
Abstract
In microglia, changes in intracellular calcium concentration ([Ca2+]i) may regulate process motility, inflammasome activation, and phagocytosis. However, while neurons and astrocytes exhibit frequent spontaneous Ca2+ activity, microglial Ca2+ signals are much rarer and poorly understood. Here, we studied [Ca2+]i changes of microglia in acute brain slices using Fluo-4–loaded cells and mice expressing GCaMP5g in microglia. Spontaneous Ca2+ transients occurred ~ 5 times more frequently in individual microglial processes than in their somata. We assessed whether microglial Ca2+ responses change in Alzheimer's disease (AD) using AppNL−G−F knock-in mice. Proximity to Aβ plaques strongly affected microglial Ca2+ activity. Although spontaneous Ca2+ transients were unaffected in microglial processes, they were fivefold more frequent in microglial somata near Aβ plaques than in wild-type microglia. Microglia away from Aβ plaques in AD mice showed intermediate properties for morphology and Ca2+ responses, partly resembling those of wild-type microglia. By contrast, somatic Ca2+ responses evoked by tissue damage were less intense in microglia near Aβ plaques than in wild-type microglia, suggesting different mechanisms underlying spontaneous vs. damage-evoked Ca2+ signals. Finally, as similar processes occur in neurodegeneration and old age, we studied whether ageing affected microglial [Ca2+]i. Somatic damage-evoked Ca2+ responses were greatly reduced in microglia from old mice, as in the AD mice. In contrast to AD, however, old age did not alter the occurrence of spontaneous Ca2+ signals in microglial somata but reduced the rate of events in processes. Thus, we demonstrate distinct compartmentalised Ca2+ activity in microglia from healthy, aged and AD-like brains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microglia, the brain-resident parenchymal macrophages, account for up to 12% of brain cells, with a particularly high density in the hippocampus [38]. They control multiple processes including neurogenesis, synapse monitoring and pruning, myelination, vasculogenesis, blood–brain barrier integrity, inflammation, and phagocytosis [56].
Microglial surveillance of the brain parenchyma, chemotactic movement of microglial processes and phagocytosis all depend on actin polymerization and subsequent cytoskeletal rearrangements. These, as well as activation, proliferation and production of inflammatory cytokines, are controlled, at least in part, by Ca2+ acting as a second messenger, and rises in [Ca2+]i have been reported to be associated with microglial functions including damage-induced chemotactic and phagocytic events [46, 50, 51, 53, 55]. In general, phagocytic cells exhibit periodic spontaneous Ca2+ transients as well as stimulus-evoked [Ca2+]i rises [9], and microglial Ca2+ activity is important for the regulation of lysosomes, which process incorporated material [63]. While ‘resting’ microglia rarely exhibit spontaneous Ca2+ transients in vivo, external pathological stimuli such as damage to nearby neurons or mechanical distortions rapidly raise their [Ca2+]i [30, 55].
Previous studies using mainly cultured microglia identified a number of ligands evoking Ca2+ responses in microglia, including ATP, ADP and UDP as endogenous stimuli [34, 40, 49] and pathology-related substances such as lipopolysaccharide (LPS) and amyloid beta (Aβ) [9]. However, Ca2+ responses are significantly different for cultured microglia compared to their counterparts embedded in the native CNS environment [9], as microglia partly lose their characteristic genetic profile under more artificial cell culture conditions [8]. In transgenic mice overexpressing Aβ, accumulation of which is a hallmark of Alzheimer’s disease (AD), altered Ca2+ signalling was detected in astrocytes and microglia electroporated with Ca2+-sensing dyes [10].
While previous research has relied mainly on Ca2+-sensitive dyes, we also took advantage of Cx3cr1CreER × GCaMP5g-IRES-tdTomato mice expressing an inducible genetically-encoded Ca2+ indicator (GECI) cross-bred with AppNL−G−F knock-in AD mice [59] to examine microglial Ca2+ activity, both spontaneous and upon inducing brain damage, in the presence and absence of AD-related amyloid plaques. This strategy allows all cells in the field of view to be captured spatially in relation to plaque pathology and abrogates the need for external manipulation to insert the Ca2+ sensor. As similar mechanisms are thought to occur during brain ageing and brain disease [3, 16, 28, 65, 67, 69], we also assessed whether the Ca2+ activity of microglia was affected differently by AD and normal ageing.
Materials and Methods
Animal procedures
Procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986 (Home Office License 70/8976). Rats (postnatal day 12–14 for work with patch-clamp loading of Ca2+-sensitive dye) and mice (postnatal day 120–130 and 300–310 for work with the GECI investigating effects of AD and ageing, respectively) were housed in open-shelf units and individually ventilated cages, respectively, with food and water ad libitum. Animals of both sexes were sacrificed by cervical dislocation followed by decapitation (for live imaging), or by an intraperitoneal overdose of pentobarbital sodium (Euthatal, 200 µg/g body weight) followed by transcardial perfusion-fixation with 4% paraformaldehyde (for immunohistochemistry).
For experiments in rats, loading of the Ca2+ indicator Fluo-4 was achieved by whole-cell patch-clamping of hippocampal microglia (see below). To study microglial properties in the initial stages of AD-related pathology, 4-month-old homozygous AppNL−G−F knock-in AD mice or wild-type (WT) littermates were used, where Aβ plaque deposition starts in the AD mice from 2 months of age [59]. For Ca2+ imaging, AppNL−G−F mice were crossed with transgenic mice expressing GCaMP5g-IRES-tdTomato [21] and Cx3cr1CreER [70], to generate offspring where microglia exhibit expression of GCaMP5g and tdTomato following tamoxifen gavage (Sigma T5648; 120 µg/g body weight for four consecutive days). Mice that were homozygous for GCaMP5g, heterozygous for Cx3cr1CreER and either WT or homozygous for AppNL−G−F were used for imaging ≥ 21 days after the first tamoxifen dose (given at P85–P105).
Genotyping
Genotyping PCR primers and their final concentrations were as follows:
App WT (5’-tgtagatgagaacttaac-3’, 0.2 µM; 5’-atctcggaagtgaagatg-3’, 0.2 µM),
App mutated (5’-atctcggaagtgaatcta-3’, 0.4 µM; 5’-cgtataatgtatgctatacgaag-3’, 0.4 µM),
Polr2a GCaMP (5’-tagacacatgccaccaaacc-3’, 0.2 µM; 5’-tctctccagcaccataactcc-3’, 0.2 µM; 5’-gatcgataaaacacatgcgtca-3’, 0.2 µM),
Cx3cr1 Cre (5’-acgcccagactaatggtgac-3’, 0.2 µM; 5’-gttaatgacctgcagccaag-3’, 0.2 µM; 5’-agctcacgactgccttcttc-3’, 0.1 µM).
Following initial denaturation of tail snip DNA for 2 min at 95°C, amplification was performed for:
36 cycles of 30 s at 95°C, 40 s at 48°C, and 45 s at 72°C (App WT),
38 cycles of 30 s at 95°C, 40 s at 53°C, and 45 s at 72°C (App mutated),
36 cycles of 30 s at 95°C, 40 s at 58°C, and 45 s at 72°C (Polr2a GCaMP),
or 36 cycles of 30 s at 95°C, 40 s at 60°C, and 45 s at 72°C (Cx3cr1 Cre).
