
Abstract
Epigenetics studies changes in gene activity without changes in the DNA sequence. Methylation is an epigenetic mechanism important in many pathways, such as biotic and abiotic stresses, cell division, and reproduction. Eragrostis curvula is a grass species reproducing by apomixis, a clonal reproduction by seeds. This work employed the MCSeEd technique to identify deferentially methylated positions, regions, and genes in the CG, CHG, and CHH contexts in E. curvula genotypes with similar genomic backgrounds but with different reproductive modes and ploidy levels. In this way, we focused the analysis on the cvs. Tanganyika INTA (4x, apomictic), Victoria (2x, sexual), and Bahiense (4x, apomictic). Victoria was obtained from the diploidization of Tanganyika INTA, while Bahiense was produced from the tetraploidization of Victoria. This study showed that polyploid/apomictic genotypes had more differentially methylated positions and regions than the diploid sexual ones. Interestingly, it was possible to observe fewer differentially methylated positions and regions in CG than in the other contexts, meaning CG methylation is conserved across the genotypes regardless of the ploidy level and reproductive mode. In the comparisons between sexual and apomictic genotypes, we identified differentially methylated genes involved in the reproductive pathways, specifically in meiosis, cell division, and fertilization. Another interesting observation was that several differentially methylated genes between the diploid and the original tetraploid genotype recovered their methylation status after tetraploidization, suggesting that methylation is an important mechanism involved in reproduction and ploidy changes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Epigenetics studies heritable gene expression changes without altering the DNA sequence (Russo 1996; Armstrong 2013). The main epigenetic mechanisms are DNA methylation, histone modifications, RNA interference, and genomic imprinting (Munshi et al. 2015). Methylation occurs when a methyl group is added to a cytosine in the 5′ position in three contexts: CG, CHH, and CHG where H represents an A, C, or T (Chen and Li 2004). Methylation is involved in gene regulation and genome stability and has an important role in many pathways, such as seed imprinting, response to biotic and abiotic stresses, and cell division (Zhang et al. 2018). More specifically, methylation and de-methylation dynamics during reproduction are fundamental for normal plant propagation (Melamed-Bessudo and Levy 2012; Underwood et al. 2018; Han et al. 2019).
One of the most widely used methods to assess DNA methylation is whole-genome bisulfite sequencing (Lister et al. 2008). However, this method requires extraordinary coverage, increasing the costs significantly. The Methylation Context Sensitive Enzyme ddRAD (MCSeEd) technique was recently developed to reduce sequencing costs (Marconi et al. 2019; Di Marsico et al. 2020). This method was successfully used to study methylation changes associated with different plant growth conditions, such as drought in Zea maize (Marconi et al. 2019), asexual reproduction of Eragrostis curvula and Paspalum rufum (Carballo et al. 2021a; Soliman et al. 2021), response to infection with the cereal pathogen Fusarium graminearum (Tini et al. 2021), and the chilling accumulation period in Prunus persica (Canton et al. 2022).
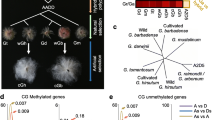
Eragrostis curvula is a C4 grass species used as forage in semiarid regions. This species is primarily investigated because of its capability to reproduce by apomixis. (Carballo et al. 2021b). Apomixis is an asexual reproduction by seeds in which the progeny is genetically identical to the maternal plant. Three main components differentiate apomixis from sexual reproduction: apomeiosis, parthenogenesis, and pseudogamy (Crane 2001). During the E. curvula embryo sac development, the meiosis is completely absent (apomeiosis), the embryo arises without fertilization (parthenogenesis), and only the polar nuclei are fertilized (pseudogamy). One advantage of using E. curvula as a model species for apomixis is that sexual and apomictic genotypes co-exist in this species. Even more, a synthetic line with a similar genetic background was created to study changes in reproductive mode and ploidy level (Cardone et al. 2006). In this way, the sexual diploid Victoria was obtained from in vitro culture of inflorescences of the apomictic tetraploid Tanganyika INTA cultivar, and the apomictic tetraploid Bahiense was obtained from the polyploidization of Victoria (Fig. 1). It is currently accepted that all seed plants have experienced at least one round of whole genome duplication in their evolutionary history (Jiao et al. 2011). After this, many species recover their diploid level due to the loss of genes during evolution. In E. curvula, the synthetic diploidization followed by polyploidization is considered a recent event compared with the ancestral duplication. This synthetic line is an ideal tool to compare genotypes with different ploidy levels and reproductive modes but with similar genomic background.

Diagram representing the recent events of diploidization of Tanganyika INTA to obtain Victoria and the tetraploidization of Victoria to obtain Bahiense
Albeit the genetic and epigenetic regulation of apomixis in E. curvula is not completely clear, several advances have been made in the last few years. Using genomics and transcriptomics approaches over sexual and apomictic genotypes, different candidate genes and regulatory pathways were found to be differentially enriched (Garbus et al. 2017; Carballo et al. 2019, 2023; Zappacosta et al. 2019; Selva et al. 2020). In addition, analyses of methylation patterns and microRNA representation in different genotypes shed light on the epigenetic regulation of apomixis (Rodrigo et al. 2017; Garbus et al. 2019; Carballo et al. 2021a; Pasten et al. 2022). Methylation studies using the MSAP technique were used in tetraploidized and diploidized lines, showing lower methylation levels in the diploid genotypes and methylation recovery after tetraploidization (Ochogavia et al. 2009). However, this technique cannot identify differentially methylated genes. The same technique in facultative apomictic E. curvula plants exposed to stress showed a positive correlation between DNA methylation changes and the percentage of sexual reproduction (Rodrigo et al. 2017). These results suggest a quantitative regulation of apomeiosis in facultative cultivars mediated by methylation under stress conditions. More recently, we reported higher levels of methylation in E. curvula tetraploid apomictic genotypes than in the sexual one with the same ploidy level (Carballo et al. 2021a). In the mentioned study, genes related to reproductive pathways, like the ubiquitin one, were differentially methylated between genotypes. However, the genetic background of the analyzed genotypes was completely different, not allowing us to distinguish whether the differences could be attributed to the reproductive mode or were associated with differences in the genotypes themselves.
Here, we employed the MCSeEd method to study methylation changes across synthetically developed E. curvula genotypes with similar genomic background but with different reproductive modes and ploidy levels. This work aimed to identify differentially methylated genes and pathways associated with reproduction and ploidy in the recent diploidized and tetraploidized E. curvula genotypes.
