
Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) anion channel and the epithelial Na+ channel (ENaC) play essential roles in transepithelial ion and fluid transport in numerous epithelial tissues. Inhibitors of both channels have been important tools for defining their physiological role in vitro. However, two commonly used CFTR inhibitors, CFTRinh-172 and GlyH-101, also inhibit non-CFTR anion channels, indicating they are not CFTR specific. However, the potential off-target effects of these inhibitors on epithelial cation channels has to date not been addressed. Here, we show that both CFTR blockers, at concentrations routinely employed by many researchers, caused a significant inhibition of store-operated calcium entry (SOCE) that was time-dependent, poorly reversible and independent of CFTR. Patch clamp experiments showed that both CFTRinh-172 and GlyH-101 caused a significant block of Orai1-mediated whole cell currents, establishing that they likely reduce SOCE via modulation of this Ca2+ release-activated Ca2+ (CRAC) channel. In addition to off-target effects on calcium channels, both inhibitors significantly reduced human αβγ-ENaC-mediated currents after heterologous expression in Xenopus oocytes, but had differential effects on δβγ-ENaC function. Molecular docking identified two putative binding sites in the extracellular domain of ENaC for both CFTR blockers. Together, our results indicate that caution is needed when using these two CFTR inhibitors to dissect the role of CFTR, and potentially ENaC, in physiological processes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
CFTR and ENaC play essential roles in ion and fluid transport in numerous epithelial tissues, dysfunction of which leads to diseases such as cystic fibrosis (CF), secretory diarrhoea and kidney disease [1]. Two drugs assumed to be relatively selective CFTR inhibitors, CFTRinh-172 and GlyH-101 [2, 3], have been important tools for defining the role of CFTR in transepithelial ion transport in vitro. Despite being widely used, these agents are not solely selective for CFTR, as they inhibit other types of Cl− channels and even affect mitochondrial function [4,5,6]. Moreover, GlyH-101 also exhibited off-target effects on voltage-gated Ca2+ and K+ channels in isolated ventricular myocytes [7]. Together, these studies suggest that these CFTR inhibitors may have more widespread off-target effects than originally appreciated. However, the potential modulatory effects of these inhibitors on epithelial cation channels to date have not been investigated. This issue is important because CFTR has been shown to modulate Ca2+ signalling in epithelial cells via multiple mechanisms, which ultimately leads to raised cytosolic Ca2+ levels and cellular dysfunction [8]. These mechanisms include altered release of Ca2+ from ER stores, as well as enhanced Ca2+ influx through Ca2+-permeable channels, such as the store-operated calcium entry (SOCE) channel Orai1 and transient receptor potential (TRP) channels [9]. Disrupted Ca2+ signalling due to the absence or malfunction of CFTR can also alter mitochondrial function, which can lead to cell injury, and ultimately activate apoptosis or necrosis [8]. In addition to effects on Ca2+ signalling, CFTR dysfunction also stimulates epithelial Na+ transport into cells, via increased ENaC activity [10]. However, the exact molecular mechanisms involved in either of these actions of CFTR on epithelial cation transport are incompletely understood. Significantly, some of the published studies implicating CFTR in the regulation of epithelial Ca2+ homeostasis have employed CFTR inhibitors to validate the role of this anion channel in Ca2+ signalling [11,12,13,14,15]. We therefore felt it was important to investigate if these CFTR inhibitors had modulatory effects on Ca2+-entry channels, both in the presence and absence of functional CFTR, as well as investigate any off-target effects directly on ENaC function. Here, we demonstrate for the first time that CFTR blockers cause a significant inhibition of SOCE, via block of the Orai1 Ca2+ influx channel, as well isoform-specific modulation of ENaC. Our results therefore highlight that care needs to be taken when assigning a specific physiological, or pathophysiological, role for CFTR in epithelial function based on the results using these two CFTR inhibitors.
Materials and methods
Cell culture
Calu-3 cells were cultured in Eagle’s minimum essential medium; HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) or in DMEM low glucose medium. All culture media were supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 1% non-essential amino-acid (NEAA) and 10% foetal bovine serum (FBS).
Transfection of cells
HEK293T cells were transfected using Lipofectamine 3000 (as per manufacturer’s instructions), with a bicistronic IRES plasmid vector encoding hOrai1 and CD8, and a plasmid encoding hStromal Interacting Molecule 1 (STIM1), at a ratio of 1/10, respectively. Control cells were mock transfected with an empty pcDNA3.1 vector (mock) and CD8 [16]. Transfected cells were visually detected by binding of anti-CD8 labelled beads [17]. All experiments were performed 48 h after transfection.
Real‐time quantitative PCR
Calu-3 and HEK293 cells were lysed for RNA extraction using the RNeasy Mini Kit (Qiagen). Reverse transcription and real-time qPCR was performed as previously described [18]. PCR primers (CFTR (Forward – AGGAGGCAGTCTGTCCTGAA; Reverse – CACTGCTGGTATGCTCTCCA), GAPDH (Forward – TGCACCACCAACTGCTTAGC; Reverse – GGCATGGACTGTGGTCATGAG)) were purchased from Integrated DNA Technologies (Leuven, Belgium).
Western blotting
Calu-3 and HEK293 cells were lysed in ice-cold RIPA buffer, and Western blot was performed as previously described [18]. A total of 30 μg of protein were loaded onto an 8% SDS-PAGE gel for electrophoresis, and protein was transferred onto a PVDF membrane (0.1 A constant for 60 min). Membranes were blocked with 5% milk (1 h at room temperature (RT)), then incubated with the primary (1:1000, overnight at 4 °C) and secondary antibody (1:5000, 1 h at RT). Primary antibody was CFFT-596, raised in mouse, from the Cystic Fibrosis Foundation’s Antibody Distribution Program.