All PCR reactions were then completed with 4 min at 72°C. All PCR products were visualized on 2.5% Tris–borate EDTA gels, and product sizes for each allele in base pairs (bp) were as follows: 500 bp (App WT), 700 bp (App mutated), 395 bp (Pol2a GCaMP WT), 272 bp (Pol2a GCaMP mutated), 151 bp (Cx3cr1 WT), and 230 bp (Cx3cr1 mutated).
Solutions
Acute brain slices (250 µm, parasagittal) containing dorsal hippocampi were prepared on a Leica VT1200S vibratome with ice-cold slicing solution containing (in mM): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 10 glucose, 1 CaCl2, 2 MgCl2 and 1 kynurenic acid. Osmolarity was adjusted to ~ 295 mOsm/kg and pH set to 7.4 when bubbled with 5% CO2/95% O2.
For live imaging, brain slices were incubated in artificial cerebrospinal fluid (aCSF) containing (in mM): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 10 glucose, 2 CaCl2, 1 MgCl, and 0.1 Na-ascorbate. Osmolarity was adjusted to ~ 295 mOsm/kg and pH was set to 7.4 with NaOH. Solutions were oxygenated with 20% O2/5% CO2/75% N2.
Immunohistochemistry
Perfusion-fixed brain slices (250 µm, parasagittal) were prepared on a Leica VT1200S vibratome, and were permeabilised and blocked for 2 h at room temperature in blocking buffer (10% normal horse serum and 0.02% Triton X-100 in phosphate-buffered saline, PBS), followed by incubation with primary antibodies (mouse anti-human Aβ 82E1 [1:500, IBL 27725], rat anti-CD68 [1:250, BioRad MCA1957], and rabbit anti-Iba1 [1:500, Synaptic Systems 234003]) in blocking buffer for 12 h at 4°C with agitation. Following PBS washes, Alexa-conjugated secondary antibodies (Invitrogen) diluted 1:1000 in blocking buffer were applied for 4 h at room temperature with agitation. Brain slices were free-floating in the solution for all steps to allow antibody penetration from both sides of the slice. Lastly, slices were washed in PBS, incubated with DAPI (Invitrogen D1306) diluted 1:50,000 in PBS, and mounted.
Analysis of microglial morphology
Confocal z-stacks of Iba1-labelled CA1 stratum radiatum microglia encompassing the entire brain slice in 0.34 µm steps were imaged on a Zeiss LSM700 microscope with a Plan-Apochromat 63×/1.4 lens. Individual cells were 3D-reconstructed using the cell tracing tool Neuron Tracing v2.0 on Vaa3D (vaa3d.org), adjusting the background threshold for each image to obtain the optimal reconstruction. Only microglial cells with somata at ~ 50 µm from the slice surface were selected for analysis to avoid cells at depths without sufficient antibody penetration (~ 100 µm in our experiments) and cells too close to the surface (which are likely to have cut-off processes). After checking the reconstructions against the raw images, they were analysed using custom-written MATLAB software (github.com/AttwellLab/Microglia). Briefly, based on Sholl analysis, concentric spheres were drawn at 5 µm intervals from an analyst-established cell centre (the soma is assumed to have a 5 µm radius to avoid misassigning differences in Iba1 signal within the soma as representing processes [41]). Cell process branching was profiled based on distance from the soma to assess ramification. For analysis of the effects of amyloid β deposition, in AppNL−G−F brain slices, both cells at 82E1-labelled Aβ plaques and > 50 µm away from them were imaged. Analysis was performed with the researcher blind to genotype and Aβ signal.
Analysis of Aβ and lysosomal burden
Immunolabelled brain slices were imaged on a Zeiss AxioScan.Z1 scanner with a Plan-Apochromat 20×/0.8 M27 lens. After background subtraction of the images by channel (using a 10-pixel rolling ball average for CD68 and Iba1 and an 80-pixel rolling ball average for Aβ) and thresholding, masks for CA1 stratum radiatum microglia at and > 50 µm away from Aβ plaques (which were Iba1+/Aβ+ and Iba1+/Aβ–, respectively) were obtained and transposed to the binarised CD68 channel to calculate the percentage of the Iba1+ area that was also labelled for CD68 in microglia located at Aβ plaques and away from them.
To identify Aβ plaques in living brain tissue, a number of stains have been developed [6, 14, 32]. However, insufficient penetration into slices, labelling of only some Aβ plaque types or non-selective binding to other proteins can hinder the interpretation of results [18, 62, 71]. Thus, in this study we relied on microglial clustering around Aβ plaques (see Supplementary Fig. 1), reported previously in AppNL−G−F mice [13, 59], as an intrinsic cue for identification of Aβ plaques (and thus of microglia at and away from them).
Fluo-4 loading of microglia by patch-clamp electrophysiology
CA1 microglia in rat hippocampal brain slices were identified by acute labelling with isolectin-B4 coupled to Alexa-594 [41] and patch-clamped using a KCl-based intracellular solution containing (mM): 130 KCl, 4 NaCl, 10 HEPES, 0.01 BAPTA, 0.01 CaCl2, 10 Na-phosphocreatine, 2 MgATP, 0.5 Na2GTP, 0.1 Fluo-4 pentapotassium salt, adjusted to a final osmolarity of 285 ± 5 mOsm/kg and a pH of 7.2. Whole-cell recordings from microglia were obtained at a depth of > 40 μm below the slice surface using borosilicate glass pipettes with a tip resistance of 3.5–5 MΩ, resulting in access resistances of < 20 MΩ that were not compensated. The average resting membrane potential of cells was −35.7 ± 2.4 mV (n = 22). Throughout imaging, cells were voltage-clamped at −30 mV.
Microglial Ca2+ imaging
CA1 stratum radiatum microglia from Cx3cr1CreER × GCaMP5g-IRES-tdTomato and Cx3cr1CreER × GCaMP5g-IRES-tdTomato × AppNL−G−F mice, or patch-clamped microglia filled with Fluo-4, were imaged on a Zeiss LSM780 or LSM710 two-photon microscope with a Plan-Apochromat 20×/1.0 lens and a Spectraphysics Mai Tai DeepSee eHP Ti:Sapphire infrared laser tuned to 920 nm at < 2% of its maximum power, which corresponded to a maximum power under the objective of 5.5 mW. Fields of view of 106 µm × 106 µm were scanned with an overall acquisition time of 25 ms/frame (pixel size 0.21 µm, 1 µs pixel dwell time). Prior to longitudinal single-plane imaging (to reduce bleaching), a z-stack (encompassing the entire cell at 1 µm steps) was acquired to confirm the morphology of the imaged microglia as visualised by tdTomato. Acquisition rates for Fluo-4 and GCaMP5g-based Ca2+ imaging were 0.2 Hz and 1 Hz, respectively.
To assess spontaneous [Ca2+]i changes, microglial cells were imaged for 5 min (1 frame/s). Recordings were re-registered with StackReg in ImageJ/FIJI and ROIs were drawn around microglial somata or individual processes; only processes ≥ 1 µm in diameter were analysed to exclude filopodia [7]. In each ROI, Ca2+ transients were analysed with custom-written MATLAB code (github.com/AttwellLab/MyelinCalcium). A locally time-smoothened baseline (100 frames of smoothing time) was generated using a piecewise cubic Hermite interpolating polynomial fit, to account for potential drifts in baseline that may arise from subtle movements or volume changes of the specimen during the time course of recording. Ca2+ transients were then defined by a detection threshold for the fractional change of fluorescence (ΔF/F > 2.25 × standard deviation of baseline points that had ΔF/F < 0.10, to exclude contributions of Ca2+ transients to the baseline), confirmed using a minimal area threshold (∫ΔF/F dt > 0.15), and manually checked to exclude false positives [37]. An example of the baseline subtraction is shown in Supplementary Fig. 2.
