
Abstract
Resistance to immunotherapy poses a significant challenge in the treatment of colorectal cancer (CRC), and the underlying mechanisms are not fully understood. Recent studies have implicated PFKFB3, a crucial glycolytic enzyme, in shaping the tumor microenvironment in CRC. Our study aimed to systematically study the role of PFKFB3 in CRC. Bioinformatic analysis revealed that PFKFB3 expression is notably elevated in CRC tissues compared to normal counterparts. In vivo experiments confirmed that suppressing PFKFB3 reduces the tumorigenesis of CRC. We identified multiple cancer-associated pathways positively correlated with high expression of PFKFB3, such as epithelial-mesenchymal transition (EMT), hypoxia, KRAS signaling, angiogenesis, PI3K/AKT/mTOR, Hedgehog, and Notch pathways. Additionally, PFKFB3 exhibited significant correlations with various immune-related pathways, including complement, IL-2/STAT5, IL-6/JAK/STAT3, IFN-α/IFN-γ, TGF-β, and TNF-α/NF-κB, as well as several immunosuppressive cell markers found in regulatory T cells (CCR8, TGFB1, STAT5B, FOXP3), M2 macrophages (CD163, VSIG4, MS4A4A), T cell exhaustion markers (CTLA-4, PDCD1, LAG3), and PD-L1. Intriguingly, increased PFKFB3 expression was observed in PD-L1 blockade-resistant patients and was associated with shorter overall survival. In a nutshell, PFKFB3 plays an important role in CRC tumorigenesis and resistance to immunotherapy. Targeting PFKFB3 inhibits tumor formation and enhances the efficacy of immunotherapy. Our findings underscore the functions of PFKFB3 in CRC, shedding light on both cancer-related and immunosuppressive pathways.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer-related mortality worldwide, but still lacking an effective understanding of its occurrence and development mechanisms [1, 2]. The five-year survival rate for patients with advanced metastatic CRC is below 15%, and immunotherapy is effective only in those with dMMR or MSI-H tumors [3,4,5,6]. Therefore, it is very important to find reliable early biomarkers for CRC and understand the molecular mechanisms behind the low response rates to immunotherapy.
Tumor cells preferentially use glycolysis for glucose metabolism, even under oxygen-rich conditions, a phenomenon known as the Warburg effect [7, 8]. In the process of glycolysis, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) plays a critical role in modulating tumor cell metabolism [9, 10]. Inhibition of glycolysis by blocking PFKFB3 in tumor endothelial cells of hepatocellular carcinoma has been shown to normalize tumor vessels and reduce metastasis by restoring vascular function [11]. Recent studies have shown that PFKFB3 promotes tumor angiogenesis in HER2+ breast cancer [12], and accelerates pancreatic ductal adenocarcinoma tumorigenesis and progression through KRAS transformation and p38γ overexpression [13]. Inhibition of PFKFB3 significantly reduces tumor growth in vivo by inducing apoptosis, autophagy, impairing DNA repair, and activating AKT/mTOR signal transduction [14]. In CRC, Ma et al. found that phosphorylation of c-Src promotes CRC development by activating PFKFB3 [15]. Dianat-Moghadam et al. [16] confirmed that LXR activation inhibits CRC stem cell growth, proliferation and migration in vitro. However, PFKFB3 operates within a complex regulatory network, and further research is needed to elucidate its mechanisms in CRC.
Recently, PFKFB3 has been found important in immune microenvironment and PD-L1-mediated immune escape in renal cancer [17, 18]. Chen et al. [19] confirmed that PFKFB3 enhances the expression of PD-L1 in monocytes surrounding hepatocellular carcinoma cells through the NF-κB signaling pathway. However, the role of PFKFB3 in the immune microenvironment of CRC is still unclear, and understanding this could reveal mechanisms underlying the poor response rates to immunotherapy in CRC.
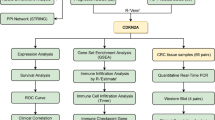
In order to study the role of PFKFB3 in CRC, we collected and analyzed transcriptome data from 491 cases of CRC and verified our findings in CRC mouse models. Additionally, the present study reveals that PFKFB3 is highly associated with an immunosuppressive microenvironment and may serve as a predictive biomarker for immunotherapy responses, as validated in a cohort of 244 patients treated with PD-L1 monoclonal antibodies (mAbs). This is the first comprehensive analysis of PFKFB3 in CRC in terms of clinical characteristics and molecular mechanisms.
