
Abstract
Epigenetic mechanisms can alter gene expression without a change in the nucleotide sequence and are increasingly recognized as important mechanisms that can generate phenotypic diversity. Most of our current knowledge regarding the origin and role of epigenetic variation comes from research on plants or mammals, often in controlled rearing conditions. Epigenetic research on birds in their natural habitats is still in its infancy, but is needed to answer questions regarding the origin of epigenetic marks and their role in phenotypic variation and evolution. Here we review the potential for studying epigenetic variation in natural bird systems. We aim to provide insights into (1) the origin of epigenetic variation, (2) the relationship between epigenetic variation and trait variation, and (3) the possible role of epigenetic variation in adaptation to changing environments. As there is currently little research on epigenetics in wild birds, we examine how findings on other taxa such as plants and mammals relate to birds. We also examine some of the pros and cons of the most commonly used methods to study patterns of DNA methylation in birds, and suggest some topics we believe need to be addressed to develop the field of wild avian epigenetics further.
Zusammenfassung
Anwendung von Epigenetik an freilebenden Vögeln
Epigenetische Mechanismen sind in der Lage die Aktivität eines Gens zu beeinflussen ohne die DNA-Sequenz zu veränderen und werden zunehmend als wichtige Mechanismen erkannt um phänotypische Diversität generieren zu können. Der größte Teil unseres derzeitigen Wissens über den Ursprung und die Rolle epigenetischer Variationen stammt aus der Erforschung von Pflanzen oder Säugetieren, oft unter kontrollierten Aufzuchtbedingungen. Die epigenetische Forschung an Vögeln in ihren natürlichen Lebensräumen steckt noch in den Kinderschuhen, ist jedoch erforderlich, um Fragen zur Herkunft der epigenetischen Merkmale und ihrer Rolle bei der Variation und Evolution des Phänotyps zu beantworten. Hier untersuchen wir das Potenzial zur Untersuchung der epigenetischen Variation in natürlichen Vogelsystemen. Wir möchten Einblicke geben in (1) den Ursprung der epigenetischen Variation, (2) die Beziehung zwischen epigenetischer Variation und Merkmalsvariation und (3) die mögliche Rolle der epigenetischen Variation bei der Anpassung an eine sich verändernde Umgebungen. Da derzeit nur wenig über die Epigenetik von Wildvögeln geforscht wird, untersuchen wir, inwiefern sich die Befunde bei anderen Taxa wie Pflanzen und Säugetieren auf Vögel auswirken. Wir untersuchen auch einige der Vor- und Nachteile der am häufigsten verwendeten Methoden zur Untersuchung von DNA-Methylierungsmustern bei Vögeln und schlagen einige Themen vor, die unserer Ansicht nach behandelt werden müssen, um das Gebiet der Epigenetik wilder Vögel weiterzuentwickeln.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epigenetics has traditionally been narrowly defined as the causal analysis of developmental processes (Waddington 1952). In recent years, the term epigenetics has been used more loosely to describe non-genetic processes affecting intergenerational phenotypic variation (e.g. Groothuis and Carere 2005) or all processes related to gene expression (Ledford et al. 2008). Epigenetics is currently often defined as the study of biochemical mechanisms that stably alter gene expression by affecting either transcription or translation without a single change in the primary nucleotide sequence of the genome (Richards 2006). Such biochemical mechanisms include molecular mechanisms like DNA methylation (Korochkin 2006), histone modification (Jaenisch and Bird 2003) and the involvement of microRNAs (Bossdorf et al. 2008), all processes known to affect gene expression (Law and Jacobsen 2010).
Since changes in gene expression are precursors or direct causes of changes in phenotypes, it is generally accepted that changes in epigenetic mechanisms alter phenotypic characteristics (Law and Jacobsen 2010). Most studies on epigenetic mechanisms investigate DNA methylation—the addition of a methyl group (–CH3) to a nucleotide, usually a cytosine (C), as the epigenetic mark. In mammals, methylation of cytosine mostly occurs in a CpG dinucleotide context, i.e. when a cytosine (C) in a nucleotide is followed by a guanine (G) separated by only a phosphate (p) group. The functionality of DNA methylation is particularly known within CpG islands (CGI), predominantly in the promoter region of genes, where methylation can interfere with the binding of proteins required for transcription initiation and can therefore negatively affect gene expression (Bird 2002). However, the functionality of DNA methylation is not always clear, for example, the function of gene body methylation is still under discussion (Bewick et al. 2016). It is also known that epigenetic factors are able to target specific cells or tissues or are only active during certain developmental stages (Christensen et al. 2009; Hoivik et al. 2011). However, general tissue processes have also been described, and cell, tissue and development specificity is therefore still under discussion (Anastasiadi et al. 2018). Besides changes in DNA methylation, the availability of DNA for transcription via states of chromatin condensation (Jaenisch and Bird 2003), chemical modification of histone and interference by microRNAs (Bossdorf et al. 2008) are also epigenetic modifications. Studies on histone modification and microRNAs are very rare in wild birds and we therefore mostly focus this review on DNA methylation.
Many studies have shown that epigenetic mechanisms can explain the causes and mechanisms of human diseases (Portela and Esteller 2010), and cell and organism development (Sasaki and Matsui 2008). More recently, a number of excellent reviews have reviewed the value of epigenetics to ecology and evolution (Bossdorf et al. 2008; Ledon-Rettig et al. 2013; Verhoeven et al. 2016), but what is the potential for epigenetic study from an ecological and evolutionary perspective? There are two main reasons why epigenetic mechanisms could be relevant to our understanding of ecological processes and evolution.
Firstly, when epigenetic changes can be induced as a response to the current local environment (Richards 2006), epigenetic mechanisms may provide an organism with the opportunity for an adaptive response to a change in the environment via phenotypic plasticity (Bossdorf et al. 2008; Jablonka and Lamb 2006; Verhoeven et al. 2016). Phenotypic plasticity is ‘the ability of a genotype to produce distinct phenotypes when exposed to different environments throughout its ontogeny’ (Pigliucci 2001, 2005). In this sense, epigenetic marks can be viewed as the mechanisms underlying phenotypic plasticity. If indeed it turns out that such environmentally induced epigenetic change can be inherited from cell to cell, this epigenetic response to ecological circumstances may affect an individual’s phenotype throughout its lifetime (Verhoeven et al. 2016). This may be specifically relevant for enduring or lasting environmental changes, such as climate change (Rey et al. 2016). The main promise for many researchers, however, lies in the possibility that such marks have the potential to be stably inherited across multiple generations. If such environmentally induced epigenetic changes can be transgenerationally inherited, this gives rise to the possibility that epigenetics can change the evolutionary potential of phenotypic traits (Bossdorf et al. 2008; Jablonka and Raz 2009; Laland et al. 2014), which is an on-going subject of discussion (Wray et al. 2014). We would like to state, however, that transgenerational inheritance has not been unequivocally shown in vertebrates.
Secondly, the functionality and significance of epigenetic processes are likely context specific. Epigenetic processes measured in captive settings might be valuable for unravelling the genomic mechanisms underlying them, but have limited power for predicting epigenetic processes in natural populations, since natural environments can be much more complex and variable than captive environments. Furthermore, once an epigenetically mediated phenotypic change has been discovered under controlled circumstances in captivity, it is impossible to mimic every aspect of a natural environment, predict what course the environment is going to take throughout time, and predict an organism’s response and fitness (Boffelli and Martin 2012; Bossdorf et al. 2008; Schrey et al. 2012). This impedes our knowledge of how the natural environment shapes epigenetic variation and how epigenetics potentially contributes to local adaptation and the evolution of phenotypic traits. Thus, although studies on captive animals can further our knowledge of the relevance of epigenetic mechanisms in wild species and give invaluable insights for a detailed mechanistic understanding of epigenetic processes, the only way to confirm findings is by assessing epigenetic mechanisms in wild populations.
Why study epigenetic effects in birds?
Most of our current knowledge on ecological epigenetics stems from plant studies (Bossdorf et al. 2008; Verhoeven et al. 2016) or research on humans or captive rodents (e.g. Jones and Takai 2001). These studies have demonstrated that although there certainly are similarities between taxa in their epigenetic mechanisms and the epigenetic landscape (Law and Jacobsen 2010) there can also be substantial differences (Hunt et al. 2013; Kvist et al. 2018; Waters et al. 2018), even within vertebrates (Jabbari et al. 1997). For example, while functional DNA methylation in plants is low throughout the genome and predominantly exists in relation to transposable element (TE) activity (Hollister and Gaut 2009; Zhang 2008), most mammal DNA methylation levels are high and variation seems to be present in promoter regions of genes and TEs, directly affecting gene expression (Suzuki and Bird 2008). Moreover, epigenetic reprogramming of DNA methylation is more drastic in vertebrates than in plants (Feng et al. 2010b; Morgan et al. 2005; Reik et al. 2001; Sasaki and Matsui 2008). In addition, for most animals, germ line segregation occurs relatively early during development, leaving fewer opportunities for environmental modification to be passed through the germ line (Jablonka and Raz 2009; Ledon-Rettig et al. 2013). Another difference between animals and plants is that, in many of the former, parents provide their offspring with an environment that is determined by their behaviour (e.g. parental care), which gives scope for a system where environments experienced by the parents affect their behaviour, thereby creating a similar environment for their offspring via parental effects (Champagne 2008). Although rarely studied, such parental effects might form the basis for a behavioural inheritance system that might not be present in plants (Jensen 2013).
There are several reasons why birds are well suited to the study of ecological epigenetics on wild animals. Many bird species have been studied to such an extent that they are model organisms for many ecological and evolutionary questions (Both et al. 2006; Charmantier et al. 2006; Grant and Grant 1996; van Oers and Mueller 2010). They are generally conspicuous, easy to observe and distributed throughout the world, experience variable natural environmental conditions, often have large broods and, since early embryogenesis and development happens outside the mother, their early rearing environment can be easily manipulated at various stages. This in combination with the fact that individual birds can be studied throughout their lives has resulted in several long-term population studies with pedigrees spanning multiple generations (see e.g. Araya-Ajoy and Dingemanse 2017; Backström et al. 2008; Bosse et al. 2017; Duckworth and Kruuk 2009; Jaari et al. 2009; Quinn et al. 2009), allowing the study of the transgenerational inheritance of epigenetic marks. The potential for transgenerational inheritance of epigenetic marks was illustrated in birds in a recent review by Guerrero-Bosagna et al. (2018), which focused on the knowledge gained from poultry, mainly Chickens. Their conclusion was that well-defined experimental design and molecular genetic analyses are needed to enhance our understanding of the function of epigenetics in birds. Moreover, their review showed that we lack an overview of how wild bird species can be used as model organisms to answer questions related to the functional role of epigenetic variation in natural situations. Such a functional approach would shed more light on the ecological role of epigenetic variation and to what extent it may contribute to local adaptation to changing environments. Moreover, while the genomic mechanisms related to how epigenetic marks may be inherited over generations can be studied under controlled circumstances, the evolutionary significance of transgenerational inheritance can only be tested in natural populations. Several bird genomes (Elgvin et al. 2017; Ellegren et al. 2012; Jarvis et al. 2014; Laine et al. 2016; Qu et al. 2013; Warren et al. 2010) have now been sequenced and sometimes even annotated to such a high degree that molecular resources have become available that allow for the functional analyses of epigenetic marks in wild bird species. In this review, we therefore aim not only to provide insight into the relationship between epigenetic variation and phenotypic variation, but also to assess the role of epigenetic variation in adaptation to changing environments.
Avian epigenetics
Epigenetic marks in birds
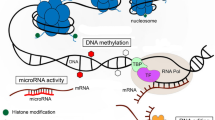
The two major questions regarding epigenetic mechanisms are: what is the origin of this variation, and what are the consequences of this for selection? Currently, epigenetic changes are believed to originate from three distinct sources and, depending on whether they are inherited across generations, may affect selection (Fig. 1).

Adjusted from EpiDiverse (2018) (colour figure online)
Origins of epigenetic variation that are heritable (passed on to future generations) and non-heritable. In blue, epigenetic variation that is under genetic control; in green, epigenetic variation that arises spontaneously as epimutations. In red, epigenetic variation that is environmentally induced. Solid lines indicate known paths in avian ecological epigenetics; dashed lines indicate hypothesized paths. Numbers in circles refer to explanations in the text (in parentheses) below.
