
Abstract
The clinical efficacy of proteasome inhibitors in the treatment of multiple myeloma has encouraged application of proteasome inhibitor containing therapeutic interventions in (pediatric) acute leukemia. Here, we summarize the positioning of bortezomib, as first-generation proteasome inhibitor, and second-generation proteasome inhibitors in leukemia treatment from a preclinical and clinical perspective. Potential markers for proteasome inhibitor sensitivity and/or resistance emerging from leukemia cell line models and clinical sample studies will be discussed focusing on the role of immunoproteasome and constitutive proteasome (subunit) expression, PSMB5 mutations, and alternative mechanisms of overcoming proteolytic stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hematological malignancies comprise of many subgroups including chronic and acute leukemia, lymphoma, and multiple myeloma (MM). In this review, we focus exclusively on acute leukemia, which can be divided into two major subgroups: acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML). In children, the majority of leukemia cases are ALL [1], while AML is more prevalent in adults with leukemia [2]. With a 5-year overall survival (OS) of 83–94% [3], the prognosis of pediatric ALL is considerably better as compared to adults (5-year OS 15–35%, depending on age) [4]. A similar difference in prognosis between children and adults is seen in AML with a 5-year OS of, respectively, 65–70% and 10–45% (depending on age) [5]. The main reasons for treatment failure in both children and adults are intrinsic or acquired drug resistance in a subset of leukemia cells that are responsible for refractory disease or the development of relapse, which have a dismal prognosis. Since the (emergence of) drug resistance is one of the limiting factors that impacts long-term efficacy of anti-leukemic drugs, the search for new anti-leukemic drugs with novel mechanisms of action is an ongoing challenge.
Most anti-leukemic drugs are targeted against DNA replication to interfere with abundant cell proliferation (Fig. 1). For leukemia cells to expand, they also rely on a very high protein turnover. In normal cells, with normal chromosome numbers and normal protein balance, protein homeostasis is maintained mainly by the ubiquitin-proteasome system (UPS) [6]. Besides rapid cell growth, leukemia cells also feature many chromosomal and molecular aberrations, including chromosomal translocations (e.g., t(8:21), Inv(16)), hypo- and hyper-diploidy, activating mutations (e.g., FLT3/ITD, cKIT), and splicing defects, the latter leading to many different protein isoforms [7, 8]. Together, this leads to an aberrant protein expression, which imposes an inherent heavy burden on the UPS. These considerations set the stage for therapeutic interventions of UPS-targeting with proteasome inhibitors (PIs) [9], of which bortezomib (BTZ) as the first prototypical PI proved successful in therapy-refractory multiple myeloma (MM) [10]. Currently, first line treatment of MM includes BTZ and extends to clinical evaluations of next-generation PIs. Updates of PI treatment in MM have been subject of several recent dedicated reviews [6, 11, 12].

Overview of cytotoxicity mechanism of chemotherapy drugs commonly used in acute leukemia treatment
This review will primarily focus on the application of PIs in acute leukemia, in particular refractory disease. Since many novel drugs with different mechanisms of action are currently available, it is crucial to select those patients for certain PI who will benefit from the treatment. Therefore, it is of clinical relevance to understand the mechanism of action of PIs in leukemia and identify parameters that can help to define (non)-responsiveness to PIs. To this end, this review provides a comprehensive overview on the molecular mechanisms of action and resistance to PIs in leukemia as well as current applications of PIs in clinical trials in leukemia patients.
2 Proteasome inhibitors in leukemia
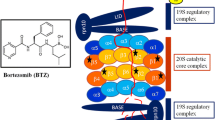
In the context of hematological cells, it is of importance to recognize that the proteasome composition is highly skewed for > 70% towards immunoproteasomes (iP) over constitutive proteasomes (cP) [13, 14]. The three catalytically active β-subunits (β1, β2, and β5) of the constitutive proteasome and the immunoproteasome counterparts β1i, β2i, and β5i harbor caspase-like, trypsin-like, and chymotrypsin-like proteolytic activity, respectively (Fig. 2).

Subunit composition and inhibitors targeting of constitutive and immunoproteasomes. a 20S core proteasome and b fully assembled immunoproteasome with various cap proteins. c Clinically active and experimental inhibitors of constitutive- and/or immunoproteasome. Adapted from Verbrugge et al. 2015 [15]
iP expression is markedly induced upon stimulation by inflammatory cytokines such as IFNγ and TNFα [16] (Fig. 2a, b). One of the primary functions of iP is to broaden the spectrum of generating antigenic peptides for presentation on MHC class I molecules [15, 17], but also additional functions for iPs have been defined, e.g., clearance of polyubiquitinated protein aggregates emerging under inflammatory oxidative stress conditions [15, 18,19,20,21,22,23] (Fig. 2c). Given the abundance of iP in leukemia cells, selective targeting of iP is an attractive treatment option [24].
BTZ and next-generation PIs [25, 26] have been evaluated in preclinical and clinical studies as potential anti-leukemia drugs. An overview of their properties is provided in Table 1. BTZ is a reversible PI primarily targeting the β5 catalytic active subunit of the proteasome. Next-generation PIs differ from BTZ by being irreversible inhibitors (e.g., carfilzomib (CFZ)), favoring oral administration (e.g., ixazomib (IXA)), attenuating hematological and neurological side effects and overcoming BTZ-associated resistance modalities [43,44,45]. Moreover, these next-generation PIs display selectivity for cP and iP, and subunits other than β5 [27, 28, 46, 47]. Initial ex vivo activity studies of BTZ and next-generation inhibitors of cP (CFZ, ONX-0912) and iP (ONX-0914) revealed considerable inter-patient variabilities but overall greater potency in ALL than AML cells [48]. Moreover, BTZ, CFZ, and ONX-0912 were 3–10-fold more potent than the iP inhibitor ONX-914. Interestingly, this study also showed that a higher ratio of immunoproteasome over constitutive proteasome levels in leukemia cells was a good indicator for PI sensitivity. An overview of selected preclinical studies of several PIs in ALL and AML cell lines and primary cells is summarized in Table 2. Overall, these data show the relevance of preclinical studies to unravel the individual specific mechanisms of action of PIs related to apoptosis induction. In addition, these data reveal promising combination strategies for improvement of successful PI therapy.
3 Markers for PI (BTZ) sensitivity/resistance in leukemia cell lines
As with any new treatment strategy, selection of patients who will benefit from the treatment is essential. With respect to PIs, studies with leukemia cell lines can help to define markers for response, long-term efficacy, and emergence of resistance to PIs. Resistance mechanisms often reveal critical processes such as targeted and compensatory mechanisms that leukemia cells harbor to overcome the effects of PIs. A large number of studies have followed the approach of exposing leukemia cells to stepwise increasing concentrations of PIs (mostly BTZ) and characterize cells with acquired resistance [68,69,70,71]. Figure 3 depicts an overview of molecular mechanisms of resistance in PI/BTZ-resistant leukemia cell lines [44, 46, 72]. The most common mechanisms are discussed below.

