
Abstract
Background
In recent years, the elucidation of splicing abnormalities as a cause of hereditary diseases has progressed. However, there are no comprehensive reports of suspected splicing variants in the CLCN5 gene in Dent disease cases. We reproduced gene mutations by mutagenesis, inserted the mutated genes into minigene vectors, and investigated the pathogenicity and onset mechanisms of these variants.
Methods
We conducted functional splicing assays using a hybrid minigene for six suspected splicing variants (c.105G>A, c.105+5G>C, c.106−17T>G, c.393+4A>G, c.517−8A>G, c.517−3C>A) in CLCN5. We extracted information on these variants from the Human Gene Mutation Database. We reproduced minigene vectors with the insertion of relevant exons with suspected splicing variants. We then transfected these minigene vectors into cultured cells and extracted and analyzed the mRNA. In addition, we conducted in silico analysis to confirm our minigene assay results.
Results
We successfully determined that five of these six variants are pathogenic via the production of splicing abnormalities. One showed only normal transcript production and was thus suspected of not being pathogenic (c.106−17T>G).
Conclusion
We found that five CLCN5 variants disrupted the original splice site, resulting in aberrant splicing. It is sometimes difficult to obtain mRNA from patient samples because of the fragility of mRNA or its low expression level in peripheral leukocytes. Our in vitro system can be used as an alternative to in vivo assays to determine the pathogenicity of suspected splicing variants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dent disease is an X-linked genetic disease of tubulopathy characterized by low-molecular-weight proteinuria, hypercalciuria, and renal calcification [1]. Approximately 60% of Dent disease cases are caused by mutations in the CLCN5 gene, which are referred to as Dent disease 1 (OMIM#300,009). In the kidney, the CLCN5 gene is involved in the synthesis of ClC-5 at proximal tubules, ascending limb of Henle’s loop, and the intercalated cells of the collecting duct [1]. ClC-5 is a type of voltage-gated chloride channel involved in regulating cell volume, membrane potential, and transepithelial transport. A mouse model of ClC-5 knockout demonstrated that impairment of the endocytic traffic is the major cause not only of tubular proteinuria, but also of hypercalciuria and consequently kidney stones [2].
In the Human Gene Mutation Database (HGMD) Professional (https://portal.biobase-international.com/hgmd/pro/start.php), most reported mutations of the CLCN5 gene are missense or nonsense mutations, followed by deletions and splicing variants located at obvious splice sites within two bases immediately before and after exons (AG-GT). However, some previously reported suspected splicing variants were outside these regions [1, 3,4,5,6]. Although cases with such mutations might exhibit splicing abnormality, this has not been proven.
RNA splicing is a process involving the removal of the intervening, noncoding sequences of genes (introns) from pre-mRNA and the joining of the protein-coding sequences (exons) together, to enable translation of mRNA into a protein. Recent research has underlined the abundance and importance of splicing variants in the etiology of inherited diseases [7,8,9,10,11]. The most effective method to determine whether the selected variants affect splicing is to analyze the mRNA extracted from the relevant tissue of patients. However, the extraction of RNA from the affected organs is sometimes difficult. In addition, the analysis of mRNA is usually difficult because of its low expression levels in peripheral leukocytes or its fragility. In silico algorithms were developed to predict splicing abnormalities and are available as tools, but they do not always predict the exact splicing abnormalities. If the appropriate material for RNA analysis is not available, an alternative method is a minigene splicing assay—an in vitro hybrid system that allows exon trapping.
Against this background, in this study, the pathogenicity of six variants in cases of Dent disease one, in which splicing abnormalities were suspected, was analyzed using in vitro (minigene assay) and in silico assays.
Materials and methods
Among the variants in the CLCN5 gene reported as the causative agents of Dent disease one, we selected six variants suspected of being associated with splicing abnormalities from HGMD Professional, which were outside the obvious splice sites (not within two bases immediately before and after exons: AG-GT). In general, variants in the acceptor and donor sites (AG-GT sites) always alter the interactions between pre-mRNA and proteins involved in intron removal. We excluded these AG-GT consensus site variants from this study. Each variant is shown in Table 1.
