
Abstract
Over the past 20 years, the introduction of new molecular techniques has given a new impetus to genetic and genomic studies of fishes. The main traits selected in the aquaculture sector conform to the polygenic model, and, thus far, effective breeding programmes based on genome-wide association studies (GWAS) and marker-assisted selection (MAS) have been applied to simple traits (e.g. disease resistance and sexual maturation of salmonids) and known Quantitative Trait Loci (QTLs). Genomic selection uses the genomic relationships between candidate loci and SNPs distributed over the entire genome and in tight linkage disequilibrium (LD) with genes that encode the traits. SNP (low and high density) arrays are used for genotyping thousands of genetic markers (single nucleotide polymorphisms, SNPs). The genomic expected breeding value (GEBV) of selection candidates is usually calculated by means of the GBLUP or ssGBLUP (single step) methods. In recent years, in several aquaculture breeding programmes, the genomic selection method has been applied to different fish and crustacean species. While routine implementation of genomic selection is now largely carried out in Atlantic salmon (Salmo salar) and rainbow trout (Oncorhynchus mykiss), it is expected that, in the near future, this method will progressively spread to other fish species. However, genomic selection is an expensive method, so it will be relevant mostly for traits of high economic value. In several studies (using different salmonid species), the accuracy of the GEBVs varied from 0.10 to 0.80 for different traits (e.g. growth rate and disease resistance) compared to traditional breeding methods based on geneology. Genomic selection applied to aquaculture species has the potential to improve selection programmes substantially and to change ongoing fish breeding systems. In the long term, the ability to use low-pass genome sequencing methods, low-cost genotyping and novel phenotyping techniques will allow genomic selection to be applied to thousands of animals directly at the farm level.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aquaculture is currently one of the fastest-growing agricultural production sectors worldwide. FAO (2019) reported that half of the fish used for human consumption were produced in the aquaculture sector, and in 2025, the expected total fish production will be approximately 200 million tons (OECD/Food and Agriculture Organization of the United Nations 2014). Among the 250 farmed fish species around the world, only 10% have been subjected to selection programmes (Teletchea and Fontaine 2014; FAO 2019). Worldwide, the current number of breeding programs for different species is approximately 60 (Abdelrahman et al. 2017; Houston 2017), sharing over 80% of the total production (FAO 2019, FAO Fisheries and Aquaculture Department 2019).
Initially, in most selection programmes, growth rate was the most-selected trait because of its generally medium heritability and ease of application. The genetic improvement rate per generation was generally 4–5 times higher than those obtained in other domestic animals (Gjerde and Korsvoll 1999; Thorgaard et al. 2002; Kause et al. 2005; Georges et al. 2019). While growth rate is one of the most economically important traits, other characteristics, such as feed conversion efficiency, carcass-quality characteristics and disease resistance, also have been gradually incorporated into selection programmes (Gjedrem et al. 2012). These traits are more difficult to select for because of lower heritability, post-mortem measurement and need for use of selection schemes based on sib selection (Yáñez et al. 2015) (which uses only the genetic variance between families) (Hill 2013). Except for a few species, such as Atlantic salmon, rainbow trout and Nile tilapia (Ponzoni et al. 2011), the availability of selected lines and the development of breeding programmes in the aquaculture sector are still in their infancy (Houston 2017). The situation is more favourable in Europe, where over 80% of the production of six main species (Atlantic salmon, rainbow trout, European seabass, gilthead seabream, carp (common carp (Cyprinus carpio) and silver carp (Hypophthalmichthys molitrix)) and turbot (Psetta maxima) derive from improved stocks (Janssen et al. 2018). There is enormous potential to improve worldwide fish production further using selection techniques. Despite the considerable diversity of production volumes, the genetic variability within species and high fish fertility allow the use of flexible breeding plans without the need for expensive multiplication centres (Houston 2017).
Quantitative trait loci (QTLs) of the main productive traits, sexual maturation, fillet characteristics, and disease resistance can be used in genetic improvement programmes through the genetic marker-assisted selection (MAS) approach. Favourable alleles can be identified using SNP arrays, Genome-Wide Association Study (GWAS) and linkage association mapping methods (Goddard and Hayes 2009). The availability of new-omics methods, such as last-generation genome sequencing techniques (e.g. long-range technologies (Pacific Biosciences and Oxford Nanopore) (Lien et al. 2016; Christensen et al. 2018a) and linked reads (Takayama et al. 2021), 10X microfluidic platform (Wang et al. 2020; Zheng et al. 2016), new genome assembly methods (long-range scaffolding, Hi-C (high-throughput chromatin conformation capture, Dovetail and optical mapping, Bionano Genomics) (Burton et al. 2013; Howe and Wood 2015; Putnam 2016; Mantere et al. 2019), transcriptome and epigenetic analysis (RNA-seq, ChIP-Seq), new genotyping techniques (high- and low-density SNP arrays) (Palti et al. 2015a) and genotyping-by-sequencing techniques (GBS, RAD-Seq), can revolutionise the aquaculture breeding sector. These new methods have the potential to identify the most important genetic variants (SNPs) that influence phenotypic expression of the main traits (Goddard and Hayes 2009; Ellegren 2014). Genetic markers can be used for various purposes, such as the analysis of the genetic variability of QTLs (Palti et al. 2015b), MAS, GWAS and genomic selection (Eggen 2012; Liu et al. 2019). The first application of MAS was carried out on Atlantic salmon for the selection of the QTL for resistance to infectious pancreatic necrosis (IPNV) (Houston et al. 2008; Moen et al. 2009). In Table 1 are reported the main definitions and abbreviations used in the text.
Continuous improvement of selective breeding
The first modern breeding programme that used family pedigree information was applied to Atlantic salmon (Salmo salar) and started in 1972 in Norway (Gjerde 1986, Gjedrem 2000; Bentsen and Olesen 2002). Initially, the programme focused only on improvement of growth rate or standard growth rate (SGR), but after obtaining positive results, late sexual maturation, carcass quality and resistance to various diseases were also included in the selection index. This programme suggested that growth rates can be improved by 10-20% per generation in most fish species (Olesen et al. 2015; Gjedrem and Rye 2018). This result can be achieved through a simple mass selection programme that does not use family information (Gjedrem et al. 2012). Such programmes have the advantage of being much less expensive, but several traits cannot be evaluated on live candidates, such as slaughter yield and disease resistance. Furthermore, the use of family information allows a more precise estimate of the genetic response at the farm level and faster progress (Sonesson et al. 2010; Vela-Avitúa et al. 2015). In various breeding programmes of Atlantic salmon and rainbow trout, 100-200 families were reared in separate tanks up to the size at which fish can be tagged (Fernández et al. 2014). In this case, the selection process has limited accuracy, as it is based on the choice of the estimated breeding values (EBVs) of the families (use of only 50% of the genetic variability and selection of related individuals) (Ødegård et al. 2011, 2014; Nirea et al. 2012).
At the end of the 1990s, a radical change in fish breeding methods was made by the introduction of a new technique for the identification of parents by means of molecular markers. This method uses the genetic markers of offspring and parents (typically 10-15 microsatellite markers or 100 SNPs). After the statistical analysis, it is possible to assign all the offspring to their parents and thus to keep track of the pedigree (Vandeputte and Haffray 2014). Today, in Europe, in relation to the six main farmed fish species, among 36 selection programmes, 12 use parental assignment as a tool for tracing the pedigree (Janssen et al. 2017). To date, however positive results using marker-assisted selection (MAS) have been obtained only for simple traits in which the main QTL is known (Jørgen et al. 2011; Yue 2014).
The mating system used in a breeding programme is a key factor because it can influence the inbreeding rate (Sonesson et al. 2012), and the selection response (Hely et al. 2013; Sonesson & Ødegård 2016). D'Agaro et al. (2007) compared different mating systems using the optimum contribution selection method (Sánchez-Molano et al. 2016; D’Agaro 2017). The results of the simulation showed that the factorial mating system allows us to obtain the lowest inbreeding rate and the highest selection response. Skaarud et al. (2014) showed that increasing the number of families from 50 to 400 and using the optimum contribution selection method increases the response to selection. Sonesson and Ødegård (2016) indicate that if the number of families increases from 100 to 1000 using 5000 SNPs, the inbreeding rate of the population decreases from 0.014 to 0.006, and the genetic gain (ΔG) is in the range 0.17–0.40. The larger number of families led to higher genetic gains because of the increased selection intensity, while the 5,000 SNPs allowed accurate prediction values to be obtained. From recent studies, it appears that 200 families are the optimal number to be used in fish breeding schemes (Skaarud et al. 2014). Skaarud et al. (2011) suggested using 8–200 offspring per family. The main traits selected in the fish breeding programmes are growth rate, morphology, disease resistance, slaughter yield and product quality; among these, substantial advances can be achieved in growth (+10–20% per generation) and disease resistance (+12.5% survival on a specific disease per generation) (Gjedrem 2000, 2012).
Genomic resources
Next-generation sequencing (NGS) refers to the set of DNA sequencing technologies that have in common the ability to sequence in parallel millions of DNA fragments. The whole process includes several steps: DNA extraction, sequencing and bio-informatic analysis.
The magnetic beads method is the most commonly used technique for DNA extraction for NGS analysis (McGaughey et al. 2018). This method greatly simplifies the extraction procedure and improves the size selection process. The choice of sequencing method depends on several factors, such as the desired coverage depth, error rate, simultaneous multiplexing and throughput capacity.
NGS technologies, also known as high-throughput sequencing (HTS) technologies, have evolved rapidly in the last two decades, offering a greater number of sequenced bases at a lower cost (Guppy et al. 2018; Robledo et al. 2018a). In 2006, second-generation NGS technology was started, which is able to perform millions of polymerisation reactions in parallel (Metzker 2010). The main platforms now available are MiSeq, HiSeq and NovaSeq from Illumina (San Diego, USA). This type of technology is based on the fragmentation of DNA and amplification in parallel with multiple reactions, obtaining short reads between 100 and 300 bp. The distance between the pairs of reads is called insert size (mate paired: 2–5 kb and paired end < 1 kb). In 2013, third-generation NGS techniques, also known as single molecule sequencing (SMS), started. SMS produces longer reads (5–50 kb), with higher costs (>2000 US$ per Gb) and higher error rates. With this method, a single DNA molecule (without enzymatic process or hybridisation) is sequenced, and no library amplification is required. The main platforms are Pacific Biosciences (PacBio) (Lien et al. 2016; Christensen et al. 2018a) and Oxford Nanopore Technologies (ONT) (Jain et al. 2016). These techniques are used mainly to complete the genomic sequence (gap filling) (since repetitive and tetrasomic regions are difficult to assemble, especially for fish) and for scaffolding sequences previously assembled using second-generation technologies (Xiao and Zhou 2020). The difficulties in assembling fish genomes derive from the recent genome duplication process in certain lineages, notably salmonids, the presence of high levels of heterozygous loci and long repetitive elements. The length of the assembled reads should be greater than the size of the haplotype blocks (ranging from 20 to 200 kb). The PacBio platform now routinely generates reads with an N50 >1 Mb. This method has recently reduced the error rate by means of a new sequencing technique called “circular consensus” and the production of high-fidelity reads of 15 kb. The software packages generally used for the bioinformatic analysis of third-generation sequencing are Canu (Koren et al. 2017) and Mecat (Xiao et al. 2017). The results obtained are subsequently cleaned and analysed with other software packages such as Racon (Vaser et al. 2017), Nanopolish (Loman et al. 2015), Pilon (Walker et al. 2014) and Busco (Simao et al. 2015). In 2016, a new third-generation mapping technology was used by Dovetail Genomics (Santa Cruz, CA, USA). This technique allows us to obtain a high-quality scaffolding process at the chromosomal level using two types of libraries (Chicago® and Dovetail™ Hi-C) generated from the chromatin proximity ligation process (Lieberman-Aiden et al. 2009; Putnam 2016). Through these new methodologies, it is possible to reconstruct long haplotypes (phasing), identify structural variations (SVs) (Edge et al. 2017) and distinguish chromosome sets (Weisenfeld et al. 2017).
New sequencing techniques have allowed the assembly of several reference genomes for salmonids (Gonzalez-Pena et al. 2016; Alhakami et al. 2017): Atlantic salmon (Gilbey et al. 2004; Davidson et al. 2010; Lien et al. 2016), rainbow trout (Berthelot et al. 2014), Chinook salmon (Oncorhynchus tshawytscha) (Christensen et al. 2018a), coho salmon (Oncorhynchus kisutch) and Artic charr (Salvelinus alpinus) (Christensen et al. 2018b). Currently, several high-density genomic maps are available for the main salmonid species: Atlantic salmon (Moen et al. 2008; Phillips et al. 2009; Lien et al. 2011; Gonen et al. 2014; Gonen et al. 2015a; Tsai et al. 2016b; Yáñez et al. 2016); coho salmon (McClelland and Naish 2008); Chinook salmon (Brieuc et al. 2014; McKinney et al. 2016); rainbow trout (Young et al. 1998; Sakamoto et al. 2000; Nichols et al. 2003; Guyomard et al. 2006, 2012; Rexroad et al. 2008; Palti et al. 2012; Silva et al. 2019); brown trout (Salmo trutta) (Gharbi et al. 2006; Leitwein et al. 2017); brook charr (Salvelinus fontinalis) (Sutherland et al. 2016) and Arctic charr (Salvelinus alpinus) (Woram et al. 2004). Several important initiatives were established, such as the GRASP (Consortium for the Genomic Research of all Salmonids) initiative (MacQueen et al. 2017) and the Salmobase platform (Samy et al. 2017). These initiatives have allowed the creation of EST (Expressed Sequence Tag) databases, gene expression platforms (microarrays) and SNP array chips. The recent publication of the PhyloFish database (Fouchécourt et al. 2019) represents another important contribution for 15 fish species. A reference genome sequence is essential for understanding all the complex characteristics in cultured and natural fish populations (Gonzalez-Pena et al. 2016).