Materials and methods
Plant material and DNA extraction
The genotypes used for methylation analyses were the apomictic Tanganyika INTA (2n = 4X = 40), Bahiense (2n = 4X = 40), and TUNS9355 (2n = 6X = 60) cultivars, and the sexual genotype Victoria (2n = 2X = 20). Tanganyika INTA, Victoria, and Bahiense were chosen based on their genomic background (Fig. 1), and TUNS9355 was selected in order to incorporate an unrelated polyploid apomictic genotype. The most plausible explanation for the emergence of the diploid genotype is that a Tanganyika INTA unreduced gamete regenerates, producing Victoria. Samples were collected from inflorescences, including stages from archespore to mature embryo sac, in order to target both apomeiosis and parthenogenesis. Three biological replicates were taken from each sample from plants growing at a CERZOS (CONICET-UNS) greenhouse (Bahía Blanca, Argentina; 38°42′ S, 62°16′ W). Genomic DNA was extracted using a cetyltrimethylammonium bromide protocol. Briefly, spikelets were frozen and ground in liquid nitrogen to obtain a fine powder by using a TissueLyser II (Qiagen). Then, 100 mg from each sample was incubated at 65 °C in preheated extraction buffer containing 100 mM Tris HCl pH 8, 1.4 M NaCl, 20 mM, EDTA pH 8, 2% (w/v) CTAB, and 0.5% (v/v) β-mercaptoethanol. Chloroform was added to reach a 2:1 ratio (buffer/chloroform), and the aqueous phase was collected after centrifugation. DNA was precipitated with one volume of isopropanol and washed with 70% (v/v) ethanol two times. Finally, the pellet was air-dried and resuspended in 50 µL of Milli-Q water containing 20 µg/mL RNase.
Library preparation
Following the MCSeEd protocol (Marconi et al. 2019; Di Marsico et al. 2020), each sample consisting of 150 ng of DNA was treated with the MseI enzyme to decrease genome complexity and with the methylation-sensitive enzymes AciI, PstI, and EcoT22I to detect methylation over the CG, CHG, and CHH contexts, respectively. After the double digestion, specific barcodes were ligated to demultiplex the samples in the downstream analysis. Then, samples were sequenced using the Illumina Hiseq X platform (Table S1). Finally, demultiplexing was carried out using the process_radtags script (Catchen et al. 2013). Reads were uploaded to NCBI database under the BioProject: PRJNA988943.
Bioinformatic analysis
Bioinformatic analysis followed the MCSeEd reference pipeline (Marconi et al. 2019, https://bitbucket.org/capemaster/mcseed/src/master/). After demultiplexing, the reads were mapped against the E. curvula Victoria genome assembly (Carballo et al. 2019), obtaining a minimum of 80% and a maximum of 97% of the mapping rate. Reads per position were normalized, and the differentially methylated positions (DMPs) were detected by using the methylkit R package (Akalin et al. 2012). As described by Marconi et al. (2019), the differentially methylated regions (DMRs) were obtained by identifying the best window for each context in which two or more positions had the same behavior (i.e., de-methylated or methylated). Differentially methylated genes (DMGs) were detected by contrasting the position of the DMRs with the Victoria genome annotation (Carballo et al. 2019). The DMGs analysis focused on the gene body and 2500 bp before and after the start and stop codons for the upstream and downstream regions, respectively. This window was chosen because in plants, gene expression regulation via methylation was previously associated with the promoter and enhancer regions located up to 2500 bp upstream and downstream, respectively (He et al. 2020, 2022; Bewick and Schmitz 2017). DMPs, DMRs, and DMGs comparisons were performed in all versus all plant materials, as follows: Victoria versus Bahiense, Victoria versus Tanganyika INTA, Victoria versus TUNS9355, Bahiense versus Tanganyika INTA, Bahiense versus TUNS9355, and Tanganyika INTA versus TUNS9355, hereafter referred as vicVSbah, vicVStan, vicVSt93, bahVStan, bahVSt93, and tanVSt93 respectively.
Differentially methylated genes
To distinguish DMGs related to the reproductive mode, the comparisons between apomictic versus sexual genotypes were specially analyzed, considering those DMGs with the same behavior in the three contrasts (vicVSbah, vicVStan, and vicVSt93). On the other hand, to detect the methylation changes related to diploidization and tetraploidization, the comparisons vicVStan and vicVSbah were also analyzed. In these comparisons, methylation and de-methylation always refer to the first genotype; for instance, in vicVStan, methylated and de-methylated DMPs, DMRs, and DMGs refer to Victoria. The genes were annotated using the string database (Szklarczyk et al. 2019). Differentially gene ontology analyses were performed using the clusterProfiler R package (Yu et al. 2012).
Results
Differentially methylated positions and regions
DNA from inflorescences of E. curvula genotypes Tanganyika INTA, Victoria, Bahiense, and TUNS9355 were extracted and sequenced in three biological replicates following the MCSeEd protocol to determine the methylation changes in CG, CHG and CHH contexts (Marconi et al. 2019). For this purpose, the Victoria genome assembly (Carballo et al. 2019) was used as a reference to identify differentially methylated positions (DMPs), differentially methylated regions (DMRs), and differentially methylated genes (DMGs). Even though Tanganyika INTA and Bahiense are facultative apomictic tetraploids and Victoria is a sexual diploid, they share a similar genomic background since Victoria was obtained from inflorescences of Tanganyika INTA through in vitro culture, and Bahiense derives from Victoria (Fig. 1) (Cardone et al. 2006). The hexaploid facultative apomict TUNS9355 was also included in the analysis to evaluate the effect of methylation in apomictic genotypes unrelated to the tetraploidized and diploidized genotypes.
By employing the methylkit pipeline (Akalin et al. 2012), a total of 42,780, 46,132, and 53,707 methylated/de-methylated positions were estimated in the Victoria genome assembly for the CG, CHG, and CHH contexts, respectively. The principal component analysis performed with these data showed a high similarity within biological replicates and differences between genotypes, thus evidencing data suitability for the downstream analysis (Figure S1). Based on the phylogenetic tree, there was no evident clustering in common for all three contexts since Victoria, TUNS9355, and Bahiense grouped for methylations/de-methylations in the CG context, while in CHG context Tanganyika INTA, Victoria, and Bahiense clustered together. Finally, for CHG context, Victoria grouped with Bahiense, and Tanganyika with TUNS9355 (Figure S2).
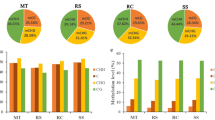
DMPs and DMRs were computed in all comparison combinations: vicVSbah, vicVStan, vicVSt93, bahVStan, bahVSt93, and tanVSt93 (Fig. 2). In these comparisons, the context with higher DMPs was CHH, followed by CHG and CG (Fig. 2, Table S2). This distribution was not reflected on DMRs since this relationship was only retained for vicVStan and bahVStan (Fig. 2, Table S3). The DMPs within DMRs were plotted in a heatmap for all the analyses, showing a high correlation between replicates and the expected differences between genotypes (Figure S3).

Number of differentially methylated and de-methylated positions (A) and regions (B) for the comparisons: Victoria versus Bahiense (vicVSbah), Victoria versus Tanganyika INTA (vicVStan), Victoria versus TUNS9355 (vicVSt93), Bahiense versus Tanganyika INTA (bahVStan), Bahiense versus TUNS9355 (bahVSt93), and Tanganyika INTA versus TUNS9355 (tanVSt93). Full-color bars represent methylated and transparent de-methylated positions/regions
In the comparisons between sexual and apomictic genotypes (i.e., vicVSbah, vicVStan, and vicVSt93), which is coincident with diploid versus polyploid genotypes, it was possible to observe more de-methylated DMPs and DMRs in the sexual genotype for all the contexts (Fig. 2). Tetraploid versus tetraploid contrasts (bahVStan) showed in Bahiense more methylated DMPs and DMRs in CG contexts, while in CHG and CHH, the number of de-methylated DMPs and DMRs was higher than Tanganyika INTA cultivar. The hexaploid versus tetraploid analysis (bahVSt93 and tanVSt93) displayed differences in DMPs and DMRs associated with the contexts. The study of DMPs in bahVSt93 showed that de-methylation in Bahiense was higher in all the contexts. The same analysis over DMRs showed more methylation in Bahiense in the CHG context. On the other hand, in the tanVSt93 comparison, the de-methylation in Tanganyika INTA was higher in CG and lower in CHG and CHH contexts in both DMPs and DMRs.