To trigger damage-evoked [Ca2+]i changes, following a baseline recording of 20 s (1 frame/s), a focal laser lesion (6 µm radius, 177.3 µs pixel dwell time) was performed in an area > 30 µm from the cell of interest, with the laser tuned to 920 nm at 80% of its maximum power. For analysis, recordings were re-registered with StackReg in ImageJ/FIJI and ΔF/F for ROIs drawn around microglial somata were calculated as (Ft − Fo)/Fo, where Ft is the fluorescence intensity at each timepoint t and Fo is the average fluorescence of the pre-lesion baseline. Peak ΔF/F values were compared for statistical analysis. To help distinguish between intensity values, a multicolour (“Fire”) lookup table was applied in ImageJ/FIJI to generate the representative images provided.
Statistics
Quantitative data are presented throughout as mean ± standard error of the mean. Normality was assessed using the D'Agostino-Pearson test. Statistical significance (defined as p < 0.05) was assessed using Mann–Whitney tests (Fig. 5B–D, Suppl. Figure 1, Suppl. Figure 4), two-tailed Wilcoxon matched-pairs signed rank test (Fig. 3E), one-way analysis of variance (ANOVA) followed by Dunn’s (Figs. 2D, 3B–D, Suppl. Figure 3) or two-way ANOVA followed by Sidak’s post-hoc tests for individual comparisons (Figs. 2E, 4B, 5F). All statistical analyses were performed in Microsoft Excel 2016 and GraphPad Prism 8.
Results
Microglia show spontaneous and damage-evoked Ca2+ rises in situ
Early studies on microglia mainly relied on cultured cells [22, 48], where gene expression is drastically changed and cells often fail to develop the complex process ramification and surveillance seen in intact microglia [8]. To overcome this limitation, while preserving the option to apply pharmacological agents in a controlled manner to modulate function, we studied microglia in situ in acute brain slices.
First, for an initial characterization of microglial Ca2+ activity in brain slices, we filled hippocampal microglia from young rats with the Ca2+ indicator Fluo-4 via the patch pipette to detect spontaneous Ca2+ transients. This experiment showed that spontaneous Ca2+ transients occur more frequently in single microglial processes than in cell somata, with individual somata and individual processes showing a spontaneous frequency of 0.04 ± 0.02 and 0.23 ± 0.04 transients/min, respectively (Fig. 1A–C; see also [64] and Discussion; comparable data recorded using GCaMP5g in WT mice are presented in Fig. 3E below). To study evoked Ca2+ responses in microglia, focal laser lesions were induced as a well-established proxy to simulate responses of microglia to brain injury, similar to focal ATP application mimicking neuronal damage [15, 25, 41]. Focal lesions lead to acute increases in microglial [Ca2+]i at short latencies (0–5 s [19]). We found that, in Fluo-4–filled microglia laser lesions triggered rapid, transient Ca2+ signals (Fig. 1D, E).

Microglia exhibit spontaneous and local damage-evoked Ca2+ activity in acute brain slices. (A) Image of hippocampal slice with microglia (and vascular basement membrane) labelled with Alexa 594-conjugated isolectin B4, showing a patch-clamped microglial cell filled with the Ca2+ indicator Fluo-4. Somatic and process regions of interest (ROIs; soma and processes 1–6) are shown. (B) Microglial [Ca2+]i time course in the ROIs shown in (A). Red circles denote peaks detected at ≥ 2.25 standard deviations from the baseline. (C) Quantification of spontaneous Ca2+ transient frequency, showing a higher rate in single processes vs. somata (n = 5 cells; see also Fig. 3E for comparable date acquired using GCaMP5g). (D) As in (A) showing nearby site of laser damage (red dotted circle). (E) A laser lesion at the site shown in (D) evokes a transient rise of [Ca2+]i. (F) Scheme depicting the genomic changes in Cx3cr1CreER × GCaMP5g-IRES-tdTomato mice to drive expression of GCaMP5g and the morphological marker tdTomato in microglia induced by tamoxifen gavage. (G) Microglia in brain slices from tamoxifen-induced Cx3cr1CreER × GCaMP5g-IRES-tdTomato mice express tdTomato (red, left panel) and show spontaneous Ca2+ activity in different regions of interest (ROIs) over time, as measured by GCaMP5g fluorescence changes (ΔF/F, “Fire” scale). White arrowheads indicate regions showing spontaneous Ca2+ activity at different timepoints (time stamps on the figures are from the time scale on panel (H); fluorescence brightness has been linearly adjusted to make the transients more visible; two persistent spots in the images represent constitutive cell fluorescence). (H) Microglial [Ca2+]i time course in the ROIs shown in (G). Red circles denote peaks detected at ≥ 2.25 standard deviations from the baseline. (I) Representative cell showing that local laser damage (red dotted circle) evokes a rapid, transient [Ca2+]i rise in tdTomato-labelled microglia. Times indicate seconds from laser lesion. (J) Quantification of somatic [Ca2+]i levels over time (ΔF/F) showing their increase upon laser lesion (vertical dashed line)

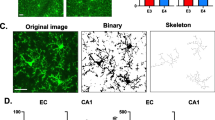
Proximity to Aβ plaques alters microglial morphology in AppNL−G−F mice. (A) Representative sagittal brain slice from a 4-month-old AppNL−G−F mouse showing Aβ plaques (82E1 antibody, green) and cell nuclei (DAPI, white) for reference. (B Quantification of the area covered by Aβ plaques in the neocortex (layer 2/3), dorsal hippocampus (CA1), cerebellum (molecular layer) and dorsal striatum of 4 AppNL−G−F mice. C Representative 3D-reconstructed App WT microglia, and AppNL−G−F microglia at or > 50 µm away from Aβ plaques in 4-month-old mice. (D–E) Sholl analysis-derived (D) total process length and (E) number of process branches at 5 μm increment distances from the soma, showing a sharp deramification of microglia at plaques (n = 53 App WT cells, 28 AppNL−G−F cells away from plaques and 42 AppNL−G−F cells at plaques from 3 mice each)

Spontaneous Ca2+ activity in microglial somata is increased near Aβ plaques. (A) Representative traces showing spontaneous Ca2+ transients (red arrowheads) in microglial somata from App WT and AppNL−G−F mice at, or > 50 µm away from, Aβ plaques. (B) Quantification of spontaneous Ca2+ transient frequency, showing a higher rate near Aβ plaques (App WT: n = 27 somata from 5 mice; AppNL−G−F away from plaques: n = 56 somata from 8 mice; AppNL−G−F at plaques: n = 43 somata from 8 mice). (C) As (A), but for individual processes. (D) As (B), but for individual processes, showing no effect of Aβ status (App WT: n = 11 processes from 3 mice; AppNL−G−F away from plaques: n = 42 processes from 6 mice; AppNL−G−F at plaques: n = 34 processes from 6 mice). (E) Paired comparisons between individual process and somatic Ca2+ transient rate, showing that, for a given microglial cell, spontaneous Ca2+ activity per process was higher in processes in App WT (n = 9 cells from 3 mice; comparable data acquired with Fluo-4 are shown in Fig. 1C) and AppNL−G−F microglia away from plaques (n = 18 cells from 6 mice), but not in microglia at Aβ plaques (n = 19 cells from 6 mice) where the somatic frequency is similar to the frequency per process in WT mice

