
Abstract
Recently, specific driver mutations were identified in chondroblastoma, giant cell tumour of bone and central cartilaginous tumours (specifically enchondroma and central chondrosarcoma), sharing the ability to induce genome-wide epigenetic alterations. In chondroblastoma and giant cell tumour of bone, the neoplastic mononuclear stromal-like cells frequently harbour specific point mutations in the genes encoding for histone H3.3 (H3F3A and H3F3B). The identification of these driver mutations has led to development of novel diagnostic tools to distinguish between chondroblastoma, giant cell tumour of bone and other giant cell containing tumours. From a biological perspective, these mutations induce several global and local alterations of the histone modification marks. Similar observations are made for central cartilaginous tumours, which frequently harbour specific point mutations in the metabolic enzymes IDH1 or IDH2. Besides an altered methylation pattern on histones, IDH mutations also induce a global DNA hypermethylation phenotype. In all of these tumour types, the mutation-driven epigenetic alterations lead to a highly altered transcriptome, resulting for instance in alterations in differentiation. These genomic alterations have diagnostic impact. Further research is needed to identify the genes and signalling pathways that are affected by the epigenetic alterations, which will hopefully lead to a better understanding of the biological mechanism underlying tumourigenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genetic make-up of tumour cells alone is insufficient to explain differences in cellular behaviour. One factor that can explain these differences is epigenetics: the stable and heritable change of gene function caused by other factors than alterations in the DNA sequence [1]. This involves mainly changes in the three-dimensional structure of DNA, which is defined by histones, nucleosomes and chromatin condensation. By altering the DNA structure, the accessibility for proteins involved in gene transcription is either enhanced or reduced, regulating gene expression.
To control DNA accessibility, several enzymes such as DNA methyltransferases, histone acetyltransferases, ubiquitin ligases and histone methyltransferases make modifications (e.g. methylation, acetylation, phosphorylation and ubiquitination) on DNA itself or on certain amino acid positions on histone tails [2]. At another level, chromatin remodelling complexes (e.g. SWI/SNF and INO80) construct, reposition or evict nucleosomes to change the packaging of the DNA [2]. Together, the dynamic and reversible epigenetic modifications define which genetic information is available for a cell and thereby regulate cellular fate and homeostasis.
Recently, it was shown that epigenetic regulatory genes are frequently mutated across several tumour types, leading to deregulation of normal gene expression patterns (e.g. silencing of tumour suppressor genes and activation of oncogenes) and thereby promotion of tumourigenesis [3]. Epigenetic alterations, unlike genetic causes of diseases, are reversible, making them interesting targets to develop novel anti-cancer therapies. In the past couple of years, several drugs targeting DNA methylation (i.e. azacitidine and decitabine) and histone acetylation (i.e. vorinostat, romidepsin and panobinostat) have been FDA approved for different haematological malignancies. Many clinical trials are ongoing to evaluate the effect of epigenetic drugs in a wide variety of tumour types, including advanced and metastatic sarcoma [4].
Bone and soft tissue tumours are a rare, heterogeneous group of mesenchymal tumours which frequently harbour epigenetic alterations. For instance, the promoter of the tumour suppressor gene PTEN is frequently hypermethylated in soft tissue sarcomas, while loss-of-function mutations in PTEN are rare in these tumours [5]. Furthermore, several bone and soft tissue tumours harbour an aberrant DNA methylation pattern across the whole genome (e.g. chondrosarcoma [6], Ewing sarcoma [7] and rhabdomyosarcoma [8]).
Deregulation of chromatin remodelling complexes is also commonly seen in sarcomas. For instance, loss of SMARCB1 is the hallmark of malignant rhabdoid tumours and epithelioid sarcomas [9, 10]. SMARCB1 is a core subunit of the SWI/SNF chromatin remodelling complex: a group of proteins involved in positioning the nucleosomes on the DNA. Furthermore, approximately 80% of all malignant peripheral nerve sheath tumours have mutations in the EED or SUZ12 subunits of the polycomb repressive complex (PRC) 2 [11]. This complex is primarily involved in maintaining the repressive tri-methylation mark on lysine 27 of histone H3 (H3K27me3) which has led to the use of an easily applicable immunohistochemical diagnostic tool [12,13,14]. Moreover, certain translocations, such as the SS18-SSX fusion in synovial sarcomas, affect epigenetics. The SS18 gene is involved in the SWI/SNF complex, while SSX1 and SSX2 are subunits of the PRC complexes [15]. Fusion of these genes leads to the formation of an altered chromatin remodelling complex which lacks the SMARCB1 subunit, resulting in transcriptional repression of tumour suppressor genes (e.g. EGR1) and transcriptional activation of oncogenes (e.g. SOX2) [15, 16].
Mesenchymal tumours can also harbour mutations in histones themselves or genes indirectly related to epigenetic regulation. This review will focus on a group of bone tumours that share mutations in genes involved in epigenetic regulation: H3 histone family member 3A and 3B (H3F3A and H3F3B) mutations in giant cell tumour of bone and chondroblastoma, respectively, and isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) mutations in central cartilaginous tumours.
Histone H3.3 variants in giant cell tumour of bone and chondroblastoma
Giant cell tumour of bone
Giant cell tumour of bone (GCTB) is a locally aggressive and rarely metastasizing neoplasm (Table 1). These tumours typically arise in the end of long bones and are predominantly formed in skeletally mature young adults between the age of 20 and 45 [17]. Although GCTB has a high recurrence rate (~ 25% of patients), malignant transformation is very rare and occurs in less than 1% of the patients [32]. Pulmonary metastases are very rare and typically slow-growing. These are thought to represent pulmonary implants that result from embolization of intravascular growths of GCTB [33].
GCTB is histologically characterized by three types of cells: the multinucleated osteoclast-like giant cells, the mononuclear macrophage-like osteoclast precursor cells and the mononuclear spindle-shaped stromal cells. The latter are considered as the neoplastic component of GCTB; these cells have the ability to form tumours in mice and can be maintained in vitro [34, 35]. The neoplastic stromal cells are of osteoblastic origin and secrete high levels of chemokines (e.g. MCP-1 (CCL2) and SDF-1 (CXCL12)) to attract the mononuclear osteoclast precursor cells to the tumour site [36]. Subsequently, M-CSF (CSF1) secreted by the neoplastic stromal cells induces the expression of the RANK (TNFRSF11A) receptor on the attracted monocytes [37]. RANK ligand (TNFSF11) expression is upregulated by the neoplastic stromal cells, resulting in monocyte differentiation and fusion and thereby formation of the characteristic large giant cells [38]. These newly formed giant cells have bone resorption properties and cause the characteristic osteolysis.