The advent of NGS technologies has facilitated the identification of a large number of DNA markers randomly distributed throughout the genome, making the use of multilocus data (multiple concatenated loci) more suitable. Numerous SNPs have recently been identified for different samonid species (Adzhubei et al. 2007; Sanchez et al. 2009; Hohenlohe et al. 2011; Houston et al. 2014, Houston 2017; Salem et al. 2012). The combined use of whole-genome sequencing and the analysis of the transcriptome (RNA-seq) (Sutherland et al. 2014; Shi et al. 2019) allows us to obtain a very large number of SNPs (He et al. 2017; Robertson et al. 2017). Note that if the goal is the identification of SNPs in non-model species, RAD-tag analysis is usually sufficient. SNPs can be analysed with SNP arrays or RAD-tag methods. The best method to genotype large numbers of SNPs is the design of SNP high-density (HD) arrays (Hoggart et al. 2008; Matukumalli et al. 2009; Ramos et al. 2009; Cheng et al. 2015).
In recent years, several basic research and applied projects on Atlantic salmon and rainbow trout were carried out. They include the development of an SNP HD array for Atlantic salmon (Houston et al. 2014), comparative whole genome analysis (WGA) of similar species (Gutierrez et al. 2018), resequencing of the rainbow trout genome (Oncorhynchus mykiss) (Gao et al. 2018) and de novo transcriptome analysis of Atlantic salmon, brown trout, and Arctic charr (Salvelinus alpinus) (Carruthers et al. 2018). Applications of SNP arrays in salmonids have been reported in several studies concerning population diversity, QTL mapping and genomic selection (Balding 2006; MacLeod et al. 2010). In the last 10 years, several SNP arrays with different marker densities and formats have become commercially available: a 130K SNP array for rainbow trout, a SNP HD array for Atlantic salmon (Houston e al. 2014; Yáñez et al. 2014) and additional SNP chips for other salmonid species (Lien et al. 2011). SNP arrays are now routinely used for genotyping rainbow trout populations and Atlantic salmon (Houston et al. 2014; Yáñez et al. 2014). Zhang et al. 2018a, 2018b) used a 57K SNP array to genotype several salmonid species. A substantial reduction in genotyping costs can be achieved by means of inference methods such as imputation. Imputation techniques allow the transfer of genomic information from an SNP HD array to a low-density chip (Marchini et al. 2007; Habier et al. 2009; Hickey et al. 2012). NGS technologies also can be used to study non-model species since, in this case, it is not necessary to have previous genomic information. In many cases, a complete genome sequence is not necessary, as the genes are organized in linkage blocks, and a high-density genetic map of the species is available. The availability of uniformly distributed genetic markers allows us to obtain the necessary coverage. Furthermore, the low-pass whole-genome sequencing method (LP-WGS) (0.1-4x) can be used as an alternative to genotyping array methods. This less-expensive technique is based on NGS with low coverage and subsequent imputation. Resequencing of specific genes (using NGS techniques) also can be used to study SNPs, INDELs (insertion–deletion mutations), CNVs (Copy Number Variations) and rearrangements in the same species with lower cost and higher coverage.
The most popular genomic methods used in selection breeding programmes are reduced-representation library (RRL) methods. These methods include the preparation of reduced libraries through the use of restriction enzymes (RAD-seq), transcriptome sequencing (RNA-seq), targeted amplicon sequencing (TAS) and hybrid enrichment with the use of probes (Gonen et al. 2015b; Robledo et al. 2017). The technical basis of RRL is the use of restriction enzymes to digest the genome and then to select the digested fragments. The advantage of RRL methods is the reduction of the cost of the analysis with high coverage (Amish et al. 2012). Genotyping errors can be eliminated by means of specific software such as Angsd (Korneliussen et al. 2014) and NgsCovar (Fumagalli et al. 2014). The most popular RRL methods are restriction-site-associated DNA sequencing (RAD-Seq) (Baird et al. 2008; Davey et al. 2013; Cruaud et al. 2014; Hand et al. 2015; Rochette et al. 2019) and genotyping by sequencing (Genotyping By Sequencing - GBS) (Elshire et al. 2011; Dodds et al. 2015; Gorjanc et al. 2015; Brouard et al. 2017). For instance, de novo RAD-Seq analysis is used in non-model species because it does not require a priori knowledge of the reference genome sequence (Gonen et al. 2015b). In addition, nGBS (normalised genotyping by sequencing) involves the use of restriction enzymes for the digestion of DNA, producing a large number of fragments for each sample. These fragments are then sequenced, producing several million reads per sample. The RAD-Seq method was used on Atlantic salmon (Houston et al. 2012) and rainbow trout by Palti et al. (2014) in selection programmes. The GBS method can be used for SNP genotyping (Elshire et al. 2011; Andrews et al. 2016; Li and Wang 2017; Robledo et al. 2017). Compared to SNP arrays, this method requires more rigorous quality control of genomic data. In fact, some spurious or missing data may lead to erroneous conclusions (Gorjanc et al. 2015; Andrews et al. 2016). These errors may occur during sequencing and alignment analyses (Nielsen et al. 2012; Davey et al. 2013; Sims et al. 2014; Verdu et al. 2016). Several studies on fish species have shown these problems in relation to high polymorphism rates and the presence of highly repetitive elements in the genomes (Lal et al. 2016; Yuan et al. 2017). Methods that are currently available for reducing the aberrant data obtained during RAD-Seq and GBS analysis are based on quality control checks (QScore ≥ 30) (Lal et al. 2016), verification of Hardy-Weinberg equilibrium, and verification of the presence of paralogous genes or SNPs with low coverage (Catchen et al. 2013; Bianco et al. 2014; Torkamaneh et al. 2017). Improving the accuracy of genomic data obtained with GBS and RAD-Seq methods is particularly important for fish populations with limited genetic resources. The interest of the GBS and RAD-Seq methods will remain high in the fish breeding sector until the cost of SNP HD arrays becomes more affordable.
Genome-wide association studies
Genome-wide association studies (GWAS) have been proposed as an effective approach for the identification of many causative mutations and genetic factors that constitute the main quantitative traits (Stram 2004; Meuwissen 2007; Witte 2010; Daetwyler et al. 2010). Unlike linkage studies, which consider the phenomenon of inheritance of chromosomal regions linked to the presence of a trait within a family, association studies consider instead the difference between the frequency of SNPs affecting the trait of interest in a population and then the incorporation of the information into the calculation of the breeding value of animals (Tsai et al. 2015). Association studies may be conducted through two approaches: direct and indirect. A direct association study is to catalog and test one by one all the possible causal mutations. However, the direct approach presents some practical problems. This strategy involves genome-wide identification of all genes (up to 30,000 genes) as well as of all SNPs. For these reasons, the use of the direct method is limited to a few cases and has almost always been replaced by the application of the indirect method. The indirect strategy avoids the need to catalog all mutations that could potentially give predisposition to a given trait and instead relies on the association between a giver phenotype and markers located near a strategic locus. These associations are obtained from studies of linkage disequilibrium (LD) between marker loci. The indirect strategy then employs a dense map of polymorphic markers to explore the genome in a systematic way.
The choice of markers differentiates further the indirect approach in two different strategies. In the first, markers are chosen very close to exon regions of known genes. The second employs markers located in large regions, virtually anywhere in the genome, thus considering the chromosomes in their entirety, including intronic regions. The choice of the marker falls on bi-allelic SNPs because of their high frequency in the animal genomes, for the low rate of mutation and for the ease with which it can be analyzed. Linkage means the presence of genes at close loci on the same chromosome.
LD is a combination of alleles at two or more loci that occurs more often than it would happen by chance. The presence of a LD thus indicates co-segregation of two markers. In generally, the LD between two SNPs decreases with the physical distance, and the extent of LD varies strongly among the regions of the genome. LD analysis is a valuable tool for fine mapping. The LD is strongly influenced by the local recombination rate, distance between the gene and markers and is correlated with other factors such as the content of bases G and C, the gene density and the presence of SINE or Alu1 repeats (Smith et al. 2005). A high degree of similarity was observed between several populations (in regions classified as high LD); this was due not only to past historical shared sequences between populations, but also by the fact that the LD may be influenced by the features of the local sequence. It is generally confirmed that while the bottlenecks of the population, geographical sub-divisions and selection tend to increase the extension of the LD in the single population, population growth and random mating tend to decrease the extension of LD in the genome (Daetwyler et al. 2010). In addition to these factors, the extension of the LD in a particular region of the genome can also be influenced by the physical characteristics of the surrounding DNA sequences. Some types of sequences, such as, for example, rich sequences of bases G and C, may be associated with higher rates of mutation, two phenomena that can directly lower the level of surrounding LD. LD is generally weaker near the ends of chromosomes, probably due to the high recombination rate in the neighborhood of these regions and is stronger around the centromeres and in other intermediate portions of each chromosome (large portions of the repeated genome), where recombination rates are on average lower. Furthermore, the LD is generally stronger in the large chromosomes, which have a recombination rate lower and weaker in the small chromosomes, that have a higher recombination. The presence of repeated sequences in a region is strongly associated with LD and this correlation is significantly increased when we consider large windows of genome. The presence of repeats LINE is associated to an increase of the level of LD while other types of repetition, in particular SINE and Alu, are strongly associated with a lower level of LD. To further characterize the relationship between LD and changes in the sequence, Smith et al. (2005) have divided the genome into quartiles according to the estimated levels of LD in each 100-kb window. The lower frequency allele (MAF) of SNPs in each of the four quartiles (corresponding to the regions of LD high, middle and bottom) were very similar. The study found that the content of G and C decreases gradually in correspondence with the increase of the degree of LD (4349 to 10,000 nucleotides of GC bases in the quartile with the low LD and 3904 to 10,000 nucleotides GC bases in the quartile with the highest LD). It was also noted that the genes are significantly more concentrated in the quartile with the highest and lowest quartiles of LD compared to the intermediate level. This suggests that while for some genes may be advantageous to be located in regions of strong LD, where no recombination takes place, other genes may be favored by the diversity of haplotypes. Smith et al. (2005) have also provided a functional annotation of each gene, depending on of their location in the quartiles with the highest and lowest LD, using the database Gene Ontology. Genes associated with immune response (including genes involved in the inflammatory response and in response to pathogens and parasites) and neurophysiological processes (including sensory perception) are often located in regions with a low degree of LD. In contrast, genes associated with response to DNA damage, cell cycle, or to the metabolism of DNA and RNA appear more often located in regions with high LD. In fact, these genes represent conserved biological processes where recombination and mutation become deleterious changes that are generally removed through natural selection. Despite the fact that LD constitutes a powerful tool for the association analysis, there are some limitations which should be taken into account. The history of recombination between pairs of SNPs can be estimated with the use of the LD measures (e.g. D’ or r2 ) (Devlin and Rish 1995). If you are using the measure D', the haplotypes are then defined as regions where the standard measure of disequilibrium D' is assumed to be 1 (or still very close to the unit value, making appropriate reference to the confidence intervals), in the absence of recombination, for all (or almost) the pairs of markers in the region. A haplotype is a particular combination of alleles at different polymorphic sites on the same chromosome observed in a population at strong LD. Haplotypes are inherited regions without substantial recombination and the level of LD is very high. In a preliminary analysis, it can be seen that the genetic variation is organized into relatively short segments in strong LD, identified as haplotype blocks, each containing a few common haplotypes separated by hot spots of recombination. You can define hot spots those segments of average length less than 10 kb, where the measure of r2 (the squared correlation between alleles present at two loci) between two closed markers never exceeds the value of 0.10. Hot spots, in essence, represent interruptions of LD, they make up less than 10% of the sequences and occur in about 50% of recombination events. These differences of the recombination rate explain, at least in part, the heterogeneity of LD values between SNPs markers along the genome of several species. The most attractive use of haplotypes is the idea that the common haplotypes can capture most of the genetic variation. A reduced number of SNPs (tag SNP), correlated with the other markers of the same haplotype, can be than used (Stram 2004).
In fish breeding, most production traits are affected by a large number of genes with possible interactions among them. As a consequence, in studies of complex traits, the single-locus analysis does not produce reliable results (Goddard 2009). Another disadvantage of the single-SNP approach is that LD could extend to a wide genomic region. In this case, the detection of the region containing the true mutation and the significant associated SNPs could be difficult (Pryce et al. 2010). A possible solution to this problem could be to fit all SNPs simultaneously by using Bayesian models (Gonzàlez-Recio and Forni 2011). Whatever the method used for GWAS, SNPs declared associated with a trait have to be validated, even if a stringent threshold is used to detect the statistical associations. The best way to validate the detected SNPs is to verify the associations in an independent population. GWAS results depend on several factors such as the actual genetic population structure, genetic characteristics of loci and LD within the population (Meuwissen 2007). LD between markers and QTL depends on the factors such as marker density, the effective size of population, and relationships between the selection candidates and the reference population (Mangin et al. 2012). Increasing the density of SNPs will improve the power of QTL detection (Correa et al. 2016). The results of numerous research studies have shown that the confidence intervals for mapped QTLs are generally large. This situation implies the presence, within the confidence intervals, of a large number of genes (1000) and, therefore, it is difficult to identify the single SNP. Second, the use of markers associated with QTLs is complicated, as this association can vary from family to family. An alternative to this method for the fine mapping of the QTL consists of using linkage analysis together with the LD in the population (Hayes et al. 2009a; Meuwissen et al. 2011).