Methods and materials
Data preparation
We acquired CRC RNA-seq data from GSE50760 [20] in GEO database (www.ncbi.nlm.nih.gov/gds/) and TCGA-COAD (portal.gdc.cancer.gov/). The GSE50760 cohort included 18 CRC tissues and paired normal tissues, while the TCGA-COAD cohort comprised 473 CRC tissues and 41 normal tissues. Expression data were normalized to transcripts per million (TPM), and data of PFKFB3 were extracted for further analysis.
Expression of PFKFB3 in colorectal cancer
We compared the expression of PFKFB3 between CRC and normal tissues. Additionally, we analyzed PFKFB3 expression across various gastrointestinal cancers, including stomach adenocarcinoma (STAD), esophageal carcinoma (ESCA), liver hepatocellular carcinoma (LIHC), pancreatic adenocarcinoma (PAAD), cholangiocarcinoma (CHOL), rectum adenocarcinoma (READ), and colon adenocarcinoma (COAD).
Then we evaluated the relevance of PFKFB3 to clinicopathological characteristics, including age (< 60 vs ≥ 60), gender (male vs female), T stage (T1-2 vs T3-4), N stage (N0 vs N+), cancer stage (stage I-IV), and prior malignancy (yes vs no). In overall survival (OS) prognosis analysis, we included 432 patients from the TCGA-COAD cohort with complete clinical information and follow-up time exceeding 30 days.
Mouse models
Construction of a colitis-associated colorectal cancer (CAC) model
C57BL/6 mice (6–8 weeks old, male, Shanghai Institute of Material Medicine, Chinese Academy of Science, China) were used in accordance with protocols approved by the Animal Care and Use Committee of Zhejiang University (NO. 26679). CAC was established using azoxymethane (AOM) and dextran sulfate sodium (DSS) as described previously [21]. Mice were intraperitoneally injected with AOM (10 mg/kg, MP Biomedicals) once, followed by 7 days on a normal diet. Subsequently, mice underwent cycles of 7 days of 2% DSS (MP Biomedicals) treatment and 14 days of recovery, for a total of 4 cycles. Control mice received water only. After sacrifice, colon tumors or tissues were collected.
Total RNA from tissues was extracted with RNA isolater reagent (#R401-01-AA, Vazyme). We utilized HiScript II 1st Strand cDNA Synthesis Kit (#R312-02, Vazyme) to reverse transcription of RNA into cDNA, MagicSYBR Mixture (#CW3008M, CWBIO) to perform quantitative real-time polymerase chain reaction (qRT-PCR). Relative expression of PFKFB3 was normalized to GAPDH. The primer sequences used for qRT-PCR are listed in Table 1.
CAC treatment
Primary CAC models were induced in C57BL/6 mice as mentioned previously and divided into two groups: solvent only (control) or receiving PFK15 (25 mg/kg, i.p., Selleck) every 3 days for 28 days, starting on the 84th day after the completion of AOM/DSS cycles. PFK15, also known as 1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one, specifically inhibits recombinant human PFKFB3 enzyme activity without affecting protein abundance [22]. And it has been confirmed that PFK15 does not inhibit other kinases or key glycolytic enzymes, including PFK-1, hexokinase, and PFKFB4, highlighting its selectivity for PFKFB3 [23]. After sacrifice, tumor number and size were recorded. The experiment was carried out in strict compliance with the regulations of animal ethics.
Functional analysis of PFKFB3
To explore the biological function of PFKFB3, we calculated the correlation between PFKFB3 and all other protein-coding genes in 473 CRC tissues from the TCGA-COAD cohort. We ranked genes according to Pearson’s correlation, and utilized “clusterProfiler” package [24] to perform Gene Set Enrichment Analysis (GSEA) [25]. Enriched pathways with an adjusted P < 0.05 were considered significant. Furthermore, to estimate the pathway activity scores in CRC, single-sample gene set enrichment analysis (ssGSEA) was conducted based on the gene expression profile [26]. By comparing these scores, we analyzed the role of the pathway of interest in disease pathogenesis, which was helpful to further understand the relationship between the pathway and PFKFB3 in CRC.