Overview of known molecular mechanisms involved in BTZ resistance. a Proteasome related resistance: relative down regulation of immunoproteasome as compared to constitutive proteasome (1) together with absolute upregulation of the constitutive proteasome (2) and mutation in the β5 subunit of the proteasome (3). b Alternative stress handling: upregulation of heat shock proteins (4) or changes in redox metabolism (5) which can prevent oxidative stress. Alternative handling ubiquitinated protein: exocytosis of ubiquitinated proteins in MARCKS-associated vesicles (6), and protein degradation through autophagy (7). c Activation pro-survival signaling: intrinsic activation of pro-survival pathways, e.g., AKT, NFκB, or MET (8) or through stimulation by direct interaction with stromal cells (9) or indirectly through soluble growth factors or interleukins (10). d Decreased apoptosis: downregulation (11) or mutation (12) of pro-apoptotic proteins. Finally, e, extrusion of BTZ via multidrug resistance efflux pump MDR1/Pgp (minor effect, more pronounced in CFZ resistance) (13). CP, constitutive proteasome; IP, immunoproteasome; MDR1, multidrug resistance protein 1; HSP, heat shock proteins; IGF1, insulin-like growth factor 1; IL, interleukin
3.1 Upregulation of proteasomal subunits
Upregulation of the primary target is a well-known response of cells exposed to drugs. Likewise, upregulation of proteasomal subunits, and the β5 subunit in particular, is frequently observed in BTZ-resistant leukemia cell lines (reviewed in [44, 46]) indicating its role in BTZ resistance. Consistently, CGH analysis of BTZ-resistant cell lines revealed amplification of the PSMB5 gene with variability in size and extent of the amplification of chromosome 6 [73]. Remarkably, immunoproteasome subunit levels are often found to be decreased in BTZ-resistant leukemia cell lines [16, 68, 71]. The resulting decrease in immuno- / constitutive proteasome ratio and alterations in subunit composition has been linked to a diminished BTZ sensitivity [74, 75]. Interferon gamma (IFNγ) is an efficient inducer of immunoproteasome expression and partly restored BTZ sensitivity in BTZ-resistant leukemia cells [16]. Beyond BTZ-resistant cells, IFNγ also enhanced BTZ sensitivity in a panel of B cell lines by 50% [76]. Together, these data underscore the importance of immuno- and constitutive proteasome subunit composition in BTZ sensitivity and resistance in leukemia cells.
3.2 PSMB5 mutations
Molecular analysis of the proteasome subunits of PI-resistant leukemia cell lines revealed several mutations in exon 2 of the PSBM5 gene encoding the highly conserved binding pocket region for PIs within the β5 subunit of the proteasome. This highly conserved region appears to be a mutation “hot spot” when cells are exposed to BTZ for a prolonged period. Figure 4 shows an overview of PSMB5 mutations in PI-resistant cell lines, including non-hematological malignancies.

All mutations result in amino acid alteration in, or in the close vicinity of, the PI binding pocket [82]. In fact, except for the A247G mutation which introduces a Thr21Ala substitution, all mutations result in amino acid alterations in the S1 pocket of the β5 subunit with Ala49 being the most affected amino acid. This specificity pocket is a highly conserved part of the subunit and responsible for recognizing the peptide bond of the substrate. It is also the site that has to be cleaved and determines the specificity as well as facilitates the binding of the P1 side chain of BTZ [82]. In silico analysis provided evidence for hindered binding of BTZ by the majority of the mutations, including the Thr21Ala amino acid alteration positioned outside of the S1 pocket but within the BTZ binding pocket [82]. The only exceptions were the Met45Ile and Met45Val substitutions, which do not directly interact with BTZ. However, Met45 is known to contribute to the specificity of the S1 pocket and upon binding, Met45 undergoes a conformational change and shifts the direction of its side chain towards Cys52 vicinity. Alterations in this amino acid might therefore hinder this conformational change and contribute as well to decreased BTZ binding. Docking the LLVY-AMC substrate in silico in the crystal structure of the β5 subunit supported the hypothesis of decreased binding of the substrate in the mutated binding pocket.
There are two strong indications that PSMB5 mutations are related to PI resistance. First, from marine biology studies, characterization of proteasome subunits of the PI Salinosporamide A (NPI-0052, Marizomib) producing actinobacterium Salinispora tropica revealed the same Ala49Val and Met45Phe mutations in the β5 subunit homologue as in BTZ-resistant leukemia cells. In S. tropica, these mutations conferred “self-resistance” to Salinosporamide A [80]. Second, acquired resistance to the immunoproteasome inhibitor in PR-924 was not associated with any mutation(s) in immunoproteasome subunits, but rather provoked PSMB5 mutations, i.e., Met45Ile, Ala49Thr, and Met45Val [29]. Taken together, inhibition of the β5 subunit is essential for the anti-leukemia effect of PIs and PSMB5 mutations emerging after prolonged PI treatment attenuate the inhibitory potency and confer resistance in leukemia cells. PSMB5 mutations have thus far not been identified in patients receiving PI therapy and it has been challenged whether these mutations hold clinical relevance for PI resistance. However, until now, longstanding treatment with the first-generation PI was hardly applied in clinical practice. This may change with the introduction of second-generation and oral PI’s allowing for long-term treatment.
3.3 Alternative protein disposal
Gene expression profiling of BTZ-resistant leukemia cells identified myristoylated alanine-rich C-kinase substrate (MARCKS) as being highly upregulated. MARCKS is an 80-kDa protein that is involved in multiple exocytosis pathways (reviewed in [83]). In fact, in BTZ-resistant leukemia cells, MARCKs protein co-localized with intracellular vesicles that contained polyubiquitinated proteins and which were formed upon exposing cells to increasing concentrations of BTZ. The ubiquitin-containing vesicles have been described before [84], and in co-cultures, it was shown that after extrusion of the vesicles, they were taken up in proteasome-proficient acceptor cells [73]. MARCKs may thus contribute to BTZ resistance by facilitating exocytosis-mediated extrusion of polyubiquitinated proteins to overcome proteolytic stress imposed by BTZ. MARCKs upregulation is not restricted to BTZ-resistant leukemia cells, but was also observed in leukemia cells with acquired resistance to second-generation PI, e.g., Salinosporamide A (Marizomib) and the immunoproteasome inhibitor PR924.
Another alternative protein disposal that is often related to drug resistance is autophagy. This process has been linked to BTZ resistance in several tumor models [85,86,87,88,89,90,91], although it was not indicated in MARCKs overexpressing PI-resistant leukemia cells [73]. Inhibition of autophagy through different mechanisms including calpain inhibitor [86], downregulation of heat shock protein B8 (HSPB8) [87], B cell lymphoma 2-interacting mediator of cell death (BIM) upregulation [90], tipifarnib [85], or (hydroxy)chloroquine [92, 93] increased BTZ sensitivity in a MM model. In addition, histone deacetylase (HDAC) inhibitors can block autophagy by disrupting aggresome formation, the process that precedes autophagy [94, 95]. Interestingly, the E3 ligase, TRAF6, links the NFkB pathway to autophagy [96] and it was shown that BTZ-resistant AML cells were sensitized by downregulating TRAF 6 [97]. Despite the potential impact of autophagy on BTZ resistance, the extent of involvement of this process in leukemia cells needs further exploration.