The GWAS method is best used when re-sequencing and reference sequences and an HD genotyping array are available. GWAS is used to identify single-nucleotide polymorphisms (SNPs) and genomic regions associated with the most important production, physiological and fitness traits. LD pruning is used to select the SNPs. Firstly, the phenotypic data should be normalized. The sample size and the number of SNPs must be adjusted to increase the level of significance. The GWAS method was initially used for the study of human diseases and allowed the identification of several SNP associated with these diseases (Visscher et al. 2017). At the beginning, the main problem with this method was the high rate of false positives. This situation derives from the very high number of statistical tests carried out and the significance threshold (Brzyski et al. 2017). Correction methods used for multiple tests include: reducion of the false discovery rate (FDR) or the Bonferroni method. Another factor that increases the number of false positives is the kinship between individuals in the population (Zhu and Yu 2009). The initial GWAS method was later replaced by the linear mixed model (LMM) which considers the kinship structure of the population (Yu et al. 2006). The population structure can be analyzed using the Structure (Pritchard et al. 2000) or DAPC (Jombart et al. 2010) softwares. Several algorithms are available for calculating the kinship matrix (Van Raden 2008) and false positives are analyzed using a fixed effect (population structure) and random effect (polygenic background) (Yu et al. 2006). The following softwares are available: Emma (Kang et al. 2008), FaSTLMM (mixed linear transformed and spectrally factored model) (Lippert et al. 2011) and Gemma (Zhou and Stephens 2012). In these methods, the computation efficiency is increased as the same variance components are not required, for all the candidate markers (Zhou and Stephens 2012). FaST-LMM uses a subset of SNPs (Lippert et al. 2011) while the Gemma software uses all markers. These methods are now available (multi-locus MLM with random SNP effect) also in the Tassel (Bradbury et al. 2007) and Gapit softwares (Tang et al. 2016).
Most of the research on fish species was carried out using individual SNPs. The alternative is to use haplotypes with 8–15 alleles. GWAS allows to detect associations between genetic markers and production, disease resistence to specific pathogens or functional phenotypes (Goddard and Hayes 2009). These strategies associated with the availability of a high-density reference sequence and a physical map of the genome allow detailed analysis of polymorphisms (Rexroad and Vallejo 2009; Vallejo et al. 2019). RNA-seq and GBS methods are also used for QTL mapping. The availability of high-density SNP arrays has enabled several GWAS studies to be carried out in the Atlantic salmon (Salmo salar) (Tsai et al. 2015) and rainbow trout (Robledo et al. 2017). GWAS was used to study and dissect complex importatant traits (Correa et al. 2015, 2016; Gonzalez-Pena et al. 2016; Tsai et al. 2016b; Gutierrez et al. 2018). QTLs identified for rainbow trout include stress response (Vallejo et al. 2009), spawning time (Colihueque et al. 2010), growth rate (Wringe et al. 2010), osmoregulation capacity (Le Bras et al. 2011), salinity tolerance (Norman et al. 2012) and for Atlantic salmon growth rate (Houston et al. 2009; Baranski et al. 2010; Gutierrez et al. 2012; Tsai et al. 2015; Yoshida et al. 2018), salinity tolerance (Norman et al. 2012), resistence to sea lice (Tsai et al. 2016a), pancreatic disease (Gonen et al. 2015a) and amoebic gill disease (Robledo et al. 2018b). In Table 2 are reported some QTLs identified for the Atlantic salmon and rainbow trout. However, these QTLs explain only a part of the total additive genetic variation of the character under consideration and have only been confirmed in some families/populations. The annual period of sexual maturity is an important trait for salmonids (Küttner et al. 2011). QTLs for sexual maturity have been identified in rainbow trout (Easton et al. 2011), Atlantic salmon (Gutierrez et al. 2014) and Artic charr (Küttner et al. 2011). One of the main QTLs for sexual maturation in Atlantic salmon was found independently in two studies (Ayllon et al. 2015; Barson et al. 2015). Incorporation of the QTL for the infectious pancreatic necrosis (IPN) in the Atlantic salmon resulted in a 47% reduction of the disease in 2009 (Houston et al. 2012; Moen et al. 2015). So far, effective breeding program based on the MAS approach were based on a simple trait in which the main QTL was known (Yue 2014). Most of the traits being selected in the aquaculture sector follow a polygenic model and therefore the study of QTLs in the era of genomic selection is likely of limited value.
Genomic selection
Recent innovations in the field of molecular biology have allowed greater efficiency and accuracy in animal selection (de los Campos et al. 2013). Meuwissen (2007) defined genomic selection the simultaneous selection of several tens or hundreds of thousands of markers, with which it is possible to cover the entire genome so densely that all genes are expected to be in LD with at least some of the markers used. In particular, genomic selection is based on the prediction of the genomic breeding values of individuals (Genomic Estimated Breeding Values, GEBV), consisting of a large number of DNA markers that are in LD with QTLs encoding traits with important economic value and that span the genome (Solberg et al. 2008; Clark et al. 2011; Börner and Reinsch 2012). The first proven methodology for implementing genomic selection was presented by Meuwissen et al. (2001). Meuwissen et al. (2016), they first introduced genomic selection as a selective strategy based on GEBV obtained by the use of molecular markers. This method implicitly recognizes the fact that quantitative traits are controlled by the segregation of a large number of loci (QTLs) and assumes that the markers are in LD with the QTLs (Calus et al. 2008; Calus and Veerkamp 2011). SNPs are located in coding and noncoding (more frequently) regions, and we assume that each quantitative trait is controlled by approximately 300-700 QTLs (Hayes et al. 2009b). Therefore, the higher the density of the markers and the stronger the level of LD between markers and the QTL, and, consequently, the proportion of genetic variation that can be explained through the markers. Genome-wide SNPs are used to calculate the genomic relationship matrix (GRM) and the estimated genomic values (GEBV) (Van Raden et al. 2009; Aguilar et al. 2011; Goddard et al. 2011; Legarra et al. 2014).
Genomic selection allows for greater accuracy in EBV estimation than traditional methods (Habier et al. 2007; Forni et al. 2011; Vallejo et al. 2017). However, the accuracy of the GBLUP estimate varies mainly in relation to the heritability of the selected trait (Daetwyler et al. 2008), allelic substitution effect for the QTL (Meuwissen et al. 2001; Goddard 2009), and the size of the training population (Daetwyler et al. 2008; Andonov et al. 2017).
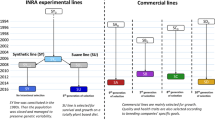
Genomic evaluation is a two-step method: (i) 1st step: the genome is divided into chromosome segments. A reference population (1000+ individuals, genotypes and phenotypes) is used to estimate the SNP or haplotype effects at each chromosome region; (ii) 2nd step: selection candidates are genotyped, and their GEBVs are calculated as the sum of segment effects. Phenotypic and pedigree data are used for the estimation of the SNP effects in the reference population (Fig. 1). Evaluations of marker effects in the reference population should be updated at each generation. The population is generally divided into reference and validation groups for the validation of statistical models (Eggen 2012; Wu et al. 2015). The population used for training must be adequate, especially for traits with low heritability. Subsequently, the effects of SNPs are validated in a population independent of the training population and verified for accuracy. All the segment effects are fitted simultaneously, reducing the bias.

Calculation of GEBV in a genomic selection scheme
If all the marker effects show a normal distribution and identical variance, the accuracy of GEBVs is equal to the traditional Best Linear Unbiased Prediction (BLUP) animal method. This method considers all markers to estimate the genetic value of the candidate for selection. These effects can also be used to select animals whose phenotype is unknown (Meuwissen et al. 2001). The BLUP method uses information from the animals' pedigrees and phenotypes to estimate breeding values (EBVs). The use of molecular markers (SNPs) provides a new source of information, which improves the accuracy of EBV prediction. We expect the selection response will be highest for traits with low heritability and in those that cannot be measured in the candidate for selection (such as disease resistance) (Taylor 2014). For markers with larger effects (e.g. longer tails) and different variances, SNP-BLUP and G-BLUP methods were first used (de Roos et al. 2011; Habier et al. 2011). The SNP-BLUP (RR-BLUP) model assumes that each SNP has a very small effect on the genetic variance of the trait. The GBLUP method computes EBVs using traditional mixed model equations, and the relationship matrix is calculated as relationship coefficients among the observed genotypes (Van Raden 2008; Hayes et al. 2009a). The G-BLUP and SNP-BLUP methods assume that all SNP effects are non-zero, small and normally distributed (Dong et al. 2016). After the GBLUP method, the ssGBLUP (single step) method was developed (Aguilar et al. 2010; Legarra et al. 2014; Yoshida et al. 2019). In this method, all the information (phenotypic, genotypic and genealogical) is simultaneously analysed (Aguilar et al. 2011) weighting the genomic and pedigree relationships. The two methods (GBLUP and ssGBLUP) assume that all SNPs are equal (weight and variance).
Subsequently, alternative methods were developed, such as BayesA, BayesB, BayesC and Lasso, using different weights of SNPs (high LD with a mutation or associated with a QTL) and the presence of QTLs) (Meuwissen et al. 2001; Habier et al. 2011; Gianola 2013; Fernando et al. 2014), WGBLUP and WssGBLUP (Snelling et al. 2011; Tiezzi and Maltecca 2015) and TABLUP (Zhang et al. 2010). Bayes A is set up at two levels. The first is similar to the SNP-BLUP model. The second is organized at the level of variance across the SNPs. In Bayes B, some SNPs are located in regions with no QTL (zero value), whereas some show a moderate or large effect because they are in linkage disequilibrium with QTLs. Lasso Bayes uses a similar model of Bayes A to estimate marker effects, but it makes a different assumption concerning the distribution of marker variance (sampling from a double exponential distribution). Bayesian methods allow us to obtain a comparable accuracy compared to traditional methods by means of a shrinkage of the segment effects based on prior QTL distribution knowledge (Habier et al. 2011).
Using SNP low-density bead chips, few differences in accuracy between GBLUP and Bayesian methods were observed (Habier et al. 2011). The genomic evaluation explains a part of the quantitative variance (between 40 and 60% depending on the traits) (Schenkel 2009). Total genetic effects are then estimated by combining genomic evaluation and polygenic effects (Fig. 2). A single or multi-step method can be used to combine genomic and polygenic information (Calus and Veerkamp 2011).

Conceptual view of genomic evaluation
Traditionally, the selection of the best animals is made using data collected over time on its own performance, records of relatives and relationships across populations. This approach means that for traits measured only in older age, the generation interval will increase. The accuracy of evaluations is almost equivalent for males and females (Schaeffer 2006). Several studies have shown some benefits of including males and females in the reference population (Fig. 3) (Sonesson and Meuwissen 2009; Toro et al. 2017). The use of genomic data keeps the response to selection constant over generations (Sonesson 2007; Sonesson and Meuwissen 2009). Solberg et al. (2008) reported that to obtain the same accuracy in genomic selection, it is necessary to use a greater number of SNPs (at least three times) than microsatellite markers and to increase the density of markers from 1Ne(effective population size)/morgan to 8Ne/morgan. For a family of 100 individuals, at least 24,000 loci are required. The response to genomic selection using a cohort of 100 families improves significantly compared to the use of single families (Khatkar et al. 2017). In this case, the cost of infrastructures used for breeding decreases significantly compared to traditional systems. Multistage selection (phenotypic + genotyping) is probably the optimal solution (Khatkar et al. 2017).

Accuracy of EBVs in the reference population for different heritabilities (adapted from D’Agaro et al. 2007)
Optimisation of fish breeding schemes using genomic selection methods
Genetic progress in fish breeding depends on several factors, such as selection intensity, generation interval and accuracy of genetic evaluations (Täubert et al. 2011; Mrode 2014). Advances in molecular genetics, which became available in recent decades, allowed us to expand, or even to rethink, the scenario of fish selection. These new discoveries have allowed us to identify the chromosome position (within a confidence interval) of quantitative trait loci (QTLs) responsible for relevant variation in several traits of economic interest and functional traits (Dekkers and Hospital 2002). For example, Ozaki et al. (2007) used microsatellite markers to identify two QTLs that influence rainbow trout (27 and 34% of the phenotypic variation) resistance to infectious pancreatic necrosis (IPN), and Rodriguez et al. (2004) developed three different linkage maps. The most famous example in fish breeding is the QTL which encodes resistance to IPN in Atlantic salmon (Houston et al. 2008; Moen et al. 2009; Moen et al. 2015). The higher frequency of the allele at this QTL induces almost total resistance to IPN, and a breeding plan was rapidly implemented in Norway. However, it appeared from these studies that only some traits are determined by important QTLs (Haidle et al. 2008; Gutierrez et al. 2012). Another successful example is the selection of populations of rainbow trout resistant to bacterial cold water disease (Bcwd) using a 57K SNP chip (Vallejo et al. 2017; Liu et al. 2018). Unfortunately, the direct use of genotypic information in MAS programmes (Dekkers 2007; Tiezzi and Maltecca 2015) or practical applications were limited. These results can be partly explained in terms of the large number of loci controlling the quantitative traits and the limited number of markers available. The reduction in the accuracy of EBVs is partly explained by the fact that QTLs are not identified directly by specific SNPs, but by the associated markers. In more recent years, new methods have been discovered for the simultaneous determination and precise genotyping of hundreds of thousands of SNP markers using new SNP HD arrays (Houston et al. 2014; Yáñez et al. 2014). SNPs, although less informative than other markers (for example, microsatellite markers), allow efficient monitoring, and, at lower cost, genetic variations among many samples (Van Tassell et al. 2008). Recombination occurring between genes can reduce the LD level at each generation. If LD (between the marker and a QTL) is present only within families (and not between families), the recombination occurring between genes can break the association (Wientjes et al. 2013). This situation makes the MAS unattractive because the LD must be verified at each generation for each family (Dekkers 2007). MAS schemes can be used only when the QTL explains a large proportion of total trait variance (as is the case for IPN resistance in Atlantic salmo (Houston et al. 2008, Laghari et al. 2014). The discovery and annotation of thousands of new genomic variants allow the production of new SNP arrays, and, in this way, the implementation of effective genomic programmes (Abdelrahman et al. 2017; Guppy et al. 2018). Currently, there are several SNP arrays available for salmonid species. Houston et al. (2014) identified approximately 132 K SNP markers in different populations of farmed and wild Atlantic salmon using a SNP array produced by Affymetrix (Santa Clara, CA, USA), and Yáñez et al. (2014) validated 160 K SNP markers using a 200 K array for Atlantic salmon (for wild and farmed populations in Europe, North America and Chile). A 57 K SNP chip (Affymetrix Axiom, Santa Clara, CA) is available for rainbow trout (Palti et al. 2014).