Role of PFKFB3 in immunosuppressive microenvironment
GSEA enrichment results showed that overexpressed PFKFB3 was positively correlated to multiple immunosuppressive pathways which were demonstrated to reshape tumor microenvironment [27,28,29,30,31], indicating that overexpressed PFKFB3 might be associated with resistance to immunotherapy and the infiltration of immunosuppressive cells. With the hypothesis, we further analyzed correlations between PFKFB3 and biomarkers from M2 macrophages (CD163, VSIG4, MS4A4A), regulatory T cells (CCR8, TGFB1, STAT5B, FOXP3), exhausted T cells (TIM-3, CTLA-4, PD-1, LAG3), and PD-L1.
To further confirm the suppressive role of PFKFB3 in immunotherapy efficacy, we collected RNA-seq data from PD-L1 blockade-treated metastatic urothelial cancer patients in the IMvigor210 cohort [32]. We compared the expression of PFKFB3 in 61 immunotherapy-sensitive patients (complete response, CR or partial response, PR) and 183 immunotherapy-resistant patients (stable disease, SD or progressive disease, PD). We also investigated whether PFKFB3 could predict OS in patients undergoing immunotherapy, using pretreatment data from Kaplan–Meier database [33], for anti-PD-L1 (atezolizumab) in urothelial tumor, and anti-PD-1 (pembrolizumab) as well as anti-CTLA-4 (ipilimumab) in all tumors.
Statistical analysis
Statistical analyses were conducted in GraphPad Prism (version 7.00) or R software (version 4.2.2). For normally distributed data, two-tailed unpaired Student's t-test was adopted, while Wilcoxon test was adopted for non-normally distributed data. One-way ANOVA was applied for multiple comparisons. Connections between PFKFB3 expression and other genes were estimated by Pearson’s correlation. OS rates were calculated by Kaplan–Meier survival analysis and examined by log-rank tests. Univariate and multivariate Cox regression analyses were used to assess the impact of clinicopathological characteristics and PFKFB3 expression on OS. P < 0.05 was considered statistically significant.
Results
PFKFB3 is overexpressed in CRC
PFKFB3 expression was significantly elevated in CRC tissues compared to normal colon tissues, as observed in both the GSE50760 (P < 0.01, Fig. 1A) and TCGA cohorts (P < 0.001, Fig. 1B), as well as in CRC mouse models (P < 0.001, Fig. 1C). From the expression landscape, PFKFB3 expression was generally increased in gastrointestinal cancers, including colon adenocarcinoma, esophageal carcinoma, stomach adenocarcinoma and cholangiocarcinoma (P < 0.05, Fig. 1D), indicating that PFKFB3 warrants further investigation into its role in cancer development and potential as a biomarker.

Expression profile of PFKFB3 in gastrointestinal cancers. PFKFB3 expression was compared between colorectal cancer tissues and normal tissues in the A GSE50760 cohort, B TCGA cohort and C mouse-derived cohort. D An overview of PFKFB3 expression across various gastrointestinal cancers. COAD, colon adenocarcinoma; READ, rectum adenocarcinoma; ESCA, esophageal carcinoma; STAD, stomach adenocarcinoma; LIHC, liver hepatocellular carcinoma; CHOL, cholangiocarcinoma; PAAD, pancreatic adenocarcinoma. mRNA expression in the GSE50760 and TCGA datasets is normalized as log2(TPM + 1)
PFKFB3 correlates with the progression of CRC and poor prognosis
Analysis of clinicopathological characteristics revealed that PFKFB3 expression was significantly higher in patients with deeper tumor invasion (P < 0.05), suggesting its potential role in promoting CRC progression. Interestingly, patients with prior malignancy showed a decreased expression of PFKFB3. The differences observed in other characteristics were not statistically significant. (Fig. 2A–F).

Correlation between PFKFB3 and clinicopathologic characteristics in CRC. A–F The expression of PFKFB3 changes in T1-2 (vs T3-4), lymph node negative (vs positive), stages (I-IV), age < 60 (vs ≥ 60), and prior malignancy (yes vs no). P < 0.05 is statistically significant
To determine the prognostic value of PFKFB3, maximum selective rank statistics (https://github.com/kassambara/survminer) were employed to identify the optimal cut-off for distinguishing CRC prognosis within the TCGA cohort (P = 0.014, HR = 1.87, 95%CI: 1.19–2.92; Fig. 3). Furthermore, Cox regression analysis confirmed that high PFKFB3 expression was a significant predictor of increased mortality risk in CRC. Even after adjusting for confounding factors, the elevated risk remained substantial (HR = 1.81, P = 0.026), reinforcing the role of PFKFB3 as an independent prognostic marker for OS. In contrast, age, gender, and prior malignancy did not significantly impact OS, while advanced T and N stages were strongly associated with poorer outcomes, highlighting their importance in risk stratification (Table 2).