When the positions of the DMR were contrasted with the Victoria genome, it was possible to observe an increased number of DMRs within intergenic areas in the CG, CHG and CHH contexts (Fig. 3). The number of DMRs in the 3´ UTR was higher than those in the 5´ UTR in all three contexts. The number of DMRs in introns and exons was linked to the methylation context; in CG, DMRs were more present in exons, while introns had a higher number of DMRs for the CHG and CHH contexts.

Number of DMRs found within intergenic, introns, exons, and UTR regions in the Victoria genome
Differentially methylated genes
DMRs within genes in the CG, CHG, and CHH contexts were analyzed for the vicVSbah, vicVStan, vicVSt93, bahVStan, bahVSt93, and tanVSt93. Since the regulation of gene expression is associated with the location of methylated positions within the gene, DMGs were classified according to whether the methylated position was located upstream, downstream or within the gene body (Table S4). Even though the total number of DMGs was similar across the upstream, gene body, and downstream regions, the distribution within each region was associated with the context. In this way, the number of DMGs in CG and CHH contexts was similar in the three regions, while the number of DMGs in the CHG context was higher in the gene body than upstream and downstream (Fig. 4). The number of methylated and de-methylated positions within DMGs was similar in all the samples in the three regions. There were no significant differences, with a minimum of 2 and a maximum of 2.37 methylated positions on average (Table S5).

Number of differentially methylated genes in CG, CHG, and CHH contexts in upstream, downstream, and gene body regions
The distribution of DMRs across the genes was computed considering 2,500 bp upstream and downstream from the start and stop position, respectively, and 1,000 bp after and before the start and stop position, respectively (Fig. 5). The distribution of DMRs in the three contexts displayed different patterns. The highest number of DMRs in the CG context was found in the area surrounding the start and stop positions, with a peak of ~ 350 DMRs, respectively. In the CHG context, the stop codon showed a peak of ~ 300 DMRs, and the gene body presented an average of 250 DMRs. In CHH, the distribution was relatively constant with 150 DMRs, except in the start positions where a depression of ~ 50 DMRs was located.

DMRs in the CG, CHG, and CHH contexts across the upstream, gene body, and downstream regions
Sexual versus apomictic analysis
The differentially methylated and de-methylated genes in common in the comparisons between sexual and apomictic genotypes (i.e., vicVSbah, vicVStan, vicVSt93) were analyzed in detail since they could shed light on the role of methylation in the regulation of the reproductive pathways (Fig. 6, Figure S4, Table S6). Methylated and de-methylated genes were computed using Victoria as a reference (i.e., methylated genes are methylated in the sexual genotype and de-methylated in the apomictic ones and vice versa). ANOVA test showed that the number of de-methylated genes in the sexual genotype was significantly higher than the methylated ones and predominantly in the CG context, except for the gene body, in which CHG presented more DMGs. Gene ontology enrichment analysis in CG, CHG, and CHH contexts in the upstream, gene body, and downstream regions showed terms such as DNA replication, DNA repair, helicase activity, and ubiquitin pathway, which were previously associated with reproduction in Eragrostis curvula (Carballo et al. 2021a, b) (Figure S5). The region being methylated/de-methylated is crucial as it can have a differential effect on gene expression (He et al. 2020, 2022; Bewick and Schmitz 2017). Even though there is no universal rule, methylation in upstream regions generally represses gene expression. Here, we observed that all the genes related to the ubiquitin pathway, such as BPM2, AFR, E3 ubiquitin-protein ligase, and GA20OX2, were de-methylated in upstream regions in the sexual genotype. The same behavior was observed for the helicase genes RECQL2 and RECQ4A. These genes prevent recombination and repair DNA during meiosis (Serra et al. 2018; Kobbe et al. 2008).

DMGs methylated or de-methylated in upstream, gene body and downstream regions in common in the comparisons between the sexual and the apomictic genotypes (vicVStan, vicVSbah, and vicVSt93)
Contrary to what happens at the upstream region, methylation in the gene body is associated with an increase in gene expression (Zhang et al. 2006; Bewick and Schmitz 2017). In this region, the genes related to ubiquitination were also found de-methylated. Other genes previously associated with reproduction and methylation, such as EMB2758 and NRPD2A, were also found in this analysis differentially methylated in the gene body (Tzafrir et al. 2004; Zhang et al. 2021). However, one of the most interesting genes methylated in the gene body was SPO11-1, which is involved in recombination during meiosis and is part of the MiMe mutant used to transform mitosis into meiosis (Grelon et al. 2001; d'Erfurth et al. 2009; Miulet et al. 2016). Another gene de-methylated in the sexual genotype in the gene body was FAR2. This gene is essential for pollen development and was found downregulated in sexual genotypes when compared with apomictic ones in Limonium spp. (Caperta et al. 2023).
Even though methylated genes in the downstream region suggest a decreased gene expression, there is evidence for and against this mechanism (He et al. 2022). As in the upstream region, the genes related to the ubiquitin pathway were mostly de-methylated here. Other genes related to replication, such as MCM6 and CDT1A were found to be methylated and de-methylated, respectively (Bell and Dutta 2002; Castellano et al. 2004).
Methylation changes in diploidized and tetraploidized genotypes
The comparisons vicVStan and vicVSbah were also particularly analyzed since Victoria was obtained from the diploidization of Tanganyika INTA and Bahiense from the tetraploidization of Victoria (Fig. 1). To assess the effects of methylation changes during diploidization and tetraploidization, DMGs detected in common in the comparisons vicVStan and vicVSbah were analyzed. The proportion of genes de-methylated in the upstream region observed in common (i.e., genes de-methylated in the diploid Victoria and methylated in both tetraploid genotypes, Bahiense and Tanganyika INTA) was higher than the methylated in the three contexts (Fig. 7). This suggests that many genes that were de-methylated during the diploidization process were methylated again after tetraploidization. In the three regions, upstream gene body and downstream, many DMGs were found involved in the ubiquitin pathway, such as E3 ubiquitin-protein ligase, RING protein, BPM2, BPM4, and SKP1-like, among others (Table S7).

Venn diagram representing the number of methylated and de-methylated genes in the comparisons vicVSbah (red) and vicVStan (black). The intersection represents genes methylated in Victoria and de-methylated in Bahiense and Tanganyika in the upper panel and vice versa in the lower panel. The number of de-methylated genes in common between the two comparisons (intersections) was higher than the methylated ones
Genes associated with reproduction and/or ploidy were found methylated and de-methylated in these comparisons. In the upstream region, RECQ4A was found de-methylated in the diploid genotype. RECQ4A is involved in the repression of crossovers during meiosis (Serra et al. 2018). On the other hand, the genes SUVH5, HAP2, and NRPD2A were found methylated in the same region. SUVH5 is a histone methyltransferase (Rajakumara et al. 2011), HAP2 is only expressed in haploid pollen and it is required for pollen tube guidance and fertilization (Valansi et al. 2017), and NRPD2A is involved in transcriptional gene silencing through RdDM pathway (Kanno et al. 2008).