The discovery in the last decades of the genomic selection approach has completely changed the method of calculation of the breeding values (GEBV) for the most important domestic livestock species (Meuwissen et al. 2016), and, potentially can do the same in the aquaculture sector (Correa et al. 2017). Recent studies have shown that the genomic selection method can be effectively used to improve some salmonid production and disease resistance traits (Yáñez et al. 2016; Yoshida et al. 2018), and the first example of a genomic selection programme was established for the genetic improvement of Atlantic salmon (Houston et al. 2014; Ødegård et al. 2014; Tsai et al. 2015). In recent years, several aquaculture breeding programmes have applied the genomic selection method: Atlantic salmon, rainbow trout, Nile tilapia and some crustacean species (Pacific whiteleg shrimp (Litopenaeus vannamei)) (Norris 2017; Zenger et al. 2017, 2019). While routine implementation of genomic selection is now largely carried out in salmonids (Yáñez et al. 2014), it can be expected that in the near future, this method will progressively spread to other fish species. However, genomic selection is an expensive method (fish genotyping costs range between 40 and 80 €/sample), so similar to the MAS method, it will be mostly relevant for traits of high economic value. Genomic selection has so far been applied for bacterial cold-water disease resistance in rainbow trout (Abdelrahman et al. 2017, Vallejo et al. 2017, Liu et al. 2018; Yoshida et al. 2018) and sea lice resistance in Atlantic salmon (Tsai et al. 2016b, 2017; Robledo et al. 2018b). In different studies, the accuracy of the GEBVs varied from 0.16 to 0.83 for different traits (+ 24% for growth traits and 22% for disease resistance) compared to traditional methods based on genealogy (Ødegård et al. 2014; Tsai et al. 2015; Robledo et al. 2018a, 2018b). Bayesian models have shown results comparable to traditional GBLUP methods (Zenger et al. 2019). Recently, Barría et al. (2018) reported an increase in accuracy compared to traditional methods of 155%.
Genomic selection can increase the selection response in the fish breeding sector (+10% for body weight) compared to traditional selection methods (Campos-Montes et al. 2013; Ødegård et al. 2014; Castillo-Juárez et al. 2015). In the future, the use of young breeders (reducing the generation interval), the reduction of the inbreeding rate (Vandeputte and Haffray 2014; Dupont-Nivet et al. 2006; Holtsmark et al. 2008) and the incorporation of dominance and epistatic effects (Dupont-Nivet et al. 2008; Nilsson et al. 2016) may further increase the genetic response (Campos-Montes et al. 2013; Castillo-Juárez et al. 2015). Improved genome annotation techniques and RNA sequencing (RNA-seq) allow the identification and characterization of new SNPs. The high fecundity and the family sizes used in fish breeding plans allow us to obtain a high selection accuracy (because of the close relationship between the reference population and selected candidates). The optimal size of the population (genotyped and phenotyped > 1000 individuals) and the population structure are important factors determining the success of the genomic selection scheme (Daetwyler et al. 2010a, 2013). A density of SNPs equal to 1000–2000 is sufficient to obtain excellent results (Zenger et al. 2019; Kriaridou et al. 2020). GEBV accuracy decreases dramatically in each generation if the relationship between the reference population and selected animals is low (Tsai et al. 2016b). Genomic selection is applied to raise fish in a common environment and thereby to reduce costly infrastructure interventions (Fernández et al. 2014).
Genomic selection using full-sib families can be performed using low-density SNP arrays to estimate GEBVs within known pedigrees (Lillehammer et al. 2013; Tsai et al. 2016b; Tsairidou et al. 2020). The use of low-density SNP arrays (GT-Seq) (Campbell et al. 2015) or fluorescence-based arrays reduces the costs of genetic analyses, particularly in aquaculture, where thousands of potential selection candidates are used (Lillehammer et al. 2013; Gutierrez et al. 2018). These technologies can be easily integrated using the imputation technique (Tsai et al. 2017; Yoshida et al. 2018; Tsairidou et al. 2020). In this case, only a sample of animals is genotyped with a SNP HD array with a significant reduction in costs (Tsai et al. 2017). Several fish breeding programmes routinely use low-density SNP arrays for parental assignment (Vandeputte and Haffray 2014); therefore, in this case, genomic selection can be applied with a small additional cost. It is worth noting that, however, the use of SNP HD arrays, complete genome sequences and amplicon genotyping allow an increase in the accuracy of the selection process. Whole-genome sequencing of selected individuals (e.g. parents) and the imputation method can improve the accuracy of the estimate (Badke et al. 2013; Browning and Browning 2016; Bilton et al. 2018). The accuracy of the imputation depends on several factors: the number of markers used, LD at the local level, minor allele frequency, and correlations between individuals (reference and selected) (Druet et al. 2010; Hickey et al. 2012). The accuracy of imputation has been measured in a few fish breeding programmes. Kijas et al. (2017) found an accuracy in Atlantic salmon of 0.89-0.97, and Tsai et al. (2017) equal to 0.90. Note that, to obtain an accurate imputation process, the precise location of genetic markers in the genome is needed. For many fish species, this information is still incomplete (Abdelrahman et al. 2017).
In fish breeding programmes, the cost of collecting phenotypic data may, in some situations, present a limitation to achievable genetic progress (Føre et al. 2018). The use of computer vision technologies such as video cameras or sensors could be helpful (Zenger et al. 2019; Saberioon et al. 2017) to monitor and to record certain production, behavioural and health characteristics of fish. Some examples are reported in the literature concerning fillet analysis by means of the hyperspectral imaging method (Saberioon et al. 2017) or camera monitoring of sea lice in Atlantic salmon farms (Føre et al. 2018). Recently, automatic food distribution systems and the use of video cameras have allowed for faster collection of phenotypic data in fish farms (McCarthy et al. 2010; Cobb et al. 2013; Saberioon et al. 2017). Machine vision systems (MVS, 2D or 3D imaging) allow the acquisition of various phenotypic measurements, such as the shape, size, weight, colour and quality of fish fillets (Schmidhuber 2015; Saberioon et al. 2017). The accuracy of the estimates is very high (≥ 0.95). Miranda and Romero (2017) measured body lengths in rainbow trout using MVS systems, and Colihueque et al. (2011, 2017) compared the fillet colour between a traditional system and a spectral model produced by MVS in Atlantic salmon and rainbow trout. Additional techniques can be used in data collection, such as hyperspectral imaging (HSI) (spectroscopy and imaging) and near-infrared (NIR) spectroscopy (Liu et al. 2013). These techniques allow us to determine the chemical characteristics (proximate analysis) and physical characteristics (qualitative characteristics of the carcass) with high precision (r > 0.8) (Saberioon et al. 2017). These technologies are automatable and non-invasive. The use of image analysis software based on neural network (ANN) algorithms can be used for the rapid and automated phenotyping of animals (Grys et al. 2017).
Prospects and challenges
Since the start of the “genomic era”, many innovations and advantages in the field of fish breeding have become available (D'Agaro 2018). On the other hand, there are some downsides. The first difficulty seems to be associated with the large amount of new data that genomic selection requires. In fact, the possibility of combining different sources of information makes the interpretation work very complex. To date, new genomic methods have enabled the integration of the information provided by molecular genetics with those hitherto available in the field by quantitative genetics. Based on these results, several breeding programs have already started to use genomic selection methods based on GEBVs.
Population genomic data sets have also provided valuable information to address many aspects of the eco-evolutionary history in several salmonid species, including the characterisation of population structure, local adaptation and genetic diversity (Crespi and Fulton 2004; Hohenlohe et al. 2011; Lamaze et al. 2012; Bourret et al. 2013; López et al. 2014; Yáñez et al. 2015; Liu et al. 2017; Lopez Dinamarca et al. 2019). Genomic information that is shared among individuals in a population can be used to construct a genomic relatedness matrix (Gienapp et al. 2017). The availability of thousands of SNPs also has allowed the development of conservation strategies in several countries (Abadía-Cardoso et al. 2013; McMahon et al. 2014; Bradbury et al. 2015; Bohling et al. 2016). Hand et al. (2015) used 10,267 SNPs to study the introgression of alleles of rainbow trout in wild populations of Cutthroat trout (Oncorhynchus clarkii lewisi) and to identify chromosomal regions affected by selection. Currently, such approaches have only rarely been implemented in brown trout (Sušnik Bajec et al. 2015). The identification of epigenetic traces in fish allows a better understanding of genotype-phenotype interactions (Gavery and Roberts 2017). New technologies allow us to understand some mechanisms, such as cytosine methylation and histone modifications. These tools allow us to understand how some active SNPs work in specific environmental conditions (Gutierrez et al. 2016) and to identify the signatures of selection (Kjærner-Semb et al. 2016). Comparative genomics and orthological analysis of wild populations can be used to identify and to analyse SNPs of interest (Emms and Kelly 2015).
In the future, additional novel technologies such as gene editing can be used to modify genes by adopting new targeted methods. These methods are based on engineered nucleases (Crispr-Cas9; Crispr/ScCas9; and Crispr-Cas12) (Zhang et al. 2018a, 2018b; Gosavi et al. 2020), which can generate a break in the double strand of DNA in a precise sequence. When cellular mechanisms repair the break, they can cause insertions or deletions that result in inactivation of the gene or replacement with an alternate sequence if an appropriate template of donor DNA is provided. The main interest in these technologies is because they allow a high success rate on targeted DNA modifications, even in homozygosity. This method can greatly reduce the time required to obtain a line with the desired characteristics, overcoming the ethical implications, as in the case of introducing a “natural” mutation from another population at the same species (Gratacap et al. 2019). At present, this editing methodology in aquaculture has been used to obtain albino and sterile Atlantic salmon (Wargelius et al. 2016), common carp with a reduced number of spines and more muscle cells (Zhong et al. 2016) and sex-reversed Nile tilapia (Li and Wang 2017). While none of these experiments have reached the commercial production stage, these modern biotechnological methods clearly provide a new impetus to the direct targeted gene modification approach. The commercial applicability in the genetic improvement of fish species will, however, depend on how these animals are considered by the regulatory authority (Abdelrahman et al. 2017).
Conclusions
The genetic improvement of fish has historically been carried on the base of estimated breeding values of animals, starting from their performance and pedigrees. Currently, genomic selection has radically changed this scenario, allowing us to estimate the genomic value of an animal even without knowing its pedigree and at birth. This information then allows us to speed up the selection process and increase the accuracy of estimates. In salmonid species, several projects have concentrated their activities on the collection of a large number of genetic markers and have increased the reference population. In recent years, the accuracy of the GEBV of selection candidates has rapidly increased. Future strategies will allow simultaneous management of the use of genomic information and control of inbreeding in these and in new species.