Intracellular Ca2+ measurements
Intracellular Ca2+ ([Ca2+]i) was measured using the Ca2+-sensitive fluorescent dye fura-2-acetoxymethylester (fura-2-AM; Thermo Fisher Scientific) using cells grown on 25-mm glass coverslips as previously described [19]. Fura-2 was excited alternately at 340 nm and 380 nm for 250 ms, and emitted light captured at 510 nm; the 340/380 emission ratio (F340/380) of fura-2 fluorescence reflects [Ca2+]i levels. Store-operated channels (SOCs) channels were activated by depleting [Ca2+]i stores with 200 nM thapsigargin (Tg), in nominally Ca2+-free conditions, and then SOCE measured by tracking changes in [Ca2+]i using a repeated extracellular Ca2+ addback protocol, alternating between a Ca2+-free solution and one containing 1 mM Ca2+. The composition of the HEPES-buffered solution was (in mM): 130 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid sodium salt (NaHEPES), 10 D-glucose, (pH 7.4). For the nominally Ca2+-free solution, CaCl2 was omitted and replaced with 1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N,N-tetraacetic acid (EGTA). SOCE amplitude was calculated as the difference between baseline F340/380 ratio (average of 30 s data) before a response and maximum peak value (average of 1–2 s of data) and calculated as peak–baseline. SOCE rate was calculated by linear regression to the initial, steepest, portion of the [Ca2+]i increase within the first few minutes after calcium addback.
Whole cell patch clamp
Cells were patch clamped after growing them on coated glass coverslips for 2 days. Patch clamp experiments were performed in the fast whole-cell configuration. Patch pipettes had an input resistance of 3–6 MΩ, when filled with a solution containing in (mM) KCl 30, K+-gluconate 95, NaH2PO4 1.2, Na2HPO4 4.8, EGTA 1, Ca2+-gluconate 0.758, MgCl2 1.034, D-glucose 5, ATP 3, pH 7.2 and Ca2+ activity 0.1 μM. The bath was perfused continuously with a standard bicarbonate-free Ringer’s solution, composed of (in mM) NaCl 145, KH2PO4 0.4, K2 HPO4 1.6, MgCl2 1, Ca-gluconate 1.3, glucose 5, pH 7.4) at a rate of 4 mL/min. The access conductance was continuously measured and was 40–100 nS. Currents were recorded with an EPC-7 patch clamp amplifier (List Medical Electronics, Germany), the LIH1600 interface and PULSE software (HEKA, Germany) as well as Chart software (AD-Instruments, Germany). Data were stored continuously on a computer hard disc and analysed using PULSE software. In regular intervals, membrane voltages (Vc) were clamped in steps of 20 mV from − 100 to + 100 mV relative to resting potential.
Plasmids and cRNA synthesis
Coding DNA sequences for human ENaC subunits (α, β, γ and δ) were present in the pTNT expression vector (Promega, UK). Plasmids were linearised with FastDigest BamH1 (ThermoFisher Scientifc, UK) and capped cRNA was generated using the T7 Ribo-MAX large-scale RNA production system (Promega) following manufacturer’s instructions. ENaC-subunits were diluted with diethyl pyrocarbonate (DEPC)-treated water to a final concentration of 10 ng/μl per ENaC subunit and combined to αβγ-ENaC or δβγ-ENaC cRNA solutions.
Heterologous expression of ENaC in Xenopus oocytes
Xenopus laevis ovaries were purchased from the European Xenopus Resource Centre (EXRC, Portsmouth, UK). The experimental procedures were approved by the Newcastle University Animal Welfare and Ethical Review Body (AWERB, project ID 630). Oocytes were isolated by incubation in collagenase and Ca2+-free solution as previously described [20]. Stage V/VI oocytes were injected (Nanoject, Drummond Scientific, Broomall, USA) with 13.8 nl of αβγ-ENaC or δβγ-ENaC cRNA, or DEPC-treated water for control experiments. Injected oocytes were incubated for 24–48 h at 16 °C in a low sodium oocyte Ringer solution containing (in mM) 80 N-methyl-D-glucamine, 10 NaCl, 1 KCl, 2 CaCl2, 2.5 sodium pyruvate, 5 HEPES (pH 7.4) and supplemented with 20 μg/ml gentamycin. All procedures for oocyte isolation, expression of human ENaC subunits (α, β, γ and δ) were exactly as previously described [20].
Two-electrode voltage-clamp recordings
Oocytes were placed in a recording chamber filled with oocyte ringer solution (ORS), containing (in mM) 90 NaCl, 1 KCl, 2 CaCl2 and 5 HEPES (pH 7.4). Chloride-coated silver wires were inserted into microelectrodes, which were pulled from borosilicate glass capillaries and filled with 1 M KCl. For reference electrodes, chloride-coated silver wires were placed into 1 M KCl/agar (3%) bridges. Oocytes were clamped to − 60 mV with an OC725B/C oocyte voltage clamp amplifier (Warner Instruments, Hamden, USA) and transmembrane currents (IM) were filtered at 1 kHz (Warner Instruments) and recorded on a strip chart recorder (Kipp&Zonen, Delft, The Netherlands). Experiments were performed at RT under continuous superfusion (5 ml/min) of oocytes with ORS.
Molecular docking
Docking experiments were carried out using the cryo-EM structure of ENaC (pdb: 6bqn) and the closed and open structures of Orai1 (pdb: 4hkr and 7kr5). Missing loops in the proteins and side chains were added via comparative structure modelling with Modeller [21]. The three-dimensional structures of CFTRinh-172, GlyH-101 and amiloride were obtained from PubChem. AutodockTools [22] was used to prepare the protein and ligand structures. Initial blind docking with subsequent targeted docking for individual binding pockets in ENaC were performed using Autodock Vina [23], and an exhaustiveness of 56.
Chemicals
All reagents, including amiloride hydrochloride hydrate, were purchased from Sigma-Aldrich (UK, Germany) unless otherwise specified. Thapsigargin (Tg), CFTRinh-172, GlyH-101 and cyclopiazonic acid (CPA) were purchased from Tocris (Abington, UK) or (Deisenhofen Germany). Dimethyl sulfoxide (DMSO) from ThermoFisher Scientific (Gloucester, UK). For all experiments, stock solutions of 20 mM CFTRinh-172, 100 mM GlyH-101 and 100 mM amiloride were prepared in DMSO. Stock solutions were diluted in bathing solutions and DMSO concentrations were kept at, or below, 0.2%.
Statistics
Statistical analysis was performed using GraphPad Prism 8 (GraphPad Software, San Diego, USA), with statistical significance indicated by an alpha value of p < 0.05. The type of analysis for each dataset, including post-hoc analysis, is indicated in the respective figure legend.
