
Abstract
Vascular endothelial cells are equipped with numerous specialized granules called Weibel-Palade bodies (WPBs). They contain a cocktail of proteins that can be rapidly secreted (3–5 min) into the vascular lumen after an appropriate stimulus such as thrombin. These proteins are ready without synthesis. Von Willebrand factor (VWF) and P-selectin are the main constituents of WPBs. Upon stimulation, release of ultralarge VWF multimers occurs and assembles into VWF strings on the apical side of endothelium. The VWF A1 domain becomes exposed in a shear-dependent manner recruiting and activating platelets. VWF is able to recruit leukocytes via direct leukocyte binding or via the activated platelets promoting NETosis. Ultralarge VWF strings are ultimately cleaved into smaller pieces by the protease ADAMTS-13 preventing excessive platelet adhesion. Under carefully performed flowing conditions and adequate dose of Shiga toxins, the toxin induces the release of ultralarge VWF multimers from cultured endothelial cells. This basic information allows insight into the pathogenesis of thrombotic thrombocytopenic purpura (TTP) and of STEC-HUS in the diarrhea phase. In TTP, ADAMTS-13 activity is deficient and systemic aggregation of platelets will occur after a second trigger. In STEC-HUS, stimulated release of WPB components in the diarrhea phase of the disease can be presumed to be the first hit in the damage of Gb3 positive endothelial cells.
Graphical abstract

A higher resolution version of the Graphical abstract is available as Supplementary information
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vascular endothelial cells line the luminal surface of the vasculature. Endothelial cells contain unique storage organelles, so called Weibel-Palade bodies (WPBs). In these WPBs two main proteins are stored, namely von Willebrand factor (VWF) and P-selectin.
WPBs are rod-shaped, elongated structures containing tubules formed by polymerized VWF and aligned parallel to the longitudinal axis of WPBs [1] (Fig. 1). Although the major component within WPBs is VWF, these vesicles also contain a number or other bioactive molecules. Expression of VWF drives the formation of WPBs [2].

von Willebrand factor in endothelial cells. Fluorescence microscopy image of cultured human endothelial monolayer (HUVEC) immunostained for VWF (green) and nuclei (blue). Electron microscopy of Weibel-Palade bodies. Courtesy of Marije Kat
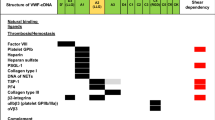
Data on the list of proteins residing in WPBs and insight into the dynamics and regulation of exocytosis will be presented with emphasis on their role in the pathogenesis of TTP and hemolytic uremic syndrome secondary to Shiga toxin-producing Escherichia coli (STEC-HUS) in the diarrhea phase.
Weibel-Palade bodies—components
Multiple components are co-stored with VWF in WPBs. VWF will be covered in a later section.
P-selectin
P-selectin is a single-chain glycoprotein with a molecular weight of 140 kDa and is stored in the α granules of platelets and WPBs, and rapidly exposed on the surface of these cells upon stimulation. Transmembrane P-selectin is present in dimers or oligomers whereas soluble P-selectin in plasma, resulting from proteolytic cleavage of transmembrane P-selectin, is in the monomeric form and unable to interact with functional ligands [3].
P-selectin binds to leucocyte P-selectin glycoprotein ligand-1 (PSGL1) so that the leucocytes become loosely attached rolling along the vessel wall, leading to firm adhesion and transmigration [4].
Following its expression on the cell membrane P-selectin is rapidly internalized, transported to early and late endosomes from where it can be recycled independent of de novo protein synthesis. The recycled P-selectin retains its adherence function [5]. In addition, P-selectin increases via PSGL1, the expression of tissue factor on monocytes [6].
CD63
After exocytosis of WPBs, the membrane glycoprotein CD63 is transported to the cellular surface where it forms clusters with P-selectin. CD63 is an essential cofactor for P-selectin function [7, 8]. CD63 is not exclusively present within WPBs but also localizes to lysosomes and late endosomes [9]. P-selectin is already included during WPB formation due to direct binding to VWF. CD63 is trafficked from endolysosomes via two-pore channel 2 (TPC2) [10]. TPC2 governs leukocyte capture on endothelium.
Endothelin
Endothelin is synthesized in human endothelial cells [11]. The constitutive pathway via small secretory vesicles maintains vascular tone [12]. Via binding to ETA and ETB receptors on vascular smooth muscle cells, an initial transient vasodilatation followed by marked sustained vasoconstriction is induced. In the regulatory pathway ET-1 is released from WPBs situated in the endothelial cells. ET-1 is generated by endothelin-converting enzyme from big ET-1. ET-1, the major endothelial isoform, is secreted on the abluminal surface, remarkably different from the exocytosis of VWF. Plasma concentrations of ET-1 do not accurately reflect ET-1 production. Apparently a small amount is passing the luminal side. The half-life of ET-1 in the healthy circulation is about 1 min [13]. The effect of ET-1 on the glomerulus has been extensively studied and will be further discussed, omitting the effect on other structures such as tubular cells. Glomerular endothelial cells are probably the principal but not unique source of ET-1 within the glomerulus. ET-1 secreted abluminally modulates podocyte and mesangial structure and function. ET-1 induces cytoskeletal (actin) remodeling in podocytes and loss of slit-diaphragm proteins such as nephrin. It increases glomerular permeability reflected in albumin excretion [14]. ET-1 expression in cultured podocytes by Shiga toxin mediates cytoskeleton rearrangements in the podocytes [15]. Podocyte activation via endothelin results in loss of endothelial surface layer (glycocalyx) [16]. In transgenic mice with GB3 expression exclusively in their podocytes, Shiga toxin induced a loss of endothelial glycocalyx with reduction of complement factor H binding [17]. This reduction has consequences for complement activation [18] and can have a role for the preferred damage of the glomeruli in STEC-HUS. Endothelial-targeted treatment has a beneficial effect in a wide range of kidney disorders [19, 20].