First, epigenetic variation might originate from environmental induction. This can either be dependent on stochastic events (such as random error) or environmental cues. This type of epigenetic variation plays a key role in phenotypic plasticity, which might enable an organism to develop an adaptive phenotype in response to environmental change (Bossdorf et al. 2008; Jablonka and Lamb 2007). Environmentally induced epigenetic variation is therefore of particular interest to avian ecologists. To what extent this induced variation (1) stably inherits via mitosis from cell to cell, or may even be inherited across multiple generations, or (2) whether it is completely erased after meiosis, still needs to be investigated in birds. There are currently few studies that have found inherited patterns of environmentally induced variation in wild animals (Feil and Fraga 2012). This is particularly the case in birds, where imprinting, a phenomenon where the expression of genes is dependent on the parent from whom the allele originates, seems to be absent, and therefore a so-called memory function may not be expected (Reik et al. 2001). While induced transgenerational epigenetic variation is typically seen as an alternative to genetic inheritance, we should also keep in mind that this source of environmental variation is not automatically independent of genetic variation (Fig. 1). For example, in the case of (3) genotype by environment interactions, the epigenetic response to environmental effects will be genotype dependent. (4) This epigenetic variation could then potentially be either inherited over one or more generations or, when it is not inherited, may contribute to natural selection within that generation, but will not affect the evolution of the expressed phenotype. More speculative is the idea that (5) such environmentally induced changes eventually become heritable (Richards 2006) via transgenerationally stable genomic reprogramming (Rapp and Wendel 2005), or (6) that they may facilitate the fixation of genetic variants via ‘genetic accommodation’ (West-Eberhard 2003). Individual introduced House Sparrows (Passer domesticus) populations were found to harbour more unmodified CpGs (no SNPs in the CpG context) and overall fewer SNPs than individuals from native populations (Hanson et al. 2018). Furthermore, the variation in the number of SNPs among CpG sites was greater in native populations. The SNP variants within or near CpG sites may indicate the genetic assimilation of initially environmentally induced epigenetic effects, although no direct evidence for this has been found. Alternatively, this might be explained by selection against certain SNPs in introduced populations (Hanson et al. 2018).
The second source of epigenetic variation lies in epigenetic mutations. Epimutations are generally expected to arise spontaneously and may inherit stably over generations (Becker et al. 2011), but can also arise as random mutations, without being heritable. To our knowledge, only one study has so far investigated epimutations in bird species in the wild by comparing differentially methylated regions between five Darwin’s finch species (Skinner et al. 2014). The authors found that epimutations were more common found than genetic mutations, confirming the hypothesis that epigenetic marks are less stable compared to DNA. The term epimutation, however, was very loosely defined in this study as regions that were differentially methylated among the species. Whether these epigenetic differences between the five species resemble stably inherited epigenetic changes (i.e. epimutations in the narrow sense) needs further investigation. Many of these epigenetic differences between species are likely related to habitat differences due to, for example, niche differentiation since, in general, epimutations are expected to be as frequent as DNA mutations (Becker et al. 2011). Potentially, epigenetic mutations may also arise randomly in the genome without being inherited by the next generation. These may affect the phenotype in this way, possibly throughout its lifetime when inherited from cell to cell, but will not affect the phenotypes of future generations.
The third origin of epigenetic variation is when changes in methylation completely depend on some underlying genetic variation, which is termed ‘obligatory epigenetic variation’ (Richards 2006). Genetic polymorphisms either within the region of variation in methylation (cis-regulation) or at distant sites (trans-regulation) are stably inherited over generations, generating stable epigenetic variation. In this case, variation in methylation can undergo selection indirectly via the genomic polymorphism underlying this variation. In plants, studies have shown that most epigenetic variation can be directly explained by genetic variation in combination with genotype-by-environment effects, suggesting that genetic variation is the main factor explaining at least inter-accession variation (Dubin et al. 2015). Determining the proportion of epigenetic variation that can be explained by these different underlying mechanisms should be an important focus of avian research.
Efforts to map DNA methylation throughout the genome in birds are generally aimed at targeting methylated cytosine, as is the case for most other organisms (Derks et al. 2016; Laine et al. 2016; Li et al. 2011, 2015; Skinner et al. 2014). Cytosine methylation in birds can occur in three different sequences: CpG, CHG or CHH (where H is A, T or C), with almost all of the methylation occurring in a CpG context (Derks et al. 2016). Non-CpG methylation (CHG or CHH cytosine methylation) also occurs in brain cells (Derks et al. 2016; Laine et al. 2016), but not in red blood cells, although it is unknown whether non-CpG methylation is brain specific or whether it also occurs in other tissues.
Genome-wide studies show that cytosine methylation in birds is not randomly distributed across the genome. Methylation levels are relatively high throughout the genome in both Chicken (Gallus gallus) (David et al. 2017; Li et al. 2011) and Great Tit (Parus major) (Derks et al. 2016), with about 70% of all CpG sites methylated at a mean level of ca. 50%, with a sharp decrease around the transcription start site (TSS) in gene promoters (Derks et al. 2016). This pattern is similar to that observed in other vertebrates (Gardiner-Garden and Frommer 1987). A whole genome DNA methylation sequence assembly for both brain and blood tissue in the Great Tit in combination with RNAseq gene expression data (Laine et al. 2016) showed that DNA methylation around a gene’s transcription start site negatively correlates with gene expression, whereas genes with promoters that were highly methylated showed lower expression levels. This pattern is also seen in other vertebrate epigenomes (Li et al. 2011). Interestingly, methylation levels within TEs in both brain and blood were also associated with RNA expression levels, which points to another function of DNA methylation (Derks et al. 2016). TE methylation has been found to be responsible for defending genes against TE activity by TE silencing both in plants (Hollister and Gaut 2009) and mammals (Whitelaw and Martin 2001). However, TE methylation differs between tissues and according to the methods used, and more research is required to make an accurate general statement on how and why TEs are hyper- or hypomethylated (Derks et al. 2016; Yi 2017). We would like to point out that these associations are present at the among-gene level, but whether this also holds for relationship between among-individual variation in gene expression and DNA methylation needs to be verified.
Birds have heteromorphic sex chromosomes and, like all organisms that have these, are confronted with the problem of gene dosage expression. For example, mammalian females have two X chromosomes (XX) while males only have one (XY). The problem of double gene expression is overcome by silencing one female X chromosome, which, in mice, is most likely done by epigenetic mechanisms such as histone modification and DNA methylation (Lyon 1961). Interestingly, this type of system has not been found in birds [where males are the homogametic sex (ZZ) and females heterogametic (ZW)], and a recent study found sex differences in gene expression and DNA methylation in both the domestic White Leghorn and the Red Jungle fowl (Natt et al. 2014). A male hypermethylated region on the Z chromosome and the zinc finger RNA binding protein gene promoter on chromosome 1 appeared to be differently expressed and methylated in the hypothalamus and thalamus between the sexes. However, there were more differences in gene expression than cytosine methylation alone could explain. This suggests that other epigenetic mechanisms may be involved in causing sex differences in gene expression (Natt et al. 2014).
Different techniques for measuring DNA methylation
As described before, we mostly focus on DNA methylation in our review. DNA methylation can be assessed using several methods that have been described in detail in other reviews (Couldrey and Cave 2014; Kurdyukov and Bullock 2016) and we will therefore not go into much detail on these in this review. The most important point for the reader is that the assumptions inherent to these methods determine the inferences that can be drawn from studies using them. We will briefly describe the most commonly used techniques that have been used to measure DNA methylation in birds, together with their pros and cons (Table 1).
Bisulphite sequencing
To date, the gold standard for assessing DNA methylation is still bisulphite sequencing. A bisulphite treatment affects only cytosine that is not methylated, and by using high-throughput sequencing techniques this method allows the estimation of methylation levels at the individual cytosine level (Frommer et al. 1992; Harris et al. 2010; Krueger et al. 2012). This method, however, greatly relies on prior knowledge of the genome of the species under study. A whole-genome bisulphite sequencing (WGBS) approach thereby gives the most complete and unbiased view of the methylation landscape throughout the whole genome, but although the cost of sequencing has decreased, it is still rather expensive (Suzuki et al. 2018).
Several techniques have been developed that make use of restriction enzymes to produce a reduced representation of the whole genome that can be further developed for bisulphite sequencing. Reduced representation bisulphite sequencing (RRBS) is a method whereby the restriction enzyme MspI cuts at (CCGG) sites, and typically regions that are CG rich are targeted. This enables one to aim for a higher proportion of sequenced CpG sites in CGIs, such as promoter regions. Size selection steps via either cutting of gels (Meissner et al. 2005) or the use of beads (Boyle et al. 2012) means that generating large fragments without CpGs can be avoided, after which bisulphite conversion takes place. RRBS has been validated for use in birds, and has already been used in several studies (Derks et al. 2016; Mäkinen et al. 2019).
Epigenotyping-by-sequencing (epiGBS) can potentially be used for a variety of species, but has been developed predominantly for non-model plant species (van Gurp et al. 2016). This reference-free RRBS approach cost-effectively and efficiently characterizes a reduced representation of the DNA both genetically and epigenetically. Part of the epiGBS protocol allows for de novo reference creation from the sequenced data, thus allowing for the use of non-model species without a reference genome. Furthermore, the price per sample is low, since there is the possibility of pooling samples after digestion and adapter ligation into one sequence library. With new adapter design (pairwise combinations of barcodes differ by a minimum of three mutational steps and length) it is possible to achieve 96 samples per sequence library. This type of low-cost reduced representation approach has the advantage of sequencing larger numbers of individuals at low cost without the need for a reference genome. However, there are limitations to this method in that it only targets a small fraction of the genome, and prior knowledge of the functionality of the targeted fragments is a great advantage.
Methyl-DNA immunoprecipitation assay
A methyl-DNA immunoprecipitation (MeDIP) assay can identify methylated CpG-rich sequences. DNA is sheared into random fragments and methylated fragments are subsequently isolated via an antibody raised against methylated cytosine (Weber et al. 2005). In other words, a random fragment of methylated cytosine can be estimated. In comparison to RRBS, this method is less biased regards CGIs and has higher coverage of non-CGI and non-genic regions (Harris et al. 2010). Moreover, sequencing is limited to the targeted methylated areas, reducing the number of false positives. Disadvantages of the method include the fact that the resolution of the outcome is on a fraction not base pair basis and regions are often quite large. A detailed functional approach is therefore needed when regions of interest have been detected.
Methylation-sensitive amplification length polymorphism
Methylation-sensitive amplification length polymorphism (MS-AFLP) is a very common method for the study of genome-wide methylation patterns in non-model species. By using this cost-efficient technique, it is possible to screen many samples at multiple loci (cytogenetic bands or regions on the chromosome) at the same time. When using MS-AFLP, a large number of random sites throughout the genome are screened, but the adjacent sequence to each site is unknown. Therefore, the screened loci are anonymous and the region or the gene influenced by methylation cannot be specified. This technique provides information on the methylation of a single cytosine per fragment. Furthermore, this technique can be used to resolve a dominant banding pattern. A band is either present or absent at each position, which makes it impossible to distinguish heterozygote epigenotypes (Schrey et al. 2013). Moreover, methylation does not follow a bimodal pattern (on or off), since it is a quantitative trait. Despite these shortcomings for bird studies, the method may prove very useful when combined with validation methods (Schrey et al. 2013).
Pyrosequencing
Pyrosequencing uses bisulphite conversion in combination with a methylation-specific polymerase chain reaction, using a forward and a reverse biotinylated primer to target a specific sequence. Pyrosequencing can be used to quantify DNA methylation at specific CpG sites based on a sequencing-by-synthesis method (Tost and Gut 2007), and is used in cases where information on a limited number of sites is needed. Targeted sites are often in the close vicinity of, or within, a candidate gene. Prior knowledge of the functionality of the gene, its exact genome sequence and the presence and functionality of CpG sites that have the potential to causally affect gene expression is a must.
Studies on avian ecological epigenetics: present and future
Environmental causes of variation in DNA methylation
Phenotypic variation is shaped by sequence variation in interaction with the environment in which genes are expressed (Feinberg 2007). We now know that epigenetic mechanisms contribute to phenotypic variation, and that these mechanisms are often active during development and can be altered in response to different environmental factors (Richards 2006), making them good candidates for the study of the mechanisms behind phenotypic plasticity. These environmental effects include chemicals that are present in the environment, like methyl donors (Weaver 2005), toxic substances (Romano et al. 2017; Wallace et al. 2018), or external factors such as incubation temperature (Vinoth et al. 2018; Yan et al. 2015) or maternal effects (Bentz et al. 2016). Here, we discuss the role of epigenetic mechanisms in plastic changes within the concept of developmental plasticity, and refer to developmental plasticity as (an) irreversible change(s) in the phenotype resulting from environmentally introduced alterations of development, like early environmental effects (Forsman 2015), parasite load and anthropogenic effects.
Early developmental effects
Some mechanisms behind environmentally induced epigenetic changes have been shown using captive rodent species (Meaney and Szyf 2005; Weaver et al. 2004). Studies in several economically important avian species have shown that maternal exposure to stressors can impact offspring methylation (Liu et al. 2018; Zimmer et al. 2017), perhaps through the deposition of steroid hormones in the yolk (Ahmed et al. 2014; Zimmer et al. 2017). These findings are supported by a study on wild Eastern Bluebirds (Sialia sialis), in which yolk testosterone concentration appeared to be positively correlated with density, nestling growth rate and nestling DNA methylation of the diencephalon estrogen receptor alpha promoter (Bentz et al. 2016). Since nestling growth rate also increased with percentage DNA methylation (Bentz et al. 2016), and avian aggression is typically affected by yolk testosterone level (Eising et al. 2006; Muller et al. 2009; Partecke and Schwabl 2008; Strasser and Schwabl 2004), it is possible that the females “prepared” their chicks for high-density conditions (high competition) through testosterone allocation, which instigated changes in DNA methylation. Unfortunately, behaviour and fitness of these individuals were not assessed, since the nestlings were sacrificed.
