
Abstract
Purpose
Formation of terminal complement complex (TCC), a downstream complement system activation product inducing inflammatory processes and cell lysis, has been identified in degenerated discs. However, it remains unclear which molecular factors regulate complement activation during disc degeneration (DD). This study investigated a possible involvement of the pro-inflammatory cytokine interleukin-1β (IL-1β) and the lysosomal protease cathepsin D (CTSD).
Methods
Disc biopsies were collected from patients suffering from DD (n = 43) and adolescent idiopathic scoliosis (AIS, n = 13). Standardized tissue punches and isolated cells from nucleus pulposus (NP), annulus fibrosus (AF) and endplate (EP) were stimulated with 5% human serum (HS) alone or in combination with IL-1β, CTSD or zymosan. TCC formation and modulation by the complement regulatory proteins CD46, CD55 and CD59 were analysed.
Results
In DD tissue cultures, IL-1β stimulation decreased the percentage of TCC + cells in AF and EP (P < 0.05), whereas CTSD stimulation significantly increased TCC deposition in NP (P < 0.01) and zymosan in EP (P < 0.05). Overall, the expression of CD46, CD55 and CD59 significantly increased in all isolated cells during culture (P < 0.05). Moreover, cellular TCC deposition was HS concentration dependent but unaffected by IL-1β, CTSD or zymosan.
Conclusion
These results suggest a functional relevance of IL-1β and CTSD in modulating TCC formation in DD, with differences between tissue regions. Although strong TCC deposition may represent a degeneration-associated event, IL-1β may inhibit it. In contrast, TCC formation was shown to be triggered by CTSD, indicating a multifunctional involvement in disc pathophysiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intervertebral disc (IVD) degeneration is perceived as the major cause of back pain [1]. The integrity of the IVD is dependent on the balance between anabolic and catabolic events [2, 3]. The IVD is defined as an immune-privileged tissue, particularly the nucleus pulposus (NP), but once the physiological barrier is disrupted, it can trigger an autoimmune response against specific proteins. Immunoglobulins against aggrecan, and collagen types I, II and V have been identified in degenerated IVDs [4]; however, this has undergone limited investigation.
IVD degeneration is characterized by a catabolic cell phenotype, with matrix degeneration, pro-inflammatory response, senescence and apoptosis [5, 6]. Matrix degeneration is mainly driven by specific enzymes including metalloproteinases and cathepsins, such as cathepsin D (CTSD) [7]. Moreover, the inflammatory response comprises an increased production of soluble mediators, such as interleukin (IL)-1β, and increased recruitment of immune cells [8,9,10]. The complement system is a crucial arm of fluid-phase innate immunity and our body’s first line of defence. It modulates and amplifies inflammation and can also significantly contribute to inflammation-mediated tissue damage [11]. Deposition of the terminal complement complex (TCC), the final complement activation product, can act as an inflammatory trigger and inducer of cell lysis [12]. In regard to IVD pathophysiology, TCC has been identified to exhibit a distinct pattern of tissue deposition [13, 14]. Recently, a positive correlation between TCC formation and the degree of IVD degeneration in human tissue has been reported, indicating an impact of the TCC in IVD degeneration [14]. Yet, pathogenetic processes of complement involvement in the development and progression of IVD degeneration remain to be elucidated. Furthermore, little is known about complement system regulation by IVD cells. However, we have recently observed that CD59 (protectin), a membrane-bound protein known to control TCC formation [15], was likewise increased in degenerated human discs [14].
We therefore hypothesize that TCC deposition plays a role in the pathomechanism of disc degeneration and can be modulated by pathogenetically relevant factors, as well as cellular complement regulators (CRegs) thereof. To address this hypothesis, TCC deposition and CRegs expression on disc cells were investigated in vitro in degenerated IVD tissue and expanded cell cultures of annulus fibrosus (AF), nucleus pulposus (NP) and endplate (EP).
Methods
Sample collection
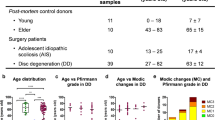
IVD tissue biopsies were collected from patients submitted to lumbar spine surgery as previously described [14]. A total of 13 patients were diagnosed with adolescent idiopathic scoliosis (AIS donors, 10 females/3 males, average age: 17 ± 5 years, age range: 13–34 years) and 43 with disc degeneration (DD donors, 26 females/17 males, average age: 57 ± 14 years, age range: 20–82 years). Detailed patient information is provided in Supplemental Table S1.