The current treatment of GCTB is curettage combined with local adjuvant therapy to fill the bone cavity (e.g. polymethylmethacrylate or cancellous bone grafts) or, if necessary, en bloc resection. If tumours are unresectable or recur, patients can be treated with denosumab, a monoclonal antibody against RANK ligand [32], or bisphosphonates [39]. Neutralization of RANK ligand will inhibit the formation of giant cells and consequently the bone resorption process. However, the neoplastic stromal cells are not affected, requiring life-long treatment and causing relapse if treatment is discontinued. This underlines the need to develop novel therapeutic strategies directly targeting the neoplastic stromal cells in GCTB.
Chondroblastoma
Chondroblastoma is a benign, cartilage-forming tumour which accounts for less than 1% of all primary bone tumours (Table 1). These tumours typically arise in skeletally immature patients ranging in age from 10 to 25 years old, predominantly in the epiphysis of long bones [18]. Chondroblastomas are successfully treated with curettage combined with bone grafting, or radiofrequency ablation. Recurrence rates are between 14 and 18%, and the development of so-called ‘benign’ pulmonary metastases is very rare. The immature cartilage cells, chondroblasts, located in growth plates are considered as the cells of origin in chondroblastoma [40], although another study suggests that these tumours deposit osteoid matrix and should be reclassified as bone-forming tumours [41]. Additionally, a variable amount of osteoclast-like giant cells is seen. Chondroblastoma differs from giant cell tumour of bone by the presence of a sclerotic rim, the relatively younger age of the patients and histological characteristics (e.g. cell type and matrix formation), although it is sometimes difficult to distinguish (Table 1).
Histone H3.3 variants
Recently, a study was published which described the presence of histone H3.3 mutations in both chondroblastoma and GCTB [24]. Interestingly, GCTB exclusively showed alterations in the H3F3A gene (92% of cases), while H3F3B was predominantly affected in chondroblastoma (95% of cases; 93% H3F3B and 7% H3F3A). H3F3A and H3F3B are paralogous genes located on different chromosomes (chromosome 1 and chromosome 17, respectively) and have a slightly different DNA sequence, but both encode for the exact same histone H3.3 protein. The altered amino acids in these genes are remarkably specific for each tumour type. GCTB specifically harbours G34W mutations or, less common, G34L variants in H3F3A, while chondroblastoma exclusively shows K36M alterations in both H3F3A and H3F3B [24]. Histone H3.3 alterations have also been described in glioma and glioblastoma; especially K27M and G34R/V alterations in H3F3A (up to 60%) [42]. Of note, histone H3.3 variants are rarely found in other tumours, making these mutations highly specific for glioma, glioblastoma, GCTB and chondroblastoma [24, 43]. This has led to development of novel diagnostic tools that either use sequencing of H3F3A and H3F3B, or immunohistochemistry using G34W and K36M mutation-specific antibodies, to distinguish between chondroblastoma, GCTB and other giant cell-rich tumours [30, 31, 44] (Fig. 1). Their high frequency of occurrence suggests an important role for these mutations in tumourigenesis, suggesting that targeting these mutations or the underlying alterations could be used as novel therapeutic strategies. Interestingly, within the tumours, the mutation was only observed in stromal-like cells and not in cells from the osteoclast lineage, indicating that these cells are the neoplastic and driving component of GCTB and chondroblastoma [24] (Fig. 1).

Histology and mutation-specific immunohistochemistry in chondroblastoma, giant cell tumour of bone and central cartilaginous tumours
Epigenetic alterations induced by histone H3.3 variants
Histone H3.3 is a well conserved replacement histone that is highly structurally related to the canonical histone H3 protein. H3.3 is the most predominant form of histone H3 in non-dividing cells, and incorporation is independent of DNA synthesis. Usually, histone H3.3 replaces the canonical histone H3 protein in the nucleosome of active genes (e.g. promoter and gene bodies), suggesting that it may be an epigenetic regulator of transcriptionally active chromatin [45]. The most frequently modified lysine residues on histone H3.3 are K4, K9, K27 and K36, which usually acquire mono-, di- or tri-methylation and acetylation marks. Of note, two histone H3.3 mutations (i.e. K27M and K36M) are exactly at the location of these lysine residues, while the G34 mutations are closely situated near the K36 position. This implies that all H3.3 mutations could hamper the formation of modification marks and thereby change the transcription of genes across the whole genome.
Several research groups have started to elucidate the epigenetic alterations caused by H3.3 mutations and the mechanisms underlying these changes in chondroblastoma and GCTB. The K36M mutation in H3F3A and H3F3B (H3K36M) causes genome-wide reduction of methylation in chondroblastoma, specifically loss of di-methylation of H3K36 (H3K36me2) at intergenic regions and H3K36me3 at gene bodies [46, 47]. Methylation of K36 is associated with active gene transcription, indicating that the H3K36M mutation inhibits gene transcription across the whole genome [48]. Pathways that are shown to be altered are chondrogenic and osteogenic differentiation (i.e. downregulation of BMP2 and RUNX2, respectively) and homologous recombination (i.e. downregulation of BRCA1 and ATRX) [46].
The reduction in methylation is caused by the inhibition of histone methyltransferases (HMTs) such as NSD1, NSD2 and SETD2. The active SET domain within these HMTs contains tyrosine residues which normally bind to a lysine residue on histone tails to deposit a methylation mark. However, in the case of a H3K36M mutation, the lysine is mutated into a methionine which results in stronger binding of HMTs to the histone tail. This causes a global reduction in the HMT levels and its enzymatic activity, leading to a genome-wide alteration of the H3K36me2/3 landscape. This higher affinity of HMTs for H3K36M was confirmed by solving the crystal structure of the SETD2-H3K36M complex and by performing pull-down assays showing an enriched interaction between HMTs and H3K36M [46, 47, 49, 50].