Postnatal behaviour in relation to DNA methylation was assessed in a study on Superb Starling (Lamprotornis superbus) chicks, in which the DNA methylation levels of the glucocorticoid receptor gene were found to correlate with among-year variation in rainfall during pre-breeding periods and with male breeding behaviour in adulthood (Rubenstein et al. 2016). These results suggest that DNA methylation marks established early in life may still influence adult behaviour (Rubenstein et al. 2016). However, it is not known if these effects were maternally induced through, for example, altered hormone deposition in the yolk, or if the effects were postnatal, environmental conditions experienced right after hatching. In this specific case, a postnatal effect could have been affected by food availability due to earlier rainfall. Another postnatal effect could be parental care, which, in relation to DNA methylation, has been especially well studied in rodents (St-Cyr and McGowan 2015; Weaver 2005; Weaver et al. 2004), but not in birds. This is particularly surprising since there is a long history of studies on parental care in ornithology (Cockburn 2006; Farmer 2000; Liker and Szekely 2005) and there are examples of the involvement of parental care in several aspects of early development (Hinde et al. 2009) that potentially affect DNA methylation in multiple ways.
Another possible postnatal factor affecting DNA methylation levels of juvenile birds is brood size. In Zebra Finch (Taeniopygia guttata) nestlings, brood size correlated positively with the level of DNA methylation in several MS-AFLP loci (Sheldon et al. 2018b). A direct cause of this could be sibling competition for food (Carere et al. 2005). As yet, it is unknown if nutritional status during early development affects DNA methylation in wild birds. Juvenile birds may also be confronted with higher parasite load when broods are large (Cantarero et al. 2013; Wenzel and Piertney 2014); this has been shown to affect DNA methylation in adult Red Grouse (Lagopus lagopus scotica). Gastrointestinal parasite load (the caecal nematode Trichostrongylus tenuis) was associated with DNA methylation at 25 MS-AFLP loci in Red Grouse populations in Scotland (Wenzel and Piertney 2014), which indicates that host-parasite interactions may at least be partly regulated at the epigenetic level (Wenzel and Piertney 2014).
Despite possible causes for this brood size effect, a difference in DNA methylation was not detected in experimentally enlarged and reduced broods (Sheldon et al. 2018b). This might be explained by a loss of variation, which is associated with the chosen approach in MS-AFLP studies. Sheldon et al. (2018b) analysed the binary haplotype-binding pattern, which shows that methylation is either present or absent in a certain fragment. In this method, slight differences in methylation remain undetected. Another explanation could be that this was not a true postnatal effect, but a prenatal effect. It could be that Zebra Finches that raise large broods have different hormone concentrations in the yolk than Zebra Finches that raise small broods, which might affect DNA methylation levels. When brood size is manipulated, this effect could remain undetected, since the hormone concentration and methylation level do not match in the manipulated brood. Interestingly, individuals that were transferred from their original brood to manipulated ones showed more hypomethylation compared to unmanipulated individuals that remained in their brood of origin, suggesting an association between manipulation and DNA methylation.
Human-induced epigenetic changes
While we often think about natural causes when discussing environmental origins of epigenetic variation, there are several examples in the literature on birds showing that human-induced changes to the environment, such as urbanization, pollution and also domestication, can directly or indirectly alter the epigenetic states of individuals or even species. In an urban Great Tit population, bolder individuals tended to have higher levels of methylation at a serotonin transporter CpG site, measured using pyrosequencing, than more timid individuals (Riyahi et al. 2015). This difference in methylation between bold and shy types was not found in a forest population (Riyahi et al. 2015). A later study found differences in DNA methylation levels and morphological features between rural and urban populations of Darwin’s finches (Geospiza fortis and Geospiza fuliginosa) (McNew et al. 2017). In contrast to genetic variation (Bonduriansky and Day 2009; Ledon-Rettig et al. 2013; Price et al. 2003), epigenetic variation may enable organisms to adjust their phenotype to match novel environments, or provide them with the ability to quickly respond to a changing environment (Jablonka and Lamb 2007; Tammen et al. 2013). Therefore, in urban environments, epigenetic mechanisms might be involved in how conditions affect the phenotypes of bird species. However, it remains to be determined if these changes are adaptive and which specific factors of the urban environment induce epigenetic changes. Also, these differences in DNA methylation could have arisen in response to the environment that the birds experienced when they were sampled, or, since they were not exposed to each environment separately, could be inherent differences between the birds.
One of the factors that has the potential to explain methylation levels between urban and rural populations is pollution. The effect of pollution was experimentally studied in wild-living Double-crested Cormorants (Phalacrocorax auritus), where they were exposed to polycyclic aromatic compounds (PACs). However, no significant difference in DNA methylation levels were found between exposed and non-exposed individuals, even though PAC levels in the liver increased with PAC exposure (Wallace et al. 2018). Nevertheless, it is possible but that differences were undetected because global DNA methylation was estimated. DNA methylation was determined by immunostaining, which compares the staining intensity of the samples to those of fully methylated and fully unmethylated DNA (Wallace et al. 2018). This method reveals a global difference in DNA methylation, but site-specific differences remain undetected. Significant differences in the effects of pollution have been found while studying circadian locomotor output cycles kaput (CLOCK) gene DNA methylation in Barn Swallow nestlings (Hirundo rustica). DNA methylation appeared to increase with individual and maternal exposure to free air particulate matter levels (Romano et al. 2017). Since the CLOCK gene is important for maintaining circadian rhythms and circannual life cycles, its methylation could severely impact fitness (Romano et al. 2017).
Environmental causes of epigenetics: future directions
The studies discussed above give very promising insights into the possible importance of methylation for individual adaption. As a next step towards understanding the ecological and adaptive relevance of DNA methylation in natural systems, variation in individual phenotypes should be linked to variation in functional DNA methylation. We thus suggest combining data on individual behavioural phenotypes and fitness with DNA methylation levels of individuals of a bird species from a natural population. Good model species for this are the Great Tit, House Sparrow and several flycatcher species, since there are numerous natural populations of these species that have been studied for years (Backström et al. 2008; Ellegren et al. 2012; Kawakami et al. 2017; Laine et al. 2016; Riyahi et al. 2017; Schrey et al. 2011, 2012; Sheldon et al. 2018a). Consequently, there are ample available data and DNA methylation can thus be associated with gene function and age (Sutherland et al. 2013), and also with variables which fluctuate annually such as climate variables and food availability. Moreover, variation in DNA methylation may be associated with important fitness components such as fledging, offspring recruitment rate, adult survival, timing of breeding and reproductive success (Gruebler and Naef-Daenzer 2008; Naef-Daenzer and Gruebler 2008, 2016; Visser and Verboven 1999). Methods that allow one to determine relationshiips between genome-wide methylation levels in functional gene regions with fitness measures such as EpiGBS or RRBS (Table 1) are preferable. Furthermore, we advise focusing on adaptive phenotypic traits that vary between individuals of the same species where expression is known to be affected by environmental factor(s). A good example of such traits is exploratory behaviour (Dingemanse et al. 2004; Naguib et al. 2011; Tinbergen and Sanz 2004; van Oers et al. 2015).
The studies described above also show that it is very hard to differentiate between correlation and causation, and to disentangle prenatal from postnatal effects when studying the causes of epigenetic marks. In order to separate the causes and effects of changes in methylation, it is essential to evaluate the consequences of methylation through careful experimental manipulation of environmental factors during early development. A split-brood cross-foster experiment is an example of one such type of experiment, in which additional factors such as food availability or brood size can be manipulated. Food availability and brood size are two naturally occurring ecologically relevant parameters during early avian development. Using a split-brood cross-foster component, the effects of different environmental factors (e.g. food deprived versus not food deprived) can be compared between siblings while also disentangling these effects from genetic effects and/or parental care.
In conclusion, (early) environmental conditions can influence (offspring) DNA methylation and this might affect the phenotype. These altered phenotypes can be adaptive changes to the environmental conditions under which they were induced. However, this can only be verified by studying the complete cascade within an individual’s lifetime.
Regulation of temporal plastic changes
Here, we consider plastic changes within the context of phenotypic flexibility defined as intra-individual and reversible changes in a phenotype that underlie seasonally expressed life history traits. Many species orchestrate their timing of life history events in circannual cycles that depend on environmental cues such as photoperiod or temperature (Dawson et al. 2001; Kumar et al. 2010; Rowan 1925; Sharp 2005). Seasonal events require an individual to undergo a combination of morphological, physiological and behavioural changes and hence the seasonal regulation of gene expression in various tissues (Stevenson 2018; Wingfield 2005). Recent studies in species of plants (Lai et al. 2018; Law and Jacobsen 2010; Shi et al. 2015; Wilschut et al. 2016), insects (Hatakeyama and Mueller 2008; Pegoraro et al. 2016), and rodents (Alvarado et al. 2015; Lynch et al. 2017; Stevenson and Prendergast 2013) show evidence for reversible epigenetic modifications, especially DNA methylation, to be involved in this. How reversible DNA methylation can shape seasonally stimulated life history events in an animal species is best studied and understood in the Siberian Hamster (Phodopus sungorus), in which short day length inhibits hypothalamic DNA methyltransferase 3a (DNMT3a) expression causing reduced promoter methylation in type III deiodinase, a gene involved in the photoperiodic regulation of reproduction (Lynch et al. 2016; Stevenson 2017; Stevenson and Prendergast 2013). Such studies in birds are currently limited. Redheaded Buntings (Emberiza bruniceps) in a migratory and non-migratory state differ in their expression level of hypothalamic DNMT3a (Sharma et al. 2018), but it remains to be established whether differential expression of DNMT3a promotes reversible methylation marks between the migratory states. In Barn Swallows increased methylation at a CpG site within the CLOCK gene is associated with earlier spring departure from the wintering area, earlier arrival at the breeding site, earlier breeding, and higher reproductive success (Saino et al. 2017). The latter study therefore provides some support that variation in DNA methylation may be of relevance for determining temporally plastic life history trait variation such as the timing of breeding in a wild bird population. Indeed, DNA methylation can show changes over short timescales, i.e. throughout the breeding season (Viitaniemi et al. 2019), and therefore facilitate phenotypic flexibility, as described above. This offers opportunities for the study of many other temporarily changing traits, such as migratory phenotypes (Merlin and Liedvogel 2019).
Role of epigenetics in regulating the timing of reproduction
The timing of reproduction is an important fitness trait in seasonally breeding birds (Catry et al. 2017; Daan et al. 1990; Perrins 1970; Thomas et al. 2001; Verhulst et al. 1995; Visser et al. 1998) and is a phenotypically plastic trait (Charmantier et al. 2008; Husby et al. 2010). Phenotypic plasticity in the timing of reproduction allows a female to seasonally express reproductive morphology and behaviour in response to changes in environmental cues such as photoperiod and temperature. While the underlying genetic basis (Gienapp et al. 2017) and regulatory mechanisms for the timing of reproduction and plasticity in the timing of reproduction are currently unknown in birds, several studies in plants and rodents suggest that epigenetic processes, such as DNA methylation, are involved. For example, in clonally reproducing dandelion, treatment with a cytosine demethylating agent altered the clonally inherited pattern of the timing of flowering (Wilschut et al. 2016). Similar findings were reported when inducing DNA methylation differences between isogenic Arabidopsis lines (Law and Jacobsen 2010); treatment resulted in differences in flowering time between the different lines. The only animal studied so far for the experimental testing of effects of DNA methylation on the timing of reproduction is the Siberian Hamster, as described by Stevenson and Prendergast (2013). Females of Nasonia vitripennis, a parasitic wasp, that were exposed to short or long day light cycles showed differences in their methylation pattern and diapause response that disappeared when DNA methylation was experimentally altered (Pegoraro et al. 2016). Although DNA methylation in insects is different from that in vertebrates (Keller et al. 2016), this result nevertheless shows the potential of DNA methylation as a mechanism effecting a photoperiodic response. The studies referred to above suggest that DNA methylation can play a role in the timing of reproduction in many different species, although this has so far been poorly explored in birds. This has been supported by temporally varying DNA methylation patterns in Great Tit individuals (Viitaniemi et al. 2019), and ongoing work indicates a correlation between DNA methylation level and a female’s reproductive stage in this species (Lindner et al., unpublished data).
Unfortunately, studies such as those described above are limited by the number of measurements required per female or by the type of tissue which needs to be collected, and this hampers our ability to simultaneously examine how changes in methylation might lead to changes in gene expression and hence a phenotypic response. Females cannot be sampled repeatedly to collect inaccessible tissues with a clear role in the timing of reproduction, i.e. the hypothalamus, liver, or gonads), and thus it cannot be determined whether differences in methylation levels indicate a flexible change in methylation marks or permanently established differences in them.