Data availability
Public available data
References
Abadía-Cardoso A, Anderson EC, Pearse DE, Garza JC (2013) Large-scale parentage analysis reveals reproductive patterns and heritability of spawn timing in a hatchery population of steelhead (Oncorhynchus mykiss). Mol Ecol 22(18):4733–4746
Abdelrahman H, El Hady M, Alcivar-Warren A, Allen S, Al-Tobasei R, Bao L (2017) Aquaculture genomics, genetics and breeding in the United States: Current status, challenges, and priorities for future research. BMC Genomics 18:191
Adzhubei AA, Vlasova AV, Hagen-Larsen H, Ruden TA, Laerdahl JK. Høyheim B (2007) Annotated expressed sequence tags (ESTs) from pre-smolt Atlantic salmon (Salmo salar) in a searchable data resource. BMC Genomics 8:209
Aguilar I, Misztal I, Johnson DL, Legarra A, Tsuruta S, Lawlor TJ (2010) Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J Dairy Sci 93:743–752
Aguilar I, Misztal I, Legarra A, Tsuruta S (2011) Efficient computation of the genomic relationship matrix and other matrices used in single-step evaluation. J Anim Breed Genet 128:422–428
Alhakami H, Mirebrahim H, Lonardi SA (2017) Comparative evaluation of genome assembly reconciliation tools. Genome Biol 18:93
Amish SJ, Hohenlohe PA, Painter S, Leary RF, Muhlfeld C, Allendorf FW, Luikart G (2012) RAD sequencing yields a high success rate for westslope cutthroat and rainbow trout species-diagnostic SNP assays. Mol Ecol Resour 12:653–660
Andonov S, Lourenco D, Fragomeni B, Masuda Y, Pocrnic I, Tsuruta S (2017) Accuracy of breeding values in small genotyped populations using different sources of external information—a simulation study. J Dairy Sci 100:395–401
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA (2016) Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 17:81
Ayllon F, Kjærner-Semb E, Furmanek T, Wennevik V, Solberg M, Dahle G (2015) The vgll3 locus controls age at maturity in wild and domesticated Atlantic Salmon (Salmo salar L.) males. PLoS Genet 11:e1005628
Badke YM, Bates RO, Ernst CW, Schwab C, Fix J, Van Tassell CP (2013) Methods of tagSNP selection and other variables affecting imputation accuracy in swine. BMC Genet 14:8
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3(10):e3376
Balding DJ (2006) A tutorial on statistical methods for population association studies. Nat Rev Genet 7:781–791
Baranski M, Moen T, Våge DI (2010) Mapping of quantitative trait loci for flesh colour and growth traits in Atlantic Salmon (Salmo salar). Genet Sel Evol 42:17–17
Barría A, Christensen KA, Yoshida GM, Correa K, Jedlicki A, Lhorente JP (2018) Genomic predictions and genome-wide association study of resistance against Piscirickettsia salmonis in Coho Salmon (Oncorhynchus kisutch) using ddRAD sequencing. G3 (Bethesda) 8:1183–1194
Barson NJ, Aykanat T, Hindar K, Baranski M, Bolstad GH, Fiske P (2015) Sex-dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature 528:405
Bentsen HB, Olesen I (2002) Designing aquaculture mass selection programs to avoid high inbreeding rates. Aquaculture 204:349–359
Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noe B (2014) The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun 5:3657
Bianco L, Cestaro A, Sargent DJ, Banchi E, Derdak S, Di Guardo M (2014) Development and validation of a 20K Single Nucleotide Polymorphism (SNP) whole genome genotyping array for apple (Malus × domestica Borkh). PLoS One 9:e110377
Bilton TP, Schofield MR, Black MA, Chagne D, Wilcox PL, Dodds KG (2018) Accounting for errors in low coverage high throughput sequencing data when constructing genetic maps using biparental outcrossed populations. Genetics. 209:65–76
Bohling J, Haffray P, Berrebi P (2016) Genetic diversity and population structure of domestic brown trout (Salmo trutta) in France. Aquaculture 462:1–9
Börner V, Reinsch N (2012) Optimising multi-stage dairy cattle breeding schemes including genomic selection using decorrelated or optimum selection indices. Genet Sel Evol 44:1
Bourret V, Kent MP, Primmer CR, Vasemägi A, Karlsson S, Hindar K (2013) SNP-array reveals genome-wide patterns of geographical and potential adaptive divergence across the natural range of Atlantic salmon (Salmo salar). Mol Ecol 22:532–551
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Bradbury IR, Hamilton LC, Rafferty S, Meerburg D, Poole R, Dempson JB, Bernatchez L (2015) Genetic evidence of local exploitation of Atlantic salmon in a coastal subsistence fishery in the Northwest Atlantic. Can J Fish Aquat Sci 72(1):83–95
Brieuc MSO, Waters CD, Seeb JE, Naish KA (2014) A dense linkage map for Chinook salmon (Oncorhynchus tshawytscha) reveals variable chromosomal divergence after an ancestral whole genome duplication event. G3 (Bethesda) 4(3):447–460
Brouard J-S, Boyle B, Ibeagha-Awemu EM, Bissonnette N (2017) Low-depth genotyping-by-sequencing (GBS) in a bovine population: strategies to maximize the selection of high quality genotypes and the accuracy of imputation. BMC Genet 18:32
Browning BL, Browning SR (2016) Genotype imputation with millions of reference samples. Am J Hum Genet 98:116–126
Brzyski D, Peterson CB, Sobczyk P, Candès EJ, Bogdan M, Sabatti C (2017) Controlling the rate of GWAS false discoveries. Genetics 205:61–75
Burton JN, Adey A, Patwardhan RP, Qiu R, Kitzman JO, Shendure J (2013) Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31:1119–1125
Calus MP, Veerkamp RF (2011) Accuracy of multi-trait genomic selection using different methods. Genet Sel Evol 43:26
Calus MP, Meuwissen THE, de Roos APW, Veerkamp RF (2008) Accuracy of genomic selection using different methods to define haplotypes. Genetics. 178:553–561
Campbell NR, Harmon SA, Narum SR (2015) Genotyping-in-Thousands by sequencing (GT-seq): a cost-effective SNP genotyping method based on custom amplicon sequencing. Mol Ecol Resour 15:855–867
Campos-Montes GR, Montaldo HH, Martínez-Ortega A, Jiménez AM, Castillo-Juárez H (2013) Genetic parameters for growth and survival traits in Pacific white shrimp Penaeus (Litopenaeus vannamei) from a nucleus population undergoing a two-stage selection program. Aquac Int 21:299–310
Carruthers M, Yurchenko AA, Augley JJ, Adams CE, Herzyk P, Elmer KR (2018) De novo transcriptome assembly, annotation and comparison of four ecological and evolutionary model salmonid fish species. BMC Genomics 19:32
Castillo-Juárez H, Campos-Montes GR, Caballero-Zamora A, Montaldo HH (2015) Genetic improvement of Pacific white shrimp Penaeus (Litopenaeus vannamei): Perspectives for genomic selection. Front Genet 6:93
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22(11):3124–3140
Cheng HH, Perumbakkam S, Pyrkosz AB, Dunn JR, Legarra A, Muir WM (2015) Fine mapping of QTL and genomic prediction using allele-specific expression SNPs demonstrates that the complex trait of genetic resistance to Marek’s disease is predominantly determined by transcriptional regulation. BMC Genomics 16:816
Christensen KA, Leong JS, Sakhrani D, Biagi CA, Minkley DR, Withler RE, Rondeau EB, Koop BF, Devlin RH (2018a) Chinook salmon (Oncorhynchus tshawytscha) genome and transcriptome. PLoS One 13(4):e0195461
Christensen KA, Rondeau EB, Minkley DR (2018b) The Arctic charr (Salvelinus alpinus) genome and transcriptome assembly. PLoS One 13:e0204076
Clark S, Hickey J, van der Werf J (2011) Different models of genetic variation and their effect on genomic evaluation. Genet Sel Evol 43:18
Cobb JN, DeClerck G, Greenberg A, Clark R, McCouch S (2013) Next-generation phenotyping: requirements and strategies for enhancing our understanding of genotype–phenotype relationships and its relevance to crop improvement. Theor Appl Genet 126:867–887
Colihueque N, Cardenas R, Ramirez L, Estay F, Araneda C (2010) Analysis of the association between spawning time QTL markers and the biannual spawning behavior in rainbow trout (Oncorhynchus mykiss). Genet Mol Biol 33:578–582
Colihueque N, Parraguez M, Estay FJ, Diaz NF (2011) Skin color characterization in rainbow trout by use of computer-based image analysis. N Am J Aquac 73:249–258
Colihueque N, Corrales O, Yáñez M (2017) Morphological analysis of Trichomycterus areolatus Valenciennes, 1846 from southern Chilean rivers using a truss-based system (Siluriformes, Trichomycteridae). ZooKeys 695:135–152
Correa K, Lhorente JP, Lopez ME (2015) Genome-wide association analysis reveals loci associated with resistance against Piscirickettsia salmonis in two Atlantic salmon (Salmo salar L.) chromosomes. BMC Genomics 16:854
Correa K, Lhorente JP, Bassini L, Lopez ME, Di Genova A, Maass A, Davidson WS, Yanez JM (2016) Genome wide association study for resistance to Caligus rogercresseyi in Atlantic salmon (Salmo salar L.) using a 50K SNP genotyping array. Aquaculture 472(S1):61–65
Correa K, Bangera R, Figueroa R, Lhorente JP, Yáñez JM (2017) The use of genomic information increases the accuracy of breeding value predictions for sea louse (Caligus rogercresseyi) resistance in Atlantic salmon (Salmo salar). Genet Sel Evol 49:15
Crespi BJ, Fulton MJ (2004) Molecular systematics of Salmonidae: combined nuclear data yields a robust phylogeny. Mol Phylogenet Evol 31:658–679
Cruaud A, Gautier M, Galan M, Foucaud J, Sauné L, Genson G, Dubois E, Nidelet S, Deuve T, Rasplus J-Y (2014) Empirical assessment of RAD sequencing for interspecific phylogeny. Mol Biol Evol 31:1272–1274
D’Agaro E (2017) New advances in NGS technologies. In: new trends in veterinary genetics. Intech Editions, London, pp 219-251
D'Agaro E (2018) Artificial intelligence used in genome analysis studies. Eurobiotech J 2(2):78–88
D’Agaro E, Woolliams J, Haley C, Lanari D (2007) Optimizing mating schemes in fish breeding. Ital J Anim Sci 6:795–796
Daetwyler HD, Villanueva B, Woolliams JA (2008) Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS One 3:e3395
Daetwyler HD, Pong-Wong R, Villanueva B, Woolliams JA (2010b) The impact of genetic architecture on genome-wide evaluation methods. Genetics 185:1021–1031
Daetwyler HD, Hickey JM, Henshall JM, Dominik S, Gredler B (2010a) Accuracy of estimated genomic breeding values for wool and meat traits in a multi-breed sheep population. Anim Prod Sci 50:1004–1010
Daetwyler HD, Calus MPL, Pong-Wong R, de los Campos G, Hickey JM (2013) Genomic prediction in animals and plants: Simulation of data, validation, reporting, and benchmarking. Genetics 193:347–365
Davey JW, Cezard T, Fuentes-Utrilla P, Eland C, Gharbi K, Blaxter ML (2013) Special features of RAD sequencing data: Implications for genotyping. Mol Ecol 22:3151–3164
Davidson WS, Koop BF, Jones SJ, Iturra P, Vidal R, Maass A (2010) Sequencing the genome of the Atlantic salmon (Salmo salar). Genome Biol 11:403
de los Campos G, Hickey JM, Pong-Wong R, Daetwyler HD, MPL C (2013) Whole genome regression and prediction methods applied to plant and animal breeding. Genetics 193:327–345
de Roos APW, Schrooten C, Veerkamp RF, van Arendonk JAM (2011) Effects of genomic selection on genetic improvement, inbreeding, and merit of young versus proven bulls. J Dairy Sci 94:1559–1156
Dekkers JC (2007) Prediction of response to marker-assisted and genomic selection using selection index theory. J Anim Breed Genet 124:331–341
Dekkers JCM, Hospital F (2002) The use of molecular genetics in the improvement of agricultural populations. Nat Rev Genet 3:22–32
Devlin B, Rish NA (1995) Comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29:311–322
Dodds KG, McEwan JC, Brauning R, Anderson RM, van Stijn TC, Kristjánsson T (2015) Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genomics 16:1047
Dong L, Xiao S, Wang Q (2016) Comparative analysis of the GBLUP, emBayesB, and GWAS algorithms to predict genetic values in large yellow croaker (Larimichthys crocea). BMC Genomics 17:460
Druet T, Schrooten C, De Roos A (2010) Imputation of genotypes from different single nucleotide polymorphism panels in dairy cattle. J Dairy Sci 93:5443–5454
Dupont-Nivet M, Vandeputte M, Haffray P, Chevassus B (2006) Effect of different mating designs on inbreeding, genetic variance and response to selection when applying individual selection in fish breeding programs. Aquaculture 252:161–170
Dupont-Nivet M, Vandeputte M, Vergnet A, Merdy O, Haffray P, Chavanne H (2008) Heritabilities and GxE interactions for growth in the European sea bass (Dicentrarchus labrax L.) using a marker-based pedigree. Aquaculture 275:81–87
Easton AA, Moghadam HK, Danzmann RG, Ferguson MM (2011) The genetic architecture of embryonic developmental rate and genetic covariation with age at maturation in rainbow trout (Oncorhynchus mykiss). J Fish Biol 78:602–623
Edge P, Vineet B, Vikas B (2017) HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res 27(5):801–812
Eggen A (2012) The development and application of genomic selection as a new breeding paradigm. Anim Front 2:10–15
Ellegren H (2014) Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol 29:51–63
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
Emms DM, Kelly S (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16:157
FAO (2019) The State of the World’s Aquatic Genetic Resources for Food and Agriculture. www.fao.org
FAO Fisheries and Aquaculture Department (2019) Fishery and Aquaculture Statistics. Global capture production 1950-2017 (FishstatJ) www.fao.org