Overall survival (OS) analysis of PFKFB3 in CRC. The expression of PFKFB3 is significantly correlated with OS in the TCGA-COAD cohort. Red denotes high expression (log2(TPM + 1) > 4.78, n = 301) of PFKFB3, and blue denotes low expression (log2(TPM + 1) ≤ 4.78, n = 131). The horizontal axis represents OS time (months), and the vertical axis represents the probability of survival. P < 0.05 is statistically significant
To further validate the prognostic power of PFKFB3, a subgroup analysis was conducted, revealing that PFKFB3 overexpression more accurately predicted outcomes in patients aged ≥ 60 years, with T3-4 stage, positive lymph nodes, and no history of malignancy (P < 0.05, Fig. S1A–F). Additionally, PFKFB3 showed a near-significant relationship with OS in stage III and female subgroups (P < 0.1). Interestingly, differentially expressed analysis showed the expression of PFKFB3 was higher in patients without prior malignancy (Fig. 2D), but overexpressed PFKFB3 indicated an undesirable prognosis (Fig. S1F).
PFKFB3 Inhibition by PFK15 suppresses CRC development
To investigate the effect of PFKFB3 inhibition on CRC tumorigenesis, mice were induced by AOM and DSS to establish CAC (Fig. 4A). Tumors were observed in both the control and the PFK15-treated groups, predominantly located in the middle to distal regions of the colon (Fig. 4B). Compared with the control group, PFK15 treatment significantly reduced the average number of tumors (9.20 ± 1.93 vs 6.33 ± 1.37, P = 0.0006, Fig. 4C) in the PFK15-treated group (n = 12) compared to the control group (n = 10). Additionally, the overall tumor load, measured as the total tumor diameter per mouse, was significantly lower after treatment (30.00 ± 6.96 vs 18.83 ± 5.04 mm, P = 0.0003, Fig. 4D).

PFK15 inhibits the development of CRC. A Schematic overview of colitis-associated colorectal cancer (CAC) induced by AOM/DSS (n = 10 for the control, n = 12 for PFK15-treated group). B Representative images of CAC tissues treated with PFK15. C Tumor count and D tumor load in each group
PFKFB3 is associated with multiple cancer-related pathways
PFKFB3 is primarily known as a glycolytic enzyme, but recent studies have highlighted its involvement in various biological processes beyond glycolysis. To comprehensively explore these roles, we performed GSEA analysis and found multiple important pathways were significantly enriched in PFKFB3 on the basis of its Pearson’s correlation value with 19,492 identified genes. The top 6 positively and negatively correlated genes are shown (Fig. S2A-B). For instance, PFKFB3 exhibited positive correlations with ITPRIP (cor = 0.7, P = 6.86E−72), RELT (cor = 0.69, P = 2.48E−68), FOSL2 (cor = 0.68, P = 1.71E−66), SOCS3 (cor = 0.67, P = 4.37E−63), SEC14L1 (cor = 0.66, P = 3.15E−61), and SSH1 (cor = 0.66, P = 5.42E−60), while showing negative correlations with TMEM261 (cor = −0.4, P = 2.22E−19), TDGF1 (cor = −0.39, P = 2.98E−18), C6orf57 (cor = −0.38, P = 5.03E−18), C10orf99 (cor = −0.38, P = 1.34E−17), ASCL2 (cor = −0.38, P = 1.43E−17), SMIM22 (cor = −0.38, P = 1.62E−17).