Calcitonin gene-related peptide (CGRP)
CGRP is the most potent microvascular vasodilator. CGRP is released from perivascular nerves. The major effects are exerted locally in the vessel wall close to its site of synthesis acting on the arterioles, causing vasodilatation and influencing the cardiovascular system. CGRP mediates this response by directly activating its receptors on the vascular smooth muscle and on endothelial cells to enhance NO production [21, 22]. Despite its potent action on the vasculature, CGRP does not play a pivotal role in the normal physiological regulation of blood pressure. Endothelin-converting enzyme (present in WPBs) mediates degradation of CGRP [23]. Is CGRP present in WPBs mitigating the effect of endothelin released from WPBs?
Osteoprotegerin (OPG)
OPG is an effective inhibitor of osteoclast differentiation and activation. Produced by osteoblasts it suppresses osteoclast formation impeding the binding of RANKL with RANK.
Endothelial cells and vascular smooth muscle cells have been demonstrated to produce OPG [24, 25]. Not all exposed platelet-binding sites on VWF secreted by endothelial cells are occupied by platelets. OPG interacts with the VWF A1 domain and is responsible for the reduced binding to platelets [26].
Angiopoetin-2 (Ang-2)
Ang-1 is constitutively expressed by many different cell types in contrast to the expression of Ang-2, which is almost exclusively expressed in endothelial cells. Ang-1 mediates Tie2 activation and is required to maintain the quiescent resting state of the endothelium. Ang-2 functions as an antagonist ligand for Tie2. It results in destabilization, rendering endothelium responsive to stimulation by inflammatory cytokines such as TNF, interleukin-1, and angiogenic cytokines (VEGF) [27] and induces permeability of endothelium.
VWF binds to Ang-2 via the VWF A1 domain, persisting after secretion [28]. Is VWF mediating the storage of Ang-2? The intracellular Ang-2 stores rapidly recover upon release. It is detectable 6 h after complete release and recovered within 16 h [29]. A repeated stimulation is possible as is also shown for P-selectin.
Ribonuclease 1 (RNase 1)
Extracellular RNAs exist in the extracellular space and are not inert molecules. They induce hyperpermeability of endothelium, and enhance adhesion and transmigration of leukocytes. Furthermore, they induce the release of cytokines from monocytes/macrophages, activate the inflammasome, and potentiate blood coagulation [30, 31]. Human ribonuclease A degrades RNAs in the extracellular space. Insight into the biological function of RNase 1 is sparse but it has a protective effect [32]. Only ribonucleases that evade RNase may be cytotoxic.
The role of a 1,3-fucosyltransferase VI (Fuct-VI) is unclear [33].
Interleukin-8, MCP-1
Resting endothelial cells do not synthesize interleukin-8 (IL-8) in significant amounts [34]. De novo synthesis of IL-8 and monocyte chemoattractant protein (MCP-1) require exposure to cytokines such as interleukin-1β, IL-4, and TNF for a prolonged period (24 h) [35].
IL-8, IL-6, MCP, tissue activator (tPA), and growth-regulated oncogen-a were found to reside in both WPBs and small punctate vesicles in human endothelial cells. It is suggested that they are missorted to WPBs. Low amounts of the de novo synthesis of IL-8 and tPA are sorted into WBPs. The storage of these proinflammatory mediators equips endothelial cells with a rapidly recruitable reservoir.
The chemokines IL-8 and MCP regulate leucocytes and monocyte movement by adhesion and extravasation including activation [36, 37].
Proteomic analysis
A proteomic screen identified novel components of endothelial cell-specific WBPs, such as IGFBP7 with a suggested role in angiogenesis [38].
Taking into account the different components of WPBs, their release will provoke fire. Can this fire be extinguished (partially) by interfering with the synthesis of VWF and blockage of P-selectin action?