Damage-evoked Ca2+ rises are reduced in microglia near Aβ plaques. (A) Representative microglia (tdTomato, red, lower panels) from App WT and AppNL−G−F mice at Aβ plaques or away (> 50 µm) from plaques. Laser damage-evoked Ca2+ rises (upper panels, shown on a “Fire” scale) are measured by GCaMP5g fluorescence changes (ΔF/F shown at peak). (B) Quantification of somatic [Ca2+]i levels over time (ΔF/F) showing their increase upon laser lesion (vertical dashed line). AppNL−G−F microglia, both at (n = 54 from 8 mice; p = 10–4) and away from Aβ plaques (n = 52 from 8 mice; p = 0.03), showed a significantly reduced damage-evoked [Ca2+]i rise compared with App WT microglia (n = 56 from 5 mice)
To build on these initial findings, we employed mice that can be induced to express GCaMP5g under the control of the Cx3cr1 promoter, which enables detection of Ca2+ activity in microglia (Fig. 1F) with an affinity (Kd 0.46 μM, ~ 30-fold dynamic range [2]) similar to Fluo-4 (Kd 0.345 μM, ~ 100-fold dynamic range [20]), and allows the Ca2+ concentration to be monitored in several microglia simultaneously. While use of organic Ca2+ indicators is very common in previous studies, these may be harmful to cells, altering their membrane potential and metabolism and causing swelling [61]. By contrast, GECIs may cause less side effects [61] and allow the study of otherwise undisturbed microglia (although all Ca2+ indicators will intrinsically buffer [Ca2+]i to some extent [45]). As in young rats, we found that microglia in GCaMP5g-expressing mice showed detectable spontaneous Ca2+ activity (Fig. 1G, H) that was 3.5-fold higher in rate in individual cell processes than in somata (as quantified below) and also exhibited local laser lesion-evoked rapid (latency ~ 1 s), transient [Ca2+]i rises (Fig. 1I, J).
Proximity to Aβ plaques alters Ca2+ signalling in activated microglia
To test whether compartmentalised Ca2+ signalling is altered in brain pathology when microglia become activated, we used AppNL−G−F mice that recapitulate amyloid plaque deposition as a pathological hallmark in AD. Amyloid burden increases progressively with age in AppNL−G−F mice [13, 59]. For a reliable assessment of amyloid pathology (i.e., avoiding individual variability at onset but also allowing analysis of microglia both close to and away from plaques, which is difficult at late times because of the high Aβ plaque density), 4-month-old mice were used to study microglia in AD mice (a common age range used across studies with this model [1, 14, 52]). At this age, Aβ plaques can be detected in multiple brain regions apart from the cerebellum in perfusion-fixed slices from AppNL−G−F mice using an antibody against human Aβ (Fig. 2A, B).
We first assessed whether microglial morphology was affected by proximity to Aβ plaques, as has been shown previously in other non-knock-in models of AD, which have a differing development of disease parameters [4, 35, 54]. While microglia were highly ramified in WT mice, their overall process length and ramification were reduced in AppNL−G−F mice (Fig. 2C–E). The total process length per cell was reduced by 13.4% in AppNL−G−F microglia > 50 μm away from Aβ plaques compared with WT microglia (not significantly different, p = 0.07) and by 64.3% in AppNL−G−F microglia at Aβ plaques compared with WT microglia (p = 10–4; Fig. 2D). Similarly, process ramification was reduced closer to Aβ plaques, with AppNL−G−F microglia showing less branched processes and less branches near the soma compared with WT cells (Fig. 2E).
Microglia are known to readily phagocytose Aβ debris [39]. In our AppNL−G−F model, engulfment of Aβ inside microglial CD68-positive lysosomes was confirmed by confocal imaging (Supplementary Fig. 1A). CD68 coverage approximately doubled in hippocampal microglia at Aβ plaques compared with cells away from plaques (p = 10–4; Supplementary Fig. 1B, C), suggesting an increase in the phagocytic capacity of microglia in the presence of Aβ. This is consistent with the increase in CD68 expression reported in plaque-proximal microglia from other AD models and in microglia from patients with AD [26, 68, 72].
Together, these results suggest that microglial responses to Aβ depend considerably on the cells’ local environment, whereby the cells closer to Aβ plaques exhibit stronger changes in morphology and lysosomal content while those further away display an intermediate phenotype closer to WT microglia. Therefore, in subsequent experiments we analysed data from cells at Aβ plaques and away from plaques separately (using a distance threshold of > 50 µm from the nearest plaque edge as the defining criterion for plaque-distant microglia).
We next examined whether the microglial morphological changes in the AppNL−G−F mice are reflected in a specific Ca2+ activity pattern. We found that the frequency of spontaneous Ca2+ transients in microglia somata was dependent on Aβ pathology and proximity to plaques (Fig. 3A, B). While somata of WT microglia exhibited a frequency of 0.09 ± 0.03 transients/min, AppNL−G−F microglia located away from Aβ plaques and at Aβ plaques had somatic transient rates of 0.25 ± 0.05 transients/min (not significantly different from WT, p = 0.1) and 0.44 ± 0.06 transients/min (p = 10–4), respectively. Of note, this effect was not seen in microglial processes, where spontaneous Ca2+ events occurred independently of AppNL−G−F genotype; the rate was 0.55 ± 0.12 transients/min per process in WT microglia, 0.58 ± 0.07 transients/min in AppNL−G−F microglia away from Aβ plaques (p = 0.9) and 0.53 ± 0.05 transients/min in plaque-associated cells (p = 0.9; Fig. 3C, D). In all of these conditions the Ca2+ transient amplitudes showed no significant differences (Supplementary Fig. 3).
Because of the different Ca2+ activity in microglial somata vs. processes when averaged across cells, we also analysed a random subset of cells calculating their individual transient rates in the somata and processes in a paired manner (averaging over processes to obtain a single mean process value for each cell: Fig. 3E). In WT mice, microglia displayed a significantly higher rate of Ca2+ transients per process than per soma (3.5-fold higher; p = 0.04; note that GCaMP5g detects a ratio of the number of transients in single processes to that in somata similar to that for Fluo-4 in Fig. 1C). This was also seen in microglia away from Aβ plaques in AppNL−G−F mice (1.8-fold higher; p = 0.02). In contrast, microglia at Aβ plaques showed a similar rate of spontaneous Ca2+ events in their processes and somata (1.1-fold higher, p = 0.6).
We compared the surface area of a typical microglial soma in a WT mouse (assumed for simplicity to be spherical and with a radius r of 7.5 µm, giving a surface area of 4πr2 = 707 µm2), with that of the ROI used for a typical microglial process segment (assumed for simplicity to be cylindrical and typically with a length l of 15 µm and an approximate radius r of 0.75 µm, giving a surface area of 2πrl = 71 µm2). Thus, the process surface area is approximately tenfold smaller than that of the soma, and this fact cannot account for the higher transient frequency per process.