In vitro assay
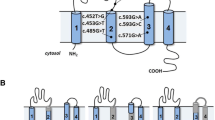
To create hybrid minigene constructs, we used the H492 vector based on the pcDNA 3.0 mammalian expression vector (Invitrogen, Carlsbad, CA, USA) (Fig. 1) that we developed previously [11,12,13,14,15]. We cloned DNA fragments containing a couple of exons and introns around the target variant in the CLCN5 gene using In-Fusion cloning methods with In-Fusion®HD Cloning Kit (Takara Bio Inc., Kusatsu, Japan), in accordance with the manufacturer’s instructions.

Schema for the hybrid minigene. The H492 vector contains two cassette exons, A and B, with a multiple cloning site. The H492 vector also contains a cytomegalovirus (CMV) enhancer–promotor and a bovine growth hormone gene (BGH) polyadenylation site. The arrows show the positions of the primers used in the RT-PCR assay
Because patients’ gDNA was not available, we initiated cloning from wild-type gDNA and then introduced mutations by site-directed mutagenesis using PrimeSTAR® Mutagenesis Basal Kit (Takara Bio Inc.), in accordance with the manufacturer’s instructions. The primers used are shown in Supplementary Table 1.
The hybrid minigenes were confirmed by sequencing and transfected into HEK293T and HeLa cells using Lipofectamine® 3000 Transfection Kit (Thermo Fisher Scientific, Waltham, MA, USA). Total RNA was extracted from cells after 36 h using the RNeasy® Plus Mini Kit (QIAGEN, Hilden, Germany).
Total RNA (1 μg) was reverse-transcribed using RNA to cDNA EcoDry™ Premix (Double Primed) (Takara Bio Inc.). PCR was performed using a forward primer corresponding to a segment upstream of exon A and reverse primer complementary to a segment downstream of exon B (Fig. 1). PCR products were analyzed by electrophoresis on a 1.5% agarose gel using a 50 bp DNA ladder and direct sequencing.
In silico assay
We predicted the splicing domain strength in each variant, using Human Splicing Finder (https://www.umd.be/HSF3/). As for potential splice sites, scores obtained using MaxEnt Scan matrix are shown in Table 1.
Results
Analysis of each cDNA revealed splicing abnormalities associated with five of the six variants (Table 1, Fig. 2, 3). We observed one transcript with a 23 bp inclusion of intron two at the end of exon two in two variants (c.105G>A and c.105+5G>C) (No. 1 and 2, Fig. 2a). In both cases, normal nucleotide sequences were not detected. We observed no change in one variant (No. 3, c.106−17T>G) (Fig. 2b). The variant c.393+4A>G led to skipping of the whole of exon 4 (No. 4, Fig. 2c). For the variant c.517−8A>G, we observed several transcripts: (1) inclusion of an intron five fragment (7 bp), (2) partial deletion (32 bp) of exon 6, (3) partial deletion (88 bp) of exon 6, and (4) exon 6 skipping (No. 5, Fig. 2d). For the variant c.517−3C>A, we observed the same transcripts as with No. 5 (No. 6, Fig. 2d), except for the inclusion of 7 bp from the intron 5 fragment (Fig. 2d).

Electrophoresis results and schematic transcript analysis results from the minigene constructs. The direct sequence is shown in Supplementary Fig.1. a Wild type (WT) exhibited a single band (full) and No. 1 and No. 2 exhibited a single band (23 bp inclusion). b Wild type (WT) and No. 3 exhibited a single band (full). c Wild type (WT) exhibited a single band (full), while No. 4 exhibited double bands (full and exon 4 skipping). d Wild type (WT) exhibited a single band (full), while No. 5 exhibited quadruple bands (7 bp inclusion, 32 bp deletion, 88 bp deletion, and exon 6 skipping). No. 6 exhibited triple bands (32 bp deletion, 88 bp deletion, and exon 6 skipping)

Schemas of the splicing patterns found in No. 1, No. 2, No. 5, and No. 6. a The splice site variants (c.105G>A and c.105+5G>C) disrupted the original donor site and resulted in the generation of an aberrant donor site that led to a 23 bp insertion. b The splice site variant (c.517−8A>G) disrupted the original acceptor site and resulted in the generation of an aberrant acceptor site that led to a 7 bp insertion, 32 bp deletion, and 88 bp deletion. c The splice site variant (c.517−3C>A) disrupted the original acceptor site and resulted in the generation of an aberrant acceptor site that led to a 32 bp deletion and an 88 bp deletion
In silico assay
We examined the change of splicing score due to each gene variant using Human Splicing Finder. The results of the MaxEnt score are shown in Table 1. There were significant decreases in the original donor site score in No. 1 (c.105G>A), No. 2 (105+5G > C), and No. 4 (c.393+4A>G), as well as in the original acceptor site score in No. 5 (c.517−8A>G) and No. 6 (c.517−3C>A). A marked decrease in the original site score was not found for No. 3 (c.106−17T>G). These results completely matched the minigene assay results.