Stimulation of release
A large, still increasing variety of diverse agonists can trigger exocytosis of WPBs [39]. Ca2+-mediated and cAMP-mediated secretagogues converge at effector pathways that control anchoring tethering vesicle fusion and actin contractility [2]. Release of WPBs has been induced by fibrin [40], thrombin, endothelin [41], C5a [42], C5b-9 [43], inflammatory cytokines [44], activated platelets [45], and heme. A continuous stimulation of release of WPBs probably occurs in TTP and STEC-HUS. Platelets express high-mobility group box 1 protein (HMGB1), and upon platelet activation, HMGB1 is exported to the cell membrane and released [46]. It induces monocyte tissue factor expression initiating coagulation, platelet activation, and NETosis [47]. In a murine model of typical HUS with increased plasma HMBGB1, administration of anti-HMBGB1 promoted amelioration of tissue damage. The Shiga toxins have an enzymatically active A moiety and non-toxic B moiety. The B moiety consists of five identical B subunits forming a pentameric ring. Each B subunit harbors three distinctive binding sites that interact with the trisaccharide moiety of the glycosphingolipid Gb3 [48]. A seminal study Nolasco et al. [49] demonstrated that Shiga toxin under flowing conditions stimulated the secretion of long VWF multimeric strings in viable human umbilical vein endothelial cells (HUVEC) and human glomerular microvascular endothelial cells (GMVEC) within 10 min. Perfused human platelets immediately adhered to these strings. In addition, Shiga toxin impairs ADAMTS-13 cleavage by binding to the A2 domain [50]. This delayed VWF cleavage may contribute to renal thrombotic microangiopathy in STEC-HUS. The B subunits of Stx1 and Stx2 in a perfusion assay stimulated the secretion of ultralarge VWF from HUVEC within 5 min [51].
More details about the exocytosis will be covered in the next section about VWF.
Role of Von Willebrand factor (VWF)
Biosynthesis, secretion, and clearance
Excellent reviews covering biosynthesis, secretion, and clearance can be found in more detail elsewhere [52,53,54,55,56]. To understand the role of VWF, however, it is important to have some background in this respect. The contribution of VWF from α granules of platelets requires platelet activation and is limited. In endothelium, in non-stimulated condition, VWF is replaced by approximately 50% after 24 h. Briefly, following synthesis of ProVWF monomers, dimers are formed in the endoplasmic reticulum and transported in vesicles to the Golgi. Once arrived at the Golgi, the propeptide is cleaved, and VWF dimers multimerize and form quanta that arrive in the transGolgi network to be packaged into WPBs. A complex pathway is followed along intraendothelial storage and trafficking combined with basal and regulated secretion.
VWF secretion occurs via three pathways: constitutive secretion of low molecular VWF primarily released at the basolateral side of the endothelium and basal and regulated secretion of high molecular VWF to the apical surface. From the large number of WPBs undergoing exocytosis upon stimulated release, ultralarge WPB multimers assemble into VWF strings on the apical side of the endothelium [57]. The large multimers have increased platelet adhesive capacity compared with the low molecular VWF [58].
Three modes of exocytosis can be distinguished. WPBs can undergo full fusion, resulting in complete release of VWF and other WPB compounds. Incomplete fusion, so called lingering kiss, occurs via small fusion pore. It allows only release of small proteins, such as IL-8, but not of VWF and P-selectin [59]. About 25% of WPBs may undergo lingering kiss fusion after stimulation. In a third mode, multiple WPBs aggregate and fuse to the membrane. The fusion of WPBs with the membrane is facilitated by SNARE complex formed by proteins which are present on vesicles (v-SNAREs) and target membranes (t-SNAREs) [2, 55]. Large WPBs undergoing full fusion exploit the contractile properties of actomyosin rings to forcibly release high molecular VWF not affecting the release of P-selectin and other cargo [55, 60].
Following release into the circulation, the half-life of VWF has a large variation ranging from 4.2 to 26 h. Individuals with blood group non-O display a longer half-life than individuals with blood group O due to different glycosylation patterns. Receptor-mediated endocytosis mainly by macrophages has an important role in the clearance of VWF [52, 61].
VWF circulates in plasma in a globular form not interacting with platelets. After vascular damage, VWF binds to collagen and uncoils in adhesive strings. This adaptation occurs also after exocytosis of WPBs at the luminal side of endothelium. The endothelial glycocalyx, via binding to heparan sulfate, anchors VWF to the vascular endothelium. VWF is essential for the capture of platelets via two receptors (Fig. 2), GP1b-IX-V and αIIbβ3, and requires flowing blood. A1 domain of VWF becomes exposed under shear stress. In static conditions no binding between VWF and the two receptors is observed [62]. The engagement of αIIβb3 occurs after initial platelet adhesion mediated by GP1b-IX-V/VWF interaction. Platelet activation is induced [63]. Activated platelets release alpha granules containing thrombospondin-1 (TSP-1). TSP-1 competes with the A2 binding site of ADAMTS-13 inhibiting VWF multimer cleavage [64]. Activated platelets expose phosphatidylserine on their membrane allowing the coagulation factor binding properties such as the prothrombinase complex. Activated platelets trigger all three pathways of complement cascade [63, 65]. Circulating VWF, in addition, acts as a chaperone for factor VIII to protect this coagulation factor from proteolytic degradation.

Domain structure of von Willebrand factor (VWF). SP signal peptide, D1–D2 propeptide; domains potentially involved in interactions relevant for inflammatory processes. GP1b and αIIbβ3 are binding sites for platelets
Outside hemostasis, VWF may play an important role in inflammatory response. Following blood vessel injury VWF escapes from plasma to subendothelium, where it comes into contact with tissue-resident macrophages. It will trigger these macrophages to adopt an M1 phenotype with consequent secretion of proinflammatory cytokines and chemokines [66].