3.4 Activation of pro-survival pathways
Analogous to resistance to many anti-leukemia drugs, PI resistance has also been associated with the activation of pro-survival pathways. The most extensively described is the association with the NFκB survival pathway (for AML reviewed in [98]). As PI result in the stability of the inhibitor of NFκB (IκB), this survival pathway is inhibited by exposure to PI. Intrinsically resistant leukemia cells (e.g., stem cells) have a constitutively activated NFκB pathway and the combination of BTZ with NFκB inhibition by the IKK inhibitor BMS-345541, enhanced the kill of AML stem cells [99]. Although the interaction between NFκB and PI resistance is clearly established for MM and other tumor types [100,101,102,103,104], the data of relevance for leukemia is limited. Besides NFκB, the pro-survival pathways via AKT/mTOR [105,106,107] and insulin-like growth factor 1 (IGF-1) [108,109,110] have also been described to confer BTZ resistance and may affect the interaction of leukemia cells with (stromal) cells in their micro-environment.
3.5 X-box binding protein 1 (XBP1)
X-box binding protein 1 (XBP1) is a transcription factor involved in the unfolded protein response. In addition, it is important for the differentiation of plasma cells. In this respect, XBP1 expression has been linked to BTZ resistance [111] and survival [112] in MM. In leukemia, this pathway may also be related to PI resistance; however, data are currently lacking. Due to the role of XBP1 in B cell differentiation, this factor may be particularly relevant for B-ALL and warrants further study.
3.6 Drug efflux transporters
Drug efflux transporters of the ATP-binding cassette protein family have been explored for their role in PI resistance. P-glycoprotein (Pgp, ABCB1), as one of its main representatives only marginally contributes to BTZ efflux [68, 113], but is of relevance for second-generation PIs such as CFZ and the immunoproteasome inhibitor ONX-0914 as these PIs are bona fide substrates for Pgp [114, 115]. Notably, BTZ can downregulate Pgp expression and this manner indirectly attenuate drug resistance [116]. There is no evidence that BTZ and other second-generation PIs are substrates for other drug efflux transporters, e.g., multidrug resistance-associated protein 1–5 (MRP1–5, ABCC1–5) or breast cancer resistance protein (BCRP, ABCG2); hence, a role in PI resistance is not anticipated [114].
4 Markers for PI (BTZ) sensitivity/resistance in primary leukemia samples
Although in vitro models are valuable tools to identify possible mechanisms of BTZ resistance, assessment of the relevance for the clinic requires validation in ex vivo studies using primary patient samples. Preferably, this is studied in add-on studies of clinical trials that include BTZ in the treatment protocol. The potential role of three markers for PI (BTZ) sensitivity and/or resistance in leukemia samples is discussed below.
4.1 Proteasome levels and subunit composition
Add-on studies of clinical trials in leukemia are limited and most data for PIs are obtained from MM studies. These studies demonstrated besides proteasome expression levels, the proteasome subunit composition is important for response [117, 118]. Although upregulation of the proteasome was not related to resistance in mantle cell lymphoma (MCL) patients [119], studies in leukemia indicated a possible correlation between higher 20S protein expression and BTZ sensitivity [120]. In particular, studies by Niewerth et al. (2013) showed that lower β5 subunit expression correlated with increased ex vivo sensitivity for proteasome inhibitors in pediatric AML and ALL samples. In addition, the sensitivity for BTZ in AML cells inversely correlated with the ratios between immunoproteasome subunits over constitutive subunits, specifically β1i/β1 and β2i/β2, and a trend for β5i/β5. ALL cells showed higher sensitivity to BTZ as compared to AML cells. Although for ALL no significant correlations were revealed with BTZ sensitivity, they featured a higher β2i/β2 ratio and trends of a higher β1i/β1 ratio as compared to AML samples. Together, these data support the notion that a relative high immuno- / constitutive proteasome ratio promotes BTZ sensitivity. These data were confirmed in AML and ALL samples obtained from two pediatric clinical COG trials (AAML07P1 and AALL07P1) in which BTZ treatment was incorporated [14, 48]. After further validation in larger studies, assessment of immunoproteasome over constitutive proteasome ratios may be used as biomarkers of response to PIs.
4.2 PSMB5 mutations
PSMB5 mutations observed in BTZ-resistant hematological cell lines were as yet not identified in clinical samples. The mutations found in the cell line models do not represent SNPs found in the general population. Wang et al. sequenced the PSMB5 gene in a large cohort of healthy persons and 61 MM patients after BTZ treatment [121]. No SNPs were found in the exon 2 of the PSMB5 gene neither in the general population nor in MM patients. Of interest, polymorphisms that influenced PSMB5 gene expression were observed, but these did not correlate with BTZ response. It should be taken into account that sequence analysis in one third of the MM patients was performed only on whole blood and not on isolated malignant plasma cells, which may have influenced the sensitivity of the analysis. In addition, it was not stated how many resistant patients were included. More recently, Lichter et al. sequenced the PSMB5 gene in blood samples of MM patients included in the APEX trial in which patients were treated with either BTZ or dexamethasone [122]. No PSMB5 mutations were identified in this group. Although sample size of this group was limited, the data suggest that PSMB5 mutations do not represent a leading cause of acquired BTZ resistance in investigated MM protocols. Whether this holds true for leukemia or MM with long-term BTZ maintenance therapy is yet to be determined. Lastly, as a preliminary account, Barrio et al. (2016) reported PSMB5 mutations in subclones of CD138+ cells of a single MM patient after a therapy [123]. Since the subclonal frequencies were low, this poses analytical challenges to detect these subclones. Also in leukemia, it is established that there is substantial oligoclonality in mutational status of cells within the leukemia. The cells with the specific mutations tendering growth advantage are probably selected to grow out and develop a (drug resistant) relapse [124], so also in leukemia patients subclonal analysis may reveal additional mutations.
4.3 MARCKS
Upregulation of MARCKs protein expression emerged as a marker for BTZ- and second-generation PI-resistant leukemia cell lines [73]. To test whether this upregulation might be a prognostic marker for clinical BTZ resistance, MARCKs expression was examined in 44 primary ALL patient samples obtained from the clinical COG trial AALL07P1 using combination chemotherapy including BTZ. A trend (p = 0.07) was seen in the inverse correlation between MARCKS expression and clinical response. Since the samples were obtained in the setting of a clinical trial using combination therapy, a direct correlation between BTZ response and MARCKS expression cannot be made. However, these findings are consistent with data from Micallef et al. who showed MARCKS protein upregulation in a small group of BTZ-resistant MM patients [125] and Yang et al. who identified MARCKS upregulation as a resistance marker in primary MM samples [126]. These studies encourage a prospective validation of the possible prognostic role of MARCKS in leukemia.