Additionally, H3K36M mutations cause redistribution of the repressive H3K27me3 histone mark and its reader complex PRC1. Upregulation of H3K27me3 has been observed in intergenic regions, while loss was observed at gene loci [47]. Consequently, re-expression of normally H3K27me3 silenced genes occurs such as Wnt6 and Sox6, genes involved in blocking mesenchymal differentiation. Thus, H3K36M mutations change the K27 and K36 methylation landscape, resulting in a highly altered transcriptome.
In contrast, the mechanism underlying the observed epigenetic changes in cells harbouring G34 mutations in H3F3A (H3F3A G34) is less clear. Although histone modifications cannot be directly made on the G34 residue, H3F3A G34 mutations may affect the deposition of marks on the K36 position, and possibly also other modification sites, due to steric hindrance. An interaction study showed that the G34 position is involved in binding SETD2 [50]. SETD2 has a narrow binding pocket at the G34 position which can only bind the smallest amino acid: glycine. Consequently, all H3F3A G34 mutations will prevent SETD2 from binding histone H3.3, leading to a decrease in H3K36me3. However, a genome-wide reduction of H3K36me2/3 was not observed in cells harbouring H3F3A G34R/V mutations, but only at sites where the mutated histone was localized [51]. The inhibitory effect on the enzymatic activity of SETD2 has also been confirmed for H3F3A G34W/L mutations in GCTB, with a concomitant decrease in H3K36me3 and, noticeably, an increase in the repressive H3K27me3 mark on the mutated histone tails [52]. However, another study found an increase of the H3K36me3 mark in GCTB harbouring H3F3A G34W/V mutations [44]. Moreover, in glioblastoma, H3F3A G34V mutations inhibit the enzymatic activity of KDM4 H3K36me3 demethylases, leading to increased H3K36me3 and H3K9me3 in histone H3.3 enriched regions [53]. This suggests that H3F3A G34 mutations can both increase and decrease the H3K36me3 mark at particular sites in the genome, leading to significant change in the transcriptome of cells. In high-grade H3F3A G34V-mutated glioma, this leads to a change in the methylation pattern of approximately 150 genes (e.g. the oncogene MYCN) [54].
For GCTB, the genes undergoing a change upon redistribution of the H3K36me3 marks as a result of the H3F3A G34W/L mutations remain to be identified. Changes that are observed in primary GCTB cells harbouring a G34W mutation are increased proliferation, migration and colony formation capacity as compared to wild-type counterparts [55, 56]. Furthermore, splicing aberrations and alternative transcription start sites are frequently observed in H3F3A G34W-mutated cells, suggesting an alteration in the RNA processing pathway [55]. Future studies are needed to elucidate which genes and signalling pathways are affected by the H3F3A G34 mutation-driven epigenetic alterations.
Therapeutic implications for epigenetic alterations induced by histone H3.3 variants
Currently, there are limited reports on in vitro or in vivo studies describing promising epigenetic therapeutic strategies for GCTB. Therapies directly targeting the mutated histones are lacking, and reversal of genome-wide epigenetic alterations is not feasible without major side-effects on the healthy, normal epigenetic landscape. However, the use of histone demethylase inhibitors, such as GSKJ4, may counteract the observed reduction or redistribution of H3K36me3 and H3K27me3. On the other hand, reduction of H3K36me3 may be a promising therapeutic target and recently it was shown that WEE1 inhibition in GCTB results in a global reduction of H3K36me3 and a decrease in proliferation [35]. Nevertheless, further research is needed to identify the genes affected by the epigenetic changes to develop targeted, effective therapeutic strategies. Additionally, most research has been performed on methylation changes across the genome, while the effect on other modification marks, such as acetylation, phosphorylation and ubiquitination, has not been examined yet. Another promising therapeutic strategy could be to stimulate the enzymatic activity of SETD2 and KDM4 or to overcome the strong binding between KDM4-H3F3A G34, but this kind of compounds are currently lacking.
IDH1 and IDH2 mutations in Enchondroma and Chondrosarcoma
Enchondroma and Chondrosarcoma
Enchondroma is a benign, cartilage producing tumour which accounts for 10–25% of all benign bone tumours [19] (Table 1). These tumours can occur at all ages and mainly affect bones of the hands and feet and long tubular bones. Normally, enchondromas are not treated, since most tumours are solitary and rarely undergo malignant transformation. Non-hereditary syndromes causing multiple enchondromas (i.e. Ollier disease and Maffucci syndrome) increase the risk of progression to chondrosarcoma to ~ 40% [57].
Chondrosarcomas are malignant, cartilage-forming tumours and make up 20% of all malignant bone tumours [20] (Table 1). These tumours usually develop during adulthood, mainly affecting the pelvis and long bones, and either arise as primary tumours (i.e. in a previously healthy bone) or as secondary tumours (i.e. in a benign precursor lesion). Conventional chondrosarcoma is the most prevalent form (85%), followed by dedifferentiated chondrosarcoma (10%). Most conventional chondrosarcomas arise in the medulla of the bone (central conventional chondrosarcoma). Histological grading is considered as the most important prognostic marker to predict the 10-year survival rate of conventional chondrosarcoma patients: 88% for atypical cartilaginous tumours and chondrosarcoma grade I (ACT/Grade I), 62% for Grade II and 26% for Grade III [23]. Furthermore, the metastatic potential of conventional chondrosarcoma also increases in higher grade tumours: 0%, 10% and 71% for ACT/Grade I, Grade II and Grade III, respectively [21]. Chondrosarcomas are intrinsically resistant towards conventional chemo- and radiotherapy, and surgery is considered as the only curative treatment option. Although mutations in for instance IDH1 and -2, COL2A1 and TP53 combined with aberrations in signalling pathways such as IHH/PTHrP, pRB and PI3K/mTOR have been identified as key molecular changes in chondrosarcoma [20], the development of novel targeted therapies has not yet been successful.
IDH1 and IDH2 mutations
Eighty-seven percent of the solitary and multiple enchondromas, ~ 50% of the primary central conventional chondrosarcomas and 86% of the secondary central chondrosarcomas harbour heterozygous point mutations in IDH1 and IDH2 [6, 25, 26]. The introduction of an IDH1 mutation alone is sufficient to induce enchondromatosis in mice [58]. Furthermore, introduction or imitation of IDH mutations in mesenchymal stem cells impairs osteogenic differentiation and promotes chondrogenic differentiation in vitro [59, 60]. Thus, IDH1 and IDH2 mutations represent an early event in the development of central cartilaginous tumours. Over time, central chondrosarcomas acquire additional mutations (e.g. COL2A1 and TP53), which probably become more important drivers of tumourigenesis than IDH mutations [61].