VWF and NETosis
VWF is able to recruit leukocytes either via direct leukocyte binding or by recruiting platelets, which in turn will attract leukocytes. VWF interacts with two distinct leucocyte receptors: PSGL-1 and various β2-integrins [67]. Activated platelets promote NET formation (neutrophil extracellular traps) via soluble factors and direct platelet–neutrophil interaction. In NET formation, neutrophils externalize their decondensed chromatin together with granule proteins (elastase, myeloperoxidase, and cathepsin) with a probable role for chromain decondensation [67, 68]. Individual NET components promote thrombin generation as illustrated by Schulz and Massberg [69]. NETs could also activate the alternative complement pathway [70]. Neutrophil activation in STEC-HUS (via elastase) mediates endothelial injury [71].
Limiting conditions for endothelial damage in culture
Cultured endothelial cells are frequently used to elucidate the pathogenesis of glomerular endothelial disorders such as STEC-HUS. Experimental approaches are needed to better mimic the in situ situation [72]:
-
Study in a flowing system is needed. In a static system the interaction of VWF with platelets is impaired and ADAMTS-13 cleaving activity is lacking.
-
There is a lack of interaction with other cell types such as the podocyte.
-
Evaluation secondary to a previous interacting factor is not correct.
-
Data obtained in HUVEC cannot be extrapolated to GMEC [73, 74]. Substantial phenotype heterogeneity exists in different cultured endothelial cells in expression of VWF, including the response to shear stress [75, 76].
The method applied by Nolasco (Sadler) approaches the clinical situation using HUVEC in a flowing system with adequate dose of STx.
In two out of four cultured glomerular microvascular endothelial cells (GMVECs), using the method of Nolasco, a decrease of intracellular VWF was observed. No effect was observed in static conditions of HUVEC and GMVEC [99].
Thrombotic thrombocytopenic purpura (TTP)
TTP is characterized by systemic aggregation of platelets within the vasculature (generally arteries and arterioles) causing microvascular thrombosis, hemolytic anemia, and thrombocytopenia. The systemic aggregation is due to lack of VWF cleaving protease ADAMTS-13. This protease is present in plasma and is also synthesized in endothelial cells. Under condition of shear stress, released VWF multimers unfold and assemble into string-like structures and bind platelets. Unfolded VWF multimers expose the VWF A2 domain, which now can be cleaved by ADAMTS-13. No effect is obtained in static conditions.
Even a complete deficiency of ADAMTS-13 is not sufficient to cause TTP [77]. A second hit is required. Infections have been recognized as triggers, although the mechanism is still not completely clear [78, 79]. The majority of patients suffering from acquired TTP develop antibodies that bind and neutralize the proteolytic activity of ADAMTS-13 and enhance its clearance [80]. The mainstay of treatment is ADAMTS-13 replacement with plasma exchange and immunosuppression [81].
Caplacizumab is a bivalent humanized immunoglobulin fragment. It binds to the A1 region of VWF and prevents platelet binding to receptor GPIb-IX-V, thus preventing the formation of platelet-rich thrombi. It has a definite role in the current guideline of management [81, 82]. Targeted ADAMTS-13 replacement therapy could help to manage acute episodes of TTP [83].
Congenital TTP is a rare disease due to severe deficiency of ADAMTS-13 caused by mutations. The patients are dependent on regular fresh plasma infusions. Recombinant ADAMTS-13 injections are an effective prophylactic therapeutic approach [84].
STEC-HUS—diarrhea phase
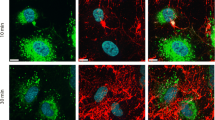
Endothelial damage occurring during the initial diarrhea phase of STEC-HUS is the first phase in the pathogenesis of this disorder. In a pivotal study, Chandler et al. [85] showed that thrombin generation and inhibition of fibrinolysis preceded renal injury. One of their explanations was the probability that circulating Shiga toxins directly injured renal cells but that the renal manifestations of this injury appeared later. The extremely high concentrations of Shiga toxin measured in the diarrhea phase of family members sustain this concept [86]. As the necessary requirements for safe collection and preservation of plasma samples were unknown, these samples were collected in ice, frozen at − 80 °C, and transported on ice. With a serum half-life of STx2 in mice of 3–3.9 min, some concentrations will remain undetected. As shown by Nolasco and Sadler [49, 51], an immediate effect of Shiga toxin (after 3–5 min) on the release of WPBs (releasing VWF) was observed in a flowing system of cultured endothelial cells (see also Fig. 3). No effect was observed after antibody application to Gb3 receptors. Angiopoetin2/angiopoetin1 ratio was increased in the preclinical phase [87]. VWF decreased size has been observed in the diarrhea phase and after onset of HUS [88]. Plasma P-selectin increased in TTP and after onset of STEC-HUS was not measured. Important data were provided by Yamamoto et al.