In the gene body, the genes GASA4, E2F1, CAS1, ULT1, FAR2, FDM1, SEC15B, OSB1, MEE44, GSL11, RECQ4A, and PC-MYB1 were found to be de-methylated, and SPO11-1 and FK were methylated in the diploid genotype and de-methylated in the polyploids. These genes are related to cell division or reproduction; however, PC-MYB1 and SPO11-1 are directly involved in these pathways. PC-MYB1 is required to maintain the ploidy level, and its mutation induces deregulation of the cell cycle and increase of the ploidy level (Haga et al. 2011). SPO11-1 is a key meiotic gene that initiates recombination and controls double-strand breaks and crossovers (Xue et al. 2018; Vrielynck et al. 2016). As in the upstream region, NRPD2A was found methylated in the gene body, reinforcing that methylation can be self-regulated since NRPD2A is involved in the RdDM pathway. In the same way, SUVH5, a methyltransferase that mediates non-CG methylation, was also found methylated in the gene body.
In the downstream region, the genes AT5G64030-like, AT3G11760-like, CDT1A, HAG1, VPS9A, and FDM1 were found de-methylated, and GSL12, EDM2, MCM6, NRPD2B, and ETG1 were methylated in the diploid genotype. Interestingly, CDT1A promotes polyploidization and is involved in the coordination of cell division (Brasil et al. 2017). Another gene related to ploidy and cell division is MCM6, which is essential to undergo a single round of replications, and it is also involved in the initiation of cell elongation at the beginning of meiosis (Vinay et al. 2023). The gene ETG1 is associated with the MCM complex and required for sister chromatid cohesion, meaning that methylation probably regulates the MCM complex at different levels in the pathway (Schubert and Shaw 2011).
Discussion
This work investigated the differentially methylated genes associated with reproduction and ploidy changes in recently diploidized and tetraploidized genotypes of E. curvula with a similar genetic background (Cardone et al. 2006). To do this, we used the MCSeEd technique to analyze differentially methylated positions, regions, and genes in plants of the related genotypes Tanganyika INTA (4 × apo), Bahiense (4 × apo), and Victoria (2 × sex) and the unrelated genotype TUNS9355 (6 × apo).
Differentially methylated and de-methylated positions were analyzed in all the CG, CHG, and CHH contexts. The genotypes were compared all against each other: vicVSbah, vicVStan, vicVSt93, bahVStan, bahVSt93, and tanVSt93. The number of DMPs between genotypes across the three DNA contexts was homogeneous for all the comparisons (Fig. 2A, Table S3). In previous works, using the same technique to compare tetraploid genotypes of E. curvula with different reproductive modes, it was found that CG and CHG had similar total numbers of DMPs in all the comparisons. At the same time, the total number of DMPs in the CHH context showed an increased number (Carballo et al. 2021a, b). In drought-stressed and control plants of Zea mays, MCSeEd detected more DMPs in CHG and CHH than in CG in stressed plants (Marconi et al. 2019). Fewer DMPs in CG means that this context is conserved after alterations such as drought stress, reproductive mode, and ploidy level. This is also consistent with what was found in Oryza sativa, comparing diploid and tetraploid genotypes (Zhang et al. 2015). In our study, the number of de-methylated positions was, in general, higher than the number of methylated ones in the six comparisons except for tanVSt93 and bahVStan in which a higher number of methylated positions were observed in CHG and CHH (tanVSt93) and CG (bahVStan). Thus, in most of the cases, higher ploidy levels correlate directly with methylation levels. This pattern in which overall methylation levels are higher in polyploids was also found in other species, like in the model species Arabidopsis thaliana when compared with its tetraploid counterpart A. arenosa (Jiang et al. 2021). In the same way, the number of methylated sites in the autotetraploid O. sativa was higher than in diploids (Zhang et al. 2015; Rao et al. 2023).
The comparisons between the related genotypes vicVSbah and vicVStan showed a lower number of DMRs than the other comparisons, suggesting that even though they have differences in terms of ploidy and reproduction, the overall methylation landscape is maintained. Although most of the DMRs are within intergenic regions, the MCSeEd method properly reduces the genome's complexity since the number of intergenic/repetitive regions does not overwhelm the analysis and produces good resolution over the regulatory regions (Fig. 3). Using the same technique, a similar distribution in CG, CHG, and CHH contexts was also observed in Z. mays and E. curvula (Marconi et al. 2019; Carballo et al. 2021a).
The DMGs were divided into upstream, gene body, and downstream regions because the impact of methylation varies depending on which gene position is affected (Bewick and Schmitz 2017; He et al. 2020, 2022). In the upstream region, methylation changes were mainly located in the CG context, in the gene body were found in CHG, and in the downstream region. However, the distribution was homogeneous in the three contexts, more differences were observed in CG (Fig. 4). The distribution of the DMRs across the genes was similar to the one previously found in E. curvula (Carballo et al. 2021a). The area surrounding the start and stop codons showed more DMRs in the CG context (Fig. 5), probably, repressing or promoting gene expression if it is methylated or de-methylated, respectively (Wang et al. 2020). In CHG, a peak of DMRs was found around the stop codon, while in CHH, a depression was observed around the start codon, followed by an increase of methylation in the gene body. Contrary to what happens upstream and downstream, gene body methylation increases gene expression (Muyle et al. 2022a; Zilberman et al. 2007).
To identify methylated/de-methylated genes involved in ploidy changes and reproduction, we focused on the DMGs shared in different comparisons. To identify genes related to reproduction, the sexual genotype was compared against the apomictic ones (vicVSbah, vicVStan, and vicVSt93). To find genes involved in ploidy changes, we took advantage of the synthetic diploidized (Victoria) and tetraploidized (Bahiense) genotypes derived from the natural tetraploid Tanganyika INTA.