Sample processing and tissue cultures
IVD tissue samples were dissected and macroscopically separated into AF, NP and EP fragments according to their morphology. From the DD group (n = 6 donors), tissue punches with about 6 mm diameter and 1 mm height were collected and cultured for 24 h in serum-free IVD medium: DMEM with low glucose supplemented with 1% penicillin–streptomycin (10,000 U/mL–10,000 μg/mL, 15,140,122, Gibco), 0.5% amphotericin B (250 μg/mL, A2812, Biochrom), 1% non-essential amino acids (Biochrom) and 1.5% 5 M NaCl/0.4 M KCl solution, at 37 °C, under reduced oxygen supply (6% O2 and 8.5% CO2) and saturated humidity. Randomly selected fragments from the three regions were enzymatically digested for 3–5 h in 0.8 mg/mL collagenase type II (Sigma-Aldrich) in IVD medium, under agitation, reduced oxygen supply (6% O2 and 8.5% CO2), 37 °C and saturated humidity. The suspension was filtered through a 100-µm filter (BD Falcon) to remove tissue debris. Cells were collected by centrifugation at 500 g for 10 min. AF, NP and EP cells were either used directly for flow cytometry analysis or seeded at a density of 3000 cells/cm2 in IVD medium with 5% heat-inactivated foetal bovine serum (FBS, Gibco). Cells were detached by exposure to 0.050% trypsin–EDTA (Gibco) for 5 min at 37 °C when reaching 70–80% confluency and expanded up to passages 2–4.
IVD tissue and cell cultures stimulation
After 24 h of culture under serum-free condition in 24-well plates with 1 mL of medium per well, the tissue punches were stimulated with different culture media (1 mL per well). Serum-free IVD medium was supplemented with: (i) 5% human serum (HS, Quidel) alone or also with (ii) 10 ng/mL IL-1β (HS + IL-1β, R&D Systems), (iii) 0.5 µg/mL CTSD (HS + CTSD, R&D Systems) or (iv) 100 µg/mL zymosan (HS + zymosan, Sigma-Aldrich). Tissues kept in serum-free IVD medium were used as control (SF group). After 6 h of stimulation, the tissue samples were fixed in 4% formalin and processed for immunohistochemistry.
Cells (5000 cells/well in 96-well plates) were cultured in SF for 24 h. Cells were then cultured for 30 min with 5% FBS, 1, 2, 5 or 10% HS and the supplements previously described for tissue cultures. The SF group was kept as control. For each donor, three experimental replicates were analysed. Afterwards, cells were fixed in 4% formalin for 15 min.
TCC and CD59 immunohistochemistry
TCC deposition and CD59 expression were assessed by avidin–biotin complex (Vector Laboratories) immunohistochemistry using NovaRED Peroxidase (HRP) Substrate Kit (Vector laboratories) as previously described [14]. Mouse anti-human C5b-9/TCC (1:250, Quidel) or mouse anti-human CD59 (1:125, Bio-Rad) was used as primary antibodies, followed by incubation with goat anti-mouse IgG (H + L), biotin-XX (1:200, Invitrogen) antibody. Isotype controls were stained with mouse IgG antibody (BioLegend). Sections were imaged with light microscopy (Zeiss). Positive cells for TCC formation (TCC +) and CD59 expression (CD59 +) were counted within a defined region and normalized to the total number of cells within the same region.
Soluble TCC ELISA
Levels of non-lytic S-protein-bound TCC, also referred to as soluble TCC [16], in tissue and cell culture supernatants were analysed using a human ELISA kit (BD Biosciences), following the manufacturer’s instructions.
Flow cytometry analysis of cellular complement regulators
After cell isolation from AF, NP and EP, red blood cells were lysed with 10 mM Tris/150 mM NH4Cl, pH 7.4, for 8 min at 37 °C. Expanded cells (passages 2–4) were trypsinized and incubated for 1 h in IVD medium. Cell surface staining was performed by incubation with the antibodies from Table 1, for 30 min on ice. Samples were analysed by flow cytometry (FACSCalibur, BD) and data evaluated with FlowJo software version 10 (Ashland).
TCC cell-based ELISA
For quantification of TCC deposition in in vitro cell cultures, a cell-based ELISA was used [17]. Briefly, after a blocking step with 5% bovine serum albumin (Sigma-Aldrich) in DPBS at 37 °C for 1 h, cells were incubated for 2 h at 37 °C with anti-C5b-9/TCC antibody (1:4000 dilution, Abcam). Anti-rabbit IgG peroxidase (1:10,000 dilution, Sigma-Aldrich) was used as secondary antibody. After 1 h of incubation at room temperature with the secondary antibody, cells were incubated with 3, 3′, 5, 5′-tetramethylbenzidine (TMB) substrate (Sigma-Aldrich) for 15 min at room temperature. Before the addition of stop solution (R&D Systems), 50 µL of the TMB solution was transferred to a new plate, to prevent cell lysis by the sulfuric acid. Absorbance values were measured at 450 nm. The cells were washed twice with PBS and imaged with light microscopy (Zeiss). DNA concentration was determined using the Quant-iT PicoGreen double-standard DNA kit (Invitrogen), following the manufacturer’s instructions.