Results
CFTRinh-172 causes a time-dependent reduction in SOCE in Calu-3 cells
Calu-3 cells are a human airway-derived cell line that expresses high levels of functional CFTR [24], which can be effectively inhibited by application of 20 µM CFTRinh-172 [2, 6, 25], which was confirmed by our own studies (Supplementary Fig. 1). To investigate the effect of CFTRinh-172 on SOCE in these cells, intracellular Ca2+ stores were first passively depleted with 200 nM thapsigargin (a SERCA pump inhibitor), in nominally Ca2+-free conditions, to activate store-operated channels (SOC). Figure 1 shows that the amplitude of SOCE, measured using a repeated Ca2+-addback protocol (see Material and Methods), was significantly reduced by pre-treating cells with 20 µM CFTRinh-172 in a time-dependent manner (24.2 ± 12.3% decrease after 10 min, p = 0.026 and 44.2 ± 10.0% after 30-min treatment, p < 0.0001 versus vehicle-treated cells). The initial rate of SOCE was also reduced for the 30 min pre-treated group (52.0 ± 16.4%, p = 0.004). Overall, the frequency of observing a greater than 10% inhibition in both amplitude and rate of SOCE by CFTRinh-172 was time-dependent (Fig. 1F; p < 0.05, chi-square test). The inhibitory effect of CFTRinh-172 was essentially irreversible up to 30 min of washout of the compound (Fig. 1D).

Time-dependent inhibition of SOCE by CFTRinh-172 in Calu-3 cells. A–D Representative Ca2+ fluorimetry traces tracking changes in [Ca2+]i following a Tg-activated, repeated Ca2+ addback protocol. Cells were exposed to the vehicle DMSO (A), or 20 µM CFTRinh-172 (CFinh172, B-D), in a nominally Ca2+ free solution for 3 (B), 10 (C), or 30 (D) min before the second SOCE was induced by adding back 1 mM Ca.2+ (black bars). The agents were removed from the perfusing solution for 15 (B, C) or 30 (D) min before the third SOCE was activated. (E) Box and whiskers summary of percentage change in SOCE peak amplitude and rate, from SOCE #1 to SOCE #2, following DMSO (control) or CFTRinh-172 treatment of different durations. One-way ANOVA with Holm-Sidak multiple comparisons tests was performed across the four groups. Boxes represent median ± 25th/75th percentiles, while whiskers represent minimum/maximum. * = p < 0.05 vs. control; # = p < 0.05 vs. 3 min. F Frequency of > 10% inhibition of SOCE amplitude and rate by DMSO or CFTRinh-172 treatment of different durations. Percentage inhibition was calculated as percentage of SOCE #2 amplitude/rate over that of SOCE #1, and the frequency of experiments with over and under 10% inhibition was tallied. A chi-square test was performed across the four groups. χ = p < 0.05 for chi-square test. n = 4–14
CFTRinh-172 and GlyH-101 inhibit SOCE in epithelial cells that do not express CFTR
To investigate if the reduction in SOCE by CFTRinh-172 was dependent on CFTR, we repeated the Ca2+-addback protocol on HEK293T cells, which do not express CFTR (Fig. 2A, B). We also tested another well-known CFTR inhibitor, GlyH-101, on SOCE. Figure 2C–F show that both CFTR inhibitors reduced SOCE amplitude and rate. Pre-treatment of HEK293 cells with 20 µM CFTRinh-172 for 30 min or 10 µM GlyH-101 for 10 min, inhibited the amplitude (50.7 ± 10.3%, p = 0.003 and 38.5 ± 8.0%, p = 0.001, respectively, vs. vehicle-treated) and rate (67.5 ± 10.5%, p = 0.002 and 65.7 ± 7.3%, p = 0.002, respectively, vs. vehicle-treated) of SOCE. These results clearly show that the inhibitory effect of these two putative CFTR inhibitors is not dependent on CFTR expression.

Distinct CFTR inhibitors reduce SOCE in HEK293T cells. A, B HEK293T cells do not express CFTR. A Raw CT values for amplification of CFTR, and the housekeeping gene GAPDH, in Calu-3 and HEK293T cell samples. Each sample was run in duplicate. B Western blot image of Calu-3 and HEK293T samples probed for CFTR expression (Band C ~ 180 kDa, fully glycosylated CFTR). C–E Representative Ca2+ fluorimetry traces tracking changes in [Ca2+]i following a Tg-activated, repeated Ca.2+ addback protocol, with pre-treatment of DMSO (C), 20 µM CFTRinh-172 for 30 min (D) or 10 µM GlyH-101 for 10 min (E) before the second SOCE was activated. F Box and whiskers summary of percentage change in SOCE peak amplitude and rate, from SOCE #1 to SOCE #2, following treatment of DMSO (control) or the CFTR inhibitors. One-way ANOVA with Holm-Sidak multiple comparisons tests was performed across the three groups. Boxes represent median ± 25th/75th percentiles, while whiskers represent minimum/maximum. * = p < 0.05 vs. control. n = 5–6
CFTRinh-172 and GlyH-101 inhibit Orai1/Stim1-mediated currents in transfected HEK293T cells
The results in Fig. 2 clearly show that both CFTR inhibitors reduced SOCE activity in a CFTR-independent manner. To gain insight into the mode of action of these two inhibitors, we investigated their effect on Orai1-mediated whole cell currents in HEK293T cells, using the patch clamp technique. Ca2+ release-activated Ca2+ (CRAC; CRACM) channels responsible for SOCE are small and difficult to detect [26, 27]. While SOCE often can be measured by fluorimetry, CRAC-mediated currents produced by endogenous Orai1/Stim1 channels cannot be resolved in the presence of physiological ion concentrations. In order to amplify CRAC, we used cells overexpressing Orai1 and Stim1 to aid measurements of CRAC currents [26], and activated them by store depletion using CPA, another SERCA pump inhibitor. CPA induced a small, but not significant, current in mock-transfected cells, which was larger in Orai1/Stim1-overexpressing cells. Figure 3A, B, D and E show current/voltage relationships of the inward currents after stimulation with CPA, and in the presence of different concentrations of CFTRinh-172 and GlyH-101. At maximal concentrations of both inhibitors, inward currents were significantly reduced in overexpressing cells, but not in non-transfected cells. Concentration-dependent inhibition of inward currents was determined by setting the inward currents at Vc = − 100 mV, in the absence of inhibitors, to 100%, and relating this to the inward currents in the presence of different inhibitor concentrations (Fig. 3C, F). While lower concentrations of each inhibitor did not affect inward currents, maximal concentrations of CFTRinh-172 and GlyH-101 clearly inhibited CPA-activated inward currents in Orai1/Stim1 overexpressing cells, suggesting block of CRAC by inhibitors of CFTR. We confirmed that both CFTR inhibitors significantly inhibited CFTR-dependent whole cell currents in transfected HEK cells (Supplementary Fig. 2).