5 Clinical trials with proteasome inhibitors in leukemia
The efficacy and safety of PIs in MM have already been extensively reviewed [127]. For leukemia, an overview of phase I/II clinical studies of PI as single agent and combination therapy in adult and pediatric leukemia is presented in Table 3. An overview of ongoing phase II/III clinical trials with BTZ (combination) therapy involving pediatric leukemia patients and adult leukemia is shown in Table 4 and Table 5, respectively. In addition, ongoing clinical trials with second-generation PIs (mainly CFZ and IXA) are depicted in Table 6. The outcome for clinical efficacy included stable disease, progressive disease, complete/partial remission, and mortality. In all studies, infections and neutropenia were common adverse drug reactions (ADRs). Neurologic ADRs were also common in all studies with BTZ, including neuropathy. Interestingly, BTZ might also have some protective effects as well, as it has been reported to prevent muscle wasting [148], which can be induced by cancer cachexia [149] but this has not yet been reported in a clinical setting. Two studies were conducted with CFZ [143, 144] and less ADRs were found in these studies including the absence of neurologic ADRs. Moreover, no dose-limiting toxicities (DLT) were found indicating a better safety profile. Importantly, the clinical response to CFZ in these studies was also better than reported for BTZ treatment.
6 Future perspectives
This overview of results of PI in leukemia reveals that treatment with PI as monotherapy may not give satisfactory clinical responses. To improve the employability of PI in leukemia, several factors are implied to be considered.
6.1 Combination strategies
For leukemia, chemotherapy commonly consists of cocktails of different drugs with different mechanisms of actions and different side effects to exert optimal treatment response with achievable dosages. For novel drugs, it is therefore essential that they can be combined with the most effective drugs currently used. Based on the mechanism of action, glucocorticoids are good candidate drugs to be combined with PI. BTZ and dexamethasone showed synergy in ex vivo combination studies in primary pediatric ALL samples [48] clinically BTZ was combined with induction therapy including dexamethasone, vincristine, PEG-asparaginase, and doxorubicin in relapsed pediatric ALL, showing promising CR rates [133, 134]. Currently, several clinical trials are ongoing (Tables 4 and 5), combining BTZ with several chemotherapy protocols which include the standard chemotherapeutics and novel strategies (heat shock protein inhibitors, HDAC inhibitors, and autophagy inhibitors). Based on preclinical data and expected toxicity profiles of the different drugs, the addition of PI to other chemotherapeutics seems a fruitful strategy. In addition, recently the nuclear transport (XPO1) inhibitor selinexor has been combined with BTZ [150] and is currently in a clinical trial for MM (NCT03110562). Whether this is also a relevant strategy for leukemia still has to be established.
6.2 Second-generation proteasome inhibitors
Despite the successful introduction of BTZ, several drawbacks such as resistance and toxic side effects led to development of second-generation proteasome inhibitors which are at several stages of clinical development (Table 6). Due to promising preclinical studies, the irreversible proteasome inhibitor CFZ has advanced rapidly into the clinic for MM as well as leukemia, and is supposed to be a promising alternative for BTZ and might even overcome BTZ resistance.
Recently, an oral formulation resembling BTZ, ixazomib (MLN9708), has emerged into the clinic, with two clinical trials investigating the efficacy of ixazomib in AML (NCT0230405 and NCT 02070458).
PR-957 (ONX 0914) and PR-924 represent members of a new class of proteasome inhibitors being directed specifically against the immunoproteasome [27, 30]. PR-924 demonstrated preclinical efficacy in leukemia and MM [29, 30]. In addition, ex vivo cytotoxicity of PR-957 was shown in primary leukemia samples [29]. Since leukemia cells, especially ALL cells, express high levels of immunoproteasome, it might be a good candidate for further clinical development in hematological malignancies.
Development of acquired resistance to the second-generation PIs [29, 31] has to be taken in account as well. Hence, combination therapy is also recommended for the treatment of leukemia with these novel PIs in order to circumvent toxicity and resistance.
6.3 Biomarkers of clinical response to proteasome inhibitors
The combination of preclinical research and ongoing clinical studies (e.g., add-on studies) will be needed to identify and confirm determinants of resistance and markers for clinical response in order to further personalize the treatment of acute leukemia with PI. Based on accumulating data, prediction of effectivity of PI lies in the composition of the proteasome, in particular the ratio between constitutive- and immunoproteasome subunits. This can either be assessed by measuring protein expression of the different subunits [14, 121] or their specific catalytic activities for which several assays are available [151, 152]. In addition, the activity assays can be used for pharmacodynamic monitoring of PI inhibition and duration of inhibition in PBMC’s [152, 153].
Since mutations in the genes encoding proteasome subunits have not been found in primary MM and acute leukemia patient samples either before or after treatment with PI, they are currently not considered potential biomarkers for resistance to proteasomes. However, when patients are treated for prolonged time periods such as the maintenance treatment of elderly MM patients with IXA, they might be acquired. Therefore, add-on studies measuring the mutational status in samples during treatment are currently performed. If mutations are identified, they may be used to monitor acquired resistance to PI. Selecting the patients that benefit from PI treatment and the recognition of PI resistance is indispensable for the optimal implementation of PI in acute leukemia treatment.
References
Terwilliger, T., & Abdul-Hay, M. (2017). Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J, 7, e577.
De Kouchkovsky, I., & Abdul-Hay, M. (2016). Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J, 6, e441.
Pui, C.-H., Carroll, W. L., Meshinchi, S., & Arceci, R. J. (2011). Biology, risk stratification, and therapy of pediatric acute leukemias: an update. Journal of Clinical Oncology, 29, 551–565.
Paul, S., Kantarjian, H., & Jabbour, E. J. (2016). Adult acute lymphoblastic leukemia. Mayo Clinic Proceedings, 91, 1645–1666.
Chaudhury, S. S., Morison, J. K., Gibson, B. E. S., & Keeshan, K. (2015). Insights into cell ontogeny, age, and acute myeloid leukemia. Experimental Hematology, 43, 745–755.
Micel, L. N., Tentler, J. J., Smith, P. G., & Eckhardt, G. S. (2013). Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. Journal of Clinical Oncology, 31, 1231–1238.
Tasian, S. K., & Hunger, S. P. (2017). Genomic characterization of paediatric acute lymphoblastic leukaemia: an opportunity for precision medicine therapeutics. British Journal of Haematology, 176, 867–882.
Prada-Arismendy, J., Arroyave, J. C., & Röthlisberger, S. (2017). Molecular biomarkers in acute myeloid leukemia. Blood Reviews, 31, 63–76.
Adams, J. (2004). The proteasome: a suitable antineoplastic target. Nature Reviews. Cancer, 4, 349–360.
Anderson, K. C. (2012). The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. Journal of Clinical Oncology, 30, 445–452.
Kapoor, P., Ramakrishnan, V., & Rajkumar, S. V. (2012). Bortezomib combination therapy in multiple myeloma. Seminars in Hematology, 49, 228–242.
Wright, J. J. (2010). Combination therapy of bortezomib with novel targeted agents: an emerging treatment strategy. Clinical Cancer Research, 16, 4094–4104.