Statistical analysis
Results are presented in box-and-whisker plots as median ± interquartile range, showing all points. Normality was assessed by Shapiro–Wilk normality test, after which statistical analysis was performed with parametric one-way ANOVA or nonparametric Kruskal–Wallis test, followed by Dunn's multiple comparison test, using GraphPad Prism 9 (GraphPad Software, Inc., USA). Statistical significance was considered for P < 0.05.
Results
TCC formation and CD59 expression in disc tissue cultures
Tissue cultures were performed to investigate whether the different stimulation factors influence IVD cells kept in their 3D microenvironment. A significantly higher percentage of TCC + cells was found in the presence of complement-competent HS in AF (2.4-fold, P < 0.01), NP (1.7-fold, P < 0.05) and EP (4.9-fold, P < 0.05) cells compared to SF, with the highest increase observed for EP (Fig. 1a; representative staining images in Supplemental Fig. S1). Addition of IL-1β resulted in lower TCC formation than in the HS group, which was significant for AF and EP (P < 0.05). CTSD stimulation contributed to an increase in TCC deposition (P < 0.05) in NP, whereas zymosan increased TCC in EP (P < 0.05). No soluble TCC was detected in the SF groups (Fig. 1b). In AF cultures, less soluble TCC was detected in supernatants from HS + IL-1β and HS + CTSD groups, in comparison with HS alone (P < 0.05). In NP tissues, higher soluble TCC was generated after zymosan stimulation (P < 0.01). No differences were found in EP. CD59 expression was assessed by immunohistochemistry (Fig. 1c; representative staining images in Supplemental Fig. S2), and it was rather low (percentage of CD59 + cells < 40%) in all conditions tested. Nevertheless, although no differences were found in AF, a significantly higher percentage of CD59 + cells was found after stimulation of NP tissues by HS + CTSD versus HS alone (2.6-fold, P < 0.05). In EP, significantly higher percentage of CD59 + cells was detected in the HS group in contrast to all other groups (P < 0.01).

Analysis of TCC formation on IVD tissue punches from annulus fibrosus (AF), nucleus pulposus (NP) and endplate (EP) tissue punches after 6 h of stimulation with serum-free medium (SF), medium containing 5% human serum (HS) alone or supplemented with IL-1β (10 ng/mL, HS + IL-1β), CTSD (CTSD, 0.5 µg/mL, HS + CTSD) or zymosan (100 µg/mL, HS + zymosan). Tissues were collected from patients diagnosed with disc degeneration. Percentage of TCC + cells in AF, NP and EP tissue punches (a). Soluble TCC (µg/mL) detected in the culture supernatants (b). Percentage of CD59 + cells in AF, NP and EP tissue punches (c). Results are presented as box-and-whisker plots as median ± interquartile range (n = 6 donors per group). *P < 0.05; **P < 0.01, repeated measures one-way ANOVA, with the Greenhouse–Geisser correction
Flow cytometry analysis of cellular complement regulators
AF, NP and EP cells from AIS and DD patients were analysed by flow cytometry for membrane-bound CRegs CD46, CD55 and CD59 (Fig. 2). Cells from AIS and DD patients were analysed at different timepoints: directly after isolation from the disc tissue (FRESH), after the first trypsinization (P0) and after expansion, in passages 2–4 (P2–4). Due to the limited number of cells, it was not possible to track the cells from the same patient along time. Overall, at the same timepoint of analysis, no differences were observed between cells from AIS and DD patients regarding the expression of the different markers. Very low CD46 expression was detected in FRESH cells (CD46 + cells < 15%, Fig. 2a), whereas at P0, higher expression of CD46 was just detected in some donors. In P2–4, a significant increase in the percentage of AF, NP and EP cells expressing CD46 was found, particularly when compared to FRESH groups (P < 0.01). The expression of CD55 (Fig. 2b) and CD59 (Fig. 2c) followed a similar pattern. For all cell types, there was a significant increase in the expression of CD55 and CD59 in P0 (P < 0.05) and P2–4 (P < 0.01), compared to FRESH samples, which ranged between 60 and 100% positivity. Interestingly, some DD donors presented lower percentage of CD59 + cells than AIS, but without overall statistical significance. Gene expression of CD46, CD55 and CD59 by AF, NP and EP cells in passages 2–4 was also analysed (Supplemental Figure S3). No differences were observed between AIS and DD groups, except for CD55 expression by NP cells, for which a higher expression was observed in DD compared to AIS (threefold, P < 0.05). When comparing the expression of each marker by cells from different regions, higher CD46 expression was found in NP cells versus AF cells from AIS patients (threefold, P < 0.01) and in NP and EP cells in comparison with AF cells from DD patients (about twofold to threefold, P < 0.05). Moreover, higher CD55 expression was observed in EP versus AF cells from DD patients (threefold, P < 0.05).