CFTRinh-172 and GlyH-101 inhibit Orai1/Stim1-mediated whole cell currents in HEK293T cells. A, B, D, E Current–voltage relationships for whole cell currents measured in cells pre-exposed to CPA (10 µM) to activate Orai1 channels, followed by CFTR inhibitors. The inhibitors were present for 3 min at each concentration tested. Cells were mock-transfected (black symbols) or overexpressed Orai1/Stim1 (red symbols). Increasing concentrations of CFTRinh-172 (CFinh172, 0.2, 2, 20 µM) or GlyH-101 (GlyH101, 0.5, 5, 50 µM) were applied in the continuous presence of CPA. Application of the highest concentration of CFTRinh-172 (20 µM) or GlyH-101 (50 µM) significantly inhibited the inward currents, but had no effect in mock-transfected cells. C, F Summary of the concentration-dependent inhibition of inward currents by CFTRinh-172 or GlyH-101. * = p < 0.05 vs. control (mock). Un-paired t-test
Molecular docking of CFTRinh-172 and GlyH-101 to the structures of Orai1 in the closed and open states (pdb: 4hkr [28] and 7kr5 [29]) did not result in a reliable prediction of the binding site. All of the possible, detected sites in the available Orai1 structures included larger unresolved areas of the structure or led to low predicted binding affinities. The best predicted binding site between the transmembrane helices TM1 and TM2, as well as TM1 of the adjacent protein chain, which was obtained in the closed Orai1 structure, comprises at least 16 amino acid residues, which are missing in the crystal structure, and is hence unreliable, as also reported for the docking of AnCoA4 to Orai1 [30]. This region is completely missing in the recent cryo-EM structure of Orai1 in the open state.
Both CFTRinh-172 and GlyH-101 affect ENaC-mediated currents in Xenopus oocytes
To investigate if the inhibitory effect of the two CFTR blockers was restricted to Ca2+-permeable channels, we studied their effect on ENaC-mediated sodium currents in Xenopus oocytes expressing either the ‘classical’ αβγ-ENaC, or oocytes expressing δβγ-ENaC. Oocytes are an excellent model system to study the effect of compounds on specific channel isoforms, and for ENaC, this is convenient given there are two isoforms and there is no suitable model for studying δβγ-ENaC. The ENaC blocker amiloride [31] was used to determine amiloride-sensitive (ΔIami), i.e. ENaC-mediated, currents before and after application of the CFTR blockers (Fig. 4). In Xenopus oocytes expressing αβγ-ENaC, 20 µM CFTRinh-172 significantly inhibited ΔIami by 17.45 ± 3.02% from 6.42 ± 0.62 µA to 5.32 ± 0.58 µA (n = 9; p = 0.0004; Student’s paired t-test; Fig. 4A). By contrast, ΔIami in δβγ-ENaC expressing oocytes significantly increased by 8.2 ± 1.13% from 7.32 ± 1.04 µA to 7.93 ± 1.15 µA in response to CFTRinh-172 (n = 9; p = 0.0019; Student’s paired t-test; Fig. 4B). GlyH-101 (10 µM) had a strong inhibitory effect on both ENaC isoforms. In Xenopus oocytes expressing αβγ-ENaC, ΔIami decreased by 49.6 ± 3.96% from 5.52 ± 0.98 µA to 2.87 ± 0.62 µA (n = 9; p = 0.0005; paired Student’s t-test; Fig. 4C). The ΔIami in δβγ-ENaC expressing oocytes decreased by 43.63 ± 2.3% from 8.99 ± 0.91 µA to 5.02 ± 0.47 µA (n = 9; p < 0.000; paired Student’s t-test; Fig. 4D). Water-injected control oocytes did not respond to amiloride, CFTRinh-172, or GlyH-101 (Fig. 4E). The transmembrane currents (IM) of water-injected control oocytes were − 0.032 ± 0.006 µA before, and − 0.03 ± 0.007 µA after exposure to CFTRinh-172 (n = 6, p > 0.999; Wilcoxon matched-pairs signed rank test), and − 0.032 ± 0.004 µA before, and − 0.027 ± 0.003 µA after exposure to GlyH-101 (n = 6, p = 0.25; Wilcoxon matched-pairs signed rank test).

CFTRinh-172 and GlyH-101 affect ENaC currents in Xenopus oocytes. A Left panel: representative current trace of a human αβγ-ENaC expressing oocyte. The application of amiloride (100 µM; ‘a’) is represented by the black bars and was used to determine amiloride-sensitive fractions of IM (ΔIami; right panel), before and after application of CFTRinh172 (20 µM; CFinh172). n = 9. B Similar experiments as shown in panel A, with human δβγ-ENaC expressing oocytes (n = 9). C/D Similar experiments as shown in panels A/B, where ΔIami were determined before and after application of GlyH-101 (GlyH101, 10 µM) n = 9 for both conditions. Student’s paired t-test was employed for all statistical analyses, with ** indicating p < 0.01 and *** indicating p < 0.001. E Left panel: Representative current trace of a water-injected control oocyte. The application of amiloride (100 µM; ‘a’) is represented by the black bars, the application of CFTR inhibitors by grey bars. Neither of the CFTR-inhibitors had any significant effect on transmembrane currents (IM). n = 6. Wilcoxon matched-pairs signed rank test was employed for statistical analyses
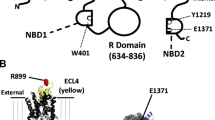
Our results suggest that both inhibitors bind to ENaC to inhibit function. We used molecular docking experiments to predict the putative binding site of the two compounds to the structure of human αβγ-ENaC (pdb: 6bqn; [32]). Initial blind docking allowed scanning for possible binding pockets in the structure. As a reference, we also docked amiloride into the same structure. For all three compounds, we identified binding of the ligands only at binding sites in the extracellular domain. In a series of subsequent targeted docking experiments with the individual binding sites as search areas, for both blockers, two putative binding sites in the ENaC α-subunit were identified, which yielded high affinity binding of CFTRinh-172 and GlyH-101 (Fig. 5A). The first binding site is located near the finger-thumb interface of ENaC, in close proximity to the reported extracellular motif WYKLHY (WYRFHY in rat), which was shown to be involved in amiloride binding [33]. Both blockers bind mostly through hydrophobic interactions to the nonpolar binding site, including residues Tyr-217, Tyr-435, Tyr-436 and His-439. Polar contacts were formed between GlyH-101 and amino acids Tyr-217 and Asn-285. The second putative binding pocket is located at the interface between two ENaC subdomains (between the ‘β-ball’ and ‘palm’). Binding of CFTRinh-172 was stabilised by hydrophobic and aromatic interactions with amino acids Ile-330, Ile-331, Val-343, Leu-391, Ala-456 Phe-459 and Trp-462, as well as hydrogen bonds with residues Ala-456, Lys-460, and Asn-532 (Fig. 5). Similarly, the best binding of GlyH-101 was also found at the same site in ENaC. The aromatic rings of GlyH-101 were accommodated at the same areas in the binding pocket and interact with the same hydrophobic residues as CFTRinh-172. Hydrogen bonds of GlyH-101 were formed with amino acids Ser-390, Leu-391, Gln-392 and Asn-532 (Fig. 5).