Fernández J, Toro MÁ, Sonesson AK, Villanueva B (2014) Optimizing the creation of base populations for aquaculture breeding programs using phenotypic and genomic data and its consequences on genetic progress. Front Genet 5:414
Fernando RL, Dekkers JCM, Garrick DJ (2014) A class of Bayesian methods to combine large numbers of genotyped and non-genotyped animals for whole-genome analyses. Genet Sel Evol 2:46–50
Føre M, Frank K, Norton T, Svendsen E, Alfredsen JA, Dempster T, Watson EH, Stahl A, Sunde LM, Schellewald C, Skøien KR, Alver MO, Berckmans D (2018) Precision fish farming: a new framework to improve production in aquaculture. Biosyst Eng 173:176–193
Forni S, Aguilar I, Misztal I (2011) Different genomic relationship matrices for single-step analysis using phenotypic, pedigree and genomic information. Genet Sel Evol 43:1–7
Fouchécourt S, Picolo F, Elis S, Lécureuil C, Thélie A, Govoroun M, Brégeon M, Papillier P, Lareyre JJ, Monget P (2019) An evolutionary approach to recover genes predominantly expressed in the testes of the zebrafish, chicken and mouse. BMC Evol Biol 19(1):137
Fumagalli M, Vieira FG, Linderoth T, Nielsen R (2014) ngsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics 30:1486–1487
Gao G, Nome T, Pearse DE, Moen T, Naish KA, Thorgaard GH, Lien S, Palti YA (2018) New single nucleotide polymorphism database for rainbow trout generated through whole genome resequencing. Front Genet 24:9–147
Gavery MR, Roberts SB (2017) Epigenetic considerations in aquaculture. PeerJ 5:e4147
Georges M, Charlier C, Hayes B (2019) Harnessing genomic information for livestock improvement. Nat Rev Genet 20:135–156
Gharbi K, Gautier A, Danzmann RG, Gharbi S, Sakamoto T, Hoyheim B (2006) A linkage map for brown trout (Salmo trutta): chromosome homeologies and comparative genome organization with other salmonid fish. Genetics 172:2405–2419
Gianola D (2013) Priors in whole-genome regression: The Bayesian alphabet returns. Genetics 194:573–596
Gienapp P, Fior S, Guillaume F, Lasky JR, Sork VL, Csilléry K (2017) Genomic quantitative genetics to study evolution in the wild. Trends Ecol Evol 32(12):897–908
Gilbey J, Verspoor E, Mclay A, Houlihan D (2004) A microsatellite linkage map for Atlantic salmon (Salmo salar). Anim Genet 35:98–105
Gjedrem T (2000) Genetic improvement of cold-water fish species. Aquac Res 31:25–33
Gjedrem T (2012) Genetic improvement for the development of efficient global aquaculture: A personal opinion review. Aquaculture 344–349:12–22
Gjedrem T, Rye M (2018) Selection response in fish and shellfish: A review. Rev Aquac 10:168–179
Gjedrem T, Robinson N, Rye M (2012) The importance of selective breeding in aquaculture to meet future demands for animal protein: A review. Aquaculture 350–353:117–129
Gjerde B (1986) Growth and reproduction in fish and shellfish. Aquaculture 57:37–55
Gjerde B, Korsvoll A (1999) Realised selection differentials for growth rate and early sexual maturity in Atlantic salmon. Aquac Eur 99:73–74
Goddard ME (2009) Genomic selection: prediction of accuracy and maximisation of long-term response. Genetica 136:245–252
Goddard ME, Hayes BJ (2009) Mapping genes for complex traits in domestic animals and their use in breeding programmes. Mature Rev Genet 10:381–391
Goddard ME, Hayes BJ, Meuwissen THE (2011) Using the genomic relationship matrix to predict the accuracy of genomic selection. J Anim Breed Genet 128:409–421
Gonen S, Lowe NR, Cezard T, Gharbi K, Bishop SC, Houston RD (2014) Linkage maps of the Atlantic salmon (Salmo salar) genome derived from RAD sequencing. BMC Genomics 15:166
Gonen S, Baranski M, Thorland I, Norris A, Grove H, Arnesen P (2015a) Mapping and validation of a major QTL affecting resistance to pancreas disease (Salmonid alphavirus) in Atlantic salmon (Salmo salar). Heredity 115:405
Gonen S, Bishop SC, Houston RD (2015b) Exploring the utility of cross-laboratory RAD-sequencing datasets for phylogenetic analysis. BMC Res Notes 8:299
Gonzalez-Pena D, Gao G, Baranski M, Moen T, Cleveland BM, Kenney PB, Vallejo RL, Palti Y, Leeds TD (2016) Genome-wide association study for identifying loci that affect fillet yield, carcass, and body weight traits in rainbow trout (Oncorhynchus mykiss). Front Genet 7:203
Gonzàlez-Recio O, Forni S (2011) Genome-wide prediction of discrete traits using bayesian regressions and machine learning. Genet Sel Evol 43:7
Gorjanc G, Cleveland MA, Houston RD, Hickey JM (2015) Potential of genotyping-by-sequencing for genomic selection in livestock populations. Genet Sel Evol 47:12
Gosavi G, Yan F, Ren B, Kuang Y, Yan D, Zhou X, Zhou H (2020) Applications of CRISPR technology in studying plant-pathogen interactions: Overview and perspective. Phytopathol Res 2:21
Gratacap RL, Wargelius A, Edvardsen RB, Houston RD (2019) Potential of genome editing to improve aquaculture breeding and production. Trends Genet 35:672–684
Grys B, Lo D, Sahin N, Kraus O, Morris Q, Boone C (2017) Machine learning and computer vision approaches for phenotypic profiling. J Cell Biol 216:65–71
Guppy JL, Jones DB, Jerry DR, Wade NM, Raadsma HW, Huerlimann R (2018) The state of ‘omics’ research for farmed penaeids: advances in research and impediments to industry utilisation. Front Genet 9:282
Gutierrez AP, Lubieniecki KP, Davidson EA, Lien S, Kent MP, Fukui S (2012) Genetic mapping of quantitative trait loci (QTL) for body-weight in Atlantic salmon (Salmo salar) using a 6.5K SNP array. Aquaculture 358–359:61–70
Gutierrez A, Lubieniecki K, Fukui S, Withler R, Swift B, Davidson W (2014) Detection of quantitative trait loci (QTL) related to grilsing and late sexual maturation in Atlantic Salmon (Salmo salar). Mar Biotechnol 16:103–110
Gutierrez AP, Yanez JM, Davidson WS (2016) Evidence of recent signatures of selection during domestication in an Atlantic salmon population. Mar Genomics 26:41–50
Gutierrez AP, Bean TP, Hooper C, Stenton CA, Sanders MB (2018) A Genome-wide association study for host resistance to ostreid herpesvirus in Pacific oysters (Crassostrea gigas). G3 (Bethesda) 8(4):1273–1280
Guyomard R, Mauger S, Tabet-Canale K, Martineau S, Genet C, Krieg F (2006) A type I and type II microsatellite linkage map of rainbow trout (Oncorhynchus mykiss) with presumptive coverage of all chromosome arms. BMC Genomics 7:302
Guyomard R, Boussaha M, Krieg F, Hervet C, Quillet E (2012) A synthetic rainbow trout linkage map provides new insights into the esalmonid whole-genome duplication and the conservation of synteny among teleosts. BMC Genet 13:15
Habier D, Fernando RL, Dekkers JCM (2007) The impact of genetic relationship information on genome-assisted breeding values. Genetics. 177:2389–2397
Habier D, Fernando RL, Dekkens JCM (2009) Genomic selection using low-density marker panels. Genetics 182:343–353
Habier D, Fernando R, Kizilkaya K, Garrick D (2011) Extension of the Bayesian alphabet for genomic selection. BMC Bioinform 12:186
Haidle L, Janssen JE, Gharbi K, Moghadam HK, Ferguson MM, Danzmann RG (2008) Determination of quantitative trait loci (QTL) for early maturation in rainbow trout (Oncorhynchus mykiss). Mar Biotechnol 10:579–592
Hand BK, Hether TD, Kovach RP, Muhlfeld CC, Amish SJ, Boyer MC, O’Rourke SM, Miller MR, Lowe WH, Hohenlohe PA, Luikart G (2015) Genomics and introgression: discovery and mapping of thousands of species-diagnostic SNPs using RAD sequencing. Curr Zool 61:146–154
Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME (2009a) Genomic selection in dairy cattle: Progress and challenges. J Dairy Sci 92:433–443
Hayes BJ, Bowman PJ, Chamberlain AJ, Verbyla KL, Goddard ME (2009b) Accuracy of genomic breeding values in multi-breed dairy cattle populations. Genet Sel Evol 41:51
He X, Houde ALS, Neff BD, Heath DD (2017) Transcriptome response of Atlantic salmon (Salmo salar) to competition with ecologically similar non-native species. Ecol Evol 8:1769–1777
Hely FS, Amer PR, Walker SP, Symonds JE (2013) Optimised parent selection and minimum inbreeding mating in small aquaculture breeding schemes: A simulation study. Animals 7:1–10
Hickey JM, Kinghorn BP, Tier B, van der Werf JH, Cleveland MA (2012) A phasing and imputation method for pedigreed populations that results in a single-stage genomic evaluation. Genet Sel Evol 44:9
Hill WG (2013) On estimation of genetic variance within families using genome-wide identity-by-descent sharing. Genet Sel Evol 45:32
Hoggart CJ, Clark TG, De Lorio M, Whittaker JC, Balding DJ (2008) Genome-wide significance for dense SNP and resequencing data. Genet Epidemiol 32(2):179–185
Hohenlohe PA, Amish SJ, Catchen JM, Allendorf FW, Luikart G (2011) Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout. Mol Ecol Resour 11(Suppl.1):117–122
Holtsmark M, Klemetsdal G, Sonesson AK, Woolliams JA (2008) Establishing a base population for a breeding program in aquaculture, from multiple subpopulations, differentiated by genetic drift: I. Effects of the number of subpopulations, heritability and mating strategies using optimum contribution selection. Aquaculture 274:232–240
Houston RD (2017) Future directions in breeding for disease resistance in aquaculture species. Rev Bras Zootec 46:545–551
Houston RD, Haley CS, Hamilton A, Guy DR, Tinch AE, Taggart JB, McAndrew BJ, Bishop SC (2008) Major quantitative trait loci affect resistance to infectious pancreatic necrosis in Atlantic salmon (Salmo salar). Genetics 178(2):1109–1115
Houston RD, Bishop SC, Hamilton A, Guy DR, Tinch AE, Taggart JB, Derayat A, McAndrew BJ, Haley CS (2009) Detection of QTL affecting harvest traits in a commercial Atlantic salmon population. Anim Genet 40:753–755
Houston RD, Davey JW, Bishop SC, Lowe NR, Mota-Velasco JC, Hamilton A (2012) Characterisation of QTL-linked and genome-wide restriction site-associated DNA (RAD) markers in farmed Atlantic salmon. BMC Genomics 13:244–244
Houston RD, Taggart JB, Cézard T, Bekaert M, Lowe NR, Downing A (2014) Development and validation of a high -density SNP genotyping array for Atlantic salmon (Salmo salar). BMC Genomics 15:90
Howe K, Wood JMD (2015) Using optical mapping data for the improvement of vertebrate genome assemblies. Gigascience 4:10
Jain M, Hugh E, Olsen B, Paten MA (2016) The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 17(1):239
Janssen K, Chavanne H, Berentsen P, Komen H (2017) Impact of selective breeding on European aquaculture. Aquaculture 472:8–16
Janssen K, Saatkamp H, Komen H (2018) Cost-benefit analysis of aquaculture breeding programs. Genet Sel Evol 50:2
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11(1):1–5
Jørgen Ø, Matthew B, Bjarne G, Trygve G (2011) Methodology for genetic evaluation of disease resistance in aquaculture species: Challenges and future prospects. Aquac Res 42:103–114
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E (2008) Efficient control of population structure in model organism association mapping. Genetics 178:1709–1723
Kause A, Ritola O, Paananen T, Wahlroos H, Mantysaari E (2005) Genetic trends in growth, sexual maturity and skeletal deformations, and rate of inbreeding in a breeding programme for rainbow trout (Oncorhynchus mykiss). Aquaculture 247:177–187
Khatkar M, Zenger K, Jones D, Prochaska J, van der Steen H, Jerry D (2017) Quantitative genomic analyses in the Pacific whiteleg shrimp (Litopenaeus vannamei). Proc Assoc Advmt Anim Breed Genet 22:121–124
Kijas J, Elliot N, Kube P, Evans B, Botwright N, King H (2017) Diversity and linkage disequilibrium in farmed Tasmanian Atlantic salmon. Anim Genet 48:237–241
Kjærner-Semb E, Ayllon F, Furmanek T (2016) Atlantic salmon populations reveal adaptive divergence of immune related genes – a duplicated genome under selection. BMC Genomics 17:610
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM (2017) Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27:722–736
Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinf 15:356
Kriaridou C, Tsairidou S, Houston RD, Robledo D (2020) Genomic prediction using low density marker panels in aquaculture: Performance across species, traits, and genotyping platforms. Front Genet 11:124
Küttner E, Moghadam HK, Skúlason S, Danzmann RG, Ferguson MM (2011) Genetic architecture of body weight, condition factor and age of sexual maturation in Icelandic Arctic charr (Salvelinus alpinus). Mol Gen Genomics 286:67–79
Laghari MY, Lashari P, Zhang Y, Sun X (2014) Identification of quantitative trait loci (QTLs) in aquaculture species. Rev Fish Sci Aquac 22:221–238
Lal MM, Southgate PC, Jerry DR, Zenger KR (2016) Fishing for divergence in a sea of connectivity: the utility of ddRADseq genotyping in a marine invertebrate, the black-lip pearl oysteinctada margaritifera. Mar Genomics 25:57–68
Lamaze FC, Sauvage C, Marie A, Garant D, Bernatchez L (2012) Dynamics of introgressive hybridization assessed by SNP population genomics of coding genes in stocked brook charr (Salvelinus fontinalis): SNP population genomics in brook charr. Mol Ecol 21:2877–2895
Le Bras Y, Dechamp N, Krieg F, Filangi O, Guyomard R, Boussaha M (2011) Detection of QTL with effects on osmoregulation capacities in the rainbow trout (Oncorhynchus mykiss). BMC Genet 12:46