To increase the number of measurements per female a more accessible tissue, like blood, should be sampled. Sampling blood allows for repeated measurements of the same individual across the breeding season, but it is not yet clear to what extent observed DNA methylation patterns in blood correlate with methylation patterns in other (reproductive) tissues. Thus, future work needs to establish how changes in red blood cell methylation patterns over the breeding season are related to changes in DNA methylation and gene expression changes in other tissues to better understand the functional role of blood DNA methylation variation on phenotypes such as the timing of reproduction.
The role of DNA and histone modifications in the brain and cognition
There is a specific research interest in defining the role of natural selection in shaping cognitive abilities (Rowe and Healy 2014). We have to understand the causes and consequences of intra-individual variation in cognitive performance to understand how natural selection shapes cognition (Boogert et al. 2018). A recent study found that cognitive ability is heritable in mice, but also highly malleable according to environmental effects (Sauce et al. 2018). Intra-individual variation in cognitive performance is thus possibly the result of constant interaction between genes and experience that may be mediated by epigenetic mechanisms (Sweatt 2019). Recent developments in the study of mammals have demonstrated that experience can lead to epigenetic alterations in the brain, altering gene expression and consequently leading to changes in behaviour and memory formation (Halder et al. 2016; Miller et al. 2008). For example, inhibition of DNMT results in learning and memory deficits and a change in the DNA methylation of several other neural plasticity genes, including the brain-derived neurotrophic factor gene (BDNF) (Feng et al. 2010a; Levenson et al. 2006).
Avian learning and memory
The results of several studies also indicate that epigenetic processes play roles in avian learning and memory. Using DNMT expression as a proxy for epigenetic potential within the hippocampus, it was found that House Sparrows with intermediate corticosterone levels had higher BDNF expression in the hippocampus than individuals with low or high levels of corticosterone (Kilvitis et al. 2018). In addition, corticosterone positively covaried with DNMT1 expression in a more recently established population, while the reverse was true in the oldest population. It was hypothesized that certain environmental conditions could induce high DNMT expression in the hippocampus facilitating neural plasticity (Kilvitis et al. 2018). Furthermore, demethylation of non-neuronal tumour cells of Zebra Finches resulted in upregulation of genes that were neurobiologically relevant and under the putative control of DNA methylation (Steyaert et al. 2016).
The role of histone modifications has also been studied in this context. Pharmacological inhibition of histone deacetylases (HDAC) in Zebra Finches contributed to the memorization of conspecific vocalizations (Phan et al. 2017). The fact that HDAC can be recruited by DNA methylation, which leads to the removal of acetyl groups resulting in constricted access to chromatin and transcriptional silencing (Day and Sweatt 2011), might indicate that DNA methylation regulates song memory formation in birds through the modulation of the structure of chromatin. This is supported by a study in rats, where inhibiting HDAC with sodium butyrate lead to the repair of impaired memory formation induced by a DNMT inhibitor (Miller et al. 2008). In addition to histone acetylation, histone methylation is also associated with learning and memory. In Zebra Finches it was shown that tutor experience, which induces the closing of the ‘critical period’ of learning, leads to differentially modified genes that are associated with histone methylation. This indicates that tutor experience causes a decreased probability of transcription by altering the epigenetic profile through post-translational modifications of histone (Kelly et al. 2018).
Natural selection and cognition
If intra-individual variations in cognitive performance are mediated by epigenetic mechanisms (Sweatt 2019), then they may play an important role in the evolution of cognition. Indeed, genes in regions of past selection showed increased CpG methylation in Great Tits, and were biased towards those involved in learning, cognition and neuronal functions in the this species (Laine et al. 2016). In addition, neuronal non-CpG methylation patterns were correlated with rates of molecular evolution (Laine et al. 2016). Assessing whether epigenetic marks underlie intra-individual variation in cognitive performance can help us to understand the role of epigenetic mechanisms in the evolution of learning and memory. By artificially selecting for cognitive performance and studying epigenetic marks in the parental generation, as well as changes in selected generations, one can assess not only the nature of these marks but also whether they have the potential to be selected upon. If bi-directional selection for cognitive performance leads to divergent epigenetic patterns, this would indicate that there is heritable variation for cognition-related epigenetic variation, most likely linked to other genetic variants.
The causes of differential cognitive performance can be studied by manipulating the early environment. Social experiences, such as maternal care and social isolation, have been linked to epigenetic changes in the brain leading to altered cognitive performance (Li et al. 2016; Roth et al. 2009). Nutritional factors can also cause epigenetic alterations. Protein deficiency, e.g. as a result of malnutrition, may reduce methionine availability and subsequent DNA methylation. Several studies have indicated the importance of methionine conversion for cognitive functioning. For example, Saunderson et al. (2016) found that DNA methylation and S-adenosylmethionine availability in the hippocampus control stress-induced gene expression and behaviour in mice, and that the offspring of mothers that had a diet deficient in methyl donors showed increased anxiety and decreased learning ability (Konycheva et al. 2011). Future studies on methylation variation in relation to cognitive performance should focus on whether genes and environment interact to influence cognitive variation, and whether this occurs through epigenetic mechanisms. This can be achieved by, for example, manipulating the environment during early development, as has been described before, when at the same time controlling for the genetic environment.
Cognitive phenotypic traits for study: reversal learning
A particularly interesting cognitive trait for selection is cognitive flexibility, the ability to cope with unexpected changes in the environment. This is generally assessed by measuring performance regards a reversal learning task, which tests how well an animal can attend to a shift in reward location and adjust its behaviour in response to this (Bonte et al. 2014; Izquierdo et al. 2017). Individual differences in reversal learning performance may be driven by variable responses and epigenetic mechanisms that involve learning and memory formation. Genetic variation between individuals may cause variable levels of enzymes that control epigenetic mechanisms, such as DNMTs, which could lead to variable functioning of memory pathways that require epigenetic alteration. Alternatively, performance variation may be due to variable feedback performance in the relevant brain regions. Individual differences in reversal learning performance may be caused by differences in sensitivity to neurotransmitter feedback during reversal learning (Klanker et al. 2015). Epigenetic marks causing the up- or downregulation of neurotransmitter synthesis enzymes, receptors or transporters (re-uptake) may lead to altered functioning of these feedback systems.
Cognitive phenotypic traits for study: innovativeness
Another possible cognitive trait for study is innovative problem-solving performance (PSP). PSP varies between individuals of the same species (Cole et al. 2011), but is rarely studied in the wild (Cole et al. 2011; Quinn et al. 2016). An individual is innovative when it solves a novel problem or solves an existing problem with an original solution (Kummer and Goodall 1985). PSP is thought to be linked to foraging strategy and personality in Great Tits (Quinn et al. 2016; Zandberg et al. 2017), and seems to be largely affected by age and environmental factors, such as habitat quality and population density in the natal environment (Quinn et al. 2016). Therefore, it is possible that environmental effects on PSP are mediated via epigenetic mechanisms such as DNA methylation. To experimentally confirm environmental effects on PSP and to assess a possible role of DNA methylation, we suggest influencing the natal environment of Great Tit chicks and assessing their PSP and DNA methylation levels later in life. PSP could be perfectly studied within (for example) split-brood cross-foster experiments described above, since it is easy to apply the method used to study PSP (Cole et al. 2011).
Inheritance, selection and evolution
Inheritance: a matter of definition
One of the central questions when studying epigenetics in an evolutionary context is whether epigenetic marks are inherited and if selection can act on this variation directly. Discussions on the inheritance of epigenetic marks often lead to great confusion. Hence, a crucial distinction must be made regards the type of inheritance that is being considered, since this has consequences for our expectations and inferences. Specifically, when discussing DNA methylation, three different ways of inheritance or epigenetic memory exist and should be identified and clarified when defining the scope of a study (Bonasio 2015).
First, there is mitotic inheritance (Zhu and Reinberg 2011), which is the replication of epigenetic marks throughout DNA replication during, for example, cell division. Most classic studies refer to mitotic inheritance as a prerequisite for epigenetic variation, and this is also the case in the definition given above. Mitotic inheritance causes early developmental effects to remain in the affected individual, with lifelong consequences.
The second way an epigenetic mark may be inherited is via meiotic inheritance, in which an epigenetic mark survives the extensive reprogramming events that are so typical in vertebrates (Feng et al. 2010b; Morgan et al. 2005). This type of inheritance is also referred to as ‘true inheritance’, since it is independent of genetic variation. It is mostly seen in plants in epialleles (Feil and Fraga 2012); in vertebrates, a few examples exist in mice, where such epialleles are stably inherited via the germ line. It was long thought that epigenetic variation was not heritable through the germ line because most epigenetic marks do not survive meiosis, gametogenesis and embryogenesis, which are processes that involve DNA demethylation (Feng et al. 2010b; Morgan et al. 2005; Reik et al. 2001; Sasaki and Matsui 2008) and restructuring of chromatin (Jablonka and Raz, 2009) in mammals. However, the majority of empirical studies to date that have shown an epigenetic mark remaining in offspring could not prove clear meiotic inheritance (Bonasio 2015). For birds, it is not clear how epigenetic marks can be inherited through the germ line, but one theory is that some ancestral epigenetic marks escaped epigenetic resetting (Brykczynska et al. 2010) and that others were reconstructed (Gapp et al. 2014; Kasowski et al. 2013; Schaefer and Nadeau 2015). There is, however, very scarce information on these processes for birds (Guerrero-Bosagna et al. 2018), which is limited to Chicken and quail.
The third way an epigenetic mark can be inherited from one generation to the next is via so-called soma-to-some inheritance (Ledon-Rettig et al. 2013), i.e. epigenetic changes are inherited through parent–offspring interactions or interactions between siblings during development. Soma refers to the fact that the epigenetic mark will not inherit via the germ line and must therefore be transmitted via a parentally induced environmental factor, with licking and grooming in rats the best example of this (Hu and Barrett 2017; Jensen 2013; Weaver et al. 2004).
One complication in the study of inherited epigenetic marks is that the presence of epigenetic similarity between two subsequent generations does not mean that this pattern has been present in and inherited via parental germ cells, an indicator of transgenerational inheritance. In birds, if the F0 generation is subjected to an environmental condition (e.g. environmental cue or stressor), then the germ cells (F1) present in these F0 individuals will also be affected by this experimental treatment. In mammals, in utero exposure of the F1 generation can also affect the germ cells (F2) of the F1 generation (Faulk and Dolinoy 2011). In birds, development of an embryo in the future (F1 generation) and its primordial germ cells (F2 generation) are directly affected by egg components, the mother, and also by her pre-laying and incubation environment. Therefore, inter-generational (from parent to offspring) or multi-generational (from parents to offspring and from grandparent to grand-offspring) effects cannot necessarily be considered as transgenerational epigenetic effects (Guerrero-Bosagna et al. 2018). Hence, even though epigenetic signals can be traced as far as the F3 generation, these might be the result of other mechanisms such as parental care by the F2 generation (Weaver et al. 2004).
Epigenetic marks may also seem to be inherited if they are a direct consequence of underlying genetic variation. In a strict sense, the epigenetic mark will therefore not be inherited directly, but will reappear each time the gene in question that is responsible for the variation in methylation is expressed. No study has investigated the way epigenetic marks might have been inherited in wild birds, but some evidence exists that there is genetic variation in underlying DNA methylation variation. In a study on on early exploratory behaviour, fast and slow exploration by Great Tits originating from a fourth generation of selection (Drent et al. 2003) showed consistent and heritable differences in exploratory behaviour (Drent et al. 2003; van Oers and Naguib 2013). Despite extensive genome-wide association and quantitative trait locus studies in both a Dutch (Santure et al. 2015) and a UK (Kim et al. 2018; Santure et al. 2015) population, no consistent candidates were found to explain substantial portions of the additive genetic variation found. However, recent research has revealed that lines selected for high and low levels of exploratory behaviour differed in their levels of DNA methylation at the Great Tit DRD4 promoter (Verhulst et al. 2016), a gene known to explain significant additive genetic variation in exploratory behaviour in a wild founder population (Fidler et al. 2007; Korsten et al. 2010; Mueller et al. 2013). Therefore, epigenetic mechanisms might be involved in functional and heritable differences in exploratory behaviour (Verhulst et al. 2016).
Future directions: multigenerational studies
Although underlying genetic variation is a crucial factor determining whether epigenetic variation is subject to evolutionary change (Furrow and Feldman 2014), to what extent genetic variation causes transgenerational consistency in wild vertebrates remains largely unknown (Daxinger and Whitelaw 2012). The genetic control of gene methylation can be unravelled by combining scans for differentially methylated sites and regions using, for example, an RRBS approach on a cross population such as an F2-cross population (van Oers et al. 2014), with WGBS on lines selected for a certain phenotypic trait. Such an approach would allow for adjustment for between-family variation in methylation and for finding differentially methylated sites throughout the genome. Furthermore, this method provides insight into which genomic features (e.g. intragenic, promoter intergenic) likely play a major role in the link between phenotypic and methylation variation. By using a control group, it is possible to assess if false positives are present due to drift and co-selection.