Next, we studied whether amyloid pathology altered lesion-evoked Ca2+ responses in microglia. While laser injury triggered a robust, rapid transient rise of [Ca2+]i in WT microglial somata, this response was impaired in the AppNL−G−F mice (Fig. 4). In AppNL−G−F microglia away from Aβ plaques, the peak Ca2+ response was reduced by 31.2% (p = 0.09), and in those at Aβ plaques the peak Ca2+ response was reduced by 77.9% (p = 10–4). Together, these data suggest that, while Aβ increases spontaneous Ca2+ activity in microglial somata, it renders microglia less able to respond to substances (such as ATP and ATP-derived metabolites) released acutely by laser-evoked brain damage. In both cases, microglia > 50 μm distant from Aβ plaques exhibited an intermediate phenotype between those at plaques and their WT counterparts.
Ageing alters microglial Ca2+ signalling differently from Aβ pathology
There is growing interest in the cellular processes of ageing and how these relate to neurodegenerative processes [3, 23, 31, 42]. Therefore, we analysed whether the changes in Ca2+ signalling that we found in microglia from AD mice may also occur in old (P300–310) WT mice.
While the frequency of spontaneous Ca2+ transients in old microglial somata was not significantly changed with age (40.0% lower than in microglia from young adult (P120–130) mice but not statistically significantly different; p = 0.46), microglial processes from old mice exhibited a significantly reduced frequency of Ca2+ transients (70.7% lower; p = 10–3; Figure 5A–D). The amplitude of the Ca2+ transients was not significantly affected (Supplementary Fig. 4). We also analysed lesion-evoked somatic Ca2+ responses and found that they were largely abolished in microglia from old mice, with the peak Ca2+ response reduced by 77.4% (p = 10–4) compared with microglia from young adult animals (Fig. 5E, F). These data suggest that normal ageing affects microglial Ca2+ activity differently from the pathological conditions of amyloid plaque deposition, reducing not only brain damage-evoked but also spontaneous Ca2+ activity in the cell processes.

Microglia from old mice exhibit reduced spontaneous and damage-evoked Ca2+ activity compared with microglia from young adult mice. (A) Representative traces showing spontaneous Ca2+ transients (red arrowheads) in microglial somata from P120–130 (young adult) and P300–310 (old) mice. (B) Quantification of spontaneous Ca2+ transient frequency, showing no difference between somata of microglia from young adult and old mice (n = 27 somata from 5 young adult mice and 30 somata from 3 old mice). (C) As (A), but for processes. (D) As (B), but for processes, showing a statistically significant decrease in spontaneous Ca2+ transient frequency per process with age (n = 11 processes from 3 young adult mice and 25 processes from 3 old mice). Data from App WT microglia in Fig. 3 are included as young adult microglia for comparison. (E) Representative microglia (tdTomato, red) from P120–130 (young adult) and P300–310 (old) mice. Laser damage-evoked Ca2+ rises are measured by GCaMP5g fluorescence changes (ΔF/F shown at peak, “Fire” scale).(F) Quantification of somatic [Ca2+]i levels over time (ΔF/F) showing their increase upon laser lesion (vertical dashed line). Microglia from old mice (n = 32 cells from 3 mice) showed a significantly reduced damage-evoked [Ca2+]i rise compared with microglia from young adult mice (n = 56 from 5 mice; p = 10–4). Data from App WT microglia in Fig. 4 are included as young adult microglia for comparison
Discussion
Intracellular Ca2+ elevations are commonly considered an indicator of cellular activity in neurons and astrocytes; however, knowledge of microglial Ca2+ signalling is still sparse despite its importance in controlling key cellular functions. While spontaneous Ca2+ transients are extremely infrequent in acutely isolated or cultured microglia from adult mice [34], in vivo experiments on OGB-1–electroporated microglia showed that ~ 20% of them displayed transients over 15 min [19], and a later study using GECI-expressing microglia found that ~ 4% of cells were active over 20 min [55]. This suggests that the environment, type of preparation and Ca2+ sensor, and the activation state may all affect spontaneous Ca2+ activity in microglia. We found that the great majority of Ca2+ transients in healthy rat or mice microglia occurred in the cells’ processes, whether studied with the soluble Ca2+ sensor Fluo-4 or with GCaMP5g. This finding, which is in agreement with previous work using GECI-expressing mice [64], implies that microglial Ca2+ signalling is finely regulated across cell compartments (as in astrocytes [60]). Ca2+ transients may fulfil different functions in the soma vs. processes, which may be related to differential expression and localisation of Ca2+ signal-transducing membrane receptors and intracellular Ca2+-dependent organelles, such as the endoplasmic reticulum or the endolysosomal compartment.
Although microglia rarely generate Ca2+ transients in the healthy brain, elevations in [Ca2+]i are much more prominent under pathological conditions such as epileptiform activity, acute brain injury, neuroinflammation, and neurodegeneration [10, 19, 55, 63, 64]. In AD, the comparatively well characterised changes in microglial morphology and biomarker expression contrast with the still poorly understood changes of Ca2+ signalling in these cells, and how these vary between plaque-proximal and plaque-remote regions and in different parts of each microglial cell. In AppNL−G−F knock-in mice, we observed that microglial deramification and increased lysosomal burden depend critically on the cells’ proximity to Aβ plaques. This is consistent with studies of transgenic models of AD, showing that changes in microglial gene expression, morphology and electrophysiological properties [13, 54, 66], as well as synapse loss [33], occur predominantly near plaques. In agreement with this, our study revealed significant differences in spontaneous and damage-evoked Ca2+ responses in the AppNL−G−F model of AD between Aβ plaque-proximal and plaque-distant microglia, with the latter presenting an intermediate phenotype closer to that of WT cells. Notably, and adding to previous work, we found that these changes were also dependent on the subcellular compartment, revealing differential effects of Aβ on Ca2+ activity in processes compared with somata. While the Ca2+ activity of microglia has been previously studied in mice modelling AD [11], this was investigated with two different transgenic mouse strains pooled together (both overexpressing App) and using a dye indicator, OGB-1, electroporated into cortical microglial cells to estimate changes in [Ca2+]i. Here, we took advantage of GCaMP5g to visualise changes in [Ca2+]i, because intrinsic GECI expression may affect cell function less than using patch-clamping or electroporation to introduce a dye into the cell (although the affinity and dynamic range of the indicator used will also affect its usefulness).
We found that proximity to Aβ plaques increased the rate of spontaneous Ca2+ transients in the somata of hippocampal microglia. This is in line with results from Brawek et al. (2014), noting an increase in the fraction of microglia with Ca2+ activity in older, transgenic amyloid-depositing mice (although that study, which analysed cortical rather than hippocampal microglia, did not observe any changes in the rate of spontaneous Ca2+ transients in the cells that exhibited them [11]). Increased Ca2+ activity in microglial cells exposed to Aβ may be due to Aβ-evoked UTP/UDP release by stressed neurons acting on microglial metabotropic P2Y6 receptors [57] or Aβ-mediated mechanotransduction mediated via PIEZO1 Ca2+-permeable ion channels involved in amyloid clearance [29, 30]. However, while the Ca2+ transient rate in microglial somata depended on their proximity to Aβ plaques, the higher rate in microglial processes did not. Given the decrease in ramification of microglia at Aβ plaques, the increased Ca2+ frequency in somata vs. processes could also reflect the membrane and Ca2+ stores of the latter being pulled back into the soma, or a redistribution of receptors underlying Ca2+ responses [11]. Of note, the higher Ca2+ activity in the soma of microglia at Aβ plaques correlates with a large increase in CD68 immunoreactivity in these cells, consistent with an increased phagocytic rate (which has been linked to microglial Ca2+ activity [63]). Indeed, in microglia this marker of mature phagocytic lysosomes is localised mainly in somatic regions [27], where it may support an increased Ca2+-dependent phagocytic activity in plaque-associated microglia.