IDH1 and IDH2 mutations also frequently occur in for instance acute myeloid leukaemia (AML), glioma and cholangiocarcinoma [62]. Most of the point mutations occur at specific arginine residues: the R132 position in IDH1 and the R140/R172 positions in IDH2. Some of these variants can be detected with the use of immunohistochemistry and mutation-specific antibodies, displaying the mosaicism for IDH mutations in central cartilaginous tumours (Fig. 1). The identification of IDH mutations in central cartilaginous tumours facilitates the diagnosis of dedifferentiated chondrosarcoma on a small biopsy, and the distinction between chondroblastic osteosarcoma and central chondrosarcoma [63]. Interestingly, the most frequent point mutations in either IDH1 or IDH2 differ among the above listed tumour types. Central chondrosarcoma and cholangiocarcinoma predominantly harbour R132C mutations in IDH1 (50%), glioma has mainly R132H mutations in IDH1 (90%) and AML has most often R140Q mutations in IDH2 (30–50%) [62]. However, none of the mutations are exclusively observed in one tumour type, suggesting a similar effect of all IDH1 and IDH2 mutations on tumourigenesis.
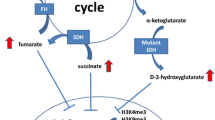
IDH1 and IDH2 are enzymes with a similar function in the tricarboxylic acid cycle (TCA cycle) where they convert isocitrate into α-ketoglutarate (α-KG) and CO2. Due to the arginine point mutations, the IDH enzymes acquire a gain-of-function, leading to additional conversion of α-KG into the oncometabolite D-2-hydroxyglutarate (D-2-HG). Of note, different IDH1 and IDH2 mutations produce variable levels of D-2-HG: R132C is a strong D-2-HG producer, while R132H and R140Q produce lower levels of the oncometabolite [62]. This could explain why some tumour types harbour specific point mutations more frequently than others, suggesting that enchondromas rely on high levels of the oncometabolite.
D-2-HG and α-KG have a high structural similarity, leading to competitive binding of α-KG-dependent enzymes by D-2-HG. Some of these α-KG-dependent enzymes are involved in maintaining the epigenetic landscape of cells, such as DNA demethylases (family of TET enzymes) and histone demethylases (family of Jumonji enzymes). IDH mutations also affect other processes within the cell, such as metabolism, cell growth signalling pathways and DNA damage repair [64], but these alterations go beyond the scope of this review.
Epigenetic alterations induced by IDH1 and IDH2 mutations
It has been shown that D-2-HG inhibits the activity of Tet Methylcytosine Dioxygenase 2 (TET2) in vitro [65]. Normally, TET2 mediates the demethylation of DNA by hydroxylation of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), which results in a DNA hypermethylation phenotype if this enzyme is inhibited by D-2-HG. Several studies confirmed the global DNA hypermethylation phenotype in several IDH-mutated tumours, including enchondroma and central chondrosarcoma [6, 66]. However, an immunohistochemistry study including 9 enchondromas and 92 central chondrosarcomas showed that the 5-mC and 5-hmC levels are highly variable and not associated with the IDH mutation status [67]. Additionally, long-term treatment of chondrosarcoma cell lines with the IDH1 mutant inhibitor AGI-5198 does not alter the DNA methylation phenotype, suggesting that the epigenetic alterations might have become independent of the IDH1 mutation [61]. Another study also observed a persistence of the aberrant DNA methylation phenotype in 25% of the loci after withdrawal of doxycycline induced expression of the IDH1 mutant in astrocytes [68]. To conclude, enchondromas and central chondrosarcomas have an altered DNA methylation phenotype, but this might have become partially static and no longer directly dependent on D-2-HG mediated inhibition of TET2.
Another group of enzymes that are inhibited by D-2-HG is the Jumonji-C domain-containing histone lysine demethylases (KDM enzymes), including the KDM4 family members which are also affected by histone H3.3 mutations [69]. Inhibition of these enzymes results in a global increase of several di- and tri-methylation marks on histone tails. Introduction of an IDH1 or IDH2 mutation in HEK293T cells leads to elevated levels of the H3K4me3, H3K9me2/3, H3K27me3, H3K36me3 and H3K79me2 histone marks [70]. An induction in H3K9me3 and H3K27me3 is also observed in glioma samples harbouring an endogenous IDH1 mutation [70]. However, in vivo studies with IDH1 mutant knock-in mice show that none or only a subset (i.e. H3K4me3, H3K36me3 and H3K79me3) of the histone methylation marks are aberrant [70, 71]. This implies that an IDH mutation alone is not enough to alter the histone methylation landscape. Similar conflicting results are also observed in enchondroma and central chondrosarcoma: an immunohistochemistry study on 101 primary tumours shows high levels of the H3K4me3, H3K9me3 and H3K27me3 histone modification marks, irrespective of the IDH1 or IDH2 mutation status [67]. Additionally, long-term AGI-5198 treatment of chondrosarcoma cell lines could not alter the expression of H3K4me3, H3K9me3 and H3K27me3 [61]. Both studies suggest that the methylation histone modification marks in enchondromas and central chondrosarcoma are regulated by additional mechanisms besides D-2-HG-dependent inhibition of the KDM enzymes.
Although the specific genes affected by IDH mutation-induced epigenetic alterations remain to be elucidated, most studies point towards reduced differentiation capacity. Both neural and haematopoietic differentiations are impaired due to aberrant histone methylation marks or DNA hypermethylation [70, 72]. In IDH1-mutated cholangiocarcinoma, reduced expression of H3K4me3 at the HNF4A promoter inhibits hepatocyte differentiation [73]. Furthermore, introduction of an IDH mutation or addition of D-2-HG impairs osteogenic differentiation and induces chondrogenic differentiation in mesenchymal stem cells [59, 60]. IDH mutations also reduce the expression of DNA repair protein ATM [74], although these findings have not yet been described in central chondrosarcoma and are contradictory to the observed chemo- and radiotherapy resistance in these tumours. Furthermore, hypermethylation of the NAPRT1 promoter seems to correlate to NAMPT inhibitor sensitivity in IDH-mutated glioma [75]. Chondrosarcoma cell lines are also sensitive to NAMPT inhibition and show a hypermethylated NAPRT1 promoter, although this phenotype is independent of the IDH mutation status [76].