In the sexual versus apomictic comparisons, many genes related to the ubiquitin pathway were found differentially methylated in the upstream, gene body, and downstream regions. The regulation of apomixis through this pathway was also mentioned in different species (Galla et al. 2017; Rodrigo et al. 2017; Selva et al. 2020; Carballo et al. 2021a). In this study, the genes related to the ubiquitin pathway were mainly de-methylated in all the regions in the sexual genotype (Table S6). Key meiotic genes such as RECQL2, RECQ4A, and SPO11-1 were also found to be differentially methylated through these comparisons. For example, the RECQ4A gene, a DNA helicase that limits meiotic recombination and its mutation, increases the number of crossovers. RECQL2 increases genomic stability, preventing non-productive recombination (Kobbe et al. 2008; Röhrig et al. 2018; Serra et al. 2018). SPO11-1 mediates the initiation of DNA double-strand breaks and was used successfully together with REC8 and OSD1 to transform mitosis into meiosis (d'Erfurth et al. 2009; Mieulet et al. 2016; Xue et al. 2018). Since RECQL2 and RECQ4A were found de-methylated in the upstream region and SPO11-1 methylated in the gene body, it seems that these genes are being expressed in the sexual genotype and repressed in the apomictic ones, meaning that the meiotic pathway is being negatively regulated at different levels in apomictic genotypes. The most plausible hypothesis is that the repression of SPO11-1 induces the decrease of double-stranded breaks, whereas the double-strand break repair machinery accomplished by RECQL2 and RECQ4A is disturbed. Regulation of DNA crossovers during meiosis through this pathway was also suggested previously, showing a negative correlation between methylation and recombination in A. thaliana (Fernandes et al. 2018). Genes associated with cell division were also found differentially methylated in the downstream region in these comparisons, like MCM6 and CDT1A which are part of the pre-replication complex responsible for the initiation of replication and prevent extra rounds of DNA replication (Brasil et al. 2017). MCM6 was associated with gynoecium sex expression in different species and was involved in sexual differentiation (Vinay et al. 2023). Recently, it was found that in A. thaliana, CDT1A is expressed in pollen sperm cells before fertilization (Voichek et al. 2023; He et al. 2023). Interestingly, mutation of CDT1A produces genotypes that, after two rounds of mitosis, produce abnormal embryo sacs with only four nuclei, which is exactly what happens in the normal E. curvula apomictic embryo sac development (Crane 2001; Domenichini et al. 2012). The CHX19 gene is methylated in the downstream region and expressed in pollen grains and tubes (Padmanaban et al. 2017). Mutation of this gene, together with CHX17 and CHX18, produces male and female abnormalities. One of these abnormalities is a single event of egg or central cell fertilization, but not both (Padmanaban et al. 2017). This phenomenon could be related to the fact that in E. curvula apomictic genotypes, only the central cell is fertilized, while the egg cell develops a parthenogenetic embryo without fertilization (Carballo et al. 2021a, b).
Another mechanism that could increase methylation in polyploid genotypes is the dosage compensation. Apomictic species are vastly polyploids and contain a dominant non-recombinant hemizygous region/s associated with their components. For instance, Pennisetum, Taraxacum, and Paspalum it was described as a single region linked to parthenogenesis (Ozias-Akins et al. 1998; Hojsgaard et al. 2011; Van Dijk et al. 2020; Underwood et al. 2022). In E. curvula, a single region was also linked to apomeiosis (Zappacosta et al. 2019). Many of the characteristics associated with these regions, such as large rearrangements, accumulation of repetitive elements, and suppression of recombination, agree with the early stage of sex chromosomes (Charlesworth 2015). As in other species, the increase in methylation could be related to the dosage compensation effect, in which gene dosage does not correlate with the gene expression level (Muyle et al. 2022b). Another effect closely related to the dosage compensation that could be associated to the increase/decrease of methylation is the parent-of-origin effect. Apomictic embryos originate exclusively by the mitotic replication of the maternal genome (Crane 2001), while sexual embryos result from the fusion of the male (sperm cell) and female (egg cell) gamete. This imbalance of genome dosage was also found to be regulated by methylation, suggesting that the changes observed in E. curvula could be related to both parent-of-origin and dosage compensation effects (Adams et al. 2000; Duszynska et al. 2013).
Ploidy changes were assessed through the comparisons vicVStan and vicVSbah. Here, it was demonstrated that many genes that changed their status during diploidization recovered the original state after tetraploidization, meaning that there is a typical methylation landscape related to each ploidy level, as was stated previously in this species (Ochogavía et al. 2009). As in the methylation assessment with contrasting reproductive modes, here, the genes RECQ4A, SPO11-1, and MCM6 were also identified as differentially methylated. In this comparison, genes specifically associated with ploidy were also found. In particular, the mutation of PC-MYB1 induces tetraploidization in A. thaliana (Haga et al. 2011). Here, we found this gene de-methylated in the gene body in the diploid and methylated in the tetraploids, probably maintaining the polyploid level in tetraploids and avoiding the tetraploidization of diploids. CDT1A is also another gene that could be related to ploidy changes. This gene was found de-methylated in the downstream region in diploids. Overexpression of this gene in A. thaliana promotes endoreduplication, which is associated with increased ploidy (Raynaud et al. 2005). Since the effect of methylation in the downstream regions is not completely understood, and either repression or promotion of this gene could lead to different hypotheses, it is not clear how this gene could regulate ploidy levels/changes (He et al. 2022). The following genes involved in cell division that might also have a function associated with increasing, decrease or maintenance of ploidy levels were identified in these comparisons: PEL1, GSL04, AT5G50340-like, E2F1, SEC15B, OSB, GSL11, FK, AT3G11760-like, VPS9A, ETG1, and AT3G11760-like. As in other studies, genes related to methylation, such as FDM1, SUVH5, NRPD2A, and EDM2, were also found differentially methylated here, suggesting that this pathway could be auto-regulated (Lei et al. 2015; Williams et al. 2015; Zhang et al. 2018; Carballo et al. 2021a).
In conclusion, here we present a comprehensive view of the methylation changes that occur across genotypes with different reproductive modes and ploidy levels and its possible effects on these characteristics. Some genetic changes introduced by the diploidization and tetraploidization processes were probably involved in the changes of the reproductive mode. For instance, the loss of a genomic portion in Victoria could have introduced rearrangements altering the reproductive pathway. However, the epigenetic landscape necessary to maintain the apomictic behavior in both Tanganyika INTA and Bahiense should also be reflected in the MCSeEd analysis in the vicVStan and vicVSbah comparisons. Even more, it is hypothesized that the genes involved in the sexual pathway are repressed by epigenetic mechanisms in apomictic genotypes (Albertini et al. 2019). Some of the genes found here, like the ones involved in meiosis, agree with this theory. The comparisons between sexual and apomictic genotypes showed a general increase of methylation in apomictic versus sexual genotypes. Also, it was possible to observe methylation changes affecting genes involved in the three components of apomixis: apomeiosis, parthenogenesis, and pseudogamy. Regarding ploidy, a general increase in methylation was associated with increases in ploidy, and genes related to changes and maintenance of ploidy were also found to be differentially methylated. Even more, many genes related to embryo sac development and ploidy found differentially methylated here, in concord with previous works. In contrast, others were not reported, opening new candidate genes and pathways potentially involved in these traits.
Data availability
The datasets presented in this study can be found in online repositories: https://www.ncbi.nlm.nih.gov/, BioProject: PRJNA988943.