Another key finding from the present study is that microglia at Aβ plaques generated reduced responses to laser lesions. This suggests that cells activated by Aβ might be less able to mount large Ca2+ responses even though they exhibited an increased rate of spontaneous transients in the absence of acute injury. In support of this hypothesis, basal Ca2+ levels are higher in microglia isolated from post-mortem AD brains than in cells from non-dementing donors, but their ATP-evoked responses are smaller [44]. Similarly, in App-overexpressing transgenic mice, chemotaxis towards local ATP is also impaired in microglia near Aβ plaques [11], suggesting that injury-evoked Ca2+ rises might also be reduced in these cells. The decreased response to laser damage–evoked nucleotide release may reflect reduced expression of P2Y12 receptors as a result of microglial activation [36, 47].
Finally, it is important to assess how changes in Ca2+ responses in AD compare to other conditions, as diverse stimuli inducing microglial activation might converge by regulating [Ca2+]i as a core signalling element. The microglial genetic signature is similar between AD and old age [3, 23, 31, 42], and accumulation of Aβ (and tau) in the disease are thought to exacerbate processes involved in ageing, such as cellular senescence, Ca2+ dyshomeostasis, and inflammation [12]. Of note, our study revealed that amyloid plaque deposition and ageing affected the Ca2+ activity of hippocampal microglia differently. In AD mice, spontaneous Ca2+ activity was increased in the somata of microglia but not in their processes, compared with age-matched, non-AD mice. By contrast, spontaneous Ca2+ activity in aged mice was unchanged in microglial somata, but decreased in processes, compared with younger adults. Interestingly and in line with the lack of observed changes in somatic Ca2+ activity with age, microglial CD68 expression is only mildly altered between young and old mice [24, 43]. Of note, Brawek et al. (2014) suggested that the fraction of microglia showing spontaneous Ca2+ activity may be increased in ageing [11], and a subsequent study revealed a bell-shaped pattern with mice of 9–11 months of age (i.e., what we refer to as old age) showing a higher rate of Ca2+ transients than both 2–4- and 18–21-month-old animals [17]. This apparent difference with our results may arise from differences between the AD mouse models used, between the brain regions studied (neocortex vs. hippocampus) or in the Ca2+ detection method used (dye electroporation vs. GECI). Although each method has its own limitations, the high sensitivity of microglia to mechanical influences mediated by channels such as PIEZO1 and TRPV4 [5, 29, 58], with differences reported even between in vivo preparations (acute vs. chronic window preparations [64]), may be a critical factor to consider along with an age-dependent increase in cellular vulnerability and thus a reduced experimental resilience to mechanical perturbations. While reduced lesion-evoked Ca2+ responses are common to microglia in both physiological ageing and AD, our study suggests that clear differences exist at the subcellular level for spontaneous Ca2+ events, the details of which should be clarified by future work. Overall, our work highlights the multifaceted functional states that microglia can adopt depending on their environment (i.e., in a healthy brain, in AD whether located at Aβ plaques or away from them, and in the aged brain), and shows that Ca2+ activity is differently regulated on a subcellular level in each case.
Data Availability
These are available from the corresponding author. All code used for analysing microglial morphological parameters and Ca2+ imaging data has been deposited in GitHub (github.com/AttwellLab). All other study data are included in the article and/or Supplementary Information.
References
Aikawa T, Ren Y, Yamazaki Y, Tachibana M, Johnson MR, Anderson CT, Martens YA, Holm ML, Asmann YW, Saito T, Saido TC, Fitzgerald ML, Bu G, Kanekiyo T (2019) ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc Natl Acad Sci U S A 116:23790–23796. https://doi.org/10.1073/pnas.1908529116
Akerboom J, Chen T-W, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderón NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R et al (2012) Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci Off J Soc Neurosci 32:13819–13840. https://doi.org/10.1523/JNEUROSCI.2601-12.2012
Alsema AM, Jiang Q, Kracht L, Gerrits E, Dubbelaar ML, Miedema A, Brouwer N, Hol EM, Middeldorp J, van Dijk R, Woodbury M, Wachter A, Xi S, Möller T, Biber KP, Kooistra SM, Boddeke EWGM, Eggen BJL (2020) Profiling microglia from Alzheimer’s Disease donors and non-demented elderly in acute human postmortem cortical tissue. Front Mol Neurosci 13:134. https://doi.org/10.3389/fnmol.2020.00134
Alzforum. Research Models. https://www.alzforum.org/research-models/search-results-compares?compare%5B%5D=1140956&compare%5B%5D=1218241&compare%5B%5D=193061. Accessed July 16, 2023
Ayata P, Schaefer A (2020) Innate sensing of mechanical properties of brain tissue by microglia. Curr Opin Immunol 62:123–130. https://doi.org/10.1016/j.coi.2020.01.003
Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT (2001) Imaging of amyloid-β deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 7:369–372
Bernier LP, Bohlen CJ, York EM, Choi HB, Kamyabi A, Dissing-Olesen L, Hefendehl JK, Collins HY, Stevens B, Barres BA, MacVicar BA (2019) Nanoscale Surveillance of the Brain by Microglia via cAMP-Regulated Filopodia. Cell Rep 27:2895-2908.e4. https://doi.org/10.1016/j.celrep.2019.05.010
Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA (2017) Diverse requirements for microglial survival, specification, and function revealed by defined-medium cultures. Neuron 94:759-773.e8. https://doi.org/10.1016/j.neuron.2017.04.043
Brawek B, Garaschuk O (2013) Microglial calcium signaling in the adult, aged and diseased brain. Cell Calcium 53:159–169. https://doi.org/10.1016/j.ceca.2012.12.003
Brawek B, Garaschuk O (2014) Network-wide dysregulation of calcium homeostasis in Alzheimer’s disease. Cell Tissue Res 357:427–438. https://doi.org/10.1007/s00441-014-1798-8
Brawek B, Schwendele B, Riester K, Kohsaka S, Lerdkrai C, Liang Y, Garaschuk O (2014) Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol 127:495–505. https://doi.org/10.1007/s00401-013-1242-2
Busche MA, Hyman BT (2020) Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci 23:1183–1193. https://doi.org/10.1038/s41593-020-0687-6
Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, Qian X, Laláková J, Kühnemund M, Voytyuk I, Wolfs L, Mancuso R, Salta E, Balusu S, Snellinx A, Munck S, Jurek A, Fernandez Navarro J, Saido TC et al (2020) Spatial transcriptomics and in situ sequencing to study Alzheimer’s Disease. Cell 182:976-991.e19. https://doi.org/10.1016/j.cell.2020.06.038