Parlati, F., Lee, S. J., Aujay, M., et al. (2009). Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood, 114, 3439–3447.
Niewerth, D., Kaspers, G. J. L., Jansen, G., et al. (2016). Proteasome subunit expression analysis and chemosensitivity in relapsed paediatric acute leukaemia patients receiving bortezomib-containing chemotherapy. Journal of Hematology & Oncology, 9, 82.
Verbrugge, S. E., Scheper, R. J., Lems, W. F., et al. (2015). Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Research & Therapy, 17, 17.
Niewerth, D., Kaspers, G. J. L., Assaraf, Y. G., et al. (2014). Interferon-γ-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. Journal of Hematology & Oncology, 7, 7.
Basler, M., Kirk, C. J., & Groettrup, M. (2012). The immunoproteasome in antigen processing and other immunological functions. Current Opinion in Immunology, 25, 74–80.
Seifert, U., Bialy, L. P., Ebstein, F., et al. (2010). Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell, 142, 613–624.
Miller, Z., Ao, L., Kim, K. B., & Lee, W. (2013). Inhibitors of the immunoproteasome: current status and future directions. Current Pharmaceutical Design, 19, 4140–4151.
Ho, Y. K., Bargagna-Mohan, P., Wehenkel, M., et al. (2007). LMP2-specific inhibitors: chemical genetic tools for proteasome biology. Chemistry & Biology, 14, 419–430.
Myung, J., Kim, K. B., Lindsten, K., et al. (2001). Lack of proteasome active site allostery as revealed by subunit-specific inhibitors. Molecular Cell, 7, 411–420.
Kuhn, D. J., Hunsucker, S. A., Chen, Q., et al. (2009). Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood, 113, 4667–4676.
de Bruin, G., Huber, E. M., Xin, B.-T., et al. (2014). Structure-based design of β1i or β5i specific inhibitors of human immunoproteasomes. Journal of Medicinal Chemistry, 57, 6197–6209.
Kuhn, D. J., & Orlowski, R. Z. (2012). The immunoproteasome as a target in hematologic malignancies. Seminars in Hematology, 49, 258–262.
Kisselev, A. F., van der Linden, W. A., & Overkleeft, H. S. (2012). Proteasome inhibitors: an expanding army attacking a unique target. Chemistry & Biology, 19, 99–115.
Huber, E. M., Basler, M., Schwab, R., et al. (2012). Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell, 148, 727–738.
Muchamuel, T., Basler, M., Aujay, M. A., et al. (2009). A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nature Medicine, 15, 781–787.
Zhou, H.-J., Aujay, M. A., Bennett, M. K., et al. (2009). Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). Journal of Medicinal Chemistry, 52, 3028–3038.
Niewerth, D., van Meerloo, J., Jansen, G., et al. (2014). Anti-leukemic activity and mechanisms underlying resistance to the novel immunoproteasome inhibitor PR-924. Biochemical Pharmacology, 89, 43–51.
Singh, A. V., Bandi, M., Aujay, M. A., et al. (2011). PR-924, a selective inhibitor of the immunoproteasome subunit LMP-7, blocks multiple myeloma cell growth both in vitro and in vivo. British Journal of Haematology, 152, 155–163.
Niewerth, D., Jansen, G., Riethoff, L. F., et al. (2014). Antileukemic activity and mechanism of drug resistance to the marine Salinispora tropica proteasome inhibitor salinosporamide A (Marizomib). Molecular Pharmacology, 86, 12–19.
Adams, J., Palombella, V. J., & Elliott, P. J. (2000). Proteasome inhibition: a new strategy in cancer treatment. Investigational New Drugs, 18, 109–121.
Almond, J. B., & Cohen, G. M. (2002). The proteasome: a novel target for cancer chemotherapy. Leukemia, 16, 433–443.
Adams, J., Behnke, M., Chen, S., et al. (1998). Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorganic & Medicinal Chemistry Letters, 8, 333–338.
Lee, D. H., & Goldberg, A. L. (1998). Proteasome inhibitors: valuable new tools for cell biologists. Trends in Cell Biology, 8, 397–403.
Kisselev, A. F., & Goldberg, A. L. (2001). Proteasome inhibitors: from research tools to drug candidates. Chemistry & Biology, 8, 739–758.
Adams, J. (2000). The proteasome: structure, function, and role in the cell. Cancer Treatment Reviews, 29(Suppl 1), 3–9.
Chauhan, D., Hideshima, T., & Anderson, K. C. (2006). A novel proteasome inhibitor NPI-0052 as an anticancer therapy. British Journal of Cancer, 95, 961–965.
Chauhan, D., Singh, A. V., Aujay, M., et al. (2010). A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood, 116, 4906–4915.
Chauhan, D., Tian, Z., Zhou, B., et al. (2011). In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clinical Cancer Research, 17, 5311–5321.
Kuhn, D. J., Chen, Q., Voorhees, P. M., et al. (2007). Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood, 110, 3281–3290.
Piva, R., Ruggeri, B., Williams, M., et al. (2008). CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood, 111, 2765–2775.
Dick, L. R., & Fleming, P. E. (2010). Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discovery Today, 15, 243–249.
Niewerth, D., Jansen, G., Assaraf, Y. G., et al. (2015). Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resistance Updates, 18, 18–35.
Ruschak, A. M., Slassi, M., Kay, L. E., & Schimmer, A. D. (2011). Novel proteasome inhibitors to overcome bortezomib resistance. Journal of the National Cancer Institute, 103, 1007–1017.
Kale, A. J., & Moore, B. S. (2012). Molecular mechanisms of acquired proteasome inhibitor resistance. Journal of Medicinal Chemistry, 55, 10317–10327.
Kirk, C. J. (2012). Discovery and development of second-generation proteasome inhibitors. Seminars in Hematology, 49, 207–214.
Niewerth, D., Franke, N. E., Jansen, G., et al. (2013). Higher ratio immune vs. constitutive proteasome level as novel indicator of sensitivity of pediatric acute leukemia cells to proteasome inhibitors. Haematologica, 98, 1896–1904.
Drexler, H. C. (1997). Activation of the cell death program by inhibition of proteasome function. Proceedings of the National Academy of Sciences of the United States of America, 94, 855–860.
Soligo, D., Servida, F., Delia, D., et al. (2001). The apoptogenic response of human myeloid leukaemia cell lines and of normal and malignant haematopoietic progenitor cells to the proteasome inhibitor PSI. British Journal of Haematology, 113, 126–135.
Vrana, J. A., & Grant, S. (2001). Synergistic induction of apoptosis in human leukemia cells (U937) exposed to bryostatin 1 and the proteasome inhibitor lactacystin involves dysregulation of the PKC/MAPK cascade. Blood, 97, 2105–2114.
Chandra, J., Niemer, I., Gilbreath, J., et al. (1998). Proteasome inhibitors induce apoptosis in glucocorticoid-resistant chronic lymphocytic leukemic lymphocytes. Blood, 92, 4220–4229.