Therapeutic implications for epigenetic alterations induced by IDH1 and IDH2 mutations
The most straightforward way to target the epigenetic alterations is direct inhibition of the IDH1 or IDH2 mutant enzyme and thereby prevention of the formation of the oncometabolite. However, in vitro experiments do not show a beneficial effect in chondrosarcoma cell lines: proliferation, migration and colony formation capacity are not affected, only at high concentrations [61, 77]. Several clinical trials are ongoing to evaluate the effect of IDH mutant inhibitors in patients, but the results for chondrosarcoma patients have not yet been published. Since several in vitro studies indicate that the observed changes might have become static and independent of the IDH mutation over time, it may be more promising to directly target these changes. To counteract the DNA hypermethylation phenotype, the use of DNA methyltransferase inhibitors, such as decitabine, could be a promising therapeutic strategy. An in vitro study shows that decitabine induces the re-expression of several epigenetically silenced tumour suppressor genes and reduces the proliferation and migration of chondrosarcoma cells [78]. Contradictory to these findings, use of decitabine in the Swarm rat chondrosarcoma model results in a more progressive phenotype [79]. These conflicting results indicate that more research is needed before epigenetic therapies can be used for central chondrosarcoma patients. For example, identification of the genes that are mainly affected by the epigenetic alterations could lead to more targeted therapeutic approaches instead of global DNA demethylation. Besides that, the non-epigenetic effects of D-2-HG could also play a major role in tumourigenesis, warranting a more wide-spread therapeutic approach to target multiple D-2-HG-affected pathways at once.
Conclusion and future perspectives
Chondroblastoma, GCTB and central cartilaginous tumours all harbour driver mutations that induce wide-spread epigenetic alterations (Fig. 2). Interestingly, these epigenetic alterations do not seem to be associated with malignant potential, as the IDH and H3.3 mutations are considered drivers only in benign (enchondroma, chondroblastoma) and locally aggressive (GCTB) neoplasms. This is in line with the fact that these epigenetic changes predominantly affect differentiation, instead of proliferation. In the case of malignant chondrosarcoma, the IDH mutation is no longer associated with tumourigenic behaviour and probably other mutations have taken over the driver function of the IDH mutation [61]. Similarly, metastases in GCTB are associated with downregulation of decorin and lumican [80]. Further research is needed to identify the genes affected by the mutation-driven epigenetic alterations in the different tumour types, as these genes probably differ between the tumour types. Moreover, it remains to be elucidated why the occurrence of certain histone H3.3 mutations is highly tumour type specific. This will hopefully lead to a better understanding of the biological mechanisms underlying the development of these bone neoplasms.

Schematic overview of the individual and shared epigenetic alterations in chondroblastoma, giant cell tumour of bone and central cartilaginous tumours
The identification of specific histone H3.3 variants in both chondroblastoma (i.e. H3K36M) and GCTB (i.e. H3F3A G34) has led to development of novel diagnostic tools. Mutant protein-specific immunohistochemistry and/or sequencing of H3F3A and H3F3B both support pathologists to differentiate between chondroblastoma, GCTB and other giant cell containing neoplasms [30, 31, 44] (Table 1). While the IDH1 R132H mutation-specific antibody is commonly used in neuropathology [81], an antibody recognizing the most common IDH1 R132C mutation in central cartilaginous tumours is not yet available but would be very helpful in distinguishing enchondroma and chondrosarcoma from their histologic mimics [63] (Table 1).
Of note, only the neoplastic stromal-like cells harbour the histone H3.3. variants, both in chondroblastoma and GCTB, which suggests a close interplay between wild-type (e.g. giant cells) and mutant cells. A similar relation between wild-type and mutant cells has been observed for central cartilaginous tumours, which display intra-neoplastic mosaicism for the IDH mutations. This makes it tempting to speculate that this close interplay between wild-type and mutant cells is essential for the development of these bone neoplasms driven by epigenetic alterations.
As only ~ 50% of the chondrosarcomas harbour IDH1 or IDH2 mutations, alternative driving mechanisms should be considered. It remains to be elucidated whether these alternative driver mechanisms induce similar epigenetic alterations as observed in IDH mutant chondrosarcomas. Also, one could speculate that the IDH wild-type cells may overgrow the IDH mutant cell population over time. While IDH mutations are essential for the initiation of enchondroma, the additional acquired mutations that take over the driver function of IDH mutations in chondrosarcoma may occur in the wild-type cells which might explain the observed IDH wild-type genotype in half of the chondrosarcomas.