Clayton K, Delpech JC, Herron S, Iwahara N, Ericsson M, Saito T, Saido TC, Ikezu S, Ikezu T (2021) Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol Neurodegener 16:18. https://doi.org/10.1186/s13024-021-00440-9
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan W-B (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758. https://doi.org/10.1038/nn1472
Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, Foreman O, Carano RAD, Chih B, Klumperman J, Bakalarski C, Hanson JE, Sheng M (2018) Changes in the synaptic proteome in tauopathy and rescue of Tau-induced synapse koss by C1q antibodies. Neuron 100:1322-1336.e7. https://doi.org/10.1016/j.neuron.2018.10.014
Del Moral MO, Asavapanumas N, Uzcátegui NL, Garaschuk O (2019) Healthy brain aging modifies microglial calcium signaling in vivo. Int J Mol Sci 20(3):589. https://doi.org/10.3390/ijms20030589
Eichhoff G, Busche MA, Garaschuk O (2008) In vivo calcium imaging of the aging and diseased brain. Eur J Nucl Med Mol Imaging 35:99–106. https://doi.org/10.1007/s00259-007-0709-6
Eichhoff G, Brawek B, Garaschuk O (2011) Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochim Biophys Acta 1813:1014–1024. https://doi.org/10.1016/J.BBAMCR.2010.10.018
Gee KR, Brown KA, Chen WN, Bishop-Stewart J, Gray D, Johnson I (2000) Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium 27:97–106. https://doi.org/10.1054/ceca.1999.0095
Gee JM, Smith NA, Fernandez FR, Economo MN, Brunert D, Rothermel M, Morris SC, Talbot A, Palumbos S, Ichida JM, Shepherd JD, West PJ, Wachowiak M, Capecchi MR, Wilcox KS, White JA, Tvrdik P (2014) Imaging activity in neurons and glia with a Polr2a-based and Cre-dependent GCaMP5G-IRES-tdTomato reporter mouse. Neuron 83:1058–1072. https://doi.org/10.1016/j.neuron.2014.07.024
Giulian D, Baker TJ (1986) Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci 6:2163–2178. https://doi.org/10.1523/jneurosci.06-08-02163.1986
Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, Marsh SE, Saunders A, Macosko E, Ginhoux F, Chen J, Franklin RJM, Piao X, McCarroll SA, Stevens B (2019) Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50:253-271.e6. https://doi.org/10.1016/j.immuni.2018.11.004
Hart AD, Wyttenbach A, Perry VH, Teeling JL (2012) Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav Immun 26:754–765. https://doi.org/10.1016/j.bbi.2011.11.006
Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan W-BB, Julius D (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 9:1512–1519. https://doi.org/10.1038/nn1805
Hemonnot-Girard A-L, Meersseman C, Pastore M, Garcia V, Linck N, Rey C, Chebbi A, Jeanneteau F, Ginsberg SD, Lachuer J, Reynes C, Rassendren F, Hirbec H (2022) Comparative analysis of transcriptome remodeling in plaque-associated and plaque-distant microglia during amyloid-β pathology progression in mice. J Neuroinflammation 19:234. https://doi.org/10.1186/s12974-022-02581-0
Hendrickx DAE, van Eden CG, Schuurman KG, Hamann J, Huitinga I (2017) Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J Neuroimmunol 309:12–22. https://doi.org/10.1016/j.jneuroim.2017.04.007
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. https://doi.org/10.1126/science.aad8373
Hu J, Chen Q, Zhu H, Hou L, Liu W, Yang Q, Shen H, Chai G, Zhang B, Chen S, Cai Z, Wu C, Hong F, Li H, Chen S, Xiao N, Wang Z-X, Zhang X, Wang B et al (2023) Microglial Piezo1 senses Aβ fibril stiffness to restrict Alzheimer’s disease. Neuron 111:15-29.e8. https://doi.org/10.1016/j.neuron.2022.10.021
Jäntti H, Sitnikova V, Ishchenko Y, Shakirzyanova A, Giudice L, Ugidos IF, Gómez-Budia M, Korvenlaita N, Ohtonen S, Belaya I, Fazaludeen F, Mikhailov N, Gotkiewicz M, Ketola K, Lehtonen Š, Koistinaho J, Kanninen KM, Hernández D, Pébay A et al (2022) Microglial amyloid beta clearance is driven by PIEZO1 channels. J Neuroinflammation 19:147. https://doi.org/10.1186/s12974-022-02486-y
Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, Thambisetty M, Montine TJ, Lee EB, Trojanowski JQ, Beach TG, Reiman EM, Haroutunian V, Wang M, Schadt E et al (2020) Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med 26:769–780. https://doi.org/10.1038/s41591-020-0815-6
Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT (2002) Imaging Aβ plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 61:797–805. https://doi.org/10.1093/jnen/61.9.797
Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL (2009) Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106:4012–4017. https://doi.org/10.1073/pnas.0811698106
Korvers L, de Andrade CA, Mersch M, Matyash V, Kettenmann H, Semtner M (2016) Spontaneous Ca2+ transients in mouse microglia. Cell Calcium 60:396–406. https://doi.org/10.1016/j.ceca.2016.09.004
Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Miller KR, Prokop S, Kettenmann H, Heppner FL (2013) Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 8:e60921. https://doi.org/10.1371/journal.pone.0060921
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H et al (2017) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47:566–581. https://doi.org/10.1016/j.immuni.2017.08.008
Krasnow AM, Ford MC, Valdivia LE, Wilson SW, Attwell D (2018) Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat Neurosci 21:24–30. https://doi.org/10.1038/s41593-017-0031-y
Lawson LJ, Perry VH, Dri P, Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39:151–170. https://doi.org/10.1016/0306-4522(90)90229-W
Lee CYD, Daggett A, Gu X, Jiang L-L, Langfelder P, Li X, Wang N, Zhao Y, Park CS, Cooper Y, Ferando I, Mody I, Coppola G, Xu H, Yang XW (2018) Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s Disease models. Neuron 97:1032-1048.e5. https://doi.org/10.1016/j.neuron.2018.02.002
Light AR, Wu Y, Hughen RW, Guthrie PB (2006) Purinergic receptors activating rapid intracellular Ca increases in microglia. Neuron Glia Biol 2:125–138. https://doi.org/10.1017/S1740925X05000323
Madry C, Kyrargyri V, Arancibia-Cárcamo IL, Jolivet R, Kohsaka S, Bryan RM, Attwell D (2018) Microglial ramification, surveillance, and interleukin-1β release are regulated by the two-pore domain K+ channel THIK-1. Neuron 97:299-312.e6. https://doi.org/10.1016/j.neuron.2017.12.002
Masuda T, Sankowski R, Staszewski O, Prinz M (2020) Microglia heterogeneity in the single-cell era. Cell Rep 30:1271–1281. https://doi.org/10.1016/j.celrep.2020.01.010
Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, NahabooSolim MA, Moens TG, Paublete RM, Ali SS, Perona M, Desai R, Smith KJ, Latcham J, Fulleylove M, Richardson JC, Hardy J, Edwards FA (2015) A Genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep 10:633–644. https://doi.org/10.1016/j.celrep.2014.12.041
McLarnon JG, Choi HB, Lue LF, Walker DG, Kim SU (2005) Perturbations in calcium-mediated signal transduction in microglia from Alzheimer’s disease patients. J Neurosci Res 81:426–435. https://doi.org/10.1002/jnr.20487
McMahon SM, Jackson MB (2018) An inconvenient truth: Calcium sensors are calcium buffers. Trends Neurosci 41:880–884. https://doi.org/10.1016/j.tins.2018.09.005