Discussion
This is the first report describing a comprehensive exploration of CLCN5 gene splicing patterns in variants reported to be pathogenic. Most of the variants showed disruption of the original splice site and the creation of new splice sites, which were shown by in vitro minigene splicing assay and confirmed by in silico analysis.
Each result can be interpreted as follows. No. 1 (c.105G>A) and No. 2 (c.105+5G>C) result in the generation of an aberrant transcript because the original donor site is disrupted, and the existing candidate new donor site in intron 2 is activated. Both of them lead to the inclusion of a 23 bp intron 2 fragment (Fig. 3a). It was also expected that No. 4 (c.393+4A>G) leads to the skipping of exon 4 due to the disruption of the original donor site of intron 4. No. 5 (c.517−8A>G) and No. 6 (c.517−3C>A) also lead to exon skipping as a result of disruption of the original acceptor site. In addition, these two variants lead to the creation of several kinds of transcript because several candidate acceptor sites are activated after the disruption of the original site. In addition, a 7 bp fragment of intron 5 is inserted because a new acceptor site is created only by No. 5 variant (c.517-8A>G) (Fig. 3b). No. 4 (c.393+4A > G), No. 5 (c.517−8A>G), and No. 6 (c.517−3C>A) lead to whole exon skipping, but No. 1 (c.105 G>A) and No. 2 (c.105+5G>C) do not. This may be due to the existence of candidate splicing sites having a large score for splicing site (Table 1, score = 10.47 in No. 1 and 2). Variant No. 3 (c.106−17T>G) was suspected of not being pathogenic, a conclusion that was derived from both in vitro and in silico splicing assays. Actually, even in the original study [3], the authors detected only normal transcript from the patient’s mRNA extracted from the peripheral leukocytes. In addition, this patient had no proteinuria but showed nephrolithiasis and hypercalciuria. This clinical information suggested that the case was unlikely to involve Dent disease.
Recently, gene targeted therapy has been developed. Splice modulation through the use of antisense oligonucleotides has emerged as an effective technique for the treatment of splicing abnormalities. The US Food and Drug Administration (FDA) approved eteplirsen (Exondys 51) as the first exon-skipping drug for the treatment of Duchenne muscular dystrophy and nusinersen (Spinraza) for spinal muscular atrophy [16]. We can also pursue the development of gene targeted therapy for cases with aberrant splicing to correct splicing patterns. The CRISPR/Cas system is also a promising tool for correcting many genetic defects [17]. For this purpose, it is critically important to determine the pathogenicity of variants of unknown significance. We believe that the results of these splicing assays are important for studying the pathogenesis and treatment of diseases.
Lourdel et al. asserted that functional studies of CLCN5 mutations have made it possible to distinguish among the three different classes of mutation [18]. Class I mutations comprise nonsense, frameshift, and aberrant mRNA splicing mutations that lead to impairment of protein synthesis. As a consequence, Class I mutations impair processing and folding and the ClC-5 mutants of this type are retained within the endoplasmic reticulum and targeted for degradation by quality control mechanisms. We speculate that the five variants examined in this study lead to the impaired function of ClC-5.
Among three of the six variants, the original paper [3, 4, 6] showed the results of mRNA sequencing. In No. 2 (c.105+5G>C) and No. 3 (c.106−17T>G), these results were identical to our minigene assay results [3, 4]. Conversely, in No. 6 (c.517−3C>A), the mRNA sequencing results identified only exon 6 skipping [4], so this result is not completely concordant with ours. This discrepancy may be derived from the difference between in vivo and in vitro minigene assays using cultured cells. We also speculate that defense mechanisms such as nonsense-mediated decay, which eliminates abnormal mRNA in vivo, may occur in the living host [19]. Because the variants examined in this study were reported in a previous study, mRNA analysis using the patients’ samples was difficult. However, in the minigene assay, variants of interest can be inserted and their effects can be examined even when patients’ samples are not available. In addition, extracting mRNA from affected organs is often difficult. Alternatively, peripheral leukocytes are usually used for in vivo mRNA analysis; however, their mRNA expression level is usually low and the mRNA is fragile. These conditions make mRNA analysis in vivo very difficult. In such cases, the minigene assay is a good tool for assessing splicing abnormalities.