—IFN-γ, TNF-α, IL-1β, IL-4, IL-6, IL-8, and IL-10 were measured in serum in the diarrhea phase. Only IL-6 and IL-8 were increased, both components of WPBs [89]. This increase in serum IL-8 was confirmed by Westerholt et al. [90]. Contribution of WPB components, stimulated by high Shiga toxin concentration, can be considered a first hit in the damage of Gb3 positive endothelial cells. In addition, serum thrombomodulin concentration, reflecting endothelial damage, was decreased [89, 91]. It is intriguing that in severe Covid-19 infection, the WPB components VWF, angiopoietin-2, and osteoprotegerin were increased in plasma [92]. Shiga toxin, via Gb3 receptor, binds to activated platelets [93], to monocytes [94], red blood cells, and doubtfully to leucocytes [95]. A transport via these cells to Gb3-positive endothelial cells is possible. An innovative concept for transfer of Shiga toxin is revealed by Stahl et al. [96]. The binding of toxin to platelets, monocytes, and red blood cells resulted in the release of extracellular vesicles. These microvesicles can be taken up not only by Gb3-positive but also by Gb3-negative cells. The recipient cell must express endogenous Gb3 for the cell to be susceptible to the toxin [97]. When incubating with glomerular endothelial cells in vitro, Stx2-containing microvesicles bound to the cells and were demonstrated within the cell after 3 h [96]. In vitro studies on cultured glomerular endothelial cells affected cell viability and inhibited protein synthesis. The effect of Stx2 microvesicles, however, was not tested in a flowing system and not taking into account a previous first hit. This first hit will influence the endothelial structure and the availability of Gb3 receptor. Is the first hit sufficient to induce severe endothelial damage, or is subsequent addition of Shiga toxin required? This question was already raised by Tarr et al. [98]. It is striking, as is shown in Fig. 3, that after a high dose of Shiga toxin in a flowing system as applied by Nolasco, the majority of VWF still remains after 15 min, as is visually illustrated by fluorescence microscopy images [99]. A repeated stimulation is possible and the WPB content can be restored. Repeated doses of DDAVP on consecutive days had approximately 30% less response than on the first day [100].

Cell lysate in static condition (left side) and in flow condition (right side) after incubation with thrombin or ST × 1. A star (*) indicates significant values
It is assumed that mechanical damage to red blood cells as a result of the formation of microthrombi induces hemolysis. In physiological conditions, cell-free hemoglobin and heme are promptly scavenged by haptoglobin and hemopexin. In hemolytic diseases, the detoxification systems are overwhelmed. In STEC-HUS heme concentration in plasma is increased, supporting a role as a secondary hit. Hemolysis is not present preceding STEC-HUS [85]. Toxicity pathways driven by hemolysis are excellently reviewed [101, 102]. Striking is the inhibition of ADAMTS-13 activity [103], degradation of heparan sulfate of the endothelial glycocalyx inducing local complement activation [104] and promoting rapid exocytosis of WPBs with membrane expression of P-selectin [105].
Conclusion
The role of the release of components from the WPBs (mainly VWF) is established in TTP due to ADAMTS-13 defect. Their role in the first phase of damage to GB3-positive cells due to high free Shiga toxin concentrations needs confirmation. Endeavors to provide more insight will indicate an effective way of prevention of STEC-HUS.
References
Rondaij M, Bierings R, Kragt A et al (2006) Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol 26:1002–1007
Schillemans M, Karampini E, Kat M et al (2019) Exocytosis of Weibel-Palade bodies:how to unpack a vascular emergency kit. J Thromb Haemost 17:6–18
Swamy S, Ueland T, Hansen JB et al (2023) Plasma levels of P-selectin and future risk of incident venous thromboembolism. J Thromb Haemost 21:2451–2460
Mitroulis I, Alexaki V, Kourtzelis I et al (2015) Leukocyte integrins: role in leucocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol Ther 147:123–135
Kameda H, Morita I, Handa M et al (1997) Re-expression of functional P-selectin molecules on the endothelial cell surface by repeated stimulation with thrombin. Br J Haematol 97:348–355
Celi A, Pellegrini G, Lorenzet R et al (1994) P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci U S A 91:9767–8771
Doyle E, Ridger V, Ferraro F et al (2011) CD63 is an essential cofactor to leukocyte recruitment by endothelial P-selectin. Blood 118:4265–4273
Ley K (2011) CD63 positions CD62P for rolling. Blood 118:4012–4013