Evolution and selection
Theory predicts that, in the case of rapid environmental changes, founding populations might not have enough genetic diversity to adapt (Allendorf and Lundquist 2003). It has therefore been suggested that epigenetic mechanisms might provide additional scope for adaptive variation. The experience of a new environment might cause a change in epigenetic patterns, and since epigenetic marks can alter gene expression, this might provide an animal with a phenotype that makes it fit better to a new environment (Liebl et al. 2013). It is unclear though whether this might apply to epigenetic variation that is independent of genetic variation.
Another role for methylation in selection and evolution is not via the influence of phenotypes, but via the characteristics of the positions in the genome where methylation variation is most functional. For example, in the genomes of two flycatcher species (Ficedula hypoleuca and Ficedula albicollis) associations between recombination hotspots and CGIs and TEs were found (Kawakami et al. 2017), indicating that CGIs, that are know to have lower methylation values, are associated with those parts of the genome that have the highest recombination. Although DNA methylation was not studied directly by Kawakami et al. (2017), DNA methylation is known to alter the accessibility of DNA via states of chromatin condensation (Jaenisch and Bird 2003). It is possible that, due to hypomethylation, the chromatin in CGIs is less condensed, which allows for binding of the transposition and recombination machinery, as seen in other species (Berglund et al. 2015; Choi et al. 2013; Comeron et al. 2012; Shilo et al. 2015). Therefore, this might be an example where epigenetic marks do not affect phenotypic variation via gene expression, but via involvement in the regulation of recombination (Kawakami et al. 2017). In Great Tit brain tissue, genes in regions that have undergone selective sweeps were found to have higher CpG methylation and lower non-CpG methylation levels compared to those that were outside of these selective sweep regions, pointing to a possible facilitating role for epigenetic variation in selection, or alternatively, that methylation variation is affected by past selection (Laine et al. 2016). The correlation between epigenetic differences (DNA methylation) and phylogenetic distance between five closely related species of Darwin’s finches (Geospiza fortis, Geospiza fuliginosa, Geospiza scandens, Camarhynchus parvulus and Platyspiza crassirostris) appeared to be positive (Skinner et al. 2014). Since phylogenetic distance can be used as a measure of evolutionary time, this suggests that DNA methylation accumulates over time. The epigenetic marks were located near gene families that were related to immunological function, colour and beak shape. The number of DNA methylations exceeded the number of genetic mutations in the form of copy number variations (Skinner et al. 2014), which suggests that the environment caused a large part of the epigenetic variation. However, this only makes sense if this epigenetic variation is independent of genetic variation. Overall, this latter study, which is not unique, indicates that DNA methylation has a great impact on the evolutionary change of a phenotype and that methylation might be involved in speciation.
Indeed, the potential role of epigenetics in evolution and selection has been quite thoroughly studied in the introduced House Sparrow. Even though methylation patterns were similar, younger populations of the House Sparrow in Kenya had greater DNA methylation at two loci compared to older populations in Florida (Schrey et al. 2012). Furthermore, epigenetic diversity appeared to be negatively correlated to genetic diversity, while a positive correlation between epigenetic diversity and inbreeding was found (Liebl et al. 2013). Since genetic diversity in relatively new populations is lower than in native and older populations (Schrey et al. 2011), this may suggest that epigenetic variation may increase phenotypic variation and therefore make up for the loss of genetic variation during the process of introduction. Again, this only is true if this epigenetic variation is independent of genetic variation.
Local epigenetic adaption
When heritable epigenetic variation translates into an increase in fitness in those environments in which it became established there is potential for local adaptation. To study this, the epigenetic differentiation between and within five native subspecies of House Sparrows from the Middle East was investigated, while using a Spanish subspecies as an out-group (Riyahi et al. 2017). These subspecies, except for one Middle Eastern subgroup (Passer domesticus bactrianus), are human commensals. The genome-wide DNA methylation variation was quite similar among the Middle Eastern subspecies. There was significant differentiation between the non-commensal subspecies and two commensal subspecies and between two Middle Eastern commensal subspecies. Furthermore, the European subspecies appeared to be differentiated from the Middle Eastern subspecies. Focusing on the Middle Eastern subspecies only, some loci showed significant differentiation based on subspecies, commensalism and sex. Furthermore, both geographical distance and standardized bill length appeared to be significantly positively correlated to the percentage of DNA methylation in the Middle Eastern subspecies. These findings in combination with the fact that most of the epigenetic differentiation could be attributed to differences within subspecies rather than among them, illustrates that a substantial proportion of the methylome is typically stable (Riyahi et al. 2017). This might mean that most of the variation observed had a genetic background, and only specific phenotypic traits can be influenced by environmental factors through methylation.
Such a specific phenotypic trait could be, for example, the functioning of the immune system. A study on Kenyan House Sparrows discovered that the expression of Toll-like receptor 4 (TLR-4), which plays a role in the immune system, varies with population age. Sparrows from older populations had lower expression of TLR-4 than individuals from younger populations (Schrey et al. 2011). The fact that genetic diversity in relatively new populations was lower than in native and older populations (Schrey et al. 2011), and since younger populations had greater DNA methylation (Schrey et al. 2012), and epigenetic diversity appeared to be negatively correlated to genetic diversity (Liebl et al. 2013), might indicate that epigenetic variation, such as DNA methylation, is responsible for this phenotypic variation. It appears that, in liver tissue of House Sparrows, DNA methylation in the TLR-4 promoter is very diverse and a good predictor of TLR-4 expression (Hanson et al. 2018; Kilvitis et al. 2016). However, the effects of differential expression remain to be studied. Furthermore, it is unknown whether this epigenetic variation is a response to environmental factors or is inherited.
In a recent study on wild Zebra Finches, levels of epigenetic and genetic diversity were compared across 15 different Australian sites (Sheldon et al. 2018a). In the analysis of the genetic data, the populations clustered into three groups that were consistent with three introduction events. There was no correlation between genetic and epigenetic pairwise site comparisons, which suggests that at least part of the variation in DNA methylation arose independently of genetic variation. Furthermore, significant epigenetic differentiation between the clusters was found. However, an opposite pattern to that of genetic differentiation was found: stronger epigenetic differentiation was found among sample sites compared to among invasion clusters. This suggests that local environmental variation is more important in explaining between-population differences in epigenetic variation than the founder diversity of an introduced population (Sheldon et al. 2018a). However, the potential environmental or ecological drivers of this variation remain to be investigated. Furthermore, the genes that were possibly influenced by differential methylation were not specified.
As mentioned above, epigenetic differentiation has been detected among subspecies of the House Sparrow in its native range (Riyahi et al. 2017). In contrast, no epigenetic differentiation was found in a Kenyan population of this species during the initial invasion stages (Liebl et al. 2013). This could be explained by a relatively small sample size in the Kenyan study, the use of blood in one study and the use of muscle tissue in the other, or the fact that the African invasion was more recent and epigenetic marks need time to stabilize and accumulate. If the latter explanation is correct, it is unclear if and how population age affects epigenetic diversity. Moreover, this indicates that comparable methods, tissues and timing are relevant factors when trying to compare epigenetic studies.
Closing remarks
We have reviewed the current status within the field of avian epigenetics and demonstrate that there is a large potential for studies on wild birds to answer many outstanding questions in ecology and evolution. It is also clear that we are facing some challenges in avian epigenetics. First, while most studies have focused on DNA methylation, mostly in the context of CGI promoters, we should keep in mind that other epigenetic mechanisms are also important (Bossdorf et al. 2008; Jaenisch and Bird 2003; Korochkin 2006). For example, studies on humans have found that changes in DNA methylation in promoter regions might not be the most informative, and that methylation variation might not act directly on the closest gene but on more distant ones (cis regulation) (Suzuki et al. 2018). Thus, observing DNA methylation changes around a putative gene might not indicate a functional change in the expression of that particular gene. This indicates the need for more validation steps in ecological studies to examine how changes in methylation relate to changes in gene expression and phenotype. This type of validation should ideally be conducted on a tissue-specific individual gene level, since general changes in tissue methylation may lead to spurious correlations that indicate that only specific genes are truly affected and differentially expressed.
It is largely unknown at which level different perturbations of DNA methylation act in birds. Methylation levels might increase or decrease genome-wide due to, for example, aging (Christensen et al. 2009) or stressful events that occur early in an individuals’s life (Sheldon et al. 2018a). But whether such changes are global or rather very local, and how many CpG sites have to be changed before a phenotypic effect arises, are open questions that need more study. The timing of sampling in epigenetic studies is therefore important (Viitaniemi et al. 2019). One benefit of working on birds is that, in contrast to mammalian erythrocytes, avian erythrocytes are nucleated. This offers a great opportunity to sample a uniform set of cells across time in the same individuals and relate such changes to observed changes in the phenotype. Although DNA methylation patterns in the blood might not reflect those of other tissues, studies on Great Tits have found a significant correlation between methylation in brain and blood tissues (Derks et al. 2016). Therefore, it is possible that avian blood cell methylation levels can be used as biomarkers for methylation in other tissues, which would greatly facilitate measuring methylation in individuals from natural populations, as most avian (long-term) studies sample blood of individuals in a population. We note, however, that this cross-tissue generality should be considered with caution, as there can be considerable variation in particular CpG sites in their methylation in different tissues, even though there is overall a strong correlation between genome-wide CpG methylation across tissues.
While there is no doubt that ecological epigenetics is an important addition to the long-standing history of functional studies on the causes and consequences of phenotypic variation, it is important to keep in mind that ecological studies are incomplete without additional laboratory studies. Laboratory studies are still needed to determine if epigenetic mechanisms causally affect gene expression, and whether these changes in gene expression also affect behaviour in the predicted way, although ideally, in the future, all of this will studied in the same natural population by, for example, releasing captive individuals with altered methylation states. Conversely, early developmental changes in phenotypes that inherit over generations do not imply stably inherited epigenetic changes. For an emerging field such as avian ecological epigenetics, it is important to produce conclusive experimental data that are followed up by functional validation studies. We anticipate that this type of study may shortly be carried out as more and more avian studies on epigenetics find interesting patterns that can be examined in more detail.