Dai, Y., Rahmani, M., & Grant, S. (2003). Proteasome inhibitors potentiate leukemic cell apoptosis induced by the cyclin-dependent kinase inhibitor flavopiridol through a SAPK/JNK- and NF-kappaB-dependent process. Oncogene, 22, 7108–7122.
Pahler, J. C., Ruiz, S., Niemer, I., et al. (2003). Effects of the proteasome inhibitor, bortezomib, on apoptosis in isolated lymphocytes obtained from patients with chronic lymphocytic leukemia. Clinical Cancer Research, 9, 4570–4577.
Dai, Y., Rahmani, M., Pei, X. Y., et al. (2004). Bortezomib and flavopiridol interact synergistically to induce apoptosis in chronic myeloid leukemia cells resistant to imatinib mesylate through both Bcr/Abl-dependent and -independent mechanisms. Blood, 104, 509–518.
Duechler, M., Shehata, M., Schwarzmeier, J. D., et al. (2005). Induction of apoptosis by proteasome inhibitors in B-CLL cells is associated with downregulation of CD23 and inactivation of Notch2. Leukemia, 19, 260–267.
Horton, T. M., Gannavarapu, A., Blaney, S. M., et al. (2006). Bortezomib interactions with chemotherapy agents in acute leukemia in vitro. Cancer Chemotherapy and Pharmacology, 58, 13–23.
Yanamandra, N., Colaco, N. M., Parquet, N. A., et al. (2006). Tipifarnib and bortezomib are synergistic and overcome cell adhesion-mediated drug resistance in multiple myeloma and acute myeloid leukemia. Clinical Cancer Research, 12, 591–599.
Houghton, P. J., Morton, C. L., Kolb, E. A., et al. (2008). Initial testing (stage 1) of the proteasome inhibitor bortezomib by the pediatric preclinical testing program. Pediatric Blood & Cancer, 50, 37–45.
Servida, F., Soligo, D., Delia, D., et al. (2005). Sensitivity of human multiple myelomas and myeloid leukemias to the proteasome inhibitor I. Leukemia, 19, 2324–2331.
Stapnes, C., Doskeland, A. P., Hatfield, K., et al. (2007). The proteasome inhibitors bortezomib and PR-171 have antiproliferative and proapoptotic effects on primary human acute myeloid leukaemia cells. British Journal of Haematology, 136, 814–828.
Kraus, M., Muller-Ide, H., Ruckrich, T., et al. (2014). Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leukemia Research, 38, 383–392.
Liu, H., Westergard, T. D., Cashen, A., et al. (2014). Proteasome inhibitors evoke latent tumor suppression programs in pro-B MLL leukemias through MLL-AF4. Cancer Cell, 25, 530–542.
Miller, C. P., Rudra, S., Keating, M. J., et al. (2009). Caspase-8 dependent histone acetylation by a novel proteasome inhibitor, NPI-0052: a mechanism for synergy in leukemia cells. Blood, 113, 4289–4299.
Miller, C. P., Ban, K., Dujka, M. E., et al. (2007). NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood, 110, 267–277.
Paulus, A., Masood, A., Miller, K. C., et al. (2014). The investigational agent MLN2238 induces apoptosis and is cytotoxic to CLL cells in vitro, as a single agent and in combination with other drugs. British Journal of Haematology, 165, 78–88.
Franke, N. E., Vink, J., Cloos, J., & Kaspers, G. J. (2008). Proteasome and protease inhibitors. In G. J. Kaspers, B. Coiffier, M. Heinrich, & E. Estey (Eds.), Innov. Leuk. Lymphoma Ther (pp. 469–489). New York: Informa Healthcare USA, Inc..
Oerlemans, R., Franke, N. E., Assaraf, Y. G., et al. (2008). Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood, 112, 2489–2499.
Lu, S., Yang, J., Chen, Z., et al. (2009). Different mutants of PSMB5 confer varying bortezomib resistance in T lymphoblastic lymphoma/leukemia cells derived from the Jurkat cell line. Experimental Hematology, 37, 831–837.
Ri, M., Iida, S., Nakashima, T., et al. (2010). Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia, 24, 1506–1512.
Franke, N. E., Niewerth, D., Assaraf, Y. G., et al. (2012). Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia, 26, 757–768.
McConkey, D. J., & Zhu, K. (2008). Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resistance Updates, 11, 164–179.
Franke, N. E., Kaspers, G. L., Assaraf, Y. G., et al. (2014). Exocytosis of polyubiquitinated proteins in bortezomib-resistant leukemia cells: a role for MARCKS in acquired resistance to proteasome inhibitors. Oncotarget, 7, 74779–74796.
Busse, A., Kraus, M., Na, I. K., et al. (2008). Sensitivity of tumor cells to proteasome inhibitors is associated with expression levels and composition of proteasome subunits. Cancer, 112, 659–670.
Fuchs, D., Berges, C., Opelz, G., et al. (2008). Increased expression and altered subunit composition of proteasomes induced by continuous proteasome inhibition establish apoptosis resistance and hyperproliferation of Burkitt lymphoma cells. Journal of Cellular Biochemistry, 103, 270–283.
Frisan, T., Levitsky, V., & Masucci, M. G. (2000). Variations in proteasome subunit composition and enzymatic activity in B-lymphoma lines and normal B cells. International Journal of Cancer, 88, 881–888.
Lu, S., Chen, Z., Yang, J., et al. (2008). Overexpression of the PSMB5 gene contributes to bortezomib resistance in T-lymphoblastic lymphoma/leukemia cells derived from Jurkat line. Experimental Hematology, 36, 1278–1284.
Verbrugge, S. E., Al, M., Assaraf, Y. G., et al. (2013). Overcoming bortezomib resistance in human B cells by anti-CD20 / rituximab-mediated complement-dependent cytotoxicity and epoxyketone-based irreversible proteasome inhibitors. Experimental Hematology & Oncology, 2, 2–12.
de Wilt, L. H. A. M., Jansen, G., Assaraf, Y. G., et al. (2012). Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochemical Pharmacology, 83, 207–217.
Kale, A. J., McGlinchey, R. P., Lechner, A., & Moore, B. S. (2011). Bacterial self-resistance to the natural proteasome inhibitor salinosporamide A. ACS Chemical Biology, 6, 1257–1264.
Suzuki, E., Demo, S., Deu, E., et al. (2011). Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS One, 6, e27996.
Groll, M., Berkers, C. R., Ploegh, H. L., & Ovaa, H. (2006). Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure, 14, 451–456.
Adler, K. B., Tuvim, M. J., & Dickey, B. F. (2013). Regulated mucin secretion from airway epithelial cells. Front Endocrinol (Lausanne), 4, 129.
Buschow, S. I., Liefhebber, J. M. P., Wubbolts, R., & Stoorvogel, W. (2005). Exosomes contain ubiquitinated proteins. Blood Cells Mol Dis, 35, 398–403.
David, E., Kaufman, J. L., Flowers, C. R., et al. (2010). Tipifarnib sensitizes cells to proteasome inhibition by blocking degradation of bortezomib-induced aggresomes. Blood, 116, 5285–5288.