Putative CFTR-inhibitor binding sites in human ENaC. Molecular docking experiments identified two potential binding pockets for CFTRinh-172 (yellow) and GlyH-101 (orange) in the human α-ENaC structure. α-ENaC is shown in blue, γ-ENaC in brown and β-ENaC in green. The structure of each ENaC subunit represents a clenched hand holding a ball of β-sheets [32]. One binding pocket is located at the ‘finger’/’ thumb’ domain interface, the second binding pocket is located between the ‘β-ball’ and ‘palm’ domains
Discussion
Our results show that the putative specific CFTR inhibitors, CFTRinh-172 and GlyH-101, exerted CFTR-independent inhibition of SOCE in human epithelial cell lines, likely through inhibition of Orai1-mediated Ca2+ entry. In Xenopus oocytes, both inhibitors also significantly reduced ENaC-mediated currents differentially based on ENaC subunit composition. Thus, our results provide strong evidence for off-target effects of these CFTR inhibitors on two distinct classes of physiologically important epithelial cation channels.
The specificity of both CFTRinh-172 and GlyH-101 has previously been questioned. Melis et al. [6] showed that, in addition to CFTR, these agents also inhibited volume-sensitive Cl− channels, and GlyH-101 inhibited Ca2+-activated Cl− channels. Melis et al. [6] also demonstrated that these agents were also cytotoxic when incubated with cells for 24 h (> 5 and 10 µM for CFTRinh-172 and GlyH-101, respectively). Moreover, GlyH-101, but not CFTRinh-172, inhibited SLC26A9, which facilitates Cl− transport in epithelial cells [4]. However, our study is the first to report off-target effects of CFTRinh-172 and GlyH-101 on SOCE in epithelial cells at concentrations used by others to inhibit CFTR. In our hands, 20 µM CFTRinh-172 significantly inhibited SOCE in both patch clamp and Ca2+ fluorimetry studies, whereas GlyH-101 inhibited SOCE in Ca2+ experiments at 10 µM, but in patch clamp experiments, while 5 µM did not, 50 µM did. The initial screening study indicated that CFTRinh-172 completely blocked CFTR currents at 5 µM [2], and the working concentration commonly used by others range from 10 [11, 13, 34, 35], 20 [25, 36,37,38] and up to 100 µM [39]. On the other hand, GlyH-101 is commonly used between 10 [40] and 50 µM [4, 41]. Therefore, the concentrations employed in the present study are within the range used by others, including 50 µM GlyH-101, despite potential cytotoxicity issues [6], and therefore, are relevant to the field in general.
In addition, we showed that CFTRinh-172 exerted a time-dependent and poorly reversible inhibition of SOCE in both Calu-3 and HEK293T cells. Using Calu-3 cells, we established that a short (3 min) pre-exposure to CFTRinh-172 had no effect on the subsequent magnitude or rate of SOCE, but longer exposures did. Because a short exposure to CFTRinh-172 is generally sufficient to inhibit CFTR in epithelial cells [2], this result suggests that inhibiting CFTR in Calu-3 cells does not directly affect SOCE. Rather, our data suggests that the inhibition of SOCE is an off-target effect on Ca2+ influx itself, which does not require CFTR. This was confirmed in HEK293T cells that do not express detectable levels of CFTR, but which demonstrated a similar level of block of SOCE by CFTRinh-172. In most epithelial cells, store depletion leads to the activation of Orai1 channels, a process initiated by ER-localised STIM1 oligomerisation, followed by STIM1 translocation and subsequent Orai1 channel opening [42]. To investigate if Orai1/STIM1 were involved in the inhibitory effects of CFTRinh-172 on SOCE, the two proteins were overexpressed in HEK293T cells. This was necessary because endogenous levels of Orai1-Ca2+ currents are difficult to detect electrophysiologically [26]. Our results showed a clear concentration-dependent reduction of Orai1-mediated currents by both CFTR inhibitors, confirming that they inhibit Orai1 channels. Both these CFTR inhibitors share chemical features with the Orai1 inhibitor AnCoA4 [30], including a central aromatic system and at least two functional groups with hydrogen bond acceptor properties, at distances of 2.5 to 3.5 Å from the aromatic system, which might suggest a similar and direct binding of the two blockers to the Orai1 channel. Unfortunately, missing areas in the two higher-resolution structures of Orai1 did not allow us to reliably identify any binding sites for CFTRinh-172 and GlyH-101 in the Orai1 channel.
An interesting aspect of the inhibitory effect of CFTRinh-172 on SOCE was the time lag to achieve inhibition. In the original study that identified the compound [2], it irreversibly inhibited CFTR current within 2 min of application. However, as shown here, a 3 min and even a 10-min exposure to CFTRinh-172 was not always sufficient to attenuate SOCE, and furthermore this inhibition was not reversed following 15–30 min of washout. This delayed effect suggests that CFTRinh-172 is unlikely to be a pore-blocker of SOC. The inhibition of SOCE may involve binding to Orai1 leading to allosteric changes in channel gating (similar to how CFTRinh-172 inhibits CFTR), or changes in the function and/or spatial localisation of the STIM1/Orai1 complex. Interestingly, CFTR itself has shown to be indirectly activated by Ca2+ (reviewed in Billet, Hanrahan [43]). Therefore, the inhibitory effects of CFTRinh-172 and GlyH-101 on CFTR-mediated anion transport may be partially attributed to the off-target downregulation of [Ca2+]i. Our recommendation would be to avoid extended pre-exposure to these inhibitors (i.e. more than 10 min to avoid potential off-target effects on SOCE complicating the interpretation of results.