In GSEA results, we found that several cancer-related pathways were closely related to PFKFB3, including epithelial-mesenchymal transition (EMT), hypoxia, upregulation of KRAS signaling pathway, angiogenesis, PI3K/AKT/mTOR pathway, Hedgehog signaling pathway and Notch signaling pathway (Fig. 5A). These pathways generally showed higher activity scores in CRC tissues with overexpressed PFKFB3 (Fig. 5B). Additionally, pathway activity scores for hypoxia, angiogenesis, Hedgehog signaling, and EMT were elevated in advanced T3-4 stages compared to earlier T1-2 stages, which may offer insights into the mechanisms by which PFKFB3 influences CRC progression (Fig. S3A). Notably, the correlation between PFKFB3 and EMT was of particular interest, as 71% of genes in the EMT pathway were positively co-expressed with PFKFB3. The top 6 positively (NOTCH2, COL5A3, ITGA5, COL6A3, NID2, and SERPINE1, cor ≥ 0.60, P < 0.001) and negatively (MEST, AREG, TGFBI, IGFBP2, SERPINE2, and MSX1, |cor|: 0.02–0.12, P: 0.008–0.61) correlated genes are shown in Fig. S3B. These findings underscore the critical role of PFKFB3 in the EMT process in CRC.

Cancer-related pathways enriched in PFKFB3. A Seven cancer-related pathways are closely associated with PFKFB3 in CRC. The short vertical lines represent the distribution of PFKFB3-related genes within the pathway among 19,492 genes. B Pathway activity scores differ between high and low PFKFB3 expression groups. P < 0.05 is statistically significant
PFKFB3 is associated with immunosuppressive microenvironment and immunotherapy resistance
The GSEA results also showed that PFKFB3 was highly correlated with immune-related pathways, such as the IL2/STAT5, IL6/JAK/STAT3, IFN-α response, IFN-γ response, TGF-β, and TNF-α/NF-κB signaling pathways (Fig. 6A–B). Some of them have been shown to strongly inhibit the anti-tumor immune response by the infiltration of Tregs [34], CD8+ T cell exhaustion [35], and PD-L1 stabilization [27]. However, there is limited research specifically addressing the role of PFKFB3 in immunosuppression, and the identified pathways offer valuable insights into PFKFB3’s involvement in immune evasion and therapy resistance.

Immune-related pathways enriched in PFKFB3. A Eight significantly enriched PFKFB3-related immunosuppressive pathways in CRC. B Pathway activity scores differ between high and low PFKFB3 expression groups. P < 0.05 is statistically significant
Further analysis revealed that PFKFB3 was highly positively correlated with markers of immunosuppressive cells, including Tregs (CCR8, FOXP3, TGFB1, STAT5B, cor: 0.35–0.5, P < 0.001), M2 macrophages (CD163, MS4A4A, VSIG4, cor: 0.43–0.57, P < 0.001) and T cell exhaustion (CTLA-4, PD-1, LAG-3, TIM-3, cor: 0.35–0.48, P < 0.001). The correlation coefficient between PFKFB3 and PD-L1 was greater than 0.5 (Fig. 7A-D). These findings also suggest that elevated PFKFB3 expression may contribute to the reshaping of the tumor immune microenvironment, potentially facilitating immune evasion in CRC.

PFKFB3 correlates with an immunosuppressive microenvironment. Correlation between PFKFB3 and immunosuppressive markers in A regulatory T cells (CCR8, FOXP3, TGFB1, STAT5B), B M2 macrophages (CD163, MS4A4A, VSIG4), C T cell exhaustion (GZMB, CTLA-4, PD-1, LAG-3, TIM-3), and D PD-L1. Correlation strength is categorized as follows: “0.00–0.29” (weak); “0.30–0.59” (moderate); “0.60–1.0” (strong). P < 0.05 is statistically significant
With these results mentioned above, we speculated that PFKFB3 might be associated with adverse immunotherapy response in CRC. Analysis of PFKFB3 expression in patients treated with PD-L1 mAbs revealed significant overexpression in PD-L1-resistant patients (Fig. 8A). When patients were categorized into three distinct immunological phenotypes, PFKFB3 could still distinguish immunotherapy response in immune-excluded patients (P < 0.05), with trends towards higher expression in immune-inflamed (P = 0.14) and immune-desert patients (P = 0.061) (Fig. 8B). Further examination of PFKFB3 expression in patients treated with PD-L1, PD-1, and CTLA-4 mAbs showed that high PFKFB3 expression was consistently associated with shorter OS (Fig. 8C–E). Specifically, in patients administrated with PD-L1 inhibitors, high PFKFB3 expression was significantly correlated with shorter OS (P = 0.00019, HR = 1.72), which validated the role of overexpressed PFKFB3 in anti-PD-L1-resistant patients (Fig. 8A). Similarly, high PFKFB3 expression was linked to worse prognosis in the PD-1 inhibitor cohort (P = 0.0004, HR = 2.05). Although the association in the CTLA-4 inhibitor cohort did not reach statistical significance (P = 0.0909), there was a trend toward shorter OS (HR = 1.66). It suggests that PFKFB3 and its correlation with these immunosuppressive pathways may play an important role in reshaping immune microenvironment to induce resistance to immunotherapy and serves as not only a marker of adverse immunotherapy response but also a potential target for improving treatment outcomes.