Legarra A, Christensen OF, Aguilar I, Misztal I (2014) Single step, a general approach for genomic selection. Livest Sci 166:54–65
Leitwein M, Guinand B, Pouzadoux J, Desmarais E, Berrebi P, Gagnaire P-A (2017) A dense brown trout (Salmo trutta) linkage map reveals recent chromosomal rearrangements in the Salmo genus and the impact of selection on linked neutral diversity. G3 (Bethesda) 7(4):1365–1376
Li YH, Wang HP (2017) Advances of genotyping-by-sequencing in fisheries and aquaculture. Rev Fish Biol Fish 27(3):535–559
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293
Lien S, Gidskehaug L, Moen T, Hayes BJ, Berg PR, Davidson WS (2011) A dense SNP-based linkage map for Atlantic salmon (Salmo salar) reveals extended chromosome homeologies and striking differences in sex-specific recombination patterns. BMC Genomics 12:615
Lien S, Koop BF, Sandve SR, Miller JR, Kent MP, Nome T (2016) The Atlantic salmon genome provides insights into rediploidization. Nature 533(7602):200–205
Lillehammer M, Meuwissen THE, Sonesson AK (2013) A low-marker density implementation of genomic selection in aquaculture using within-family genomic breeding values. Genet Sel Evol 45:39
Lippert C, Listgarten J, Liu Y, Kadie CM, Davidson RI, Heckerman D (2011) FaST linear mixed models for genome-wide association studies. Nat Methods 8:833–835
Liu D, Zeng X-A, Sun D-W (2013) NIR spectroscopy and imaging techniques for evaluation of fish quality—a review. Appl Spectrosc Rev 48:609–628
Liu L, Ang KP, Elliott JAK, Kent MP, Lien S, MacDonald D, Boulding EG (2017) A genome scan for selection signatures comparing farmed Atlantic salmon with two wild populations: testing colocalization among outlier markers, candidate genes, and quantitative trait loci for production traits. Evol Appl 10:276–296
Liu S, Vallejo RL, Evenhuis JP, Martin KE, Hamilton A, Gao G (2018) Retrospective evaluation of marker-assisted selection for resistance to bacterial cold water disease in three generations of a commercial rainbow trout breeding population. Front Genet 9:286
Liu G, Dong L, Gu L, Han Z, Zhang W, Fang M, Wang Z (2019) Evaluation of genomic selection for seven economic traits in yellow drum (Nibea albiflora). Mar Biotechnol 21:806–812
Loman NJ, Quick J, Simpson JT (2015) A complete bacterial genome assembled de novo using only Nanopore sequencing data. Nat Methods 12(8):733–735
Lopez Dinamarca ME, Linderoth T, Norris A, Lhorente JP, Neira R, Jáñez JM (2019) Multiple selection signatures in farmed Atlantic salmon adapted to different environments across hemispheres. Front Genet 10:90
López ME, Neira R, Yáñez JM (2014) Applications in the search for genomic selection signatures in fish. Front Genet 5:458
MacLeod IM, Hayes BJ, Chamberlain AJ, McPartlan HC, Goddard ME (2010) Power of a genome scan to detect and locate quantitative trait loci in cattle using dense single nucleotide polymorphisms. J Anim Breed Genet 127( 2:133–142
Macqueen DJ, Primmer CR, FAASG Consortium (2017) Functional Annotation of All Salmonid Genomes (FAASG): An international initiative supporting future salmonid research, conservation and aquaculture. BMC Genomics 18:484
Mangin B, Siberchicot A, Nicolas S, Doligez A, This P, Cierco-Ayrolles C (2012) Novel measures of linkage disequilibrium that correct the bias due to population structure and relatedness. Heredity 108:285–291
Mantere T, Kersten S, Hoischen A (2019) Long-read sequencing emerging in medical genetics. Front Genet 10:426
Marchini J, Howie B, Myer S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39(7):906–913
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP (2009) Development and characterization of a high density SNP genotyping assay for cattle. PLoS One 4(4):e5350
McCarthy CL, Hancock NH, Raine SR (2010) Applied machine vision of plants: a review with implications for field deployment in automated farming operations. Intell Serv Robot 3:209–217
McClelland EK, Naish KA (2008) A genetic linkage map for coho salmon (Oncorhynchus kisutch). Anim Genet 39:169–179
McGaughey KD, Yilmaz-Swenson T, Elsayed NM, Cruz DA, Rodriguez RR, Kritzer MD (2018) Comparative evaluation of a new magnetic bead-based DNA extraction method from fecal samples for downstream next-generation 16S rRNA gene sequencing. PLoS One 13(8):e0202858
McKinney GJ, Seeb LW, Larson WA, Gomez-Uchida D, Limborg MT, Brieuc MSO (2016) An integrated linkage map reveals candidate genes underlying adaptive variation in Chinook salmon (Oncorhynchus tshawytscha). Mol Ecol Resour 16(3):769–783
McMahon BJ, Höglund E, Höglund J (2014) How and why should we implement genomics into conservation? Evol Appl 7 (9:999–1007
Metzker ML (2010) Sequencing technologies — the next generation. Nat Rev Genet 11(1):31–46
Meuwissen THE (2007) Genomic selection: marker assisted selection on a genome-wide scale. J Anim Breed Genet 124:321–322
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker map. Genetics 157:819–829
Meuwissen THE, Hayes BJ, Goddard ME (2011) Genomic selection: A paradigm shift in animal breeding. Anim Front 6:6–14
Meuwissen T, Hayes B, Goddard M (2016) Genomic selection: A paradigm shift in animal breeding. Anim Front 6(1):6–14
Miranda JM, Romero M (2017) A prototype to measure rainbow trout’s length using image processing. Aquac Eng 76:41–49
Moen T, Hayes B, Baranski M, Berg PR, Kjoglum S, Koop BF (2008) A linkage map of the Atlantic salmon (Salmo salar) based on EST-derived SNP markers. BMC Genomics 9:223
Moen T, Baranski M, Sonesson AK, Kjøglum S (2009) Confirmation and fine-mapping of a major QTL for resistance to infectious pancreatic necrosis in Atlantic salmon (Salmo salar): Population-level associations between markers and trait. BMC Genomics 10:368
Moen T, Torgersen J, Santi N, Davidson WS, Baranski M, Ødegård J (2015) Epithelial cadherin determines resistance to infectious pancreatic necrosis virus in Atlantic Salmon. Genetics 200:1313–1326
Mrode R (2014) Linear models for the prediction of animal breeding values, 3rd edn. CABI publishing, Wallingford, UK
Nichols KM, Young WP, Danzmann RG, Robison BD, Rexroad C, Noakes M (2003) A consolidated linkage map for rainbow trout (Oncorhynchus mykiss). Anim Genet 34:102–115
Nielsen R, Korneliussen T, Albrechtsen A, Li Y, Wang J (2012) SNP calling, genotype calling, and sample allele frequency estimation from new-generation sequencing data. PLoS One 7:e37558
Nilsson J, Backström T, Stien LH, Carlberg H, Jeuthe H, Magnhagen C (2016) Effects of age and rearing environment on genetic parameters of growth and body weight and heritability of skin pigmentation in Arctic charr (Salvelinus alpinus L.). Aquaculture 453:67–72
Nirea KG, Sonesson AK, Woolliams JA, Meuwissen THE (2012) Strategies for implementing genomic selection in family-based aquaculture breeding schemes: Double haploid sib test populations. Genet Sel Evol 44:30–30
Norman JD, Robinson M, Glebe B, Ferguson MM, Danzmann RG (2012) Genomic arrangement of salinity tolerance QTLs in salmonids: a comparative analysis of Atlantic salmon (Salmo salar) with Arctic charr (Salvelinus alpinus) and rainbow trout (Oncorhynchus mykiss). BMC Genomics 13:420
Norris A (2017) Application of genomics in salmon aquaculture breeding programs: Who knows where the genomic revolution will lead us? Marine Genom 36:13–15
Ødegård J, Baranski M, Gjerde B, Gjedrem T (2011) Methodology for genetic evaluation of disease resistance in aquaculture species: Challenges and future prospects. Aquac Res 42:103–114
Ødegård J, Moen T, Santi N, Korsvoll SA, Kjoglum S, Meuwissen TH (2014) Genomic prediction in an admixed population of Atlantic salmon (Salmo salar). Front Genet 5:402
OECD/Food and Agriculture Organization of the United Nations (2014) OECD-FAO Agricultural Outlook 2014. OECD Publishing, Paris
Olesen I, Bentsen HB, Phillips M, Ponzoni RW (2015) Can the global adoption of genetically improved farmed fish increase beyond 10%, and how? J Mar Sci Eng 3:240–266
Ozaki A, Khoo SK, Yoshiura Y, Ototake M, Sakamoto T, Dijkstra JM (2007) Identification of additional quantitative trait loci (QTL) responsible for susceptibility to infectious pancreatic necrosis virus in rainbow trout. Fish Pathol 42:131–140
Palti Y, Genet C, Gao G, Hu Y, You F, Boussaha M (2012) A second generation integrated map of the rainbow trout (Oncorhynchus mykiss) genome: analysis of conserved synteny with model fish genomes. Mar Biotechnol 14:343–357
Palti Y, Gao G, Miller MR, Vallejo RL, Wheeler PA, Quillet E (2014) A resource of single-nucleotide polymorphisms for rainbow trout generated by restriction-site associated DNA sequencing of doubled haploids. Mol Ecol Resour 14:588–596
Palti Y, Gao G, Liu S, Kent MP, Lien S, Miller MR, Moen T (2015a) The development and characterization of a 57K single nucleotide polymorphism array for rainbow trout. Mol Ecol Resour 15(3):662–672
Palti Y, Silverstein JT, Wieman H, Phillips JG, Barrows FT, Parsons JE (2015b) Detection and validation of QTL affecting bacterial cold water disease resistance in rainbow trout using restriction-site associated DNA sequencing. PLoS One 10:e0138435
Phillips R, Keatley K, Morasch M, Ventura A, Lubieniecki K, Koop B (2009) Assignment of Atlantic salmon (Salmo salar) linkage groups to specific chromosomes: conservation of large syntenic blocks corresponding to whole chromosome arms in rainbow trout (Oncorhynchus mykiss). BMC Genet 10:46
Ponzoni RW, Nguyen NH, Khaw HL, Hamzah A, Bakar KR, Yee HY (2011) Genetic improvement of Nile tilapia (Oreochromis niloticus) with special reference to the work conducted by the WorldFish Center with the GIFT strain. Rev Aquac 3(1):27–41
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pryce JE, Bolormaa S, Chamberlain SJ, Bowman PJ, Savin K, Goddard ME, Hayes BJ (2010) A validated genome-wide association study in two dairy cattle breeds for milk production and fertility traits using variable length haplotypes. J Dairy Sci 93:3331–3345
Putnam NH (2016) Chromosome-scale shotgun assembly using an in vitro method for longrange linkage. Genome Res 26(3):342–350
Ramos AM, Crooijmans RPMA, Affara NA, Amaral AJ, Archibald AL, Beever JE (2009) Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One 4(8):e6524
Rexroad CE, Vallejo RL (2009) Estimates of linkage disequilibrium and effective population size in rainbow trout. BMC Genet 10:83
Rexroad CE, Palti Y, Gahr SA, Vallejo RL (2008) A second-generation genetic map for rainbow trout (Oncorhynchus mykiss). BMC Genet 9:74
Robertson FM, Gundappa MK, Grammes F (2017) Lineage specific rediploidization is a mechanism to explain time-lags between genome duplication and evolutionary diversification. Genome Biol 18:111
Robledo D, Palaiokostas C, Bargelloni L, Martínez P, Houston R (2017) Applications of genotyping by sequencing in aquaculture breeding and genetics. Rev Aquac 10(3):670–682
Robledo D, Matika O, Hamilton A, Houston RD (2018a) Genome-wide association and genomic selection for resistance to amoebic gill disease in Atlantic salmon. G3 (Bethesda) 8:1195–1203
Robledo D, Gutierrez AP, Barria A, Yanez JM, Houston RD (2018b) Gene expression response to sea lice in Atlantic salmon skin: An RNA-Seq comparison between resistant and susceptible animals. Front Genet 9:287
Rochette NC, Rivera-Colon AG, Catchen JM (2019) Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol Ecol 28(21):4737–4754
Rodriguez MF, Lapatra S, Williams S, Famula T, May B (2004) Genetic marker sassociated with resistance to infectious hematopoietic necrosis in rainbow and steelhead trout (Oncorhynchus mykiss) back crosses. Aquaculture 241:93–115
Saberioon M, Gholizadeh A, Cisar P, Pautsina A, Urban J (2017) Application of machine vision systems in aquaculture with emphasis on fish: State-of-the-art and key issues. Rev Aquac 9:369–387
Sakamoto T, Danzmann RG, Gharbi K, Howard P, Ozaki A, Khoo SK (2000) A microsatellite linkage map of rainbow trout (Oncorhynchus mykiss) characterized by large sex-specific differences in recombination rates. Genetics 155:1331–1345
Salem M, Vallejo RL, Leeds TD, Palti Y, Liu S, Sabbagh A, Rexroad CE 3rd, Yao J (2012) RNA-seq identifies SNP markers for growth traits in rainbow trout. PLoS One 7(5):e36264
Samy JKA, Mulugeta TD, Nome T, Sandve SR, Grammes F, Kent MP, Lien S, Våge DI (2017) SalmoBase: an integrated molecular data resource for salmonid species. BMC Genomics 18:482
Sanchez C, Smith T, Wiedmann R, Vallejo R, Salem M, Yao J (2009) Single nucleotide polymorphism discovery in rainbow trout by deep sequencing of a reduced representation library. BMC Genomics 10:559
Sánchez-Molano E, Pong-Wong R, Banos G (2016) Genomic-based optimum contribution in conservation and genetic improvement programs with antagonistic fitness and productivity traits. Front Genet 7:25
Schaeffer LR (2006) Strategy for applying genome-wide selection in dairy cattle. J Anim Breed Genet 123(4):218–23
Schenkel FS (2009) Reliability of genomic predictions for North American Holstein bulls. J Dairy Sci 92:16–24
Schmidhuber J (2015) Deep learning in neural networks: An overview. Neural Netw 61:85–117