Our findings also have a broader relevance to the putative role of CFTR in Ca2+ signalling in CF-affected cells. Previous reports have established that CF cells have enhanced SOCE [13, 44], and/or TRP-mediated Ca2+ entry [45], which has been linked to up-regulation of inflammatory pathways such as IL-8 secretion [13, 46] and a reduced ability to kill bacteria by neutrophils [47]. These findings have stimulated much interest in understanding the underlying molecular mechanisms for these effects of CFTR dysfunction on Ca2+ homeostasis. However, in some cases, conclusions have been based on the results using CFTR blockers [12,13,14]. Furthermore, the fact that CFTR inhibitors were capable of blocking SOCE, it is conceivable that putative specific SOC blockers could have off-target effects on CFTR which, as far as we are aware, has not to date been investigated.
In addition to off-target effects of CFTR inhibitors on Ca2+ transport, we also observed modulatory effects of the CFTR inhibitors on Na+ transport through ENaC. CFTR is often co-expressed with ENaC in epithelial tissues and the activity of both ion channels facilitates transepithelial ion secretion or absorption, respectively. This is best illustrated in airway epithelia, where CFTR-mediated Cl− and HCO3− secretion, together with ENaC-mediated Na+ absorption maintain the composition and volume of airway surface liquid [1]. Off-target effects of CFTR blockers on ENaC are therefore important when dissecting the contributions of these ion channels to transepithelial ion transport processes. To assess CFTR activity in airway epithelia, ENaC is often inhibited by amiloride to enhance the electrical gradient facilitating transepithelial anion secretion [48]. Any off-target effects of CFTR blockers on ENaC remain therefore undetectable using such protocols. ENaC is a heterotrimeric ion channel [32] which has two functional isoforms, depending on the channel’s subunit composition. ‘Classical’ αβγ-ENaCs are present in many sodium-absorbing epithelia [49], but the physiology of δβγ-ENaC remains unclear [50]. Using heterologous expression of human ENaC isoforms in Xenopus oocytes, we found that both CFTR blockers affected ENaC activity. While GlyH-101 strongly inhibited αβγ-ENaC and δβγ-ENaC, the effect of CFTRinh-172 was dependent on the ENaC subunit composition. While αβγ-ENaC was inhibited by CFTRinh-172, δβγ-ENaC was stimulated. Using molecular docking, we identified that both CFTR blockers, could bind to two putative binding sites in the extracellular domain of ENaC. The position of the first binding site agrees well with amiloride binding site identified in the related acid-sensing ion channel (ASIC1) [51]. Mutational studies of the ENaC WYKLHY (WYRFHY in rat) motif, which is in close proximity to the predicted first binding site, resulted in a loss of amiloride binding to ENaC [33]. In addition, mutagenesis of amino acids Gly-525, Gly-537 or Ser-583, which contribute to the second identified binding pocket for CFTRinh-172 and GlyH-101, were shown earlier to decrease channel blocking by amiloride [31, 52]. Experiments with these CFTR blockers in combination with mutagenesis of residues in the putative binding pockets, could provide functional evidence to support the structural predictions, which is worth exploring in the future. Currently, there is no structural information on δβγ-ENaC available, preventing us to hypothesise putative interaction sites between this channel isoform and the CFTR inhibitors. Nevertheless, while both ENaC isoforms are inhibited by amiloride, isoform-specific activation/inhibition by various compounds has been reported [53]. Structural differences between the ENaC isoforms likely account for the subunit-specific channel modulation by CFTRinh-172.
In conclusion, our data indicate that caution is needed when interpreting results using these two CFTR inhibitors, especially after prolonged exposure to the compounds, or when utilising calcium mobilising agonists that lead to activation of SOCE, and when investigating CFTR or ENaC function.
Data availability
The data that support the findings of this study are available from the corresponding author (m.a.gray@newcastle.ac.uk) upon reasonable request.
References
Saint-Criq V, Gray MA (2017) Role of CFTR in epithelial physiology. Cell Mol Life Sci 74:93–115. https://doi.org/10.1007/s00018-016-2391-y
Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest 110:1651–1658. https://doi.org/10.1172/JCI16112
Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS (2004) Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol 124:125–137. https://doi.org/10.1085/jgp.200409059
Bertrand CA, Zhang R, Pilewski JM, Frizzell RA (2009) SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol 133:421–438. https://doi.org/10.1085/jgp.200810097
Kelly M, Trudel S, Brouillard F, Bouillaud F, Colas J, Nguyen-Khoa T, Ollero M, Edelman A, Fritsch J (2010) Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. J Pharmacol Exp Ther 333:60–69. https://doi.org/10.1124/jpet.109.162032
Melis N, Tauc M, Cougnon M, Bendahhou S, Giuliano S, Rubera I, Duranton C (2014) Revisiting CFTR inhibition: a comparative study of CFTRinh -172 and GlyH-101 inhibitors. Br J Pharmacol 171:3716–3727. https://doi.org/10.1111/bph.12726
Barman PP, Choisy SC, Gadeberg HC, Hancox JC, James AF (2011) Cardiac ion channel current modulation by the CFTR inhibitor GlyH-101. Biochem Biophys Res Commun 408:12–17. https://doi.org/10.1016/j.bbrc.2011.03.089
Madacsy T, Pallagi P, Maleth J (2018) Cystic fibrosis of the pancreas: the role of CFTR Channel in the regulation of intracellular Ca(2+) Signaling and mitochondrial function in the exocrine pancreas. Front Physiol 9:1585. https://doi.org/10.3389/fphys.2018.01585