Expression of PFKFB3 in patients with different immunotherapy responses. Differentially expressed analysis of PFKFB3 in A total CR/PR versus SD/PD patients, and B CR/PR versus SD/PD patients within three immunological phenotypes (inflamed, excluded, and desert). CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease. The role of PFKFB3 in predicting OS in patients receiving immunotherapy, including C atezolizumab (anti-PD-L1), D pembrolizumab (anti-PD-1), and E ipilimumab (anti-CTLA-4). P < 0.05 is statistically significant
Discussion
In the present study, we found that the expression of PFKFB3 increased in CRC and patients with characteristics of T3-4 (vs T1-2). Prognostic analysis showed that overexpressed PFKFB3 is associated with shorter OS and serves as an independent prediction factor. Elevated PFKFB3 expression was also consistently observed in other gastrointestinal cancers, including esophageal carcinoma, stomach adenocarcinoma, and cholangiocarcinoma. The inhibition of PFKFB3 by PFK15 effectively suppressed tumorigenesis in CAC model. Enrichment analysis comprehensively revealed several underlying mechanisms related to PFKFB3. Notably, PFKFB3 was found overexpressed in PD-L1 mAbs-resistant patients and associated with shorter OS in those receiving immunotherapy, which might be associated with multiple immunosuppressive-related pathways.
Previous studies on PFKFB3 mainly focused on glycolysis, but recent studies indicate that PFKFB3 also directly interacts with various signaling pathways, regulating downstream cellular functions. In the result of GSEA, we found that overexpressed PFKFB3 is significantly positively correlated with multiple cancer-related pathways, including hypoxia, KRAS signaling, angiogenesis and PI3K/AKT/mTOR signaling, and EMT. These pathways are crucial for cell proliferation, resistance, and migration [36,37,38,39,40]. For instance, PFKFB3 has been shown to enhance hepatoma cell proliferation via hypoxia-inducible factor 1 (HIF-1α) accumulation [41] and to promote the occurrence of pancreatic cancer mediated by KRAS transformation, which can be disrupted by inhibiting p38γ and PFKFB3. During the process, PFKFB3 is phosphorylated at S467 by p38γ, thereby linking the Warburg effect to KRAS signaling [13]. Angiogenesis is necessary to tumor progression, providing fundamental supports for nutrients transportation and metastasis. LT-α secreted by lymphocytes strengthens the PFKFB3-mediated glycolytic flux and accelerates tube formation of human umbilical vein endothelial cells (HUVECs), depending on the TNFR/NF-κB signaling pathway. Conversely, PFKFB3 inhibitor reduces the density of CD31-positive blood vessels [42]. These results suggest that PFKFB3 may act as a common downstream effector in multiple pathways, underscoring its importance in cancer biology. This aligns with our GSEA findings and highlights the potential for further research regarding PFKFB3 in CRC.