References
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A (2009) An operational definition of epigenetics. Genes Dev 23:781–783
Dawson MA, Kouzarides T (2012) Cancer epigenetics: from mechanism to therapy. Cell 150:12–27
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G (2014) Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505:495–501
Que Y, Xiao W, Xu B, Wen X, Weng D, Zhang X (2018) The changing landscape of phase II/III metastatic sarcoma clinical trials - analysis of ClinicalTrials.gov. BMC Cancer 18:1251
Yin L, Cai WJ, Liu CX, Chen YZ, Hu JM, Jiang JF, Li HA, Cui XB, Chang XY, Zhang WJ, Sun K, Li F (2013) Analysis of PTEN methylation patterns in soft tissue sarcomas by MassARRAY spectrometry. PLoS One 8:e62971
Pansuriya TC, van Eijk R, d’Adamo P, van Ruler MA, Kuijjer ML, Oosting J, Cleton-Jansen AM, van Oosterwijk JG, Verbeke SL, Meijer D, van Wezel T, Nord KH, Sangiorgi L, Toker B, Liegl-Atzwanger B, San-Julian M, Sciot R, Limaye N, Kindblom LG, Daugaard S, Godfraind C, Boon LM, Vikkula M, Kurek KC, Szuhai K, French PJ, Bovée JV (2011) Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet 43:1256–1261
Sheffield NC, Pierron G, Klughammer J, Datlinger P, Schönegger A, Schuster M, Hadler J, Surdez D, Guillemot D, Lapouble E, Freneaux P, Champigneulle J, Bouvier R, Walder D, Ambros IM, Hutter C, Sorz E, Amaral AT, de Álava E, Schallmoser K, Strunk D, Rinner B, Liegl-Atzwanger B, Huppertz B, Leithner A, de Pinieux G, Terrier P, Laurence V, Michon J, Ladenstein R, Holter W, Windhager R, Dirksen U, Ambros PF, Delattre O, Kovar H, Bock C, Tomazou EM (2017) DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat Med 23:386–395
Mahoney SE, Yao Z, Keyes CC, Tapscott SJ, Diede SJ (2012) Genome-wide DNA methylation studies suggest distinct DNA methylation patterns in pediatric embryonal and alveolar rhabdomyosarcomas. Epigenetics 7:400–408
Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–206
Sullivan LM, Folpe AL, Pawel BR, Judkins AR, Biegel JA (2013) Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol 26:385–392
Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, Qin LX, Viale A, Singer S, Zheng D, Berger MF, Chen Y, Antonescu CR, Chi P (2014) PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 46:1227–1232
Cleven AH, Al Sannaa GA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, Rubin BP, de Vries MW, Watson KL, Torres KE, Wang WL, van Duinen SG, Hogendoorn PC, Lazar AJ, Bovée JV (2016) Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol 29:582–590
Schaefer IM, Fletcher CD, Hornick JL (2016) Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol 29:4–13
Prieto-Granada CN, Wiesner T, Messina JL, Jungbluth AA, Chi P, Antonescu CR (2016) Loss of H3K27me3 expression is a highly sensitive marker for sporadic and radiation-induced MPNST. Am J Surg Pathol 40:479–489
Kadoch C, Crabtree GR (2013) Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153:71–85
Lubieniecka JM, de Bruijn DR, Su L, van Dijk AH, Subramanian S, van de Rijn M, Poulin N, van Kessel AG, Nielsen TO (2008) Histone deacetylase inhibitors reverse SS18-SSX-mediated polycomb silencing of the tumor suppressor early growth response 1 in synovial sarcoma. Cancer Res 68:4303–4310
Athanasou NA, Bansal M, Forsyth R, Reid RP, Sapi Z (2013) Giant cell tumour of bone. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds) WHO classification of Tumours of soft tissue and bone, 4th edn. IARC Press, Lyon, pp 321–324
Kilpatrick SE, Romeo S (2013) Chondroblastoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds) WHO classification of tumours of soft tissue and bone, 4th edn. IARC Press, Lyon, pp 262–263
Lucas DR, Bridge JA (2013) Chondromas: enchondroma, periosteal chondroma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds) WHO classification of Tumours of soft tissue and bone, 4th edn. IARC Press, Lyon, pp 252–254
Hogendoorn PCW, Bovée JVMG, Nielsen GP (2013) Chondrosarcoma (grades I-III), including primary and secondary variants and periosteal chondrosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds) WHO classification of Tumours soft tissue and bone, 4th edn. IARC Press, Lyon, pp 264–268
Evans HL, Ayala AG, Romsdahl MM (1977) Prognostic factors in chondrosarcoma of bone. A clinicopathologic analysis with emphasis on histologic grading. Cancer 40:818–831
Nota SPFT, Braun Y, Schwab JH, van Dijk CN, Bramer JAM (2015) The identification of prognostic factors and survival statistics of conventional central chondrosarcoma. Sarcoma 2015:1–11
van Praag Veroniek VM, Rueten-Budde AJ, Ho V, Dijkstra PDS; Study group Bone and Soft tissue tumours (WeBot), Fiocco M, van de Sande MAJ (2018) Incidence, outcomes and prognostic factors during 25 years of treatment of chondrosarcomas. Surg Oncol 27:402–408
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, Nik-Zainal S, Martin S, McLaren S, Goodie V, Robinson B, Butler A, Teague JW, Halai D, Khatri B, Myklebost O, Baumhoer D, Jundt G, Hamoudi R, Tirabosco R, Amary MF, Futreal PA, Stratton MR, Campbell PJ, Flanagan AM (2013) Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45:1479–1482
Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, McCarthy S, Fantin VR, Straley KS, Lobo S, Aston W, Green CL, Gale RE, Tirabosco R, Futreal A, Campbell P, Presneau N, Flanagan AM (2011) Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet 43:1262–1265
Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O'Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, Futreal A, Tirabosco R, Flanagan AM (2011) IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 224:334–343
Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, Marshall J, O’Meara S, Davies H, Nik-Zainal S, Beare D, Butler A, Gamble J, Hardy C, Hinton J, Jia MM, Jayakumar A, Jones D, Latimer C, Maddison M, Martin S, McLaren S, Menzies A, Mudie L, Raine K, Teague JW, Tubio JM, Halai D, Tirabosco R, Amary F, Campbell PJ, Stratton MR, Flanagan AM, Futreal PA (2013) Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet 45:923–926
Totoki Y, Yoshida A, Hosoda F, Nakamura H, Hama N, Ogura K, Yoshida A, Fujiwara T, Arai Y, Toguchida J, Tsuda H, Miyano S, Kawai A, Shibata T (2014) Unique mutation portraits and frequent COL2A1 gene alteration in chondrosarcoma. Genome Res 24:1411–1420