Approximately 9% of all mutations reported in the HGMD are splicing mutations (18,761/208,368) (HGMD database, accessed on October 10, 2017), although this number may well be an underestimate. Here, we assessed only variants suspected of causing splicing abnormalities. However, even variants in exons can cause splicing abnormalities. Studies for the detection of splicing abnormalities have been conducted for a long time, but it is still difficult to accurately predict abnormal splicing caused by genetic variants [20]. Recently, the number of genes identified as being responsible for inherited kidney diseases has increased rapidly. Genetic disorders are present in any part of the renal and urinary tract. Recently, using minigene assay, we have detected splicing abnormalities in the COL4A5 gene as a cause of glomerular disease [11, 21]. In this report, we summarize splicing abnormalities associated with Dent disease 1, one of the most common renal tubulopathies. Next, we plan to analyze splicing abnormalities in the OCRL gene causing Lowe syndrome and in the GLA gene causing Fabry disease, using the same approach. Other inherited kidney diseases are also targets for this approach. Cases suspected of having inherited kidney diseases are less likely to undergo renal biopsy than other diseases. As a result, the affected organ, the kidney itself, cannot be used for splicing analysis. However, analysis using minigene assay that does not require biological samples and may provide a comprehensive understanding of splicing abnormalities in inherited kidney diseases. Further analysis is thus required to further elucidate the mechanism of splicing.
In conclusion, this study provides proof of splicing abnormalities associated with some suspected variants in the CLCN5 gene. The incidence of genetic disorders due to splicing abnormalities has been underestimated, and further comprehensive studies should be conducted to clarify the contribution of splicing abnormalities to the onset of disease. In this study, we have focused on Dent disease, but we have also conducted a comprehensive approach for Alport syndrome [11, 21]. In future work, we should apply minigene assay to other common inherited kidney diseases such as Lowe syndrome and Fabry disease.
Change history
15 March 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10157-021-02041-8
References
Mansour-Hendili L, Blanchard A, Le Pottier N, Roncelin I, Lourdel S, Treard C, et al. Mutation update of the CLCN5 gene responsible for Dent disease 1. Hum Mutat. 2015;36(8):743–52. https://doi.org/10.1002/humu.22804.
Piwon N, Gunther W, Schwake M, Bosl MR, Jentsch TJ. ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature. 2000;408(6810):369–73. https://doi.org/10.1038/35042597.
Forino M, Graziotto R, Tosetto E, Gambaro G, D'Angelo A, Anglani F. Identification of a novel splice site mutation of CLCN5 gene and characterization of a new alternative 5' UTR end of ClC-5 mRNA in human renal tissue and leukocytes. J Hum Genet. 2004;49(1):53–60. https://doi.org/10.1007/s10038-003-0108-1.
Minamikawa S, Nozu K, Nozu Y, Yamamura T, Taniguchi-Ikeda M, Nakanishi K, et al. Development of ultra-deep targeted RNA sequencing for analyzing X-chromosome inactivation in female Dent disease. J Hum Genet. 2018;63(5):589–95. https://doi.org/10.1038/s10038-018-0415-1.
Tosetto E, Ceol M, Mezzabotta F, Ammenti A, Peruzzi L, Caruso MR, et al. Novel mutations of the CLCN5 gene including a complex allele and A 5' UTR mutation in Dent disease 1. Clin Genet. 2009;76(4):413–6. https://doi.org/10.1111/j.1399-0004.2009.01212.x.
Cox JP, Yamamoto K, Christie PT, Wooding C, Feest T, Flinter FA, et al. Renal chloride channel, CLCN5, mutations in Dent's disease. J Bone Miner Res. 1999;14(9):1536–42. https://doi.org/10.1359/jbmr.1999.14.9.1536.
Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018;59(3):253–68. https://doi.org/10.1007/s13353-018-0444-7.
Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet. 2005;42(10):737–48. https://doi.org/10.1136/jmg.2004.029538.