Additionally, we found that PFKFB3 was closely related to the tumor microenvironment. Enrichment analysis revealed significant correlations between PFKFB3 and immune-related pathways, including IL-2/STAT5, IL-6/JAK/STAT3, IFN-α, IFN-γ, TNF-α/NF-κB, and TGF-β signaling. These pathways play crucial roles in reshaping immunosuppressive microenvironment. Previous studies have demonstrated that the IL-2/STAT5 pathway is essential for maintaining homeostasis and migration ability of Tregs [28, 34], as well as mediating CD8+ T cell exhaustion [31, 35]. An increased number of Tregs has been linked to a reduced response rate to CTLA-4 mAbs in patients with advanced cancer [43], although such negative effects can be reversed by TGF-β pathway inhibitor galunisertib which restores sensitivity to PD-L1 blockade in CRC mice [30]. A low proportion of Tregs within the tumor donated a 30% to 40% complete regression rate in CRC mice treated with a combination of endoglin antibodies and PD-1 inhibition [44]. Additionally, Yalcin et al. [45] identified PFKFB3 as a key TGF-β1 effector, promoting snail expression, invasion, and glycolysis of CRC cells. TGF-β1 also induces the expression of PFKFB3 and glycolysis in glioblastoma T98G cells by activating the PI3K/AKT signaling pathway [46]. Both TNF-α/NF-κB and JAK/STAT3 signaling pathways have been shown to promote and stabilize the expression of PD-L1, driving cancer immunosuppression and evading T cell surveillance [27, 29]. Therefore, we may speculate that PFKFB3 is vital in creating an inhibitory immune microenvironment.
Further analysis showed that PFKFB3 is highly correlated with markers of suppressive immune cells (Tregs, M2 macrophages, T cell exhaustion) and PD-L1, indicating that PFKFB3 might promote tumor immune evasion and affect the efficacy of immunotherapy. In the IMvigor210 cohort, we observed a close relationship between PFKFB3 expression and response to PD-L1 mAbs immunotherapy, with higher PFKFB3 levels in PD-L1 mAbs-resistant patients compared to those who were sensitive to the treatment. Supporting this, previous studies, such as that by Chen et al [19], [32] reported that soluble factors derived from hepatocellular carcinoma, like hyaluronan fragments, can induce the upregulation of PFKFB3 in monocytes of the peritumoral region, leading to increased PD-L1 expression via the PFKFB3-NF-κB signaling pathway. Notably, the upregulation of PD-L1 could be effectively blocked by the PFKFB3 inhibitor 3PO. Therefore, combining a PFKFB3 inhibitor with PD-L1 mAb holds promise for improving the response rate and prolonging overall survival in CRC patients undergoing immunotherapy.
In conclusion, PFKFB3 plays an important role in the tumorigenesis and progression of CRC through cancer-related pathways, and the combination of PFKFB3 inhibitor and immunotherapy presents a promising strategy for enhancing treatment response in CRC.
Data availability
The datasets in the present study are open to public, and corresponding accession numbers are GSE50760 and TCGA-COAD (https://portal.gdc.cancer.gov/).
References
Sung H, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Wu Q, et al. Survival outcomes of stage I colorectal cancer: development and validation of the ACEPLY model using two prospective cohorts. BMC Med. 2023. https://doi.org/10.1186/s12916-022-02693-7.
Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA. 2021;325(7):669–85.
Dekker E, et al. Colorectal cancer. Lancet. 2019;394(10207):1467–80.
Le DT, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–13.
Cervantes A, et al. Metastatic colorectal cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(1):10–32.
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
Zhong X, et al. Warburg effect in colorectal cancer: the emerging roles in tumor microenvironment and therapeutic implications. J Hematol Oncol. 2022. https://doi.org/10.1186/s13045-022-01358-5.
Okar DA, et al. PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem Sci. 2001;26(1):30–5.
Dou Q, et al. PFKFB3-mediated pro-glycolytic shift in hepatocellular carcinoma proliferation. Cell Mol Gastroenterol Hepatol. 2023;15(1):61–75.
Matsumoto K, et al. Inhibition of glycolytic activator PFKFB3 suppresses tumor growth and induces tumor vessel normalization in hepatocellular carcinoma. Cancer Lett. 2021;500:29–40.
O’Neal J, et al. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res Treat. 2016;160(1):29–40.
Wang F, et al. p38γ MAPK is essential for aerobic glycolysis and pancreatic tumorigenesis. Cancer Res. 2020;80(16):3251–64.
Xiao Y, et al. Inhibition of PFKFB3 induces cell death and synergistically enhances chemosensitivity in endometrial cancer. Oncogene. 2021;40(8):1409–24.
Batista A, et al. IRE1alpha regulates macrophage polarization, PD-L1 expression, and tumor survival. PLoS Biol. 2020;18(6):e3000687.
Dianat-Moghadam H, et al. Modulation of LXR signaling altered the dynamic activity of human colon adenocarcinoma cancer stem cells in vitro. Cancer Cell Int. 2021;21(1):100.