Cell surface detection of membrane-bound complement regulators in annulus fibrosus (AF), nucleus pulposus (NP) and endplate (EP) freshly isolated cells (FRESH), at passage 0 (P0) and after cell expansion (P2–4), from adolescent idiopathic scoliosis (AIS) and disc degeneration (DD) patient groups. Percentage of CD46 + (a), CD55 + (b) and CD59 + (c) cells. Results are presented in box-and-whisker plots as median ± interquartile range (n = 4–10 donors per group). * P < 0.05; ** P < 0.01; *** P < 0.001, Kruskal–Wallis test
TCC formation in IVD cell cultures
Cells in P2–4 were stimulated with different concentrations of HS and compared with standard IVD cell culture medium containing 5% heat-inactivated FBS (Fig. 3). Both cellular TCC deposition (Fig. 3a) and soluble TCC formation (Fig. 3b) increased with increasing complement-competent HS in a concentration-dependent manner (P < 0.05). In cell cultures with 5% heat-inactivated FBS, TCC formation was not detected in the supernatants similar to SF. Again, higher cellular and soluble TCC formation was found in cells stimulated with 5% HS (P < 0.05), but with no differences between AIS and DD groups.

Analysis of TCC formation on isolated annulus fibrosus (AF), nucleus pulposus (NP) and endplate (EP) cells after culture with different concentrations of human serum (HS) for 30 min. Cellular TCC deposition after cell culture with medium supplemented with 1, 2, 5 and 10% HS, respectively, and of cells from adolescent idiopathic scoliosis (AIS) and disc degeneration (DD) patients cultured with 5% heat-inactivated foetal bovine serum (5% FBS) or 5% human serum (5% HS). Data were normalized to serum-free medium (SF) group (a). Soluble TCC (µg/mL) detected in the culture supernatants (b). Results are presented in box-and-whisker plots as median ± interquartile range (n = 4–12 donors per group). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, ordinary one-way ANOVA
Since AF, NP and EP cells isolated from AIS and DD patients behaved similarly after isolation and expansion (as shown in Figs. 2, 3 and supplemental Figure S3), Fig. 4 shows data from both AIS and DD patient-derived cells. In cell culture experiments, stimulation with IL-1β, CTSD or zymosan did not alter TCC deposition in comparison with HS alone (Fig. 4a); however, significantly higher deposition was observed for HS in all cell types (P < 0.01), for HS + CTSD in AF (P < 0.05) and NP (P < 0.001) and for HS + zymosan in AF (P < 0.05) compared to SF. More soluble TCC was detected in supernatants of AF and NP cells stimulated with HS + zymosan versus HS alone (P < 0.01), whereas less TCC was found in supernatants of EP cells stimulated with HS + IL-1β (P < 0.05, Fig. 4b).

Analysis of TCC formation on isolated annulus fibrosus (AF), nucleus pulposus (NP) and endplate (EP) cells after 30 min of stimulation with medium containing 5% human serum (HS) alone or supplemented with IL-1β (10 ng/mL, HS + IL-1β), CTSD (CTSD, 0.5 µg/mL, HS + CTSD) or zymosan (100 µg/mL, HS + zymosan). Serum-free medium cultures (SF) were kept as control. Cellular TCC deposition normalized to SF group (a). Soluble TCC (µg/mL) detected in the culture supernatants (b). Results are presented as box-and-whisker plots as median ± interquartile range (n = 6–10 donors per group). # P < 0.05; ## < 0.01; ### P < 0.001 (significant differences with respect to SF group); *P < 0.05; **P < 0.01 (significant differences with respect to HS group), Kruskal–Wallis test
Discussion
To investigate whether TCC formation can be activated in vitro and to identify possible modulating factors known to be involved in disc degeneration, such as IL-1β and CTSD, human IVD tissue and isolated cell cultures were conducted. IL-1β is a key pro-inflammatory factor involved in DD [18]. CTSD is a matrix-degrading enzyme previously identified in human degenerated disc tissues [7] that increases during catabolic activity, such as in DD [19, 20]. In the present study, an additional group stimulated with zymosan was included as positive control. This polysaccharide derived from yeast cell walls has been shown to robustly activate the complement system through the alternative pathway [21] and to cause inflammation in different tissues [22,23,24].