Lewandowska MA. The missing puzzle piece: splicing mutations. Int J Clin Exp Pathol. 2013;6(12):2675–82.
Montes M, Sanford BL, Comiskey DF, Chandler DS. RNA splicing and disease: animal models to therapies. Trends Genet. 2019;35(1):68–87. https://doi.org/10.1016/j.tig.2018.10.002.
Horinouchi T, Nozu K, Yamamura T, Minamikawa S, Nagano C, Sakakibara N, et al. Determination of the pathogenicity of known COL4A5 intronic variants by in vitro splicing assay. Sci Rep. 2019;9(1):12696. https://doi.org/10.1038/s41598-019-48990-9.
Nozu K, Iijima K, Kawai K, Nozu Y, Nishida A, Takeshima Y, et al. In vivo and in vitro splicing assay of SLC12A1 in an antenatal salt-losing tubulopathy patient with an intronic mutation. Hum Genet. 2009;126(4):533–8. https://doi.org/10.1007/s00439-009-0697-7.
Yamamura T, Nozu K, Miyoshi Y, Nakanishi K, Fujimura J, Horinouchi T, et al. An in vitro splicing assay reveals the pathogenicity of a novel intronic variant in ATP6V0A4 for autosomal recessive distal renal tubular acidosis. BMC Nephrol. 2017;18(1):353. https://doi.org/10.1186/s12882-017-0774-4.
Yamamura T, Nozu K, Ueda H, Fujimaru R, Hisatomi R, Yoshida Y, et al. Functional splicing analysis in an infantile case of atypical hemolytic uremic syndrome caused by digenic mutations in C3 and MCP genes. J Hum Genet. 2018;63(6):755–9. https://doi.org/10.1038/s10038-018-0436-9.
Nakanishi K, Okamoto T, Nozu K, Hara S, Sato Y, Hayashi A, et al. Pair analysis and custom array CGH can detect a small copy number variation in COQ6 gene. Clin Exp Nephrol. 2019;23(5):669–75. https://doi.org/10.1007/s10157-018-1682-z.
Lim KRQ, Yokota T. Invention and early history of exon skipping and splice modulation. Methods Mol Biol. 2018;1828:3–30. https://doi.org/10.1007/978-1-4939-8651-4_1.
Cai L, Fisher AL, Huang H, Xie Z. CRISPR-mediated genome editing and human diseases. Genes Dis. 2016;3(4):244–51. https://doi.org/10.1016/j.gendis.2016.07.003.
Lourdel S, Grand T, Burgos J, Gonzalez W, Sepulveda FV, Teulon J. ClC-5 mutations associated with Dent's disease: a major role of the dimer interface. Pflugers Arch. 2012;463(2):247–56. https://doi.org/10.1007/s00424-011-1052-0.
Kurihara Y, Matsui A, Hanada K, Kawashima M, Ishida J, Morosawa T, et al. Genome-wide suppression of aberrant mRNA-like noncoding RNAs by NMD in Arabidopsis. Proc Natl Acad Sci USA. 2009;106(7):2453–8. https://doi.org/10.1073/pnas.0808902106.
Ohno K, Takeda JI, Masuda A. Rules and tools to predict the splicing effects of exonic and intronic mutations. Wiley Interdiscip Rev RNA. 2018;9(1):e1451. https://doi.org/10.1002/wrna.1451.
Horinouchi T, Nozu K, Yamamura T, Minamikawa S, Omori T, Nakanishi K, et al. Detection of splicing abnormalities and genotype-phenotype correlation in X-linked alport syndrome. J Am Soc Nephrol. 2018;29(8):2244–54. https://doi.org/10.1681/ASN.2018030228.
Acknowledgements
This study was supported by Grants-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Subject ID: 18K15713 to China Nagano, 17H04189 to Kazumoto Iijima, and 19K08726 to Kandai Nozu). The authors thank Edanz (www.edanzediting.co.jp) for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Review Board of Kobe University Graduate School of Medicine (IRB approval number 301) and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised due to a Retrospective Open Access order.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Inoue, T., Nagano, C., Matsuo, M. et al. Functional analysis of suspected splicing variants in CLCN5 gene in Dent disease 1. Clin Exp Nephrol 24, 606–612 (2020). https://doi.org/10.1007/s10157-020-01876-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10157-020-01876-x