Zhang YX, van Oosterwijk JG, Sicinska E, Moss S, Remillard SP, van Wezel T, Bühnemann C, Hassan AB, Demetri GD, Bovée JV, Wagner AJ (2013) Functional profiling of receptor tyrosine kinases and downstream signaling in human chondrosarcomas identifies pathways for rational targeted therapy. Clin Cancer Res 19:3796–3807
Amary F, Berisha F, Ye H, Gupta M, Gutteridge A, Baumhoer D, Gibbons R, Tirabosco R, O'Donnell P, Flanagan AM (2017) H3F3A (histone 3.3) G34W immunohistochemistry. Am J Surg Pathol 41:1059–1068
Amary MF, Berisha F, Mozela R, Gibbons R, Guttridge A, O'Donnell P, Baumhoer D, Tirabosco R, Flanagan AM (2016) The H3F3 K36M mutant antibody is a sensitive and specific marker for the diagnosis of chondroblastoma. Histopathology 69:121–127
van der Heijden L, Dijkstra PDS, van de Sande MAJ, Kroep JR, Nout RA, van Rijswijk C, Bovée JV, Hogendoorn PC, Gelderblom H (2014) The clinical approach toward Giant cell tumor of bone. Oncologist 19:550–561
Itkin B, Straminsky S, De Ronato G, Lewi D, Marantz A, Bardach A (2018) Prognosis of metastatic giant cell tumor of bone in the pre-denosumab era. A systematic review and a meta-analysis. Jpn J Clin Oncol 48:640–652
Forsyth RG, De Boeck G, Baelde JJ, Taminiau AH, Uyttendaele D, Roels H, Praet MM, Hogendoorn PC (2009) CD33+ CD14- phenotype is characteristic of multinuclear osteoclast-like cells in giant cell tumor of bone. J Bone Miner Res 24:70–77
Lübbehüsen C, Lüke J, Seeling C, Mellert K, Marienfeld R, von Baer A, Schultheiss M, Möller P, Barth TFE (2019) Characterization of three novel H3F3A-mutated giant cell tumor cell lines and targeting of their Wee1 pathway. Sci Rep 9:6458
Liao TS, Yurgelun MB, Chang SS, Zhang HZ, Murakami K, Blaine TA, Parisien MV, Kim W, Winchester RJ, Lee FY (2005) Recruitment of osteoclast precursors by stromal cell derived factor-1 (SDF-1) in giant cell tumor of bone. J Orthop Res 23:203–209
Maggiani F, Forsyth R, Hogendoorn PCW, Krenacs T, Athanasou NA (2011) The immunophenotype of osteoclasts and macrophage polykaryons. J Clin Pathol 64:701–705
Roux S, Amazit L, Meduri G, Guiochon-Mantel A, Milgrom E, Mariette X (2002) RANK (receptor activator of nuclear factor kappa B) and RANK ligand are expressed in giant cell tumors of bone. Am J Clin Pathol 117:210–216
Balke M, Campanacci L, Gebert C, Picci P, Gibbons M, Taylor R, Hogendoorn P, Kroep J, Wass J, Athanasou N (2010) Bisphosphonate treatment of aggressive primary, recurrent and metastatic giant cell tumour of bone. BMC Cancer 10:462
Romeo S, Bovée JVMG, Jadnanansing NAA, Taminiau AHM, Hogendoom PCW (2004) Expression of cartilage growth plate signalling molecules in chondroblastoma. J Pathol 202:113–120
Aigner T, Loos S, Inwards C, Perris R, Perissinotto D, Unni KK, Kirchner T (1999) Chondroblastoma is an osteoid-forming, but not cartilage-forming neoplasm. J Pathol 189:463–469
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Zhang J, Baker SJ, St. Jude Children's Research Hospital–Washington University Pediatric Cancer Genome Project (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253
Presneau N, Baumhoer D, Behjati S, Pillay N, Tarpey P, Campbell PJ, Jundt G, Hamoudi R, Wedge DC, Loo PV, Hassan AB, Khatri B, Ye H, Tirabosco R, Amary MF, Flanagan AM (2015) Diagnostic value of H3F3A mutations in giant cell tumour of bone compared to osteoclast-rich mimics. J Pathol Clin Res 1:113–123
Cleven AH, Höcker S, Briaire-de Bruijn I, Szuhai K, Cleton-Jansen AM, Bovée JV (2015) Mutation analysis of H3F3A and H3F3B as a diagnostic tool for giant cell tumor of bone and chondroblastoma. Am J Surg Pathol 39:1576–1583
Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver R, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CD (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140:678–691
Fang D, Gan H, Lee JH, Han J, Wang Z, Riester SM, Jin L, Chen J, Zhou H, Wang J, Zhang H, Yang N, Bradley EW, Ho TH, Rubin BP, Bridge JA, Thibodeau SN, Ordog T, Chen Y, van Wijnen AJ, Oliveira AM, Xu RM, Westendorf JJ, Zhang Z (2016) The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 352(80):1344–1348
Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, Wunder JS, Dickson BC, Lundgren SM, Jani KS, De Jay N, Papillon-Cavanagh S, Andrulis IL, Sawyer SL, Grynspan D, Turcotte RE, Nadaf J, Fahiminiyah S, Muir TW, Majewski J, Thompson CB, Chi P, Garcia BA, Allis CD, Jabado N, Lewis PW (2016) Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352(80):844–849
Wagner EJ, Carpenter PB (2012) Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13:115–126
Zhang Y, Shan CM, Wang J, Bao K, Tong L, Jia S (2017) Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep 7:43906
Yang S, Zheng X, Lu C, Li GM, Allis CD, Li H (2016) Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev 30:1611–1616
Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340(80):857–861
Shi L, Shi J, Shi X, Li W, Wen H (2018) Histone H3.3 G34 mutations alter histone H3K36 and H3K27 methylation in Cis. J Mol Biol 430:1562–1565
Voon HPJ, Udugama M, Lin W, Hii L, Law RHP, Steer DL, Das PP, Mann JR, Wong LH (2018) Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat Commun 9:3142
Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, Bax DA, Carvalho D, Taylor KR, Vinci M, Bajrami I, McGonnell I, Lord CJ, Reis RM, Hargrave D, Ashworth A, Workman P, Jones C (2013) Histone H3.3 mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov 3:512–519
Lim J, Park JH, Baude A, Yoo Y, Lee YK, Schmidt CR, Park JB, Fellenberg J, Zustin J, Haller F, Krücken I, Kang HG, Park YJ, Plass C, Lindroth AM (2017) The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing. Sci Rep 7:13459
Fellenberg J, Sähr H, Mancarella D, Plass C, Lindroth AM, Westhauser F, Lehner B, Ewerbeck V (2019) Knock-down of oncohistone H3F3A-G34W counteracts the neoplastic phenotype of giant cell tumor of bone derived stromal cells. Cancer Lett 448:61–69
Bovée JVMG, Alman BA (2013) Enchondromatosis: Ollier disease and Maffucci syndrome. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F (eds) WHO classification of Tumours of soft tissue and bone, 4th edn. IARC Press, Lyon, pp 376–378