Schroder J, Lullmann-Rauch R, Himmerkus N et al (2009) Deficiency of the tetraspanin CD63 associated with kidney pathology but normal lysosomal function. Mol Cell Biol 29:1083–1094
Goretzko J, Pauels I, Heitzig N et al (2023) P-selectin-dependent leukocyte adhesion is governed by endolysosomal two-pore channel 2. Cell Rep 42:113501
Davenport A, Hyndman K, Dhaun N et al (2016) Endothelin. Pharmacol Rev 68:357–418
Haynes W, Webb D (1994) Contribution of endogenous generation of endothelin-1 to vascular tone. Lancet 344:852–854
Dhaun N, Webb D, Kluth D (2012) Endothelin-1 and the kidney-beyond BP. Br J Pharmacol 167:720–731
Dolinina J, Rippe A, Oberg C (2019) Sustained, delayed, and small increments in glomerular permeability to macromolecules during systemic ET-1 infusion mediated via the ETA receptor. Am J Physiol Renal Physiol 316:F1173–F2117
Morigi M, Buelli S, Zanchi C et al (2006) Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am J Pathol 169:1965–1975
Ebefors K, Wiener R, Yu L et al (2019) Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int 96:957–970
Bowen E, Hurcombe J, Barrington F et al (2023) Shiga toxin targets the podocyte causing hemolytic uremic syndrome through endothelial complement activation. Med 4:761–777
Boels M, Lee D, van den Berg B et al (2013) The endothelial glycocalyx as a potential modifier of the hemolytic uremic syndrome. Eur J Intern Med 24:503–507
Kohan D, Barton M (2014) Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int 86:896–904
Benigni A, Buelli S, Kohan D (2021) Endothelin-targeted new treatments for proteinuric and inflammatory glomerular diseases: focus on the added value to anti-renin-angiotensin system inhibition. Pediatr Nephrol 36:763–775
Russell F, King S, Smillie SJ et al (2014) Calcitonin gene–related peptide: physiology and pathophysiology. Physiol Rev 94:1099–1142
Russo A, Hay D (2023) CGRP physiology, pharmacology, and therapeutic targets :migraine and beyond. Physiol Rev 103:1565–1644
Hartopo A, Emoto N, Vignon-Zellweger N et al (2013) Endothelin-converting enzyme-1 gene ablation attenuates pulmonary fibrosis via CGRP-cAMP/EPAC1 pathway. Am J Resp Cell Mol Biol 48:465–476
Van Campenhout A, Golledge J (2009) Osteoprotegerin, vascular calcification and atherosclerosis. Atherosclerosis 204:321–329
Anandarajah A (2008) Role of RANKL in bone disease. Trends Endocrinol Metab 20:88–94
Wohner N, Sebastian S, Muczynski V et al (2022) Osteoprotegerin modulates platelet adhesion to von Willebrand factor during release from endothelial cells. J Thromb Haemost 20:755–766
Fiedler U, Augustin H (2006) Angiopoetins: a link between angiogenesis and inflammation. Trends immunol 27:552–558
Mobayen G, Smith K, Ediriwickrema K (2023) von Willebrand factor binds to angiopoetin-2 within endothelial cells and after release from Weibel-Palade bodies. J Thromb Haemost 21:1802–1812
Fiedler U, Scharpfendecker M, Koldi S (2004) The TieFASB J-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood 103:4150–4156
Gansler J, Preissner K, Fisher S (2014) Influence of proinflammatory stimuli on the expression of vascular ribonuclease 1 in endothelial cells. FASEB J 28:752–760
Garnet E, Raines R (2022) Emerging biological functions of ribonuclease 1 and angiogenin. Crit Rev Biochem Mol Biol 57:244–260
Dickson KA, Haigis MC, Raines RT (2005) Riobonuclease inhibitor: structure and function. Prog Nucleic Acid Res Mol Biol 80:249–374
Schnyder-Candrian S, Borsig L, Moser R et al (2000) Localisation of α1,3-fucosyltransferase VI in Weibel-Palade bodies of human endothelial cells. Proc Natl Acad Sci U S A 97:8369–8374
Bierings R, van den Biggelaar M, Kragt A et al (2007) Effficiency of von Willebrand factor-mediated targeting of IL-8 into Weibel-Palade bodies. J Thromb Haemost 5:2512–2519
Knipe L, Meli A, Hewlett L et al (2010) A revised model for the secretion of tPA and cytokines from cultured endothelial cells. Blood 116:2183–2191
Adams D, Lloyd A (1997) Chemokines: leucocyte recruitment and activation cytokines. Lancet 349:490–495
Luster A (1998) Chemokines-chemotactic cytokines that mediate inflammation. N Engl J Med 338:436–444
Van Breevoort D, van Agtmaal E, Dragt B et al (2012) Proteomic screen identifies IGFBP7 as a novel component of endothelial cell-specific Weibel-Palade bodies. J Proteomic Res 11:2925–2936
Lowenstein C, Morrell C, Yamakuchi M (2005) Regulation of Weibel-Palade body exocytosis. Trends Cardiovasc Med 15:302–308
Ribes J, Francis C, Wagner D (1987) Fibrin induces release of von Willebrand factor from endothelial cells. J Clin Invest 79:117–123
Leitner G, Schmetterer L, Kapiotis S et al (2010) Effects of endothelin-1 and phenylephrine on plasma levels of von Willebrand factor and protein S. Thromb Res 125:e5–e8
Aiello S, Gastoldi S, Galbusera M et al (2022) C5a and C5aR1 are key drivers of microvascular platelet aggregation in clinical entities spanning from aHus to Covid-19. Blood Adv 6:866–881
Hattori R, Hamilton K, Rodger P et al (1989) Complement proteins C5b–9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem 264:9053–9060