References
Ahmed AA, Ma WQ, Ni YD, Zhou Q, Zhao RQ (2014) Embryonic exposure to corticosterone modifies aggressive behavior through alterations of the hypothalamic pituitary adrenal axis and the serotonergic system in the Chicken. Horm Behav 65:97–105. https://doi.org/10.1016/j.yhbeh.2013.12.002
Allendorf FW, Lundquist LL (2003) Introduction: population biology, evolution, and control of invasive species. Conserv Biol 17:24–30. https://doi.org/10.1046/j.1523-1739.2003.02365.x
Alvarado S, Mak T, Liu S, Storey KB, Szyf M (2015) Dynamic changes in global and gene-specific DNA methylation during hibernation in adult thirteen-lined ground squirrels, Ictidomys tridecemlineatus. J Exp Biol 218:1787–1795. https://doi.org/10.1242/jeb.116046
Anastasiadi D, Esteve-Codina A, Piferrer F (2018) Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 11:37. https://doi.org/10.1186/s13072-018-0205-1
Araya-Ajoy YG, Dingemanse NJ (2017) Repeatability, heritability, and age-dependence of seasonal plasticity in aggressiveness in a wild passerine bird. J Anim Ecol 86:227–238. https://doi.org/10.1111/1365-2656.12621
Backström N, Karaiskou N, Leder EH, Gustafsson L, Primmer CR, Qvarnstrom A, Ellegren H (2008) A gene-based genetic linkage map of the Collared Flycatcher (Ficedula albicollis) reveals extensive synteny and gene-order conservation during 100 million years of avian evolution. Genetics 179:1479–1495. https://doi.org/10.1534/genetics.108.088195
Becker C, Hagmann J, Muller J, Koenig D, Stegle O, Borgwardt K, Weigel D (2011) Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480:245–249. https://doi.org/10.1038/nature10555
Bentz AB, Sirman AE, Wada H, Navara KJ, Hood WR (2016) Relationship between maternal environment and DNA methylation patterns of estrogen receptor alpha in wild Eastern Bluebird (Sialia sialis) nestlings: a pilot study. Ecol Evol 6:4741–4752. https://doi.org/10.1002/ece3.2162
Berglund J, Quilez J, Arndt PF, Webster MT (2015) Germ line methylation patterns determine the distribution of recombination events in the dog genome. Genome Biol Evol 7:522–530. https://doi.org/10.1093/gbe/evu282
Bewick AJ et al (2016) On the origin and evolutionary consequences of gene body DNA methylation. Proc Natl Acad Sci USA 113:9111–9116. https://doi.org/10.1073/pnas.1604666113
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21. https://doi.org/10.1101/gad.947102
Boffelli D, Martin DIK (2012) Epigenetic inheritance: a contributor to species differentiation? DNA Cell Biol 31:S-11–S-16. https://doi.org/10.1089/dna.2012.1643
Bonasio R (2015) The expanding epigenetic landscape of non-model organisms. J Exp Biol 218:114–122. https://doi.org/10.1242/jeb.110809
Bonduriansky R, Day T (2009) Nongenetic inheritance and its evolutionary implications. In: Annual review of ecology evolution and systematics, vol 40. Annual Reviews, Palo Alto, pp 103–125. https://doi.org/10.1146/annurev.ecolsys.39.110707.173441
Bonte E, Kemp C, Fagot J (2014) Age effects on transfer index performance and executive control in Baboons (Papio papio). Front Psychol 5:7. https://doi.org/10.3389/fpsyg.2014.00188
Boogert NJ, Madden JR, Morand-Ferron J, Thornton A (2018) Measuring and understanding individual differences in cognition. Philos Trans R Soc B Biol Sci 373:10. https://doi.org/10.1098/rstb.2017.0280
Bossdorf O, Richards CL, Pigliucci M (2008) Epigenetics for ecologists. Ecol Lett 11:106–115
Bosse M et al (2017) Recent natural selection causes adaptive evolution of an avian polygenic trait. Science 358:365–368. https://doi.org/10.1126/science.aal3298
Both C, Bouwhuis S, Lessells CM, Visser ME (2006) Climate change and population declines in a long-distance migratory bird. Nature 441:81–83
Boyle P et al (2012) Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. https://doi.org/10.1186/gb-2012-13-10-r92
Brykczynska U et al (2010) Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nat Struct Mol Biol 17:679–687. https://doi.org/10.1038/nsmb.1821
Cantarero A, Lopez-Arrabe J, Rodriguez-Garcia V, Gonzalez-Braojos S, Ruiz-de-Castaneda R, Redondo AJ, Moreno J (2013) Factors affecting the presence and abundance of generalist ectoparasites in nests of three sympatric hole-nesting bird species. Acta Ornithol 48:39–54. https://doi.org/10.3161/000164513x669982
Carere C, Drent PJ, Koolhaas JM, Groothuis TGG (2005) Epigenetic effects on personality traits: early food provisioning and sibling competition. Behaviour 142:1329–1355
Catry T, Moreira F, Alcazar R, Rocha PA, Catry I (2017) Mechanisms and fitness consequences of laying decisions in a migratory raptor. Behav Ecol 28:222–232. https://doi.org/10.1093/beheco/arw150
Champagne FA (2008) Epigenetic mechanisms and the transgenerational effects of maternal care. Front Neuroendocrinol 29:386–397. https://doi.org/10.1016/j.yfrne.2008.03.003
Charmantier A, Perrins C, McCleery RH, Sheldon BC (2006) Evolutionary response to selection on clutch size in a long-term study of the Mute Swan. Am Nat 167:453–465. https://doi.org/10.1086/499378
Charmantier A, McCleery RH, Cole LR, Perrins C, Kruuk LEB, Sheldon BC (2008) Adaptive phenotypic plasticity in response to climate change in a wild bird population. Science 320:800–803. https://doi.org/10.1126/science.1157174
Choi KH et al (2013) Arabidopsis meiotic crossover hot spots overlap with H2A.Z nucleosomes at gene promoters. Nat Genet 45:1327
Christensen BC et al (2009) Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CPG island context. PLoS Genet. https://doi.org/10.1371/journal.pgen.1000602
Cockburn A (2006) Prevalence of different modes of parental care in birds. Proc R Soc B Biol Sci 273:1375–1383. https://doi.org/10.1098/rspb.2005.3458
Cole EF, Cram DL, Quinn JL (2011) Individual variation in spontaneous problem-solving performance among wild Great Tits. Anim Behav 81:491–498. https://doi.org/10.1016/j.anbehav.2010.11.025
Comeron JM, Ratnappan R, Bailin S (2012) The many landscapes of recombination in Drosophila melanogaster. PLoS Genet 8:21. https://doi.org/10.1371/journal.pgen.1002905
Couldrey C, Cave V (2014) Assessing DNA methylation levels in animals: choosing the right tool for the job. Anim Genet 45:15–24. https://doi.org/10.1111/age.12186
Daan S, Dijkstra C, Tinbergen JM (1990) Family-planning in the kestrel (Falco tinnunculus)—the ultimate control of covariation of laying date and clutch size. Behaviour 114:83–116. https://doi.org/10.1163/156853990x00068
David SA, Mersch M, Foissac S, Collin A, Pitel F, Coustham V (2017) Genome-wide epigenetic studies in Chicken: a review. Epigenomes. https://doi.org/10.3390/epigenomes1030020
Dawson A, King VM, Bentley GE, Ball GF (2001) Photoperiodic control of seasonality in birds. J Biol Rhythms 16:365–380. https://doi.org/10.1177/074873001129002079
Daxinger L, Whitelaw E (2012) Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet 13:153–162. https://doi.org/10.1038/nrg3188
Day JJ, Sweatt JD (2011) Epigenetic mechanisms in cognition. Neuron 70:813–829. https://doi.org/10.1016/j.neuron.2011.05.019
Derks MFL, Schachtschneider KM, Madsen O, Schijlen E, Verhoeven KJF, van Oers K (2016) Gene and transposable element methylation in Great Tit (Parus major) brain and blood. BMC Genomics 17:13. https://doi.org/10.1186/s12864-016-2653-y
Dingemanse NJ, Both C, Drent PJ, Tinbergen JM (2004) Fitness consequences of avian personalities in a fluctuating environment. Proc R Soc B Biol Sci 271:847–852. https://doi.org/10.1098/rspb.2004.2680
Drent PJ, van Oers K, van Noordwijk AJ (2003) Realized heritability of personalities in the great tit (Parus major). Proc R Soc B Biol Sci 270:45–51. https://doi.org/10.1098/rspb.2002.2168
Dubin MJ et al (2015) DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 4:23. https://doi.org/10.7554/eLife.05255
Duckworth RA, Kruuk LEB (2009) Evolution of genetic integration between dispersal and colonization ability in a bird. Evolution 63:968–977
Eising CM, Muller W, Groothuis TGG (2006) Avian mothers create different phenotypes by hormone deposition in their eggs. Biol Lett 2:20–22
Elgvin TO et al (2017) The genomic mosaicism of hybrid speciation. Sci Adv. https://doi.org/10.1126/sciadv.1602996
Ellegren H et al (2012) The genomic landscape of species divergence in Ficedula flycatchers. Nature 491:756–760. https://doi.org/10.1038/nature11584
EpiDiverse (2018) Concepts in ecological epigenetics. https://www.epidiverse.eu/en/training/summer-schools. Accessed 24 Dec 2018
Farmer CG (2000) Parental care: the key to understanding endothermy and other convergent features in birds and mammals. Am Nat 155:326–334. https://doi.org/10.1086/303323
Faulk C, Dolinoy DC (2011) Timing is everything. The when and how of environmentally induced changes in the epigenome of animals. Epigenetics 6:791–797. https://doi.org/10.4161/epi.6.7.16209
Feil R, Fraga MF (2012) Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet 13:97–109. https://doi.org/10.1038/nrg3142
Feinberg AP (2007) Phenotypic plasticity and the epigenetics of human disease. Nature 447:433–440. https://doi.org/10.1038/nature05919
Feng J et al (2010a) Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13:423–430. https://doi.org/10.1038/nn.2514
Feng S, Jacobsen SE, Reik W (2010b) Epigenetic reprogramming in plant and animal development. Science 330:622–627. https://doi.org/10.1126/science.1190614
Fidler AE, van Oers K, Drent PJ, Kuhn S, Mueller JC, Kempenaers B (2007) Drd4 gene polymorphisms are associated with personality variation in a passerine bird. Proc R Soc Lond Ser B Biol Sci 274:1685–1691
Forsman A (2015) Rethinking phenotypic plasticity and its consequences for individuals, populations and species. Heredity 115:276–284. https://doi.org/10.1038/hdy.2014.92
Frommer M et al (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA 89:1827–1831. https://doi.org/10.1073/pnas.89.5.1827
Furrow RE, Feldman MW (2014) Genetic variation and the evolution of epigenetic regulation. Evolution 68:673–683. https://doi.org/10.1111/evo.12225
Gapp K et al (2014) Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci 17:667–669. https://doi.org/10.1038/nn.3695
Gardiner-Garden M, Frommer M (1987) CpG Islands in vertebrate genomes. J Mol Biol 196:261–282. https://doi.org/10.1016/0022-2836(87)90689-9
Gienapp P, Laine VN, Mateman AC, van Oers K, Visser ME (2017) Environment-dependent genotype–phenotype associations in avian breeding time. Front Genet 8:9. https://doi.org/10.3389/fgene.2017.00102
Grant BR, Grant PR (1996) Cultural inheritance of song and its role in the evolution of Darwin’s finches. Evolution 50:2471–2487
Groothuis TGG, Carere C (2005) Avian personalities: characterization and epigenesis. Neurosci Biobehav Rev 29:137–150
Gruebler MU, Naef-Daenzer B (2008) Fitness consequences of pre- and post-fledging timing decisions in a double-brooded passerine. Ecology 89:2736–2745. https://doi.org/10.1890/07-0786.1
Guerrero-Bosagna C, Morisson M, Liaubet L, Rodenburg TB, de Haas EN, Kost’al L, Pitel F (2018) Transgenerational epigenetic inheritance in birds. Environ Epigenet 4:dvy008. https://doi.org/10.1093/eep/dvy008
Halder R et al (2016) DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci 19:102. https://doi.org/10.1038/nn.4194
Hanson HE, Kilvitis HJ, Schrey AW, Martin LB (2018) Epigenetic potential in native and introduced populations of House Sparrows. Integr Comp Biol 58:E88–E88
Harris RA et al (2010) Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol 28:1097–1105. https://doi.org/10.1038/nbt.1682
Hatakeyama D, Mueller U (2008) Seasonal changes of methylation status in promoter region of a MAPK on honeybee genomic DNA. Comp Biochem Physiol B Biochem Mol Biol 151:457. https://doi.org/10.1016/j.cbpb.2008.09.040
Hinde CA, Buchanan KL, Kilner RM (2009) Prenatal environmental effects match offspring begging to parental provisioning. Proc R Soc B Biol Sci 276:2787–2794. https://doi.org/10.1098/rspb.2009.0375
Hoivik EA et al (2011) DNA methylation of intronic enhancers directs tissue-specific expression of steroidogenic factor 1/adrenal 4 binding protein (SF-1/Ad4BP). Endocrinology 152:2100–2112. https://doi.org/10.1210/en.2010-1305
Hollister JD, Gaut BS (2009) Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res 19:1419–1428. https://doi.org/10.1101/gr.091678.109
Hu J, Barrett RDH (2017) Epigenetics in natural animal populations. J Evol Biol 30:1612–1632. https://doi.org/10.1111/jeb.13130
Hunt BG, Glastad KM, Yi SV, Goodisman MAD (2013) The function of intragenic DNA methylation: insights from insect epigenomes. Integr Comp Biol 53:319–328. https://doi.org/10.1093/icb/ict003
Husby A, Nussey DH, Visser ME, Wilson AJ, Sheldon BC, Kruuk LEB (2010) Contrasting patterns of phenotypic plasticity in reproductive traits in two Great Tit (Parus major) populations. Evolution 64:2221–2237. https://doi.org/10.1111/j.1558-5646.2010.00991.x