Overall, the results clearly indicate that human IVD cells are not able to completely protect themselves against complement attack in culture. Exposure to HS alone increased TCC deposition on isolated IVD cells (AF, NP and EP) in a dose-dependent manner. HS contains all the necessary components (C5b–C9) for TCC formation [25]. Its TCC-forming capacity was proven in an erythrocyte lysis test (Supplemental Figure S3). Despite enhanced expression levels of CRegs on isolated IVD cells, TCC deposition was significantly increased after stimulation with 5% HS. Therefore, this concentration was considered sufficient and selected for further experiments. In a previous study, fixed human IVD tissues presented higher TCC deposition in several patients diagnosed with DD compared to AIS, which was significantly higher in NP tissue, most likely due to differences in immune cell infiltration [14].
Modulation of TCC was not similar between cell and organ cultures. In monolayer cultures, no significant effects of the different medium supplements on TCC formation by AF, NP or EP cells were detected after 30 min of incubation. A longer stimulation period of 6 h was investigated in the organ cultures in order to ensure exposure of disc cells to complement factors present in HS in their native environment. Moreover, passage-dependent changes in the expression profiles of CRegs were observed, which might alter the response of IVD cells in monolayer compared to their three-dimensional tissue environment. Interestingly, TCC deposition on AF and EP tissues was significantly decreased (with the co-stimulation) in the presence of IL-1β. It is a key molecule produced by IVD cells during degeneration [18] and is responsible to promote the activation of immune cells including macrophages [26, 27] and the expression of matrix-degrading enzymes, such as MMP-3 [28]. Moreover, IL-1β is an early mediator of the inflammatory response involved in a variety of cellular activities, including cell proliferation, differentiation and apoptosis [29]. It has also been shown to induce production of the central complement component C3 in vitro by a human intestinal epithelial cell line [30].
Concerning sources of IL-1β, the activation of the complement system, and subsequent generation of anaphylatoxins C3a and C5a, has been shown to promote its production [31, 32]. For instance, C3a is known to increase IL-1β secretion in human tenocytes [33] and monocytes and regulates the inflammasome activation [34]. Likewise, C5a is able to induce the synthesis of IL-1β by monocytes and macrophages [35]. However, whether the production of pro-inflammatory molecules contributes to the TCC formation has not yet been investigated. Regulation can occur at different stages of the complement activation cascade by membrane-bound CRegs such as CD46, CD55 or CD59 or fluid-phase proteins such as vitronectin, clusterin or factor-I. IL-1β and TNF-α have been described to upregulate the expression of CD46, CD55 and CD59 by chondrocytes in vitro after 24 h [36]. Since TCC activation is a very rapid process, we selected short stimulation times of 30 min for cell culture and 6 h for tissue culture studies. Therefore, effects of IL-1β involving induction of regulatory protein expression probably cannot explain the observed inhibitory influence on TCC deposition in the experimental setting. Interestingly, IL-1β is known to bind to both, C3b and alpha2-macroglobulin [37]; thus, complement modulation via a direct interaction could be more likely relevant [38]. The protein complex formation of IL-1β, C3b and alpha2-macroglobulin might impair further processing of C3b and/or activate the protease inhibitor activity of alpha2-macroglobulin. However, further mechanistic studies will be needed to understand the complex interplay between pro-inflammatory molecules—especially IL-1β—and the complement activation cascade.
In NP tissue, TCC deposition increased in the presence of CTSD. Besides other proteases, CTSD has been described as a potential complement activator by direct cleavage of C5 and subsequent generation of functional C5a [39] which is accompanied by generation of C5b as initial component of TCC formation. Moreover, this endo-protease contributes to the cleavage of cartilaginous matrix components, such as aggrecan [40]. CTSD has been previously identified in degenerated IVDs. It was also colocalized with cells from the cleft between the cartilaginous EP and vertebral body, indicating its association with endplate separation and AF disorganization [7]. Furthermore, the presence of the tissue renin–angiotensin system (tRAS) components, including CTSD, has been recently associated with inflammation and subsequent degeneration of IVD tissue [19, 20]. Since CTSD can replace renin as a renin-like enzyme to generate angiotensin I and angiotensin II, which interacts with the angiotensin II receptors to induce pro-inflammatory signals [19], CTSD-mediated induction of tRAS might be plausible. The results described in the present manuscript further indicate that CTSD may also be involved in the activation of the terminal part of the complement cascade.