References
Adams S, Vinkenoog R, Spielman M, Dickinson HG, Scott RJ (2000) Parent-of-origin effects on seed development in Arabidopsis thaliana require DNA methylation. Development 127(11):2493–2502. https://doi.org/10.1242/dev.127.11.2493
Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE (2012) methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13:1–9. https://doi.org/10.1186/gb-2012-13-10-r87
Albertini E, Barcaccia G, Carman JG, Pupilli F (2019) Did apomixis evolve from sex or was it the other way around? J Exp Bot 70(11):2951–2964. https://doi.org/10.1093/jxb/erz109
Armstrong L (2013) Epigenetics. Garland science, New York
Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Ann Rev Biochem 71:333–374. https://doi.org/10.1146/annurev.biochem.71.110601.135425
Bewick AJ, Schmitz RJ (2017) Gene body DNA methylation in plants. Curr Opin Plant Biol 36:103–110. https://doi.org/10.1016/j.pbi.2016.12.007
Brasil JN, Costa CN, Cabral LM, Ferreira PC, Hemerly AS (2017) The plant cell cycle: pre-replication complex formation and controls. Genet Mol Biol 40:276–291. https://doi.org/10.1590/1678-4685-GMB-2016-0118
Canton M, Forestan C, Marconi G, Carrera E, Bonghi C, Varotto S (2022) Evidence of chromatin and transcriptional dynamics for cold development in peach flower bud. New Phytol 236:974–988. https://doi.org/10.1111/nph.18393
Caperta AD, Fernandes I, Conceição SI, Marques I, Róis AS, Paulo OS (2023) Ovule transcriptome analysis discloses deregulation of genes and pathways in sexual and apomictic Limonium species (Plumbaginaceae). Genes 14:901. https://doi.org/10.3390/genes14040901
Carballo J, Santos BA, Zappacosta D, Garbus I, Selva JP, Gallo CA, Díaz A, Albertini E, Caccamo M, Echenique V (2019) A high-quality genome of Eragrostis curvula grass provides insights into Poaceae evolution and supports new strategies to enhance forage quality. Sci Rep UK 9:1–5. https://doi.org/10.1038/s41598-019-46610-0
Carballo J, Zappacosta D, Marconi G, Gallardo J, Di Marsico M, Gallo CA, Caccamo M, Albertini E, Echenique V (2021a) Differential methylation patterns in apomictic vs. sexual genotypes of the diplosporous grass Eragrostis curvula. Plants 10:946. https://doi.org/10.3390/plants10050946
Carballo J, Zappacosta D, Selva JP, Caccamo M, Echenique V (2021b) Eragrostis curvula, a model species for diplosporous apomixis. Plants 10:1818. https://doi.org/10.3390/plants10091818
Carballo J, Bellido AM, Selva JP, Zappacosta D, Gallo CA, Albertini E, Caccamo M, Echenique V (2023) From tetraploid to diploid, a pangenomic approach to identify genes lost during synthetic diploidization of Eragrostis curvula. Front Plant Sci 14:1133986. https://doi.org/10.3389/fpls.2023.1133986
Cardone S, Polci P, Selva JP, Mecchia M, Pessino S, Hermann P, Cambi V, Voigt P, Spangenberg G, Echenique V (2006) Novel genotypes of the subtropical grass Eragrostis curvula for the study of apomixis (diplospory). Euphytica 151:263–272. https://doi.org/10.1007/s10681-006-9156-x
Castellano MD, Boniotti MB, Caro E, Schnittger A, Gutierrez C (2004) DNA replication licensing affects cell proliferation or endoreplication in a cell type–specific manner. Plant Cell 16:2380–2393. https://doi.org/10.1105/tpc.104.022400
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140. https://doi.org/10.1111/mec.12354
Charlesworth D (2015) Plant contributions to our understanding of sex chromosome evolution. New Phytol 208(1):52–65. https://doi.org/10.1111/nph.13497
Chen T, Li E (2004) Structure and function of eukaryotic DNA methyltransferases. Curr Top Dev Biol 60:55–89. https://doi.org/10.1016/S0070-2153(04)60003-2/
Crane CF (2001) Classification of apomictic mechanisms. In: Savidan Y, Carman JG, Dresselhaus T (eds) The flowering of apomixis: from mechanisms to genetic engineering. CIMMYT IRD, European Commission DG VI (FAIR), Mexico, pp 24–43
d’Erfurth I, Jolivet S, Froger N, Catrice O, Novatchkova M, Mercier R (2009) Turning meiosis into mitosis. PLoS Biol 7:e1000124. https://doi.org/10.1371/journal.pbio.1000124
Di Marsico M, Cerruti E, Comino C, Porceddu A, Acquadro A, Capomaccio S, Marconi G, Albertini E (2020) MCSeEd (Methylation Context Sensitive Enzyme ddRAD): a new method to analyze DNA methylation. In: Spillane C, McKeown P (eds) Plant epigenetics and epigenomics: methods and protocols. Springer, New York, pp 47–64
Domenichini S, Benhamed M, De Jaeger G, Van De Slijke E, Blanchet S, Bourge M, De Veylder L, Bergounioux C, Raynaud C (2012) Evidence for a role of Arabidopsis CDT1 proteins in gametophyte development and maintenance of genome integrity. Plant Cell 24:2779–2791. https://doi.org/10.1105/tpc.112.100156
Duszynska D, McKeown PC, Juenger TE, Pietraszewska-Bogiel A, Geelen D, Spillane C (2013) Gamete fertility and ovule number variation in selfed reciprocal F 1 hybrid triploid plants are heritable and display epigenetic parent-of-origin effects. New Phytol 198(1):71–81. https://doi.org/10.1111/nph.12147
Fernandes JB, Séguéla-Arnaud M, Larchevêque C, Lloyd AH, Mercier R (2018) Unleashing meiotic crossovers in hybrid plants. Proc Natl Acad Sci USA 115:2431–2436. https://doi.org/10.1073/pnas.171307811
Galla G, Zenoni S, Avesani L, Altschmied L, Rizzo P, Sharbel TF, Barcaccia G (2017) Pistil transcriptome analysis to disclose genes and gene products related to aposporous apomixis in Hypericum perforatum L. Front Plant Sci 8:79. https://doi.org/10.3389/fpls.2017.00079
Garbus I, Romero JR, Selva JP, Pasten MC, Chinestra C, Carballo J, Zappacosta DC, Echenique V (2017) De novo transcriptome sequencing and assembly from apomictic and sexual Eragrostis curvula genotypes. PLoS ONE 12:e0185595. https://doi.org/10.1371/journal.pone.0185595
Garbus I, Selva JP, Pasten MC, Bellido AM, Carballo J, Albertini E, Echenique V (2019) Characterization and discovery of miRNA and miRNA targets from apomictic and sexual genotypes of Eragrostis curvula. BMC Genomics 20:839. https://doi.org/10.1186/s12864-019-6169-0
Grelon M, Vezon D, Gendrot G, Pelletier G (2001) AtSPO11-1 is necessary for efficient meiotic recombination in plants. EMBO J 20:589–600. https://doi.org/10.1093/emboj/20.3.589
Haga N, Kobayashi K, Suzuki T, Maeo K, Kubo M, Ohtani M, Mitsuda N, Demura T, Nakamura K, Jürgens G, Ito M (2011) Mutations in MYB3R1 and MYB3R4 cause pleiotropic developmental defects and preferential down-regulation of multiple G2/M-specific genes in Arabidopsis. Plant Physiol 157:706–717. https://doi.org/10.1104/pp.111.180836
Han Q, Bartels A, Cheng X, Meyer A, An YQ, Hsieh TF, Xiao W (2019) Epigenetics regulates reproductive development in plants. Plants 8:564. https://doi.org/10.3390/plants8120564