Izquierdo A, Brigman JL, Radke AK, Rudebeck PH, Holmes A (2017) The neural basis of reversal learning: an updated perspective. Neuroscience 345:12–26. https://doi.org/10.1016/j.neuroscience.2016.03.021
Jaari S, Li MH, Merila J (2009) A first-generation microsatellite-based genetic linkage map of the Siberian Jay (Perisoreus infaustus): insights into avian genome evolution. BMC Genomics 1:1. https://doi.org/10.1186/1471-2164-10-1
Jabbari K, Caccio S, de Barros JPP, Desgres J, Bernardi G (1997) Evolutionary changes in CpG and methylation levels in the genome of vertebrates. Gene 205:109–118. https://doi.org/10.1016/s0378-1119(97)00475-7
Jablonka E, Lamb JR (2006) Evolutionary epigenetics. In: Fox CW, Wolf JB (eds) Evolutionary genetics: concepts and case studies. Oxford University Press, Oxford, pp 252–264
Jablonka E, Lamb MJ (2007) Precis of evolution in four dimensions. Behav Brain Sci 30:353. https://doi.org/10.1017/s0140525x07002221
Jablonka E, Raz G (2009) Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q Rev Biol 84:131–176
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33:245–254. https://doi.org/10.1038/ng1089
Jarvis ED et al (2014) Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346:1320–1331. https://doi.org/10.1126/science.1253451
Jensen P (2013) Transgenerational epigenetic effects on animal behaviour. Prog Biophys Mol Biol 113:447. https://doi.org/10.1016/j.pbiomolbio.2013.01.001
Jones PA, Takai D (2001) The role of DNA methylation in mammalian epigenetics. Science 293:1068–1070. https://doi.org/10.1126/science.1063852
Kasowski M et al (2013) Extensive variation in chromatin states across humans. Science 342:750–752. https://doi.org/10.1126/science.1242510
Kawakami T, Mugal CF, Suh A, Nater A, Burri R, Smeds L, Ellegren H (2017) Whole-genome patterns of linkage disequilibrium across flycatcher populations clarify the causes and consequences of fine-scale recombination rate variation in birds. Mol Ecol 26:4158–4172. https://doi.org/10.1111/mec.14197
Keller TE, Han P, Yi SV (2016) Evolutionary transition of promoter and gene body DNA methylation across invertebrate–vertebrate boundary. Mol Biol Evol 33:1019–1028. https://doi.org/10.1093/molbev/msv345
Kelly TK, Ahmadiantehrani S, Blattler A, London SE (2018) Epigenetic regulation of transcriptional plasticity associated with developmental song learning. Proc R Soc B Biol Sci 285:8. https://doi.org/10.1098/rspb.2018.0160
Kilvitis HJ, Martin LB, Schrey AW (2016) Epigenetic regulation of toll-like receptor 4 expression as a facilitator of invasiveness in Kenyan House Sparrows (Passer domesticus). Integr Comp Biol 56:E111
Kilvitis HJ, Ardia DR, Thiam M, Martin LB (2018) Corticosterone is correlated to mediators of neural plasticity and epigenetic potential in the hippocampus of Senegalese House Sparrows (Passer domesticus). Gen Comp Endocrinol 269:177–183. https://doi.org/10.1016/j.ygcen.2018.09.014
Kim JM et al (2018) A high-density SNP chip for genotyping Great Tit (Parus major) populations and its application to studying the genetic architecture of exploration behaviour. Mol Ecol Resour 18:877–891. https://doi.org/10.1111/1755-0998.12778
Klanker M, Sandberg T, Joosten R, Willuhn I, Feenstra M, Denys D (2015) Phasic dopamine release induced by positive feedback predicts individual differences in reversal learning. Neurobiol Learn Mem 125:135–145. https://doi.org/10.1016/j.nlm.2015.08.011
Konycheva G, Dziadek MA, Ferguson LR, Krageloh CU, Coolen MW, Davison M, Breier BH (2011) Dietary methyl donor deficiency during pregnancy in rats shapes learning and anxiety in offspring. Nutr Res 31:790–804. https://doi.org/10.1016/j.nutres.2011.09.015
Korochkin LI (2006) What is epigenetics? Russ J Genet 42:958–965. https://doi.org/10.1134/s102279540609002x
Korsten P et al (2010) Association between DRD4 gene polymorphism and personality variation in Great Tits: a test across four wild populations. Mol Ecol 19:832–843. https://doi.org/10.1111/j.1365-294x.2009.04518.x
Krueger F, Kreck B, Franke A, Andrews SR (2012) DNA methylome analysis using short bisulfite sequencing data. Nat Methods 9:145–151. https://doi.org/10.1038/nmeth.1828
Kumar V, Wingfield JC, Dawson A, Ramenofsky M, Rani S, Bartell P (2010) Biological clocks and regulation of seasonal reproduction and migration in birds. Physiol Biochem Zool 83:827–835. https://doi.org/10.1086/652243
Kummer H, Goodall J (1985) Conditions of innovative behavior in primates. Philos Trans R Soc Lond Ser B Biol Sci 308:203–214. https://doi.org/10.1098/rstb.1985.0020
Kurdyukov S, Bullock M (2016) DNA methylation analysis: choosing the right method. Biology. https://doi.org/10.3390/biology5010003
Kvist J, Gonçalves Athanàsio C, Shams Solari O, Brown JB, Colbourne JK, Pfrender ME, Mirbahai L (2018) Pattern of DNA methylation in daphnia: evolutionary perspective. Genome Biol Evol 10:1988–2007. https://doi.org/10.1093/gbe/evy155
Lai YS et al (2018) Temperature and photoperiod changes affect cucumber sex expression by different epigenetic regulations. BMC Plant Biol 18:13. https://doi.org/10.1186/s12870-018-1490-3
Laine VN et al (2016) Evolutionary signals of selection on cognition from the Great Tit genome and methylome. Nat Commun 7:9. https://doi.org/10.1038/ncomms10474
Laland K et al (2014) Does evolutionary theory need a rethink?—POINT Yes, urgently. Nature 514:161–164. https://doi.org/10.1038/514161a
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204–220. https://doi.org/10.1038/nrg2719
Ledford H et al (2008) Disputed definitions. Nature 455:1023–1028. https://doi.org/10.1038/4551023a
Ledon-Rettig CC, Richards CL, Martin LB (2013) Epigenetics for behavioral ecologists. Behav Ecol 24:311–324. https://doi.org/10.1093/beheco/ars145
Levenson JM et al (2006) Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 281:15763–15773. https://doi.org/10.1074/jbc.m511767200
Li Q et al (2011) Genome-wide mapping of DNA methylation in Chicken. PLoS One 6:e19428
Li JX et al (2015) Genome-wide DNA methylome variation in two genetically distinct Chicken lines using MethylC-seq. BMC Genomics 16:13. https://doi.org/10.1186/s12864-015-2098-8
Li M, Du W, Shao F, Wang WW (2016) Cognitive dysfunction and epigenetic alterations of the BDNF gene are induced by social isolation during early adolescence. Behav Brain Res 313:177–183. https://doi.org/10.1016/j.bbr.2016.07.025
Liebl AL, Schrey AW, Richards CL, Martin LB (2013) Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integr Comp Biol 53:351–358. https://doi.org/10.1093/icb/ict007
Liker A, Szekely T (2005) Mortality costs of sexual selection and parental care in natural populations of birds. Evolution 59:890–897
Liu L et al (2018) Transgenerational transmission of maternal stimulatory experience in domesticated birds. FASEB J 32:7002–7017. https://doi.org/10.1096/fj.201800762RR
Lynch EWJ, Coyle CS, Lorgen M, Campbell EM, Bowman AS, Stevenson TJ (2016) Cyclical DNA methyltransferase 3a expression is a seasonal and estrus timer in reproductive tissues. Endocrinology 157:2469–2478. https://doi.org/10.1210/en.2015-1988
Lynch EWJ, Coyle CS, Stevenson TJ (2017) Photoperiodic and ovarian steroid regulation of histone deacetylase 1, 2, and 3 in Siberian Hamster (Phodopus sungorus) reproductive tissues. Gen Comp Endocrinol 246:194–199. https://doi.org/10.1016/j.ygcen.2016.12.008
Lyon MF (1961) Gene action in the X-chromosome of the Mouse (Mus musculus L.). Nature 190:372–373. https://doi.org/10.1038/190372a0
Mäkinen H, Viitaniemi HM, Visser ME, Verhagen I, van Oers K, Husby A (2019) Temporally replicated DNA methylation patterns in Great Tit using reduced representation bisulfite sequencing. Sci Data (in press)
McNew SM, Beck D, Sadler-Riggleman I, Knutie SA, Koop JAH, Clayton DH, Skinner MK (2017) Epigenetic variation between urban and rural populations of Darwin’s finches. BMC Evol Biol 17:14. https://doi.org/10.1186/s12862-017-1025-9
Meaney MJ, Szyf M (2005) Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci 7:103–123
Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R (2005) Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 33:5868–5877. https://doi.org/10.1093/nar/gki901
Merlin C, Liedvogel M (2019) The genetics and epigenetics of animal migration and orientation: birds, butterflies and beyond. J Exp Biol. https://doi.org/10.1242/jeb.191890
Miller CA, Campbell SL, Sweatt JD (2008) DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem 89:599–603. https://doi.org/10.1016/j.nlm.2007.07.016
Morgan HD, Santos F, Green K, Dean W, Reik W (2005) Epigenetic reprogramming in mammals. Hum Mol Genet 14:R47–R58. https://doi.org/10.1093/hmg/ddi114
Mueller JC et al (2013) Haplotype structure, adaptive history and associations with exploratory behaviour of the DRD4 gene region in four Great Tit (Parus major) populations. Mol Ecol 22:2797–2809. https://doi.org/10.1111/mec.12282
Muller W, Dijkstra C, Groothuis TGG (2009) Maternal yolk androgens stimulate territorial behaviour in Black-headed Gull chicks. Biol Lett 5:586–588. https://doi.org/10.1098/rsbl.2009.0283
Naef-Daenzer B, Gruebler MU (2008) Post-fledging range use of Great Tit Parus major families in relation to chick body condition. Ardea 96:181–190. https://doi.org/10.5253/078.096.0204
Naef-Daenzer B, Gruebler MU (2016) Post-fledging survival of altricial birds: ecological determinants and adaptation. J Field Ornithol 87:227–250. https://doi.org/10.1111/jofo.12157
Naguib M, Florcke C, van Oers K (2011) Effects of social conditions during early development on stress response and personality traits in Great Tits (Parus major). Dev Psychobiol 53:592–600. https://doi.org/10.1002/dev.20533
Natt D, Agnvall B, Jensen P (2014) Large sex differences in Chicken behavior and brain gene expression coincide with few differences in promoter DNA-methylation. PLoS One 9:12. https://doi.org/10.1371/journal.pone.0096376
Partecke J, Schwabl H (2008) Organizational effects of maternal testosterone on reproductive behavior of adult House Sparrows. Dev Neurobiol 68:1538–1548
Pegoraro M, Bafna A, Davies NJ, Shuker DM, Tauber E (2016) DNA methylation changes induced by long and short photoperiods in Nasonia. Genome Res 26:203–210. https://doi.org/10.1101/gr.196204.115
Perrins CM (1970) The timing of birds’ breeding seasons. Ibis 112:242–255
Phan ML, Gergues MM, Mahidadia S, Jimenez-Castillo J, Vicario DS, Bieszczad KM (2017) HDAC3 inhibitor RGFP966 modulates neuronal memory for vocal communication signals in a songbird model. Front Syst Neurosci 11:12. https://doi.org/10.3389/fnsys.2017.00065
Pigliucci M (2001) Phenotypic plasticity: beyond nature and nurture. Syntheses in ecology and evolution. Johns Hopkins University Press, Baltimore
Pigliucci M (2005) Evolution of phenotypic plasticity: where are we going now? Trends Ecol Evol 20:481–486. https://doi.org/10.1016/j.tree.2005.06.001
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28:1057–1068. https://doi.org/10.1038/nbt.1685
Price TD, Qvarnstrom A, Irwin DE (2003) The role of phenotypic plasticity in driving genetic evolution. Proc Biol Sci 270:1433–1440. https://doi.org/10.1098/rspb.2003.2372
Qu Y et al (2013) Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nat Commun. https://doi.org/10.1038/ncomms3071
Quinn JL, Patrick SC, Bouwhuis S, Wilkin TA, Sheldon BC (2009) Heterogeneous selection on a heritable temperament trait in a variable environment. J Anim Ecol 78:1203–1215
Quinn JL, Cole EF, Reed TE, Morand-Ferron J (2016) Environmental and genetic determinants of innovativeness in a natural population of birds. Philos Trans R Soc Lond B Biol Sci. https://doi.org/10.1098/rstb.2015.0184
Rapp RA, Wendel JF (2005) Epigenetics and plant evolution. New Phytol 168:81–91. https://doi.org/10.1111/j.1469-8137.2005.01491.x
Reik W, Dean W, Walter J (2001) Epigenetic reprogramming in mammalian development. Science 293:1089–1093
Rey O, Danchin E, Mirouze M, Loot C, Blanchet S (2016) Adaptation to global change: a transposable element-epigenetics perspective. Trends Ecol Evol 31:514–526. https://doi.org/10.1016/j.tree.2016.03.013
Richards EJ (2006) Opinion—inherited epigenetic variation—revisiting soft inheritance. Nat Rev Genet 7:395–401
Riyahi S, Sanchez-Delgado M, Calafell F, Monk D, Senar JC (2015) Combined epigenetic and intraspecific variation of the DRD4 and SERT genes influence novelty seeking behavior in Great Tit Parus major. Epigenetics 10:516–525. https://doi.org/10.1080/15592294.2015.1046027
Riyahi S, Vilatersana R, Schrey AW, Node HG, Aliabadian M, Senar JC (2017) Natural epigenetic variation within and among six subspecies of the House Sparrow, Passer domesticus. J Exp Biol 220:4016–4023. https://doi.org/10.1242/jeb.169268