Of note, the CReg CD59 expression did not follow a specific pattern compared to TCC formation. However, the duration of HS exposition was very short. In an injury model of cartilage explants, an increase in gene expression of CD59 and clusterin—another TCC inhibitor—was measured after 4 days of HS stimulation [41]. Interestingly, CD55 and CD59 were shown to be expressed in disc cells (even freshly isolated) and rapidly increased under culture with 5% heat-inactivated FBS. At protein level, CD46 did not seem to be present in freshly isolated cells and increased slower than the other complement regulatory factors CD55 and CD59. These results indicate for the first time that IVD cells differentially express CRegs supporting some functional relevance of the complement system in IVD tissues.
Based on findings for osteoarthritis [42], inhibition of TCC formation could be a promising therapeutic strategy against DD. This work presents substantial new information on the cell biological connection between IL-1β, CTSD and TCC formation in IVD tissue, which might open new perspectives to identify more effective DD therapeutic targets. We propose CTSD as an interesting candidate. Its inhibition might not only directly protect from degradation of extracellular matrix components but also attenuate inflammation and tissue degeneration induced by tRAS, as well as from complement activation associated processes. With respect to TCC involvement in disc degeneration, further studies are important to better understand the complexity of underlying mechanisms of its regulation and potential contribution to inflammation and cell death.
Conclusion
Our findings indicate that TCC deposition can spontaneously occur in vitro in the presence of HS in a concentration-dependent manner. This confirms that human disc cells are not completely protected against TCC formation, although they express a certain capacity of CRegs. Our results also show that CTSD, an aspartic acid protease which plays a role in tRAS and complement activation, increases TCC deposition in NP tissue. In contrast, IL-1β seems to inhibit complement activation in AF and EP to a certain extent. Elucidation of the complex mechanisms of complement modulation in DD and its interplay with other pathogenetic processes holds great promise for identification of novel therapeutic targets.
References
Cheung KMC, Karppinen J, Chan D, Ho DWH, Song YQ, Sham P et al (2009) Prevalence and pattern of lumbar magnetic resonance imaging changes in a population study of one thousand forty-three individuals. Spine Phila Pa 1976 34:934–940. https://doi.org/10.1097/BRS.0b013e3181a01b3f
Duance VC, Crean JK, Sims TJ, Avery N, Smith S, Menage J et al (1998) Changes in collagen cross-linking in degenerative disc disease and scoliosis. Spine Phila Pa 1976 23:2545–2551. https://doi.org/10.1097/00007632-199812010-00009
Pokharna HK, Phillips FM (1998) Collagen crosslinks in human lumbar intervertebral disc aging. Spine Phila Pa 1976 23:1645–168. https://doi.org/10.1097/00007632-199808010-00005
Capossela S, Schläfli P, Bertolo A, Janner T, Stadler BM, Pötzel T et al (2014) Degenerated human intervertebral discs contain autoantibodies against extracellular matrix proteins. Eur Cell Mater 27:251–263. https://doi.org/10.22203/ecm.v027a18
Adams MA, Roughley PJ (2006) What is intervertebral disc degeneration, and what causes it? Spine Phila Pa 1976 31:2151–2161. https://doi.org/10.1097/01.brs.0000231761.73859.2c
Vergroesen PP, Kingma I, Emanuel KS, Hoogendoorn RJW, Welting TJ, van Royen BJ et al (2015) Mechanics and biology in intervertebral disc degeneration: a vicious circle. Osteoarthr Cartil 23:1057–1070. https://doi.org/10.1016/j.joca.2015.03.028
Ariga K, Yonenobu K, Nakase T, Kaneko M, Okuda S, Uchiyama Y et al (2001) Localization of cathepsins D, K, and L in degenerated human intervertebral discs. Spine Phila Pa 1976 26:2666–2672. https://doi.org/10.1097/00007632-200112150-00007
Molinos M, Almeida CR, Caldeira J, Cunha C, Gonçalves RM, Barbosa MA (2015) Inflammation in intervertebral disc degeneration and regeneration. J R Soc Interface 12:20141191. https://doi.org/10.1098/rsif.2014.1191
Risbud MV, Shapiro IM (2014) Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol 10:44–56. https://doi.org/10.1038/nrrheum.2013.160