Shi KP, Dong SL, Zhou YG, Li Y, Gao QF, Sun DJ (2019) RNA-seq reveals temporal differences in the transcriptome response to acute heat stress in the Atlantic salmon (Salmo salar). Comp Biochem Physiol 30:169–178
Silva RMO, Evenhuis JP, Vallejo RL, Gao G, Martin KE, Leeds TD, Palti Y, Lourenco DAL (2019) Whole-genome mapping of quantitative trait loci and accuracy of genomic predictions for resistance to columnaris disease in two rainbow trout breeding populations. Genet Sel Evol 1:42
Simao F, Waterhouse R, Ioannidis P, Kriventseva E, Zdobnov E (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP (2014) Sequencing depth and coverage: Key considerations in genomic analyses. Nat Rev Genet 15:121
Skaarud A, Woolliams JA, Gjøen HM (2011) Strategies for controlling inbreeding in fish breeding programs: An applied approach using optimum contribution (OC) procedures. Aquaculture 311:110–114
Skaarud A, Woolliams JA, Gjøen HM (2014) Optimising resources and management of genetic variation in fish-breeding schemes with multiple traits. Aquaculture. 420–421:133–138
Smith AV, Thomas DJ, Munro HM, Abecasis GR (2005) Sequence features in regions of weak and strong linkage disequilibrium. Genome Res 15(11):1519–1534
Snelling WM, Allan MF, Keele JW, Kuehn LA, Thallman RM, Bennett GL (2011) Partial-genome evaluation of postweaning feed intake and efficiency of crossbred beef cattle. J Anim Sci 89:1731–1741
Solberg TR, Sonesson AK, Woolliams JA, Meuwissen THE (2008) Genomic selection using different marker types and densities. J Anim Sci 86:2447–2454
Sonesson AK (2007) Within-family marker-assisted selection for aquaculture species. Genet Sel Evol 39:301–317
Sonesson AK, Meuwissen TH (2009) Testing strategies for genomic selection in aquaculture breeding programs. Genet Sel Evol 41:37
Sonesson AK, Ødegård J (2016) Mating structures for genomic selection breeding programs in aquaculture. Genet Sel Evol 48:46
Sonesson AK, Meuwissen THE, Goddard ME (2010) The use of communal rearing of families and DNA pooling in aquaculture genomic selection schemes. Genet Sel Evol 42:41
Sonesson AK, Woolliams JA, Meuwissen THE (2012) Genomic selection requires genomic control of inbreeding. Genet Sel Evol 44:27–27
Stram DO (2004) Tag SNP selection for association studies. Genet Epidemiol 27:365–374
Sušnik Bajec S, Pustovrh G, Jesenšek D, Snoj A (2015) Population genetic SNP analysis of marble and brown trout in a hybridization zone of the Adriatic watershed in Slovenia. Biol Conserv 184:239–250
Sutherland B, Koczka K, Yasuike M, Jantzen S, Yazawa R, Koop B (2014) Comparative transcriptomics of Atlantic Salmo salar, chum Oncorhynchus keta and pink salmon O. gorbuscha during infections with salmon lice Lepeophtheirus salmonis. BMC Genomics:15, 200
Sutherland BJG, Gosselin T, Normandeau E, Lamothe M, Isabel N, Audet C (2016) Salmonid chromosome evolution as revealed by a novel method for comparing RADseq linkage maps. Genome Biol Evol 8(12):3600–3617
Takayama J, Tadaka S, Yano K (2021) Construction and integration of three de novo Japanese human genome assemblies toward a population-specific reference. Nat Commun 12:226
Tang Y, Liu X, Wang J, Li M, Wang Q, Tian F, Zhang Z (2016) GAPIT version 2: An enhanced integrated tool for genomic association and prediction. Plant Genome 9
Täubert H, Rensing S, Reinhardt F (2011) Comparing conventional and genomic breeding programs with ZPLAN+. Interbull Bulletin 44:162–168
Taylor JF (2014) Implementation and accuracy of genomic selection. Aquaculture 420–421(Suppl.1):S8–S14
Thorgaard GH, Bailey GS, Williams D, Buhler DR, Kaattari SL, Ristow SS (2002) Status and opportunities for genomics research with rainbow trout. Comp Biochem Physiol 133:609–646
Tiezzi F, Maltecca C (2015) Accounting for trait architecture in genomic predictions of US Holstein cattle using a weighted realized relationship matrix. Genet Sel Evol 47:24
Torkamaneh D, Laroche J, Bastien M, Abed A, Belzile F (2017) Fast-GBS: A new pipeline for the efficient and highly accurate calling of SNPs from genotyping-by-sequencing data. BMC Bioinf 18:5
Toro MA, Saura M, Fernandez J, Villanueva B (2017) Accuracy of genomic within-family selection in aquaculture breeding programmes. J Anim Breed Genet 134:256–263
Tsai H-Y, Hamilton A, Tinch AE, Guy DR, Gharbi K, Stear MJ (2015) Genome-wide association and genomic prediction for growth traits in juvenile farmed Atlantic salmon using a high density SNP array. BMC Genomics 16:969
Tsai H-Y, Hamilton A, Tinch AE, Guy DR, Bron JE, Taggart JB (2016a) Genomic prediction of host resistance to sea lice in farmed Atlantic salmon populations. Genet Sel Evol 48:47
Tsai HY, Robledo D, Lowe NR, Bekaert M, Taggart JB, Bron JE, Houston RD (2016b) Construction and annotation of a high density SNP linkage map of the Atlantic salmon (Salmo salar) genome. G3 (Bethesda) 6:2173–2179
Tsai H-Y, Matika O, Edwards SM, Antolín-Sánchez R, Hamilton A, Guy DR (2017) Genotype imputation to improve the cost-efficiency of genomic selection in farmed Atlantic salmon. G3 (Bethesda) 7:1377–1383
Tsairidou S, Hamilton A, Robledo D, Bron JE, Houston RD (2020) Optimizing low-cost genotyping and imputation strategies for genomic selection in Atlantic salmon. G3 (Bethesda) 10:581–590
Vallejo RL, Rexroad CE, Silverstein JT, Janss LLG, Weber GM (2009) Evidence of major genes affecting stress response in rainbow trout using Bayesian methods of complex segregation analysis. J Anim Sci 87:3490–3505
Vallejo RL, Leeds TD, Gao G, Parsons JE, Martin KE, Evenhuis JP (2017) Genomic selection models double the accuracy of predicted breeding values for bacterial cold water disease resistance compared to a traditional pedigree-based model in rainbow trout aquaculture. Genet Sel Evol 49:17
Vallejo RL, Cheng H, Fragomeni BO, Shewbridge KL, Gao G, MacMillan JR, Towner R, Palti Y (2019) Genome-wide association analysis and accuracy of genome-enabled breeding value predictions for resistance to infectious hematopoietic necrosis virus in a commercial rainbow trout breeding population. Genet Sel Evol 51:47
Van Raden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Van Raden PM, Van Tassell CP, Wiggans GR, Sonstegard TS, Schnabel RD (2009) Reliability of genomic predictions for North American Holstein bulls. J Dairy Sci 92:16–24
Van Tassell CP, Smith TPL, Matukumalli LK, Taylor JF, Schnabel RD (2008) SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods 5:247–252
Vandeputte M, Haffray P (2014) Parentage assignment with genomic markers: a major advance for understanding and exploiting genetic variation of quantitative traits in farmed aquatic animals. Front Genet 5:432
Vaser R, Sović I, Nagarajan N, Šikić M (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27(5):737–746
Vela-Avitúa S, Meuwissen TH, Luan T, Ødegård J (2015) Accuracy of genomic selection for a sib-evaluated trait using identity-by-state and identity-by-descent relationships. Genet Sel Evol 47:9
Verdu CF, Guichoux E, Quevauvillers S, De Thier O, Laizet YH, Delcamp A (2016) Dealing with paralogy in RADseq data: In silico detection and single nucleotide polymorphism validation in Robinia pseudoacacia L. Ecol Evol 6:7323–7333
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J (2017) 10 years of GWAS discovery: Biology, function, and translation. Am J Hum Genet 101:5–22
Walker BJ, Abee IT, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK (2014) Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9(11):e112963
Wang H, Yang Z, Li X, Huang D, Yu S, He J (2020) Single-cell in vivo imaging of cellular circadian oscillators in zebrafish. PLoS Biol 18(3):e3000435
Wargelius A, Leininger S, Skaftnesmo KO, Kleppe L, Andersson E, Taranger GL, Schulz RW, Edvardsen RB (2016) Dnd knockout ablates germ cells and demonstrates germ cell-independent sex differentiation in Atlantic salmon. Sci Rep 6:21284
Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB (2017) Direct determination of diploid genome sequences. Genome Res 27:757–767
Wientjes YCJ, Veerkamp RF, Calus MPL (2013) The effect of linkage disequilibrium and family relationships on the reliability of genomic prediction. Genetics. 193:621–631
Witte JS (2010) Genome-wide association studies and beyond. Annu Rev Public Health 31:9–20
Woram RA, Mcgowan C, Stout JA, Gharbi K, Ferguson MM, Hoyheim B (2004) A genetic linkage map for Arctic charr (Salvelinus alpinus): Evidence for higher recombination rates and segregation distortion in hybrid versus pure strain mapping parents. Genome 47:304–315
Wringe BF, Devlin RH, Ferguson MM, Moghadam HK, Sakhrani D, Danzmann RG (2010) Growth-related quantitative trait loci in domestic and wild rainbow trout (Oncorhynchus mykiss). BMC Genet 11:63
Wu X, Lund MS, Sun D, Zhang Q, Su G (2015) Impact of relationships between test and training animals and among training animals on reliability of genomic prediction. J Anim Breed Genet 132:366–375
Xiao T, Zhou W (2020) The third generation sequencing: The advanced approach to genetic diseases. Transl Pediatr 9(2):163–173
Xiao CL, Chen Y, Xie SQ (2017) MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat Methods 14:1072–1074
Yáñez JM, Houston RD, Newman S (2014) Genetics and genomics of disease resistance in salmonid species. Front Genet 5:415
Yáñez JM, Newman S, Houston RD (2015) Genomics in aquaculture to better understand species biology and accelerate genetic progress. Front Genet 6:128
Yáñez JM, Naswa S, López M, Bassini L, Correa K, Gilbey J (2016) Genome-wide single nucleotide polymorphism discovery in Atlantic salmon (Salmo salar): Validation in wild and farmed American and European populations. Mol Ecol Resour 16:1002–1011
Yoshida GM, Carvalheiro R, Lhorente JP, Correa K, Figueroa R, Ross D, Houstond RD, Yáñez JM (2018) Accuracy of genotype imputation and genomic predictions in a two-generation farmed Atlantic salmon population using high-density and low-density SNP panels. Aquaculture 491:147–154
Yoshida GM, Carvalheiro R, Rodriguez FH, Lhorente JP, Yanez JM (2019) Single-step genomic evaluation improves accuracy of breeding value predictions for resistance to infectious pancreatic necrosis virus in rainbow trout. Genomics 111:127–132
Young WP, Wheeler PA, Coryell VH, Keim P (1998) A detailed linkage map of rainbow trout produced using doubled haploids. Genetics 148:839–850
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yuan J, Gao Y, Zhang X, Wei J, Liu C, Li F (2017) Genome sequences of marine shrimp (Exopalaemon carinicauda Holthuis) provide insights into genome size evolution of Caridea. Mar Drugs 15:213
Yue GH (2014) Recent advances of genome mapping and marker-assisted selection in aquaculture. Fish Fish 15:376–396
Zenger KR, Khatkar MS, Jerry DR, Raadsma HW (2017) The next wave in selective breeding: implementing genomic selection in aquaculture. Proc Assoc Advmt Anim Breed Genet 22:105–112
Zenger KR, Khatkar MS, Jones DB, Khalilisamani N, Jerry DR, Raadsma HW (2019) Genomic selection in aquaculture: application, limitations and opportunities with special reference to marine shrimp and pearl oysters. Front Genet 9:693
Zhang Z, Liu J, Ding X, Bijma P, de Koning D-J, Zhang Q (2010) Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS One 5:e12648
Zhang H-Y, Zhao Z-X, Xu J, Xu P, Bai Q-L, Yang S-Y (2018a) Population genetic analysis of aquaculture salmonid populations in China using a 57K rainbow trout SNP array. PLoS One 13(8):e0202582
Zhang Y, Massel K, Godwin ID, Gao C (2018b) Applications and potential of genome editing in crop improvement. Genome Biol 19:210
Zheng Z, Yang Y, Zeng Y, He M (2016) A microfluidic ExoSearch chip formultiplexed exosome detection towards blood-based ovarian cancer diagnosis. Lab Chip 16(3):489–96
Zhong Z, Niu P, Wang M (2016) Targeted disruption of sp7 and myostatin with CRISPR-Cas9 results in severe bone defects and more muscular cells in common carp. Sci Rep 6:22953
Zhou X, Stephens M (2012) Genome-wide efficient mixed model analysis for association studies. Nat Genet 44:821–824
Zhu C, Yu J (2009) Nonmetric multidimensional scaling corrects for population structure in association mapping with different sample types. Genetics 182:875–888
Zuo S, Karami AM, Ødegård J, Mathiessen H, Marana MH, Jaafar R (2020) Immune gene expression and genome-wide association analysis in rainbow trout with different resistance to Yersinia ruckeri infection. Fish Shellfish Immunol 106:441–450
Acknowledgements
The authors acknowledge funding for this study from the University of Udine (Reseach Grant: START—UP, DI4A, University of Udine, GENSAL—Genomic analysis of salmonid species).
Code availability
Not applicable
Funding
Open access funding provided by Università degli Studi dell'Insubria within the CRUI-CARE Agreement. University of Udine
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Handling Editor: Gavin Burnell
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Innovations in disease control and diagnosis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
D’Agaro, E., Favaro, A., Matiussi, S. et al. Genomic selection in salmonids: new discoveries and future perspectives. Aquacult Int 29, 2259–2289 (2021). https://doi.org/10.1007/s10499-021-00747-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10499-021-00747-w