Bernardo A, Ball C, Nolasco L et al (2004) Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood 104:100–106
Dole V, Bergmeier W, Mitchell H et al (2005) Activated platelets induce Weibel-Palade-body secretion and leucocyte rolling in vivo: role of P-selectin. Blood 106:2334–2339
Pawlinsly R (2016) Platelet HJMGB1:the venous clot coordinator. Blood 18:2376–2378
Maeda R, Kawasaki Y, Kume Y (2019) Involvement of high mobiliyy group box 1 in the pathogenesis of severe hemolytic uremic syndrome in a murine model. Am J Physiol Renal Physiol 317:1420-F1429
Bergan J, Lingelem A, Simm R (2012) Shiga toxins. Toxicon 60:1085–1107
Nolasco L, Turner N, Bernardo et al (2005) Hemolytic uremic syndrome-associated shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand multimers. Blood 106:4199–4209
Lo N, Turner N, Cruz M et al (2013) Interaction of shiga toxin with the A-domains and multimers of von Willebrand factor. J Biol Chem 288:33118–33123
Huang J, Motto D, Bundle D et al (2010) Shiga toxin B subunits induce VWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood 116:3653–3659
Lenting P, Christophe O, Denis C (2015) Von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood 125:2019–2028
Leebeek FW, Eikelboom JC (2016) Von Willebrand’s disease. N Engl J Med 375:2067–2080
Naβ J, Terglane J, Gerke V (2021) Weibel-Palade bodies: unique secretory organelles of endothelial cells that control blood vessel homeostasis. Front Cell Dev Biol 9:813995
El-Mansi S, Nightingale T (2021) Emerging mechanisms to modulate VWF release form endothelial cells. Int J Biochem Cell Biol 131:105900
Kat M, Margadant C, Voorberg J et al (2022) Dispatch and delivery at the ER-GOLGI interface: how endothelial cells tune their hemostatic response. FEBS J 289:6863–6870
Lopes da Silva M, Cutler D (2016) Von Willebrand factor multimerization and the polarity of secretory pathways in endothelial cell. Blood 128:277–285
Federici A, Bader R, Pagani M et al (1989) Binding of von Willebrand factor to glycoproteins Ib and IIb/IIIa complex: affinity is related to multimeric size. Br J Haematol 73:93–99
Babich V, Meli A, Knipe L et al (2008) Selective release of molecules from Weibel-Palade bodies during lingering kiss. Blood 111:5282–5290
El-Mansi S, Robinson C, Kostelnik K et al (2023) Proximity proteomics identifies septin and PAK2 as decisive regulators of actomyosin-mediated expulsion of von Willebrand factor. Blood 141:930–944
Denis C, Lenting P (2018) VWF clearance :it’s glycomplicated. Blood 131:842–843
Bryckaert M, Rosa J, Penis C et al (2015) of von Willebrand factor and platelets. Cell Mol Life Sci 72:307–326
Peerschke E, Yin W, Ghebrehiwet B (2010) Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol 47:2170–2175
De Ceunynck K, De Meyer S, Vanhoorelbeke K (2013) Unwinding the von Willebrand factor strings puzzle. Blood 121:270–277
Nording H, Langer H (2018) Complement links platelets to innate immunity. Semin Immunol 37:43–52
Drakeford C, Aguila S, Roche F et al (2022) von Willebrand factor links primary hemostasis to innate immunity. Nat Commun 13:6320
Kawecki C, Lenting P, Denis C (2017) Von Willebrand factor and inflammation. J Thromb Haemost 15:1285–1294
Herre M, Cedervall J, Mackman N et al (2023) Neutrophil extracellular traps in the pathology of cancer and other inflammatory diseases. Physiol Rev 103:277–312
Schulz C, Massberg S (2017) Demystifying the prothrombotic role of NETs. Blood 129:925–926
Wang H, Wang C, Zhao M (2015) Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol 181:518–527
Fitzpatrick M, Shah Y, Filler G et al (1992) Neutrophil activation in the haemolytic uraemic syndrome: free and complexed elastase in plasma. Pediatr Nephrol 6:50–53
Aman J, Weijers E, Van Nieuw AG et al (2016) Using cultured endothelial cells to study endothelial barrier dysfunction: challenges and opportunities. Am J Physiol Lung Cell Mol Physiol 311:L453–L466
Luxen M, Zwiers P, Meester F et al (2023) Unique miRNome and transcriptome profils underlie microvascular heterogeneity in mouse kidney. Am J Physiol Renal Physiol 325:F299–F316
Van Hinsbergh V (2012) Endothelium-role in regulation of coagulation and inflammation. Semin Immunopathol 34:93–106
Hough C, Notley C, Mo A et al (2022) Heterogeneity and reciprocity of FVIII and VWF expression and the response to shear stress in cultured endothelial cells. J Thromb Haemost 20:2507–2518
Mojiri A, Alavi P, Carrillo M et al (2019) Endothelial cells of different organs exhibit heterogeneity in von Willebrand factor expression in response to hypoxia. Atherosclerosis 282:1–10
Banno F, Kokame K, Okuda T et al (2006) Complete deficiency in ADAMTS13 is prothrombotic, but is alone not sufficient to cause thrombotic thrombocytopenic purpura. Bood 107:3161–3166
Thoreau B, von Tokarski F, Bauvois A et al (2021) Infection in patients with suspected thrombotic microangiopathy based on clinical presentation. Clin J Am Soc Nephrol 16:1355–1364