Teixeira GQ, Gonçalves RM, Barobosa MA (2018) Immunomodulation in degenerated intervertebral disc. In: Gonçalves RM, Barbosa MA (eds) Gene and cell delivery for intervertebral disc degeneration, 1st edn. CRC Press, Boca Raton, p 48
Ricklin D, Lambris JD (2013) Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol 190:3831–3838. https://doi.org/10.4049/jimmunol.1203487
Morgan BP (2016) The membrane attack complex as an inflammatory trigger. Immunobiology 221:747–751. https://doi.org/10.1016/j.imbio.2015.04.006
Grönblad M, Habtemariam A, Virri J, Seitsalo S, Vanharanta H, Guyer RD (2003) Complement membrane attack complexes in pathologic disc tissues. Spine Phila Pa 1976 28:114–118. https://doi.org/10.1097/00007632-200301150-00004
Teixeira GQ, Yong Z, Goncalves RM, Kuhn A, Riegger J, Brisby H et al (2020) Terminal complement complex formation is associated with intervertebral disc degeneration. Eur Spine J 30:217–226. https://doi.org/10.1007/s00586-020-06592-4
Ruiz-Argüelles A, Llorente L (2007) The role of complement regulatory proteins (CD55 and CD59) in the pathogenesis of autoimmune hemocytopenias. Autoimmun Rev 6:155–161. https://doi.org/10.1016/j.autrev.2006.09.008
Johnson E, Berge V, Høgåsen K (1994) Formation of the terminal complement complex on agarose beads: further evidence that vitronectin (complement S-protein) inhibits C9 polymerization. Scand J Immunol 39:281–285. https://doi.org/10.1111/j.1365-3083.1994.tb03372.x
Jeon H, Lee JS, Yoo S, Lee MS (2014) Quantification of complement system activation by measuring C5b–9 cell surface deposition using a cell-ELISA technique. J Immunol Methods 415:57–62. https://doi.org/10.1016/j.jim.2014.09.002
Johnson ZI, Schoepflin ZR, Choi H, Shapiro IM, Risbud MV (2015) Disc in flames: roles of TNF-α and IL-1β in intervertebral disc degeneration. Eur Cells Mater 30:104–117. https://doi.org/10.22203/ecm.v030a08
Morimoto R, Akeda K, Iida R, Nishimura A, Tsujii M, Obata S et al (2013) Tissue renin-angiotensin system in the intervertebral disc. Spine Phila Pa 1976 38:E129–E136. https://doi.org/10.1097/BRS.0b013e31827b8c89
Li Z, Wystrach L, Bernstein A, Grad S, Alini M, Richards RG et al (2020) The tissue-renin-angiotensin-system of the human intervertebral disc. Eur Cell Mater 40:115–132. https://doi.org/10.22203/eCM.v040a07
Smith MC, Pensky J, Naff GB (1982) Inhibition of zymosan-induced alternative complement pathway activation by concanavalin A. Infect Immun 38:1279–1284. https://doi.org/10.1128/IAI.38.3.1279-1284.1982
Rahat MA, Brod V, Amit-Cohen BC, Henig O, Younis S, Bitterman H (2016) Oxygen mitigates the inflammatory response in a model of hemorrhage and zymosan-induced inflammation. Shock 45:198–208. https://doi.org/10.1097/SHK.0000000000000492
Mazur-Bialy AI, Pocheć E (2016) HMGB1 inhibition during zymosan-induced inflammation: the potential therapeutic action of riboflavin. Arch Immunol Ther Exp Warsz 64:171–176. https://doi.org/10.1007/s00005-015-0366-6
Lima JBM, Veloso CC, Vilela FC, Giusti-Paiva A (2017) Prostaglandins mediate zymosan-induced sickness behavior in mice. J Physiol Sci 67:673–679. https://doi.org/10.1007/s12576-016-0494-8
Ivanov PA, Faktor MI, Karpova NS, Cheremnykh EG, Brusov OS (2016) Complement-mediated death of ciliate tetrahymena pyriformis caused by human blood serum. Bull Exp Biol Med 160:775–778. https://doi.org/10.1007/s10517-016-3307-4
Yoshida M, Nakamura T, Sei A, Kikuchi T, Takagi K, Matsukawa A (2005) Intervertebral disc cells produce tumor necrosis factor alpha, interleukin-1beta, and monocyte chemoattractant protein-1 immediately after herniation: an experimental study using a new hernia model. Spine Phila Pa 1976 30:55–61. https://doi.org/10.1097/01.brs.0000149194.17891.bf
Zhang Q, Wang H, Mao C, Sun M, Dominah G, Chen L et al (2018) Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol Immunol 94:27–35. https://doi.org/10.1016/j.molimm.2017.12.011
Nagase H, Visse R, Murphy G (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69:562–573. https://doi.org/10.1016/j.cardiores.2005.12.002
Lin LR, Liu W, Zhu XZ, Chen YY, Gao ZX, Gao K et al (2018) Treponema pallidum promotes macrophage polarization and activates the NLRP3 inflammasome pathway to induce interleukin-1β production. BMC Immunol 19:28. https://doi.org/10.1186/s12865-018-0265-9