Escalante, A. M., McGrath, R. T., Karolak, M. R., et al. (2013). Preventing the autophagic survival response by inhibition of calpain enhances the cytotoxic activity of bortezomib in vitro and in vivo. Cancer Chemotherapy and Pharmacology, 71, 1567–1576.
Hamouda, M.-A., Belhacene, N., Puissant, A., et al. (2014). The small heat shock protein B8 (HSPB8) confers resistance to bortezomib by promoting autophagic removal of misfolded proteins in multiple myeloma cells. Oncotarget, 5, 6252–6266.
Hoang, B., Benavides, A., Shi, Y., et al. (2009). Effect of autophagy on multiple myeloma cell viability. Molecular Cancer Therapeutics, 8, 1974–1984.
Milani, M., Rzymski, T., Mellor, H. R., et al. (2009). The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with bortezomib. Cancer Research, 69, 4415–4423.
Chen, S., Zhang, Y., Zhou, L., et al. (2014). A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood, 124, 2687–2697.
Milan, E., Perini, T., Resnati, M., et al. (2015). A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy, 11, 1161–1178.
Jarauta, V., Jaime, P., Gonzalo, O., et al. (2016). Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Letters, 382, 1–10.
Baranowska, K., Misund, K., Starheim, K. K., et al. (2016). Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget, 7, 70845–70856.
Hideshima, T., Bradner, J. E., Wong, J., et al. (2005). Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proceedings of the National Academy of Sciences, 102, 8567–8572.
Nawrocki, S. T., Carew, J. S., Pino, M. S., et al. (2006). Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Research, 66, 3773–3781.
Sriram, S. M., Han, D. H., & Kim, S. T. (2011). Partners in crime: ubiquitin-mediated degradation and autophagy. Science Signaling, 4, jc4.
Fang, J., Rhyasen, G., Bolanos, L., et al. (2012). Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloid leukemia depend on autophagy-mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood, 120, 858–867.
Zhou, J., Ching, Y. Q., & Chng, W.-J. (2015). Aberrant nuclear factor-kappa B activity in acute myeloid leukemia: from molecular pathogenesis to therapeutic target. Oncotarget, 6, 5490–5500.
Bosman, M. C. J., Schuringa, J. J., Quax, W. J., & Vellenga, E. (2013). Bortezomib sensitivity of acute myeloid leukemia CD34(+) cells can be enhanced by targeting the persisting activity of NF-κB and the accumulation of MCL-1. Experimental Hematology, 41, 530–538.e1.
Markovina, S., Callander, N. S., O’Connor, S. L., et al. (2010). Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-κB activity in myeloma cells. Molecular Cancer, 9, 176.
Markovina, S., Callander, N. S., O’Connor, S. L., et al. (2008). Bortezomib-resistant nuclear factor-κB activity in multiple myeloma cells. Molecular Cancer Research, 6, 1356–1364.
Hideshima, T., Ikeda, H., Chauhan, D., et al. (2009). Bortezomib induces canonical nuclear factor-κB activation in multiple myeloma cells. Blood, 114, 1046–1052.
Jung, H. J., Chen, Z., Fayad, L., et al. (2012). Bortezomib-resistant nuclear factor κB expression in stem-like cells in mantle cell lymphoma. Experimental Hematology, 40, 107–118.e2.
Yang, D. T., Young, K. H., Kahl, B. S., et al. (2008). Prevalence of bortezomib-resistant constitutive NF-kappaB activity in mantle cell lymphoma. Molecular Cancer, 7, 40.
Kim, A., Park, S., Lee, J.-E., et al. (2012). The dual PI3K and mTOR inhibitor NVP-BEZ235 exhibits anti-proliferative activity and overcomes bortezomib resistance in mantle cell lymphoma cells. Leukemia Research, 36, 912–920.
Liu, C.-Y., Shiau, C.-W., Kuo, H.-Y., et al. (2013). Cancerous inhibitor of protein phosphatase 2A determines bortezomib-induced apoptosis in leukemia cells. Haematologica, 98, 729–738.
Que, W., Chen, J., Chuang, M., & Jiang, D. (2012). Knockdown of c-Met enhances sensitivity to bortezomib in human multiple myeloma U266 cells via inhibiting Akt/mTOR activity. APMIS, 120, 195–203.
Kuhn, D. J., Berkova, Z., Jones, R. J., et al. (2012). Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood, 120, 3260–3270.
Maiso, P., Ocio, E. M., Garayoa, M., et al. (2008). The insulin-like growth factor-I receptor inhibitor NVP-AEW541 provokes cell cycle arrest and apoptosis in multiple myeloma cells. British Journal of Haematology, 141, 470–482.
Wu, K.-D., Zhou, L., Burtrum, D., et al. (2006). Antibody targeting of the insulin-like growth factor I receptor enhances the anti-tumor response of multiple myeloma to chemotherapy through inhibition of tumor proliferation and angiogenesis. Cancer Immunology, Immunotherapy, 56, 343–357.
Leung-Hagesteijn, C., Erdmann, N., Cheung, G., et al. (2013). Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell, 24, 289–304.
Gambella, M., Rocci, A., Passera, R., et al. (2014). High XBP1 expression is a marker of better outcome in multiple myeloma patients treated with bortezomib. Haematologica, 99, e14–e16.
Minderman, H., Zhou, Y., O’Loughlin, K. L., & Baer, M. R. (2007). Bortezomib activity and in vitro interactions with anthracyclines and cytarabine in acute myeloid leukemia cells are independent of multidrug resistance mechanisms and p53 status. Cancer Chemotherapy and Pharmacology, 60, 245–255.
Verbrugge, S. E., Assaraf, Y. G., Dijkmans, B. A., et al. (2012). Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with. The Journal of Pharmacology and Experimental Therapeutics, 341, 174–182.
Ao, L., Wu, Y., Kim, D., et al. (2012). Development of peptide-based reversing agents for p-glycoprotein-mediated resistance to carfilzomib. Molecular Pharmaceutics, 9, 2197–2205.
O’Connor, R., Ooi, M. G., Meiller, J., et al. (2013). The interaction of bortezomib with multidrug transporters: implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemotherapy and Pharmacology, 71, 1357–1368.
Shaughnessy Jr., J. D., Qu, P., Usmani, S., et al. (2011). Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with total therapy 3. Blood, 118, 3512–3524.
Shuqing, L., Jianmin, Y., Chongmei, H., et al. (2011). Upregulated expression of the PSMB5 gene may contribute to drug resistance in patient with multiple myeloma when treated with bortezomib-based regimen. Experimental Hematology, 39, 1117–1118.
Weniger, M. A., Rizzatti, E. G., Perez-Galan, P., et al. (2011). Treatment-induced oxidative stress and cellular antioxidant capacity determine response to bortezomib in mantle cell lymphoma. Clinical Cancer Research, 17, 5101–5112.
Matondo, M., Bousquet-Dubouch, M. P., Gallay, N., et al. (2010). Proteasome inhibitor-induced apoptosis in acute myeloid leukemia: a correlation with the proteasome status. Leukemia Research, 34, 498–506.