Cauchois R, Muller R, Lagarde M et al (2023) Is endothelial activation a critical event in thrombotic thrombocytopenic purpura? J Clin Med 12:758
Underwood M, Alswan F, Thomas M et al (2023) Autoabtibodies enhance ADAMTS-13 clearance in patients with immune thrombotic thrombocytopenic purpura. J Thromb Haemost 21:1544–1552
Subhan M, Scully M (2022) Advances in the management of TTP. Blood Rev 55:100945
Scully M, Rayment R, Clark A et al (2023) A British society for haematology guideline: diagnosis and management of thrombotic thrombocytopenic purpura and thrombotic microangiopathies. Br J Haematol 203:546–563
Moroniti J, Vrbensky J, Nazy I et al (2024) Targeted ADAMTS-13 replacement therapy for thrombotic thrombocytopenic purpura. J Thromb Haemost 22:896–904
Scully M, Antun A, Cataland S et al (2024) Recombinant ADAMTS13 in congenital thrombotic thrombocytopenic purpura. N Engl J Med 390:1584–1596
Chandler W, Jelacic S, Boster D et al (2002) Prothrombotic coagulation abnormalities preceding the hemolytic uremic syndrome. N Engl J Med 346:23–32
He X, Quinones B, Te Loo M et al (2015) Serum shiga toxin2 values in patients during acute phase of diarrhoea-associated Haemolytic uraemic syndrome. Acta Paediatr 104:564–568
Page A, Tarr P, Watkins S et al (2013) Dysregulation of angiopoeitin1 and 2 in Escherichia coli 0157:H7 infection and hemolytic-uremic syndrome. J infect Dis 208:929–933
Tai H, Chandler W, Sarode R et al (2001) Von Willebrand factor and Von Willebrand factor-cleaving metalloprotease activity in Escherichia coli 0157:H7-associated hemolytic uremic syndrome. Pediatr Res 49:653–659
Yamamoto T, Nagayama K, Satomura K et al (2000) Increased serum Il-10 and endothelin levels in hemolytic uremic syndrome caused by Escherichia coli 0157. Nephron 84:326–332
Westerholt S, Hartung T, Tollens M et al (2002) Inflammatory and immunological parameters in children with haemolytic uremic sundrome (hus) and gastroenteritis-pathophysiological and diagnostic clues. Cytokine 12:823–827
Fernandez G, Te Loo M, van der Velden T et al (2003) Decrease of thrombomodulin contributes to the procoagulant state of endothelium in hemolytic uremic syndrome. Pediatr Nephrol 18:1066–1068
Karampini E, Fogarty H, Elliot S et al (2023) Endothelial cel activation, Weibel-Palade body secretion, and enhanced angiogenesis in severe COVID-19. Res Pract Thromb Haemost 7:e10085
Ghosh S, Polanowska-Grabowska R, Fujii J et al (2004) Shiga toxin binds to activated platelets. J Thromb Haemost 2:499–506
Geelen J, van der Velden T, van den Heuvel et al (2007) Interaction of shiga-like toxin with human peripheral monocytes. Pediatr Nephrol 22:1181–1187
Geelen J, van der Velden T, Te Loo M et al (2007) Lack of specific binding of Shiga-like toxin( verocytotoxin) and non-specific interaction of Shiga-lik toxin antibody with human polymorphonuclear leucocytes. Nephrol Dial Transplant 22:749–755
Stahl A, Arvidsson I, Johansson K et al (2015) A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathogen 11:e1004619
Johansson K, Willysson A, Kristoffersson A et al (2020) Shiga toxin-bearing microvescles exert a cytotoxic effect on recipient cells only when the cells express the toxin receptor. Front Cell Infect Microbiol 10:212
Tarr P, Gordon C, Chandler W (2005) Shiga-toxin producing Escherichia coli and Haemolytic uraemic syndrome. Lancet 365:1073–1086
Geelen J, van den Biggelaar M, Linssen P et al (2014) The effect of Shiga toxin on Weibel-Palade bodies in primary human endothelial cells. Nephron Extra 4:101–107
Manucci PM, Bettega D, Cattaneo M (1992) Patterns of development of tachyphylaxis in patients with hemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Br J Haematol 82:87–93
Van Avondt K, Nur E, Zeerleder S (2019) Mechanisms of haemolysis-induced kidney injury. Nat Rev 15:671–692
Vallelian F, Buehler P, Schaer D (2022) Hemolysis, free hemoglobin toxicity, and scavenger protein therapeutics. Blood 140:1837–1844
Studt J, Kremer Hovinga J, Antoine G et al (2005) Fatal thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibition of ADAMTS13 activity by hemoglobine. Blood 105:542–544
Laboux T, Maanaoui M, Allain F et al (2023) Hemolysis is associated with altered heparan sulfate of the endothelial glycocalyx and with local complement activation in thrombotic microangiopathies. Kidney Int 104:353–366
Frimat M, Tabarin F, Dimitov J et al (2013) Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 122:282–292
Acknowledgements
I gratefully acknowledge the permission to use Fig. 1 by Marije Kat. Sebastian Quiroz Monnens took care of the figures.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Monnens, L. Weibel-Palade bodies: function and role in thrombotic thrombocytopenic purpura and in diarrhea phase of STEC-hemolytic uremic syndrome. Pediatr Nephrol (2024). https://doi.org/10.1007/s00467-024-06440-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00467-024-06440-3