Melendez AJ, Tay HK (2008) Phagocytosis: A repertoire of receptors and Ca2+ as a key second messenger. Biosci Rep 28:287–298. https://doi.org/10.1042/BSR20080082
Mildner A, Huang H, Radke J, Stenzel W, Priller J (2017) P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 65:375–387. https://doi.org/10.1002/glia.23097
Möller T (2002) Calcium signaling in microglial cells. Glia 40:184–194. https://doi.org/10.1002/glia.10152
Möller T, Kann O, Verkhratsky A, Kettenmann H (2000) Activation of mouse microglial cells affects P2 receptor signaling. Brain Res 853:49–59. https://doi.org/10.1016/s0006-8993(99)02244-1
Monif M, Reid CA, Powell KL, Smart ML, Williams DA (2009) The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci Off J Soc Neurosci 29:3781–3791. https://doi.org/10.1523/JNEUROSCI.5512-08.2009
Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 109:11282–11287. https://doi.org/10.1073/pnas.1117765109
Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Sethi H, Attwell D (2019) Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365:aav9518. https://doi.org/10.1126/science.aav9518
Nunes P, Demaurex N (2010) The role of calcium signaling in phagocytosis. J Leukoc Biol 88:57–68. https://doi.org/10.1189/jlb.0110028
Plescher M, Seifert G, Hansen JN, Bedner P, Steinhäuser C, Halle A (2018) Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer’s disease mouse model. Glia 66(7):1464–1480. https://doi.org/10.1002/glia.23318
Pozner A, Xu B, Palumbos S, Gee JMJM, Tvrdik P, Capecchi MR (2015) Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC::G5-tdT reporter mouse. Front Mol Neurosci 8:12. https://doi.org/10.3389/fnmol.2015.00012
Prinz M, Jung S, Priller J (2019) Microglia biology: One century of evolving concepts. Cell 179:292–311. https://doi.org/10.1016/j.cell.2019.08.053
Puigdellívol M, Allendorf DH, Brown GC (2020) Sialylation and Galectin-3 in Microglia-Mediated Neuroinflammation and Neurodegeneration. Front Cell Neurosci 14:1–11. https://doi.org/10.3389/fncel.2020.00162
Redmon SN, Yarishkin O, Lakk M, Jo A, Mustafic E, Tvrdik P, Krizaj D (2021) TRPV4 channels mediate the mechanoresponse in retinal microglia. Glia 69:1563–1582. https://doi.org/10.1002/glia.23979
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC (2014) Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci 17:661–663. https://doi.org/10.1038/nn.3697
Semyanov A, Henneberger C, Agarwal A (2020) Making sense of astrocytic calcium signals — from acquisition to interpretation. Nat Rev Neurosci 21:551–564. https://doi.org/10.1038/s41583-020-0361-8
Smith NA, Kress BT, Lu Y, Chandler-Militello D, Benraiss A, Nedergaard M (2018) Fluorescent Ca2+ indicators directly inhibit the Na,K-ATPase and disrupt cellular functions. Sci Signal 11:. https://doi.org/10.1126/scisignal.aal2039
Tejera D, Heneka MT (2019) In vivo phagocytosis analysis of amyloid beta. Methods Mol Biol 2034:287–292. https://doi.org/10.1007/978-1-4939-9658-2_21
Umpierre AD, Li B, Ayasoufi K, Zhao S, Xie M, Thyen G, Hur B, Zheng J, Liang Y, Wu Z, Yu X, Sung J, Johnson AJ, Li Y, Wu L-J (2023) Microglial P2Y6 calcium signaling promotes phagocytosis and shapes neuroimmune responses in epileptogenesis. BioRxiv. 2023.06.12.544691. https://doi.org/10.1101/2023.06.12.544691
Umpierre AD, Bystrom LL, Ying Y, Liu YU, Worrell G, Wu L-J (2020) Microglial calcium signaling is attuned to neuronal activity in awake mice. Elife 9:e56502. https://doi.org/10.7554/eLife.56502
Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, Yu J, Perez-Torres C, Frouin A, Wilton DK, Funk K, DeMasters BK, Jiang X, Bowen JR, Mennerick S, Robinson JK, Garbow JR, Tyler KL, Suthar MS et al (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534:538–543. https://doi.org/10.1038/nature18283
Wendt S, Maricos M, Vana N, Meyer N, Guneykaya D, Semtner M, Kettenmann H (2017) Changes in phagocytosis and potassium channel activity in microglia of 5xFAD mice indicate alterations in purinergic signaling in a mouse model of Alzheimer’s disease. Neurobiol Aging 58:41–53. https://doi.org/10.1016/j.neurobiolaging.2017.05.027
Werneburg S, Jung J, Kunjamma RB, Ha SK, Luciano NJ, Willis CM, Gao G, Biscola NP, Havton LA, Crocker SJ, Popko B, Reich DS, Schafer DP (2020) Targeted complement inhibition at synapses prevents microglial synaptic engulfment and synapse loss in demyelinating disease. Immunity 52:167-182.e7. https://doi.org/10.1016/j.immuni.2019.12.004
Woollacott IOC, Toomey CE, Strand C, Courtney R, Benson BC, Rohrer JD, Lashley T (2020) Microglial burden, activation and dystrophy patterns in frontotemporal lobar degeneration. J Neuroinflammation 17:1–27. https://doi.org/10.1186/s12974-020-01907-0
Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, Srinivasan K, Huntley MA, Wang Y, Wang TM, Hedehus M, Barck KH, Stark M, Ngu H, Foreman O, Meilandt WJ, Elstrott J, Chang MC, Hansen DV et al (2019) Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep 28:2111-2123.e6. https://doi.org/10.1016/j.celrep.2019.07.060
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91. https://doi.org/10.1016/j.immuni.2012.12.001
Yuan P, Grutzendler J (2016) Attenuation of β-amyloid deposition and neurotoxicity by chemogenetic modulation of neural activity. J Neurosci 36:632–641. https://doi.org/10.1523/JNEUROSCI.2531-15.2016
Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA et al (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26:131–142. https://doi.org/10.1038/s41591-019-0695-9
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by European Research Council (BrainEnergy) and Wellcome Investigator Awards (099222) to DA, a Wellcome Trust four-year PhD studentship to PI and an Alzheimer Forschung Initiative grant (21072) to CM. For the purpose of open access, the authors have applied a CC-BY public licence to any Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to the conception or design of the work. PI, RBJ and CM acquired or analysed data. PI, RBJ and CM prepared Fig. 1. PI prepared Figs. 2, 3, 4 and 5. PI wrote the main manuscript text and all authors edited it. All authors reviewed the manuscript critically for important intellectual content and approved the final version for submission. DA obtained funding for the laboratory in which the research was conducted.
Corresponding authors
Ethics declarations
Ethical Approval
Animal procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986 (Home Office License 70/8976) after review by UCL's internal review board and by the UK Government Home Office.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Izquierdo, P., Jolivet, R.B., Attwell, D. et al. Amyloid plaques and normal ageing have differential effects on microglial Ca2+ activity in the mouse brain. Pflugers Arch - Eur J Physiol 476, 257–270 (2024). https://doi.org/10.1007/s00424-023-02871-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-023-02871-3