Hirata M, Sasaki M, Cairns RA, Inoue S, Puviindran V, Li WY, Snow BE, Jones LD, Wei Q, Sato S, Tang YJ, Nadesan P, Rockel J, Whetstone H, Poon R, Weng A, Gross S, Straley K, Gliser C, Xu Y, Wunder J, Mak TW, Alman BA (2015) Mutant IDH is sufficient to initiate enchondromatosis in mice. Proc Natl Acad Sci 112:2829–2834
Jin Y, Elalaf H, Watanabe M, Tamaki S, Hineno S, Matsunaga K, Woltjen K, Kobayashi Y, Nagata S, Ikeya M, Kato T Jr, Okamoto T, Matsuda S, Toguchida J (2015) Mutant idh1 dysregulates the differentiation of mesenchymal stem cells in association with gene-specific histone modifications to cartilage- and bone-related genes. PLoS One 10:e0131998
Suijker J, Baelde HJ, Roelofs H, Cleton-Jansen A-M, Bovée JVMG (2015) The oncometabolite D-2-hydroxyglutarate induced by mutant IDH1 or −2 blocks osteoblast differentiation in vitro and in vivo. Oncotarget 6:
Suijker J, Oosting J, Koornneef A, Struys EA, Salomons GS, Schaap FG, Waaijer CJ, Wijers-Koster PM, Briaire-de Bruijn IH, Haazen L, Riester SM, Dudakovic A, Danen E, Cleton-Jansen AM, van Wijnen A, Bovée JV (2015) Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget 6:12505–12519
Molenaar RJ, Radivoyevitch T, Maciejewski JP, van Noorden CJF, Bleeker FE (2014) The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim Biophys Acta - Rev Cancer 1846:326–341
de Andrea CE, San-Julian M, Bovée JVMG (2017) Integrating morphology and genetics in the diagnosis of cartilage tumors. Surg Pathol Clin 10:537–552
Molenaar RJ, Maciejewski JP, Wilmink JW, Van Noorden CJF (2018) Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 37:1949–1960
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, Gomez V, Hergovich A, Tirabosco R, Fernanda Amary M, Baumhoer D, Jundt G, Ross MT, Flanagan AM, Beck S (2013) Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun 4:2166
Cleven AHG, Suijker J, Agrogiannis G, Briaire-de Bruijn IH, Frizzell N, Hoekstra AS, Wijers-Koster PM, Cleton-Jansen AM, Bovée JVMG (2017) IDH1 or −2 mutations do not predict outcome and do not cause loss of 5-hydroxymethylcytosine or altered histone modifications in central chondrosarcomas. Clin Sarcoma Res 7:8
Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, Zheng Y, el-Amine N, Haddock S, Nanjangud G, LeKaye H, Brennan C, Cross J, Huse JT, Kelleher NL, Osten P, Thompson CB, Chan TA (2018) Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet 50:62–72
Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough M, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12:463–469
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O'Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483:474–478
Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J, ten A, Snow B, Ueda T, Inoue S, Yamamoto K, Ko M, Rao A, Yen KE, Su SM, Mak TW (2012) D-2-hydroxyglutarate produced by mutant Idh1 perturbs collagen maturation and basement membrane function. Genes Dev 26:2038–2049
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A (2010) Leukemic IDH1 and IDH2 mutations result in a Hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567
Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, Miltiadous O, Walesky C, Deshpande V, Zhu AX, Hezel AF, Yen KE, Straley KS, Travins J, Popovici-Muller J, Gliser C, Ferrone CR, Apte U, Llovet JM, Wong KK, Ramaswamy S, Bardeesy N (2014) Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 513:110–152
Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, Lemonnier F, Kron KJ, Cescon DW, Hao Z, Lind EF, Takayama N, Planello AC, Shen SY, Shih AH, Larsen DM, Li Q, Snow BE, Wakeham A, Haight J, Gorrini C, Bassi C, Thu KL, Murakami K, Elford AR, Ueda T, Straley K, Yen KE, Melino G, Cimmino L, Aifantis I, Levine RL, de Carvalho DD, Lupien M, Rossi DJ, Nolan GP, Cairns RA, Mak TW (2016) Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell 30:337–348
Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, He T, Shi X, Gerszten RE, Zhang Y, Yeh JJ, Curry WT, Zhao D, Sundaram S, Nigim F, Koerner MVA, Ho Q, Fisher DE, Roider EM, Kemeny LV, Samuels Y, Flaherty KT, Batchelor TT, Chi AS, Cahill DP (2015) Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 28:773–784
Peterse EFP, van den Akker BEWM, Niessen B, Oosting J, Suijker J, de Jong Y, Danen EHJ, Cleton-Jansen AM, Bovée JVMG (2017) NAD synthesis pathway interference is a viable therapeutic strategy for chondrosarcoma. Mol Cancer Res 15:1714–1721
Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A, Hu G, Tinoco G, Bodamer O, Trent JC (2015) Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS One 10
Bui C, Ouzzine M, Talhaoui I, Sharp S, Prydz K, Coughtrie MW, Fournel-Gigleux S (2009) Epigenetics: methylation-associated repression of heparan sulfate 3- O -sulfotransferase gene expression contributes to the invasive phenotype of H-EMC-SS chondrosarcoma cells. FASEB J 24:436–450
Hamm CA, Xie H, Costa FF, Vanin EF, Seftor EA, Sredni ST, Bischof J, Wang D, Bonaldo MF, Hendrix MJ, Soares MB (2009) Global demethylation of rat chondrosarcoma cells after treatment with 5-Aza-2′-deoxycytidine results in increased tumorigenicity. PLoS One 4:e8340
Lieveld M, Bodson E, De Boeck G, Nouman B, Cleton-Jansen AM, Korsching E, Benassi MS, Picci P, Sys G, Poffyn B, Athanasou NA, Hogendoorn PC, Forsyth RG (2014) Gene expression profiling of giant cell tumor of bone reveals downregulation of extracellular matrix components decorin and lumican associated with lung metastasis. Virchows Arch 465:703–713
Capper D, Reuss D, Schittenhelm J, Hartmann C, Bremer J, Sahm F, Harter PN, Jeibmann A, von Deimling A (2011) Mutation-specific IDH1 antibody differentiates oligodendrogliomas and oligoastrocytomas from other brain tumors with oligodendroglioma-like morphology. Acta Neuropathol 121:241–252
Author information
Authors and Affiliations
Contributions
All authors contributed equally and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Venneker, S., Szuhai, K., Hogendoorn, P.C.W. et al. Mutation-driven epigenetic alterations as a defining hallmark of central cartilaginous tumours, giant cell tumour of bone and chondroblastoma. Virchows Arch 476, 135–146 (2020). https://doi.org/10.1007/s00428-019-02699-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-019-02699-2