared to controls and AD [16].
Overall, the present study shows a decrease in protein degradation in FTLD-associated disorders nfvPPA and svPPA when compared to the AD group (lvPPA) and controls. We also see a trend to an increase in the levels of the proteins measured in the lvPPA group similar to that seen in typical amnestic AD. These results suggest a dysfunction of endo-lysosomal and ubiquitin systems in the FTD spectrum that will lead to a decrease in the degradation of proteins and possible accumulation.
There are a number of limitations of the study. We did not have access to detailed behavioral or neuropsychometry data within the cohort and it would be useful for future studies to investigate the correlation of clinical features with endo-lysosomal proteins and ubiquitin levels. The presence of co-morbidities such as systemic disease, mood disorders, and cerebrovascular disease (including for the latter, the presence of white matter hyperintensities on MRI) was also not evaluated: their effect on lysosomal-associated protein levels would be important to investigate in further analyses. While each group was of similar disease duration (time since symptom onset), participants were on average around 3–5 years into their illness. It would, therefore, be helpful to study both people very early in their clinical syndrome as well as to investigate longitudinal change in endo-lysosomal proteins and ubiquitin in PPA to understand the temporal relationship within the disease.
Conclusions
This study highlights the complex endo-lysosomal system in the different variants of PPA and shows clear differences between those with AD and FTLD pathology. Our results establish a baseline for further study of the role of endo-lysosomal and ubiquitin proteins in PPA with the potential role of lysosomal dysfunction as a therapeutic target in these sporadic disorders, an important area of future research.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Marshall CR, Hardy CJD, Volkmer A, Russell LL, Bond RL, Fletcher PD, Clark CN, Mummery CJ, Schott JM, Rossor MN, Fox NC, Crutch SJ, Rohrer JD, Warren JD (2018) Primary progressive aphasia: a clinical approach. J Neurol 265:1474–1490
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014
Graff-Radford J, Duffy JR, Strand EA, Josephs KA (2012) Parkinsonian motor features distinguish the agrammatic from logopenic variant of primary progressive aphasia. Parkinsonism Relat Disord 18:890–892
Kremen SA, Mendez MF, Tsai PH, Teng E (2011) Extrapyramidal signs in the primary progressive aphasias. Am J Alzheimers Dis Other Demen 26:72–77
Hodges JR, Patterson K (2007) Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol 6:1004–1014
Henry ML, Gorno-Tempini ML (2010) The logopenic variant of primary progressive aphasia. Curr Opin Neurol 23:633–637
Goetzl EJ, Boxer A, Schwartz JB, Abner EL, Petersen RC, Miller BL, Kapogiannis D (2015) Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85:40–47
Boman A, Svensson S, Boxer A, Rojas JC, Seeley WW, Karydas A, Miller B, Kagedal K, Svenningsson P (2016) Distinct lysosomal network protein profiles in Parkinsonian syndrome cerebrospinal fluid. J Parkinsons Dis 6:307–315
Parnetti L, Balducci C, Pierguidi L, De Carlo C, Peducci M, D’Amore C, Padiglioni C, Mastrocola S, Persichetti E, Paciotti S, Bellomo G, Tambasco N, Rossi A, Beccari T, Calabresi P (2009) Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in Dementia with Lewy Bodies. Neurobiol Dis 34:484–486
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Schwartz AL, Ciechanover A (2009) Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol 49:73–96
Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H, Sardone V, Mitchell JC, Rogelj B, Rubinsztein DC, Shaw CE (2014) Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci 127:1263–1278
Jiang S, Bhaskar K (2020) Degradation and transmission of tau by autophagic-endolysosomal networks and potential therapeutic targets for tauopathy. Front Mol Neurosci 13:586731
Blennow K, Zetterberg H, Fagan AM (2012) Fluid biomarkers in Alzheimer disease. Cold Spring Harb Perspect Med. 2(9):a006221
Brinkmalm G, Sjödin S, Simonsen AH, Hasselbalch SG, Zetterberg H, Brinkmalm A, Blennow K (2018) A parallel reaction monitoring mass spectrometric method for analysis of potential CSF biomarkers for Alzheimer’s disease. Proteomics Clin Appl 12:1700131
Sjödin S, Brinkmalm G, Öhrfelt A, Parnetti L, Paciotti S, Hansson O, Hardy J, Blennow K, Zetterberg H, Brinkmalm A (2019) Endo-lysosomal proteins and ubiquitin CSF concentrations in Alzheimer’s and Parkinson’s disease. Alzheimers Res Ther 11:82
MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26:966–968
Zaidi N, Maurer A, Nieke S, Kalbacher H (2008) Cathepsin D: a cellular roadmap. Biochem Biophys Res Commun 376:5–9
Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824:68–88
Schwagerl AL, Mohan PS, Cataldo AM, Vonsattel JP, Kowall NW, Nixon RA (1995) Elevated levels of the endosomal-lysosomal proteinase cathepsin D in cerebrospinal fluid in Alzheimer disease. J Neurochem 64:443–446
Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, Krainc D (2017) Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet 26:4861
Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MNG (2006) Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis. J Mol Biol 359:913–929
Kolter T, Sandhoff K (2010) Lysosomal degradation of membrane lipids. FEBS Lett 584:1700–1712
Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S (2007) LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J 26:313–324
Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10:623–635
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH (2009) Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35:385–398
Armstrong A, Mattsson N, Appelqvist H, Janefjord C, Sandin L, Agholme L, Olsson B, Svensson S, Blennow K, Zetterberg H, Kågedal K (2014) Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromolecular Med 16:150–160
Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2(5):a006270
Müller UC, Zheng H (2012) Physiological functions of APP family proteins. Cold Spring Harb Perspect Med. 2(2):a006288
McMahon HT, Boucrot E (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12:517–533
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479
Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81:203–229
Blennow kaj T, Davidsson P, Wallin A, Gottfries CG, Svennerholm L, (1994) Ubiquitin in cerebrospinal fluid in Alzheimer’s disease and vascular dementia. Int Psychogeriatr 6:13–22
Acknowledgements
We thank the research participants for their contribution to the study.
Funding
The Dementia Research Centre is supported by Alzheimer’s Research UK, Alzheimer’s Society, Brain Research UK, and The Wolfson Foundation. This work was supported by the NIHR UCL/H Biomedical Research Centre, the Leonard Wolfson Experimental Neurology Centre (LWENC) Clinical Research Facility, and the UK Dementia Research Institute, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. ASE is funded by Race Against Dementia fellowship, supported by Alzheimer’s Research UK (ARUK-RADF2021A-003). ASE is also supported by the UK Dementia Research Institute which receives its funding from DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. IJS is supported by the Alzheimer’s Association. KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-930351, #AF-939721 and #AF-968270), Hjärnfonden, Sweden (#FO2017-0243 and #ALZ2022-0006), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986 and #ALFGBG-965240), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), the National Institute of Health (NIH), USA, (grant #1R01AG068398-01), and the Alzheimer’s Association 2021 Zenith Award (ZEN-21-848495). HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712 and #101053962), Swedish State Support for Clinical Research (#ALFGBG-71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376-C, #ADSF-21-831381-C and #ADSF-21-831377-C), the Bluefield Project, the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2022-0270), the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), the European Union Joint Programme—Neurodegenerative Disease Research (JPND2021-00694), and the UK Dementia Research Institute at UCL (UKDRI-1003). JDR has received funding from an MRC Clinician Scientist Fellowship (MR/M008525/1) and an NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH); his work is also supported by the MRC UK GENFI grant (MR/M023664/1), the Bluefield Project and the JPND GENFI-PROX grant (2019-02248).
Author information
Authors and Affiliations
Contributions
AS-E, IJS, and JDR: analyzed and interpreted the data and wrote the initial draft of the manuscript. SS: measured the samples under supervision of JG, AB, KB, and HZ. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Passage Bio, Pinteon Therapeutics, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). JDR has served on medical advisory boards and consultancy for Alector, Arkuda Therapeutics, Wave Life Sciences, and Prevail Therapeutics. Consultancy for UCB, AC Immune, Astex Pharmaceuticals, Biogen, Takeda and Eisai. KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, BioArctic, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Ono Pharma, Pharmatrophix, Prothena, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, outside the work presented in this paper. ASE, SS, JG, AB, and IJS have nothing to disclose.
Ethical approval
The London Queen Square Ethics committee approved the study. The study complies with the Declaration of Helsinki.
Consent for publication
All participants provided written informed consent at enrollment including consent to publication.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Swift, I.J., Sjödin, S., Gobom, J. et al. Differential patterns of lysosomal dysfunction are seen in the clinicopathological forms of primary progressive aphasia. J Neurol 271, 1277–1285 (2024). https://doi.org/10.1007/s00415-023-12063-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-12063-9