Grebert C, Becq F, Vandebrouck C (2019) Focus on TRP channels in cystic fibrosis. Cell Calcium 81:29–37. https://doi.org/10.1016/j.ceca.2019.05.007
Mall MA (2020) ENaC inhibition in cystic fibrosis: potential role in the new era of CFTR modulator therapies. The European respiratory journal 56https://doi.org/10.1183/13993003.00946-2020
Antigny F, Norez C, Becq F, Vandebrouck C (2008) Calcium homeostasis is abnormal in cystic fibrosis airway epithelial cells but is normalized after rescue of F508del-CFTR. Cell Calcium 43:175–183. https://doi.org/10.1016/j.ceca.2007.05.002
Antigny F, Norez C, Dannhoffer L, Bertrand J, Raveau D, Corbi P, Jayle C, Becq F, Vandebrouck C (2011) Transient receptor potential canonical channel 6 links Ca2+ mishandling to cystic fibrosis transmembrane conductance regulator channel dysfunction in cystic fibrosis. Am J Respir Cell Mol Biol 44:83–90. https://doi.org/10.1165/rcmb.2009-0347OC
Balghi H, Robert R, Rappaz B, Zhang X, Wohlhuter-Haddad A, Evagelidis A, Luo Y, Goepp J, Ferraro P, Romeo P, Trebak M, Wiseman PW, Thomas DY, Hanrahan JW (2011) Enhanced Ca2+ entry due to Orai1 plasma membrane insertion increases IL-8 secretion by cystic fibrosis airways. FASEB J 25:4274–4291. https://doi.org/10.1096/fj.11-187682
Bertrand J, Dannhoffer L, Antigny F, Vachel L, Jayle C, Vandebrouck C, Becq F, Norez C (2015) A functional tandem between transient receptor potential canonical channels 6 and calcium-dependent chloride channels in human epithelial cells. Eur J Pharmacol 765:337–345. https://doi.org/10.1016/j.ejphar.2015.08.005
Henry CO, Dalloneau E, Perez-Berezo MT, Plata C, Wu Y, Guillon A, Morello E, Aimar RF, Potier-Cartereau M, Esnard F, Coraux C, Bornchen C, Kiefmann R, Vandier C, Touqui L, Valverde MA, Cenac N, Si-Tahar M (2016) In vitro and in vivo evidence for an inflammatory role of the calcium channel TRPV4 in lung epithelium: Potential involvement in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 311:L664-675. https://doi.org/10.1152/ajplung.00442.2015
Li P, Rubaiy HN, Chen GL, Hallett T, Zaibi N, Zeng B, Saurabh R, Xu SZ (2019) Mibefradil, a T-type Ca(2+) channel blocker also blocks Orai channels by action at the extracellular surface. Br J Pharmacol 176:3845–3856. https://doi.org/10.1111/bph.14788
Jurman ME, Boland LM, Liu Y, Yellen G (1994) Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotech 17:876–881
Lin J, Taggart M, Borthwick L, Fisher A, Brodlie M, Sassano MF, Tarran R, Gray MA (2021) Acute cigarette smoke or extract exposure rapidly activates TRPA1-mediated calcium influx in primary human airway smooth muscle cells. Sci Rep 11:9643. https://doi.org/10.1038/s41598-021-89051-4
Boese SH, Aziz O, Simmons NL, Gray MA (2004) Kinetics and regulation of a Ca2+-activated Cl- conductance in mouse renal inner medullary collecting duct cells. Am J Physiol Renal Physiol 286:F682-692. https://doi.org/10.1152/ajprenal.00123.2003
Gettings SM, Maxeiner S, Tzika M, Cobain MRD, Ruf I, Benseler F, Brose N, Krasteva-Christ G, Vande Velde G, Schonberger M, Althaus M (2021) Two functional epithelial sodium channel isoforms are present in rodents despite pronounced evolutionary pseudogenization and exon fusion. Mol Biol Evol 38:5704–5725. https://doi.org/10.1093/molbev/msab271
Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. https://doi.org/10.1006/jmbi.1993.1626
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Shen BQ, Finkbeiner WE, Wine JJ, Mrsny RJ, Widdicombe JH (1994) Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl- secretion. Am J Physiol 266:L493-501
Chen L, Bosworth CA, Pico T, Collawn JF, Varga K, Gao Z, Clancy JP, Fortenberry JA, Lancaster JR Jr, Matalon S (2008) DETANO and nitrated lipids increase chloride secretion across lung airway cells. Am J Respir Cell Mol Biol 39:150–162. https://doi.org/10.1165/rcmb.2008-0005OC
Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP (2006) Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat Cell Biol 8:771–773. https://doi.org/10.1038/ncb1435
Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312:1220–1223. https://doi.org/10.1126/science.1127883
Hou X, Pedi L, Diver MM, Long SB (2012) Crystal structure of the calcium release-activated calcium channel Orai. Sci 338:1308–1313. https://doi.org/10.1126/science.1228757
Hou X, Outhwaite IR, Pedi L, Long SB (2020) Cryo-EM structure of the calcium release-activated calcium channel Orai in an open conformation. Elife 9https://doi.org/10.7554/eLife.62772
Sadaghiani AM, Lee SM, Odegaard JI, Leveson-Gower DB, McPherson OM, Novick P, Kim MR, Koehler AN, Negrin R, Dolmetsch RE, Park CY (2014) Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem Biol 21:1278–1292. https://doi.org/10.1016/j.chembiol.2014.08.016
Kashlan OB, Sheng S, Kleyman TR (2005) On the interaction between amiloride and its putative alpha-subunit epithelial Na+ channel binding site. J Biol Chem 280:26206–26215. https://doi.org/10.1074/jbc.M503500200
Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I (2018) Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife 7https://doi.org/10.7554/eLife.39340
Kelly O, Lin C, Ramkumar M, Saxena NC, Kleyman TR, Eaton DC (2003) Characterization of an amiloride binding region in the alpha-subunit of ENaC. Am J Physiol Renal Physiol 285:F1279-1290. https://doi.org/10.1152/ajprenal.00094.2003
Banga A, Flaig S, Lewis S, Winfree S, Blazer-Yost BL (2014) Epinephrine stimulation of anion secretion in the Calu-3 serous cell model. Am J Physiol Lung Cell Mol Physiol 306:L937-946. https://doi.org/10.1152/ajplung.00190.2013
Kim D, Kim J, Burghardt B, Best L, Steward MC (2014) Role of anion exchangers in Cl- and HCO3- secretion by the human airway epithelial cell line Calu-3. Am J Physiol Cell Physiol 307:C208-219. https://doi.org/10.1152/ajpcell.00083.2014