Romano A et al (2017) Methylation of the circadian Clock gene in the offspring of a free-living passerine bird increases with maternal and individual exposure to PM10. Environ Pollut 220:29–37. https://doi.org/10.1016/j.envpol.2016.08.060
Roth TL, Lubin FD, Funk AJ, Sweatt JD (2009) Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry 65:760–769. https://doi.org/10.1016/j.biopsych.2008.11.028
Rowan W (1925) Relation of light to bird migration and developmental changes. Nature 115:494
Rowe C, Healy SD (2014) Measuring variation in cognition. Behav Ecol 25:1287–1292. https://doi.org/10.1093/beheco/aru090
Rubenstein DR, Skolnik H, Berrio A, Champagne FA, Phelps S, Solomon J (2016) Sex-specific fitness effects of unpredictable early life conditions are associated with DNA methylation in the avian glucocorticoid receptor. Mol Ecol 25:1714–1728. https://doi.org/10.1111/mec.13483
Saino N et al (2017) Migration phenology and breeding success are predicted by methylation of a photoperiodic gene in the Barn Swallow. Sci Rep 7:10. https://doi.org/10.1038/srep45412
Santure AW et al (2015) Replicated analysis of the genetic architecture of quantitative traits in two wild Great Tit populations. Mol Ecol 24:6148–6162. https://doi.org/10.1111/mec.13452
Sasaki H, Matsui Y (2008) Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet 9:129–140. https://doi.org/10.1038/nrg2295
Sauce B et al (2018) The impact of environmental interventions among Mouse siblings on the heritability and malleability of general cognitive ability. Philos Trans R Soc B Biol Sci 373:9. https://doi.org/10.1098/rstb.2017.0289
Saunderson EA et al (2016) Stress-induced gene expression and behavior are controlled by DNA methylation and methyl donor availability in the dentate gyrus. Proc Natl Acad Sci USA 113:4830–4835. https://doi.org/10.1073/pnas.1524857113
Schaefer S, Nadeau JH (2015) The genetics of epigenetic inheritance: modes, molecules, and mechanisms. Q Rev Biol 90:381–415. https://doi.org/10.1086/683699
Schrey AW et al (2011) Broad-scale latitudinal patterns of genetic diversity among native European and introduced House Sparrow (Passer domesticus) populations. Mol Ecol 20:1133–1143. https://doi.org/10.1111/j.1365-294x.2011.05001.x
Schrey AW et al (2012) Epigenetic variation may compensate for decreased genetic variation with introductions: a case study using House Sparrows (Passer domesticus) on two continents. Genet Res Int 2012:979751. https://doi.org/10.1155/2012/979751
Schrey AW et al (2013) Ecological epigenetics: beyond MS-AFLP. Integr Comp Biol 53:340–350. https://doi.org/10.1093/icb/ict012
Sharma A, Singh D, Malik S, Gupta NJ, Rani S, Kumar V (2018) Difference in control between spring and autumn migration in birds: insight from seasonal changes in hypothalamic gene expression in captive buntings. Proc R Soc B Biol Sci 285:10. https://doi.org/10.1098/rspb.2018.1531
Sharp PJ (2005) Photoperiodic regulation of seasonal breeding in birds. Ann N Y Acad Sci 1040:189–199. https://doi.org/10.1196/annals.1327.024
Sheldon EL, Schrey A, Andrew SC, Ragsdale A, Griffith SC (2018a) Epigenetic and genetic variation among three separate introductions of the House Sparrow (Passer domesticus) into Australia. R Soc Open Sci 5:11. https://doi.org/10.1098/rsos.172185
Sheldon EL, Schrey AW, Ragsdale AK, Griffith SC (2018b) Brood size influences patterns of DNA methylation in wild Zebra Finches (Taeniopygia guttata). Auk 135:1113–1122. https://doi.org/10.1642/auk-18-61.1
Shi J, Dong A, Shen WH (2015) Epigenetic regulation of rice flowering and reproduction. Front Plant Sci 5:13. https://doi.org/10.3389/fpls.2014.00803
Shilo S, Melamed-Bessudo C, Dorone Y, Barkai N, Levy AA (2015) DNA crossover motifs associated with epigenetic modifications delineate open chromatin regions in Arabidopsis. Plant Cell 27:2427–2436. https://doi.org/10.1105/tpc.15.00391
Skinner MK, Gurerrero-Bosagna C, Haque MM, Nilsson EE, Koop JAH, Knutie SA, Clayton DH (2014) Epigenetics and the evolution of Darwin’s finches. Genome Biol Evol 6:1972–1989. https://doi.org/10.1093/gbe/evu158
St-Cyr S, McGowan PO (2015) Programming of stress-related behavior and epigenetic neural gene regulation in mice offspring through maternal exposure to predator odor. Front Behav Neurosci 9:10. https://doi.org/10.3389/fnbeh.2015.00145
Stevenson TJ (2017) Circannual and circadian rhythms of hypothalamic DNA methyltransferase and histone deacetylase expression in male Siberian Hamsters (Phodopus sungorus). Gen Comp Endocrinol 243:130–137. https://doi.org/10.1016/j.ygcen.2016.11.011
Stevenson TJ (2018) Epigenetic regulation of biological rhythms: an evolutionary ancient molecular timer. Trends Genet 34:90–100. https://doi.org/10.1016/j.tig.2017.11.003
Stevenson TJ, Prendergast BJ (2013) Reversible DNA methylation regulates seasonal photoperiodic time measurement. Proc Natl Acad Sci USA 110:16651–16656. https://doi.org/10.1073/pnas.1310643110
Strasser R, Schwabl H (2004) Yolk testosterone organizes behavior and male plumage coloration in House Sparrows (Passer domesticus). Behav Ecol Sociobiol 56:491–497. https://doi.org/10.1007/s00265-004-0810-9
Steyaert S et al (2016) A genome-wide search for eigenetically regulated genes in zebra finch using MethylCap-seq and RNA-seq. Sci Rep 6:16. https://doi.org/10.1038/srep20957
Sutherland WJ et al (2013) Identification of 100 fundamental ecological questions. J Ecol 101:58–67. https://doi.org/10.1111/1365-2745.12025
Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9:465–476. https://doi.org/10.1038/nrg2341
Suzuki M et al (2018) Whole-genome bisulfite sequencing with improved accuracy and cost. Genome Res 28:1364–1371. https://doi.org/10.1101/gr.232587.117
Sweatt JD (2019) The epigenetic basis of individuality. Curr Opin Behav Sci 25:51–56. https://doi.org/10.1016/j.cobeha.2018.06.009
Tammen SA, Friso S, Choi SW (2013) Epigenetics: the link between nature and nurture. Mol Aspects Med 34:753–764. https://doi.org/10.1016/j.mam.2012.07.018
Thomas DW, Blondel J, Perret P, Lambrechts MM, Speakman JR (2001) Energetic and fitness costs of mismatching resource supply and demand in seasonally breeding birds. Science 291:2598–2600. https://doi.org/10.1126/science.1057487
Tinbergen JM, Sanz JJ (2004) Strong evidence for selection for larger brood size in a Great Tit population. Behav Ecol 15:525–533. https://doi.org/10.1093/beheco/arh045
Tost J, Gut IG (2007) DNA methylation analysis by pyrosequencing. Nat Protoc 2:2265–2275. https://doi.org/10.1038/nprot.2007.314
van Gurp TP, Wagemaker N, Wouters B, Vergeer P, Ouborg JNJ, Verhoeven KJF (2016) epiGBS: reference-free reduced representation bisulfite sequencing. Nat Methods 13:322–324. https://doi.org/10.1038/nmeth.3763
van Oers K, Mueller JC (2010) Evolutionary genomics of animal personality. Philos Trans R Soc Lond Ser B 365:3991–4000
van Oers K, Naguib M (2013) Avian personality. In: Carere C, Maestripieri D (eds) Animal personalities: behavior, physiology, and evolution. The University of Chicago Press, Chicago/London, pp 66–95
van Oers K et al (2014) Replicated high-density genetic maps of two Great Tit populations reveal fine-scale genomic departures from sex-equal recombination rates. Heredity 112:307–316. https://doi.org/10.1038/hdy.2013.107
van Oers K, Kohn GM, Hinde CA, Naguib M (2015) Parental food provisioning is related to nestling stress response in wild Great Tit nestlings: implications for the development of personality. Front Zool 12:10. https://doi.org/10.1186/1742-9994-12-s1-s10
Verhoeven KJF, vonHoldt BM, Sork VL (2016) Epigenetics in ecology and evolution: what we know and what we need to know. Mol Ecol 25:1631–1638. https://doi.org/10.1111/mec.13617
Verhulst S, Vanbalen JH, Tinbergen JM (1995) Seasonal decline in reproductive success of the Great Tit—variation in time or quality. Ecology 76:2392–2403. https://doi.org/10.2307/2265815
Verhulst EC, Mateman AC, Zwier MV, Caro SP, Verhoeven KJF, van Oers K (2016) Evidence from pyrosequencing indicates that natural variation in animal personality is associated with DRD4 DNA methylation. Mol Ecol 25:1801–1811. https://doi.org/10.1111/mec.13519
Viitaniemi HM, Verhagen I, Visser ME, Honkela A, van Oers K, Husby A (2019) Seasonal variation in genome-wide DNA methylation patterns and the onset of seasonal timing of reproduction in Great Tits. Genome Biol Evol 11:970–983
Vinoth A, Thirunalasundari T, Shanmugam M, Uthrakumar A, Suji S, Rajkumar U (2018) Evaluation of DNA methylation and mRNA expression of heat shock proteins in thermal manipulated Chicken. Cell Stress Chaperones 23:235–252. https://doi.org/10.1007/s12192-017-0837-2
Visser ME, Verboven N (1999) Long-term fitness effects of fledging date in Great Tits. Oikos 85:445–450
Visser ME, van Noordwijk AJ, Tinbergen JM, Lessells CM (1998) Warmer springs lead to mistimed reproduction in Great Tits (Parus major). Proc R Soc B Biol Sci 265:1867–1870. https://doi.org/10.1098/rspb.1998.0514
Waddington CH (1952) The epigenetics of birds. University Press, Cambridge
Wallace SJ, de Solla SR, Thomas PJ, Harner T, Eng A, Langlois VS (2018) Airborne polycyclic aromatic compounds contribute to the induction of the tumour-suppressing P53 pathway in wild double-crested cormorants. Ecotoxicol Environ Saf 150:176–189. https://doi.org/10.1016/j.ecoenv.2017.12.028
Warren WC et al (2010) The genome of a songbird. Nature 464:757–762. https://doi.org/10.1038/nature08819
Waters SA et al (2018) Landscape of DNA methylation on the marsupial x. Mol Biol Evol 35:431–439. https://doi.org/10.1093/molbev/msx297
Weaver ICG (2005) Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci 25:11045–11054. https://doi.org/10.1523/JNEUROSCI.3652-05.2005
Weaver ICG et al (2004) Epigenetic programming by maternal behavior. Nat Neurosci 7:847–854. https://doi.org/10.1038/nn1276
Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D (2005) Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37:853–862. https://doi.org/10.1038/ng1598
Wenzel MA, Piertney SB (2014) Fine-scale population epigenetic structure in relation to gastrointestinal parasite load in Red Grouse (Lagopus lagopus scotica). Mol Ecol 23:4256–4273. https://doi.org/10.1111/mec.12833
West-Eberhard MJ (2003) Developmental plasticity and evolution. Oxford University Press, New York
Whitelaw E, Martin DIK (2001) Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat Genet 27:361–365. https://doi.org/10.1038/86850
Wilschut RA, Oplaat C, Snoek LB, Kirschner J, Verhoeven KJF (2016) Natural epigenetic variation contributes to heritable flowering divergence in a widespread asexual dandelion lineage. Mol Ecol 25:1759–1768. https://doi.org/10.1111/mec.13502
Wingfield JC (2005) Flexibility in annual cycles of birds: implications for endocrine control mechanisms. J Ornithol 146:291–304. https://doi.org/10.1007/s10336-005-0002-z
Wray GA, Hoekstra HE, Futuyma DJ, Lenski RE, Mackay TFC, Schluter D, Strassmann JE (2014) Does evolutionary theory need a rethink?—COUNTERPOINT No, all is well. Nature 514:161
Yan XP et al (2015) Evidence in duck for supporting alteration of incubation temperature may have influence on methylation of genomic DNA. Poult Sci 94:2537–2545. https://doi.org/10.3382/ps/pev201
Yi SV (2017) Insights into epigenome evolution from animal and plant methylomes. Genome Biol Evol 9:3189–3201. https://doi.org/10.1093/gbe/evx203
Zandberg L, Quinn JL, Naguib M, van Oers K (2017) Personality-dependent differences in problem-solving performance in a social context reflect foraging strategies. Behav Process 134:95–102. https://doi.org/10.1016/j.beproc.2016.09.007
Zhang X (2008) The epigenetic landscape of plants. Science 320:489–492. https://doi.org/10.1126/science.1153996
Zhu B, Reinberg D (2011) Epigenetic inheritance: uncontested? Cell Res 21:435–441. https://doi.org/10.1038/cr.2011.26
Zimmer C, Larriva M, Boogert NJ, Spencer KA (2017) Transgenerational transmission of a stress-coping phenotype programmed by early-life stress in the Japanese quail. Sci Rep 7:19. https://doi.org/10.1038/srep46125
Acknowledgements
We thank Koen Verhoeven for helpful discussions and comments on earlier versions of the manuscript. This work was supported by a Research Council of Norway grant (239974) to A. H.; an NWO open competition grant (ALWOP.314) to K. V. O. and B. S.; and the KNAW Research Fund (round 2017) to K. V. O. and K. H.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Wink.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the Topical Collection 27th International Ornithological Congress, Vancouver, Canada, 19–26 August 2018.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sepers, B., van den Heuvel, K., Lindner, M. et al. Avian ecological epigenetics: pitfalls and promises. J Ornithol 160, 1183–1203 (2019). https://doi.org/10.1007/s10336-019-01684-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10336-019-01684-5