Moon MR, Parikh AA, Pritts TA, Kane C, Fischer JE, Salzman AL et al (2000) Interleukin-1beta induces complement component C3 and IL-6 production at the basolateral and apical membranes in a human intestinal epithelial cell line. Shock 13:374–378. https://doi.org/10.1097/00024382-200005000-00005
Yang S, Li L, Zhu L, Zhang C, Li Z, Guo Y et al (2019) Aucubin inhibits IL-1bβ- or TNF-α-induced extracellular matrix degradation in nucleus pulposus cell through blocking the miR-140-5p/CREB1 axis. J Cell Physiol 234:13639–13648. https://doi.org/10.1002/jcp.28044
Batra R, Suh MK, Carson JS, Dale MA, Meisinger TM, Fitzgerald M et al (2018) IL-1β (interleukin-1β) and TNF-α (tumor necrosis factor-α) impact abdominal aortic aneurysm formation by differential effects on macrophage polarization. Arterioscler Thromb Vasc Biol 38:457–463. https://doi.org/10.1161/ATVBAHA.117.310333
Busch C, Girke G, Kohl B, Stoll C, Lemke M, Krasnici S et al (2013) Complement gene expression is regulated by pro-inflammatory cytokines and the anaphylatoxin C3a in human tenocytes. Mol Immunol 53:363–373. https://doi.org/10.1016/j.molimm.2012.09.001
Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Köhl J et al (2013) C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 122:3473–3481. https://doi.org/10.1182/blood-2013-05-502229
Schindler R, Gelfand JA, Dinarello CA (1990) Recombinant C5a stimulates transcription rather than translation of interleukin-1 (IL-1) and tumor necrosis factor: translational signal provided by lipopolysaccharide or IL-1 itself. Blood 76:1631–1638. https://doi.org/10.1182/blood.V76.8.1631.1631
Hyc A, Osiecka-Iwan A, Strzelczyk P, Moskalewski S (2003) Effect of IL-1beta, TNF-alpha and IL-4 on complement regulatory protein mRNA expression in human articular chondrocytes. Int J Mol Med 11:91–94. https://doi.org/10.3892/ijmm.11.1.91
Terai I, Kobayashi K, Matsushita M, Fujita T, Matsuno K (1995) α2-Macroglobulin binds to and inhibits mannose-binding protein-associated serine protease. Int Immunol 7:1579–1584. https://doi.org/10.1093/intimm/7.10.1579
Borth W, Urbanski A, Prohaska R, Susanj M, Luger TA (1990) Binding of recombinant interleukin-1β to the third complement component and α2-macroglobulin after activation of serum by immune complexes. Blood 75:2388–2395. https://doi.org/10.1182/blood.V75.12.2388.2388
Huber-Lang M, Denk S, Fulda S, Erler E, Kalbitz M, Weckbach S et al (2012) Cathepsin D is released after severe tissue trauma in vivo and is capable of generating C5a in vitro. Mol Immunol 50:60–65. https://doi.org/10.1016/j.molimm.2011.12.005
Handley CJ, Mok MT, Ilic MZ, Adcocks C, Buttle DJ, Robinson HC (2001) Cathepsin D cleaves aggrecan at unique sites within the interglobular domain and chondroitin sulfate attachment regions that are also cleaved when cartilage is maintained at acid pH. Matrix Biol 20:543–553. https://doi.org/10.1016/s0945-053x(01)00168-8
Riegger J, Huber-Lang M, Brenner RE (2020) Crucial role of the terminal complement complex in chondrocyte death and hypertrophy after cartilage trauma. Osteoarthr Cartil 28:685–697. https://doi.org/10.1016/j.joca.2020.01.004
Wang Q, Rozelle AL, Lepus CM, Scanzello CR, Song JJ, Larsen DM et al (2011) Identification of a central role for complement in osteoarthritis. Nat Med 17:1674–1679. https://doi.org/10.1038/nm.2543
Acknowledgements
The authors of the present study wish to acknowledge Dr. Helga Joos, Dr. Taryn Saggese and Mrs. Iris Baum for their technical support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the German Research Foundation (NE 549/6–1, BR 919/12–1), German Academic Exchange Service, Conselho de Reitores das Universidades Portuguesas and the Alexander von Humboldt Foundation. Jana Riegger is supported by the European Social Fund and by the Ministry of Science, Research and Arts Baden-Württemberg.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Informed consent for sample collection was obtained from all individual patients, and the study was approved by the local ethical committees (208/15 Ulm University, Germany; S-051/2016 Heidelberg University, Germany).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Teixeira, G.Q., Yong, Z., Kuhn, A. et al. Interleukin-1β and cathepsin D modulate formation of the terminal complement complex in cultured human disc tissue. Eur Spine J 30, 2247–2256 (2021). https://doi.org/10.1007/s00586-021-06901-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00586-021-06901-5