Baudouin-Legros M, Colas J, Moriceau S, Kelly M, Planelles G, Edelman A, Ollero M (2012) Long-term CFTR inhibition modulates 15d-prostaglandin J2 in human pulmonary cells. Int J Biochem Cell Biol 44:1009–1018. https://doi.org/10.1016/j.biocel.2012.03.010
Bijvelds MJC, Roos FJM, Meijsen KF, Roest HP, Verstegen MMA, Janssens HM, van der Laan LJW, de Jonge HR (2022) Rescue of chloride and bicarbonate transport by elexacaftor-ivacaftor-tezacaftor in organoid-derived CF intestinal and cholangiocyte monolayers. J cyst fibros : off J Eur Cyst Fibros Soc 21:537–543. https://doi.org/10.1016/j.jcf.2021.12.006
Sharma J, Du M, Wong E, Mutyam V, Li Y, Chen J, Wangen J, Thrasher K, Fu L, Peng N, Tang L, Liu K, Mathew B, Bostwick RJ, Augelli-Szafran CE, Bihler H, Liang F, Mahiou J, Saltz J, Rab A, Hong J, Sorscher EJ, Mendenhall EM, Coppola CJ, Keeling KM, Green R, Mense M, Suto MJ, Rowe SM, Bedwell DM (2021) A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat Commun 12:4358. https://doi.org/10.1038/s41467-021-24575-x
Baniak N, Luan X, Grunow A, Machen TE, Ianowski JP (2012) The cytokines interleukin-1beta and tumor necrosis factor-alpha stimulate CFTR-mediated fluid secretion by swine airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 303:L327-333. https://doi.org/10.1152/ajplung.00058.2012
Garnett JP, Hickman E, Tunkamnerdthai O, Cuthbert AW, Gray MA (2013) Protein phosphatase 1 coordinates CFTR-dependent airway epithelial HCO3- secretion by reciprocal regulation of apical and basolateral membrane Cl(-)-HCO3- exchangers. Br J Pharmacol 168:1946–1960. https://doi.org/10.1111/bph.12085
Muanprasat C, Wongborisuth C, Pathomthongtaweechai N, Satitsri S, Hongeng S (2013) Protection against oxidative stress in beta thalassemia/hemoglobin E erythrocytes by inhibitors of glutathione efflux transporters. PLoS ONE 8:e55685. https://doi.org/10.1371/journal.pone.0055685
Prakriya M, Lewis RS (2015) Store-Operated Calcium Channels. Physiol Rev 95:1383–1436. https://doi.org/10.1152/physrev.00020.2014
Billet A, Hanrahan JW (2013) The secret life of CFTR as a calcium-activated chloride channel. J Physiol 591:5273–5278. https://doi.org/10.1113/jphysiol.2013.261909
Philippe R, Antigny F, Buscaglia P, Norez C, Becq F, Frieden M, Mignen O (2015) SERCA and PMCA pumps contribute to the deregulation of Ca2+ homeostasis in human CF epithelial cells. Biochim Biophys Acta 1853:892–903. https://doi.org/10.1016/j.bbamcr.2015.01.010
Vachel L, Norez C, Jayle C, Becq F, Vandebrouck C (2015) The low PLC-delta1 expression in cystic fibrosis bronchial epithelial cells induces upregulation of TRPV6 channel activity. Cell Calcium 57:38–48. https://doi.org/10.1016/j.ceca.2014.11.005
Ribeiro CM, Paradiso AM, Schwab U, Perez-Vilar J, Jones L, O’Neal W, Boucher RC (2005) Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J Biol Chem 280:17798–17806. https://doi.org/10.1074/jbc.M410618200
Robledo-Avila FH, Ruiz-Rosado JD, Brockman KL, Kopp BT, Amer AO, McCoy K, Bakaletz LO, Partida-Sanchez S (2018) Dysregulated calcium homeostasis in cystic fibrosis neutrophils leads to deficient antimicrobial responses. J Immunol 201:2016–2027. https://doi.org/10.4049/jimmunol.1800076
Haq IJ, Althaus M, Gardner AI, Yeoh HY, Joshi U, Saint-Criq V, Verdon B, Townshend J, O’Brien C, Ben-Hamida M, Thomas M, Bourke S, van der Sluijs P, Braakman I, Ward C, Gray MA, Brodlie M (2021) Clinical and molecular characterization of the R751L-CFTR mutation. Am J Physiol Lung Cell Mol Physiol 320:L288–L300. https://doi.org/10.1152/ajplung.00137.2020
Rotin D, Staub O (2021) Function and regulation of the epithelial Na(+) channel ENaC. Compr Physiol 11:2017–2045. https://doi.org/10.1002/cphy.c200012
Wichmann L, Althaus M (2020) Evolution of epithelial sodium channels: current concepts and hypotheses. Am J Physiol Regul Integr Comp Physiol 319:R387–R400. https://doi.org/10.1152/ajpregu.00144.2020
Baconguis I, Bohlen CJ, Goehring A, Julius D, Gouaux E (2014) X-ray structure of acid-sensing ion channel 1-snake toxin complex reveals open state of a Na(+)-selective channel. Cell 156:717–729. https://doi.org/10.1016/j.cell.2014.01.011
Kellenberger S, Gautschi I, Schild L (2003) Mutations in the epithelial Na+ channel ENaC outer pore disrupt amiloride block by increasing its dissociation rate. Mol Pharmacol 64:848–856. https://doi.org/10.1124/mol.64.4.848
Ji HL, Zhao RZ, Chen ZX, Shetty S, Idell S, Matalon S (2012) delta ENaC: a novel divergent amiloride-inhibitable sodium channel. Am J Physiol Lung Cell Mol Physiol 303:L1013-1026. https://doi.org/10.1152/ajplung.00206.2012
Acknowledgements
We would like to thank Julie Taggart for training on Western blotting techniques.
Funding
This work was funded by a Newcastle University JJ Hunter Bequest Scholarship (JL, MJT, MAG) and the Cystic Fibrosis Trust (SRC013: MAG, KK).
Author information
Authors and Affiliations
Contributions
MT, KK, MA and MAG conceived, supervised and designed experiments; JL, SMG and KT performed experiments; RS provided new tools and reagents; MP performed in silico simulation studies; JL, SG and KT analysed data; JL, KK, MP, MA and MAG wrote the manuscript; MT revised the manuscript for critically important intellectual content.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Animal ethics
The experimental procedures were approved by the Newcastle University Animal Welfare and Ethical Review Body (AWERB, project ID 630).
Consent for publication
All authors consent for publication.
Competing interests
The authors declare no competing interests/
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, J., Gettings, S.M., Talbi, K. et al. Pharmacological inhibitors of the cystic fibrosis transmembrane conductance regulator exert off-target effects on epithelial cation channels. Pflugers Arch - Eur J Physiol 475, 167–179 (2023). https://doi.org/10.1007/s00424-022-02758-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-022-02758-9