He C, Zhang HY, Zhang YX, Fu P, You LL, Xiao WB, Wang ZH, Song HY, Huang YJ, Liao JL (2020) Cytosine methylations in the promoter regions of genes involved in the cellular oxidation equilibrium pathways affect rice heat tolerance. BMC Genomics 21:1–6. https://doi.org/10.1186/s12864-020-06975-3
He L, Huang H, Bradai M, Zhao C, You Y, Ma J, Zhao L, Lozano-Durán R, Zhu JK (2022) DNA methylation-free Arabidopsis reveals crucial roles of DNA methylation in regulating gene expression and development. Nat Commun 13:1335. https://doi.org/10.1038/s41467-022-28940-2
He L, Fan Y, Zhang Z, Wei X, Yu J (2023) Identifying genes associated with female flower development of Phellodendron amurense Rupr. using a transcriptomics approach. Genes 14:661. https://doi.org/10.3390/genes14030661
Hojsgaard DH, Martínez EJ, Acuña CA, Quarin CL, Pupilli F (2011) A molecular map of the apomixis-control locus in Paspalum procurrens and its comparative analysis with other species of Paspalum. Theor Appl Genet 123:959–971. https://doi.org/10.1007/s00122-011-1639-z
Jiang X, Song Q, Ye W, Chen ZJ (2021) Concerted genomic and epigenomic changes accompany stabilization of Arabidopsis allopolyploids. Nat Ecol Evol 5:1382–1393. https://doi.org/10.1038/s41559-021-01523-y
Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE (2011) Ancestral polyploidy in seed plants and angiosperms. Nature 473(7345):97–100. https://doi.org/10.5061/dryad.8546
Kanno T, Bucher E, Daxinger L, Huettel B, Böhmdorfer G, Gregor W, Kreil DP, Matzke M, Matzke AJ (2008) A structural-maintenance-of-chromosomes hinge domain–containing protein is required for RNA-directed DNA methylation. Nat Genet 40:670–675. https://doi.org/10.1038/ng.119
Kobbe D, Blanck S, Demand K, Focke M, Puchta H (2008) AtRECQ2, a RecQ helicase homologue from Arabidopsis thaliana, is able to disrupt various recombinogenic DNA structures in vitro. Plant J 55:397–405. https://doi.org/10.1111/j.1365-313X.2008.03511.x
Lei M, Zhang H, Julian R, Tang K, Xie S, Zhu JK (2015) Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc Natl Acad Sci USA 112:3553–3557. https://doi.org/10.1073/pnas.1502279112
Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133:523–536. https://doi.org/10.1016/j.cell.2008.03.029
Marconi G, Capomaccio S, Comino C, Acquadro A, Portis E, Porceddu A, Albertini E (2019) Methylation content sensitive enzyme ddRAD (MCSeEd): a reference-free, whole genome profiling system to address cytosine/adenine methylation changes. Sci Rep UK 9:14864. https://doi.org/10.1038/s41598-019-51423-2
Melamed-Bessudo C, Levy AA (2012) Deficiency in DNA methylation increases meiotic crossover rates in euchromatic but not in heterochromatic regions in Arabidopsis. Proc Natl Acad Sci USA 109:E981-988. https://doi.org/10.1073/pnas.11207421
Mieulet D, Jolivet S, Rivard M, Cromer L, Vernet A, Mayonove P, Pereira L, Droc G, Courtois B, Guiderdoni E, Mercier R (2016) Turning rice meiosis into mitosis. Cell Res 26:1242–1254. https://doi.org/10.1038/cr.2016.117
Munshi A, Ahuja YR, Bir B (2015) Epigenetic mechanisms in plants: an overview. In: Dev T (ed) Plant biology and biotechnology: volume II: plant genomics and biotechnology. Springer, New Delhi, pp 265–278. https://doi.org/10.1007/978-81-322-2283-5_12
Muyle A, Marais GA, Bačovský V, Hobza R, Lenormand T (2022a) Dosage compensation evolution in plants: theories, controversies and mechanisms. Philos Trans R Soc Lond 377(1850):20210222. https://doi.org/10.1098/rstb.2021.0222
Muyle AM, Seymour DK, Lv Y, Huettel B, Gaut BS (2022b) Gene body methylation in plants: mechanisms, functions, and important implications for understanding evolutionary processes. Genome Biol Evol 14:evac038. https://doi.org/10.1093/gbe/evac038
Ochogavía AC, Cervigni G, Selva JP, Echenique VC, Pessino SC (2009) Variation in cytosine methylation patterns during ploidy level conversions in Eragrostis curvula. Plant Mol Biol 70:17–29
Ozias-Akins P, Roche D, Hanna WW (1998) Tight clustering and hemizygosity of apomixis-linked molecular markers in Pennisetum squamulatum implies genetic control of apospory by a divergent locus that may have no allelic form in sexual genotypes. Proc Natl Acad Sci 95:5127–5132. https://doi.org/10.1073/pnas.95.9.5127
Padmanaban S, Czerny DD, Levin KA, Leydon AR, Su RT, Maugel TK, Zou Y, Chanroj S, Cheung AY, Johnson MA, Sze H (2017) Transporters involved in pH and K+ homeostasis affect pollen wall formation, male fertility, and embryo development. J Exp Bot 68:3165–3178. https://doi.org/10.1093/jxb/erw483
Pasten MC, Carballo J, Gallardo J, Zappacosta D, Selva JP, Rodrigo JM, Echenique V, Garbus I (2022) A combined transcriptome-miRNAome approach revealed that a kinesin gene is differentially targeted by a novel miRNA in an apomictic genotype of Eragrostis curvula. Front Plant Sci 13:1012682. https://doi.org/10.3389/fpls.2022.1012682
Rajakumara E, Law JA, Simanshu DK, Voigt P, Johnson LM, Reinberg D, Patel DJ, Jacobsen SE (2011) A dual flip-out mechanism for 5mC recognition by the Arabidopsis SUVH5 SRA domain and its impact on DNA methylation and H3K9 demethylation in vivo. Gene Dev 25:137–152. https://doi.org/10.1101/gad.1980311
Rao X, Ren J, Wang W, Chen R, Xie Q, Xu Y, Li D, Song Z, He Y, Cai D, Yang P (2023) Comparative DNA-methylome and transcriptome analysis reveals heterosis-and polyploidy-associated epigenetic changes in rice. Crop J 11:427–437. https://doi.org/10.1016/j.cj.2022.06.011
Raynaud C, Perennes C, Reuzeau C, Catrice O, Brown S, Bergounioux C (2005) Cell and plastid division are coordinated through the prereplication factor AtCDT1. Proc Natl Acad Sci USA 102:8216–8221. https://doi.org/10.1073/pnas.0502564102
Rodrigo JM, Zappacosta DC, Selva JP, Garbus I, Albertini E, Echenique V (2017) Apomixis frequency under stress conditions in weeping lovegrass (Eragrostis curvula). PLoS ONE 12:e0175852. https://doi.org/10.1371/journal.pone.0175852
Röhrig S, Dorn A, Enderle J, Schindele A, Herrmann NJ, Knoll A, Puchta H (2018) The RecQ-like helicase HRQ1 is involved in DNA crosslink repair in Arabidopsis in a common pathway with the Fanconi anemia-associated nuclease FAN1 and the postreplicative repair ATPase RAD5A. New Phytol 218:1478–1490. https://doi.org/10.1111/nph.15109
Russo VE, Martienssen RA, Riggs AD (1996) Epigenetic mechanisms of gene regulation. Cold Spring Harbor Laboratory Press
Schubert I, Shaw P (2011) Organization and dynamics of plant interphase chromosomes. Trends Plant Sci 16:273–281. https://doi.org/10.1016/j.tplants.2011.02.002