Larionova I, et al. PFKFB3 overexpression in monocytes of patients with colon but not rectal cancer programs pro-tumor macrophages and is indicative for higher risk of tumor relapse. Front Immunol. 2023. https://doi.org/10.3389/fimmu.2022.1080501.
Yu Y, et al. Glucose metabolism involved in PD-L1-mediated immune escape in the malignant kidney tumour microenvironment. Cell Death Discov. 2021;7(1):15.
Chen DP, et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J Hepatol. 2019;71(2):333–43.
Kim SK, et al. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol Oncol. 2014;8(8):1653–66.
Han W, et al. Orally deliverable nanotherapeutics for the synergistic treatment of colitis-associated colorectal cancer. Theranostics. 2019;9(24):7458–73.
Wang L, et al. CYLD deficiency enhances metabolic reprogramming and tumor progression in nasopharyngeal carcinoma via PFKFB3. Cancer Lett. 2022;532:215586.
Clem BF, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. 2013;12(8):1461–70.
Yu G, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50.
Cui K, et al. Comprehensive characterization of the rRNA metabolism-related genes in human cancer. Oncogene. 2020;39(4):786–800.
Lim SO, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30(6):925–39.
Song M, et al. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. 2018;562(7727):423–8.
Ma X, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30(1):143–56.
Holmgaard RB, et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer. 2018;6(1):47.
Liu Y, et al. IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. 2021;22(3):358–69.
Mariathasan S, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–8.
Kovacs SA, Fekete JT, Gyorffy B. Predictive biomarkers of immunotherapy response with pharmacological applications in solid tumors. Acta Pharmacol Sin. 2023;44(9):1879–89.
Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. 2023;41(3):450–65.
Lutz V, et al. IL18 receptor signaling regulates tumor-reactive CD8+ T-cell exhaustion via activation of the IL2/STAT5/mTOR pathway in a pancreatic cancer model. Cancer Immunol Res. 2023;11(4):421–34.
Han J, et al. PFKFB3 was overexpressed in gastric cancer patients and promoted the proliferation and migration of gastric cancer cells. Cancer Biomark. 2017;18(3):249–56.
Cantelmo AR, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell. 2016;30(6):968–85.
La Belle Flynn A, et al. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun. 2019;10(1):3668.
Yamamoto T, et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun. 2014;5:3480.
Jingtai Z, et al. Targeting Aurora-A inhibits tumor progression and sensitizes thyroid carcinoma to Sorafenib by decreasing PFKFB3-mediated glycolysis. Cell Death Dis. 2023;14(3):224.
Hu L, et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1alpha/PFKFB3/PFK1 pathway. Life Sci. 2019;239:116966.
Yang JG, et al. Lymphotoxin-alpha promotes tumor angiogenesis in HNSCC by modulating glycolysis in a PFKFB3-dependent manner. Int J Cancer. 2019;145(5):1358–70.
Coutzac C, et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat Commun. 2020;11(1):2168.
Schoonderwoerd MJA, et al. Targeting endoglin-expressing regulatory T cells in the tumor microenvironment enhances the effect of PD1 checkpoint inhibitor immunotherapy. Clin Cancer Res. 2020;26(14):3831–42.
Yalcin A, et al. 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for transforming growth factor beta1-enhanced invasion of Panc1 cells in vitro. Biochem Biophys Res Commun. 2017;484(3):687–93.
Rodriguez-Garcia A, et al. TGF-beta1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017;284(20):3437–54.
Funding
This work was supported by the National Natural Science Foundation of China (81772543), Ministry of Science and Technology of the People's Republic of China (2020YFA0803300, Y.F), Hebei Health Commission Project (20240287), Hebei Natural Science Foundation (H2024104001), and Baoding Technology Project (2241ZF046).
Author information
Authors and Affiliations
Contributions
Conceptualization and supervision: H.P, J.S. and Y.F. Investigation and analysis: S.L., R.Z., S.S., Y.J. and J.Z.. Experiment: Y.H. Original draft: S.L., R.Z. and Y.H. Review and editing: H.P, J.S. and Y.F. All authors approved the submission.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, S., Zhao, R., Han, Y. et al. Identification of PFKFB3 as a key factor in the development of colorectal cancer and immunotherapy resistance. Clin Exp Med 24, 219 (2024). https://doi.org/10.1007/s10238-024-01479-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10238-024-01479-w