Selva JP, Zappacosta D, Carballo J, Rodrigo JM, Bellido A, Gallo CA, Gallardo J, Echenique V (2020) Genes modulating the increase in sexuality in the facultative diplosporous grass Eragrostis curvula under water stress conditions. Genes 11:969. https://doi.org/10.3390/genes11090969
Serra H, Lambing C, Griffin CH, Topp SD, Nageswaran DC, Underwood CJ, Ziolkowski PA, Séguéla-Arnaud M, Fernandes JB, Mercier R, Henderson IR (2018) Massive crossover elevation via combination of HEI10 and recq4a recq4b during Arabidopsis meiosis. Proc Natl Acad Sci USA 115:2437–2442. https://doi.org/10.1073/pnas.1713071115
Soliman M, Podio M, Marconi G, Di Marsico M, Ortiz JP, Albertini E, Delgado L (2021) Differential epigenetic marks are associated with apospory expressivity in diploid hybrids of Paspalum rufum. Plants 10:793. https://doi.org/10.3390/plants10040793
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl Acids Res 47:D607–D613. https://doi.org/10.1093/nar/gky1131
Tini F, Beccari G, Marconi G, Porceddu A, Sulyok M, Gardiner DM, Albertini E, Covarelli L (2021) Identification of putative virulence genes by DNA methylation studies in the cereal pathogen Fusarium graminearum. Cells 10:1192. https://doi.org/10.3390/cells10051192
Tzafrir I, Pena-Muralla R, Dickerman A, Berg M, Rogers R, Hutchens S, Sweeney TC, McElver J, Aux G, Patton D, Meinke D (2004) Identification of genes required for embryo development in Arabidopsis. Plant Physiol 135:1206–1220. https://doi.org/10.1104/pp.104.045179
Underwood CJ, Choi K, Lambing C, Zhao X, Serra H, Borges F, Simorowski J, Ernst E, Jacob Y, Henderson IR, Martienssen RA (2018) Epigenetic activation of meiotic recombination near Arabidopsis thaliana centromeres via loss of H3K9me2 and non-CG DNA methylation. Genome Res 28:519–531. https://doi.org/10.1101/gr.227116.117
Underwood CJ, Vijverberg K, Rigola D, Okamoto S, Oplaat C, Camp RH, Radoeva T, Schauer SE, Fierens J, Jansen K, Mansveld S (2022) A PARTHENOGENESIS allele from apomictic dandelion can induce egg cell division without fertilization in lettuce. Nat Genet 54(1):84–93. https://doi.org/10.1038/s41588-021-00984-y
Valansi C, Moi D, Leikina E, Matveev E, Graña M, Chernomordik LV, Romero H, Aguilar PS, Podbilewicz B (2017) Arabidopsis HAP2/GCS1 is a gamete fusion protein homologous to somatic and viral fusogens. J Cell Biol 216:571–581. https://doi.org/10.1083/jcb.201610093
Van Dijk PJ, Op den Camp R, Schauer SE (2020) Genetic dissection of apomixis in dandelions identifies a dominant parthenogenesis locus and highlights the complexity of autonomous endosperm formation. Genes 11(9):961. https://doi.org/10.3390/genes11090961
Vinay ND, Matsumura H, Munshi AD, Ellur RK, Chinnusamy V, Singh A, Iquebal MA, Jaiswal S, Jat GS, Panigrahi I, Gaikwad AB (2023) Molecular mapping of genomic regions and identification of possible candidate genes associated with gynoecious sex expression in bitter gourd. Front Plant Sci 14:1071648. https://doi.org/10.3389/fpls.2023.1071648
Voichek Y, Hurieva B, Michaud C, Schmücker A, Vergara Z, Desvoyes B, Gutierrez C, Nizhynska V, Jaegle B, Borg M, Berger F (2023) Cell-cycle status of male and female gametes during Arabidopsis reproduction. bioRxiv. 2023–02. https://doi.org/10.1101/2023.02.22.529524
Vrielynck N, Chambon A, Vezon D, Pereira L, Chelysheva L, De Muyt A, Mézard C, Mayer C, Grelon M (2016) A DNA topoisomerase VI–like complex initiates meiotic recombination. Science 351:939–943. https://doi.org/10.1126/science.aad5196
Wang G, Li H, Meng S, Yang J, Ye N, Zhang J (2020) Analysis of global methylome and gene expression during carbon reserve mobilization in stems under soil drying. Plant Physiol 183:1809–1824. https://doi.org/10.1104/pp.20.00141
Williams BP, Pignatta D, Henikoff S, Gehring M (2015) Methylation-sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLoS Genet 11:e1005142. https://doi.org/10.1371/journal.pgen.1005142
Xue M, Wang J, Jiang L, Wang M, Wolfe S, Pawlowski WP, Wang Y, He Y (2018) The number of meiotic double-strand breaks influences crossover distribution in Arabidopsis. Plant Cell 30:2628–2638. https://doi.org/10.1105/tpc.18.00531
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287. https://doi.org/10.1089/omi.2011.0118
Zappacosta D, Gallardo J, Carballo J, Meier M, Rodrigo JM, Gallo CA, Selva JP, Stein J, Ortiz JP, Albertini E, Echenique V (2019) A high-density linkage map of the forage grass Eragrostis curvula and localization of the diplospory locus. Front Plant Sci 10:918. https://doi.org/10.3389/fpls.2019.00918
Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, Ecker JR (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126:1189–1201. https://doi.org/10.1016/j.cell.2006.08.003
Zhang J, Liu Y, Xia EH, Yao QY, Liu XD, Gao LZ (2015) Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc Natl Acad Sci USA 112:E7022-7029. https://doi.org/10.1073/pnas.1515170112
Zhang H, Lang Z, Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Bio 19:489–506. https://doi.org/10.1038/s41580-018-0016-z
Zhang Y, Shi C, Fu W, Gu X, Qi Z, Xu W, Xia G (2021) Arabidopsis MED18 interaction with RNA pol IV and V subunit nrpd2a in transcriptional regulation of plant immune responses. Front Plant Sci 12:692036. https://doi.org/10.3389/fpls.2021.692036
Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39(1):61–69. https://doi.org/10.1038/ng1929
Acknowledgements
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie Grant Agreement No 101007438. This project has received funding from the European Union’s Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie Grant Agreement No 872417. This project has received funding from CONICET (PIP 1220200101905CO) and Universidad Nacional del Sur (PGI UNS 24/A261).
Funding
Open access funding provided by Università degli Studi di Perugia within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
EV, AE, ZD, and CJ contributed to conceptualization; CJ, AA, HF, MG, AE, BM, and ZD provided methodology; CJ, AA, HF, PMC, AE, ZD, and EV performed formal analysis and investigation; EV, CJ, ZD, and AA performed writing—original draft preparation; EV, AE, CJ, ZD, and AA performed writing—review and editing; EV and AE performed funding acquisition; EV and AE provided resources; EV and AE performed supervision.
Corresponding authors
Additional information
Communicated by Stewart Gillmor.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carballo, J., Achilli, A., Hernández, F. et al. Differentially methylated genes involved in reproduction and ploidy levels in recent diploidized and tetraploidized Eragrostis curvula genotypes. Plant Reprod 37, 133–145 (2024). https://doi.org/10.1007/s00497-023-00490-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00497-023-00490-7