Wang, L., Kumar, S., Fridley, B. L., et al. (2008). Proteasome beta subunit pharmacogenomics: gene resequencing and functional genomics. Clinical Cancer Research, 14, 3503–3513.
Lichter, D. I., Danaee, H., Pickard, M. D., et al. (2012). Sequence analysis of β-subunit genes of the 20S proteasome in patients with relapsed multiple myeloma treated with bortezomib or dexamethasone. Blood, 120, 4513–4516.
Barrio, S., Stühmer, T., Teufel, E., et al. (2016). Parallel evolution of multiple PSMB5 mutations in a myeloma patient treated with bortezomib. Blood, 128, 3282.
Bachas, C., Schuurhuis, G. J., Reinhardt, D., et al. (2014). Clinical relevance of molecular aberrations in paediatric acute myeloid leukaemia at first relapse. British Journal of Haematology, 166, 902–910.
Micallef, J., Dharsee, M., Chen, J., et al. (2010). Applying mass spectrometry based proteomic technology to advance the understanding of multiple myeloma. Journal of Hematology & Oncology, 3, 13.
Yang, Y., Chen, Y., Saha, M. N., et al. (2015). Targeting phospho-MARCKS overcomes drug resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia, 29, 715–726.
Scott, K., Hayden, P. J., Will, A., et al. (2016). Bortezomib for the treatment of multiple myeloma. In K. Scott (Ed.), Cochrane Database Syst. Rev (p. CD010816). Chichester: John Wiley & Sons, Ltd.
Orlowski, R. Z., Stinchcombe, T. E., Mitchell, B. S., et al. (2002). Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. Journal of Clinical Oncology, 20, 4420–4427.
Cortes, J., Thomas, D., Koller, C., et al. (2004). Phase I study of bortezomib in refractory or relapsed acute leukemias. Clinical Cancer Research, 10, 3371–3376.
Orlowski, R. Z., Voorhees, P. M., Garcia, R. A., et al. (2005). Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood, 105, 3058–3065.
Horton, T. M., Pati, D., Plon, S. E., et al. (2007). A phase 1 study of the proteasome inhibitor bortezomib in pediatric patients with refractory leukemia: a Children’s Oncology Group study. Clinical Cancer Research, 13, 1516–1522.
Attar, E. C., De Angelo, D. J., Supko, J. G., et al. (2008). Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clinical Cancer Research, 14, 1446–1454.
Messinger, Y., Gaynon, P., Raetz, E., et al. (2010). Phase I study of bortezomib combined with chemotherapy in children with relapsed childhood acute lymphoblastic leukemia (ALL): a report from the therapeutic advances in childhood leukemia (TACL) consortium. Pediatric Blood & Cancer, 55, 254–259.
Messinger, Y. H., Gaynon, P. S., Sposto, R., et al. (2012). Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood, 120, 285–290.
Lancet, J. E., Duong, V. H., Winton, E. F., et al. (2011). A phase I clinical-pharmacodynamic study of the farnesyltransferase inhibitor tipifarnib in combination with the proteasome inhibitor bortezomib in advanced acute leukemias. Clinical Cancer Research, 17, 1140–1146.
Attar, E. C., Johnson, J. L., Amrein, P. C., et al. (2013). Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. Journal of Clinical Oncology, 31, 923–929.
Walker, A. R., Klisovic, R., Johnston, J. S., et al. (2013). Pharmacokinetics and dose escalation of the heat shock protein inhibitor 17-allyamino-17-demethoxygeldanamycin in combination with bortezomib in relapsed or refractory acute myeloid leukemia. Leukemia & Lymphoma, 54, 1996–2002.
Blum, W., Schwind, S., Tarighat, S. S., et al. (2012). Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood, 119, 6025–6031.
Attar, E. C., Amrein, P. C., Fraser, J. W., et al. (2013). Phase I dose escalation study of bortezomib in combination with lenalidomide in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Leukemia Research, 37, 1016–1020.
Howard, D. S., Liesveld, J., Phillips, G. L., et al. (2013). A phase I study using bortezomib with weekly idarubicin for treatment of elderly patients with acute myeloid leukemia. Leukemia Research, 37, 1502–1508.
Walker, A. R., Klisovic, R. B., Garzon, R., et al. (2014). Phase I study of azacitidine and bortezomib in adults with relapsed or refractory acute myeloid leukemia. Leukemia & Lymphoma, 55, 1304–1308.
Walker, A. R., Wang, H., Walsh, K., et al. (2016). Midostaurin, bortezomib and MEC in relapsed/refractory acute myeloid leukemia. Leukemia & Lymphoma, 57, 2100–2108.
Wartman, L. D., Fiala, M. A., Fletcher, T., et al. (2016). A phase I study of carfilzomib for relapsed or refractory acute myeloid and acute lymphoblastic leukemia. Leukemia & Lymphoma, 57, 728–730.
Awan, F. T., Flynn, J. M., Jones, J. A., et al. (2015). Phase I dose escalation trial of the novel proteasome inhibitor carfilzomib in patients with relapsed chronic lymphocytic leukemia and small lymphocytic lymphoma. Leukemia & Lymphoma, 56, 2834–2840.
Horton, T. M., Perentesis, J. P., Gamis, A. S., et al. (2014). A phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: a report from the Children’s Oncology Group. Pediatric Blood & Cancer, 61, 1754–1760.
Ishitsuka, K., Utsunomiya, A., Katsuya, H., et al. (2015). A phase II study of bortezomib in patients with relapsed or refractory aggressive adult T-cell leukemia/lymphoma. Cancer Science, 106, 1219–1223.
Royer, B., Minvielle, S., Diouf, M., et al. (2016). Bortezomib, doxorubicin, cyclophosphamide, dexamethasone induction followed by stem cell transplantation for primary plasma cell leukemia: a prospective phase II study of the Intergroupe Francophone du Myélome. Journal of Clinical Oncology, 34, 2125–2132.
Araujo, K. P. C., Bonuccelli, G., Duarte, C. N., et al. (2013). Bortezomib (PS-341) treatment decreases inflammation and partially rescues the expression of the dystrophin-glycoprotein complex in GRMD dogs. PLoS One, 8, e61367.
Penna, F., Bonetto, A., Aversa, Z., et al. (2016). Effect of the specific proteasome inhibitor bortezomib on cancer-related muscle wasting. Journal of Cachexia, Sarcopenia and Muscle, 7, 345–354.
Turner, J. G., Kashyap, T., Dawson, J. L., et al. (2016). XPO1 inhibitor combination therapy with bortezomib or carfilzomib induces nuclear localization of IκBα and overcomes acquired proteasome inhibitor resistance in human multiple myeloma. Oncotarget, 7, 78896–78909.
de Bruin, G., Xin, B. T., Kraus, M., et al. (2016). A set of activity-based probes to visualize human (immuno)proteasome activities. Angewandte Chemie (International Ed. in English), 55, 4199–4203.
Lee, S. J., Levitsky, K., Parlati, F., et al. (2016). Clinical activity of carfilzomib correlates with inhibition of multiple proteasome subunits: application of a novel pharmacodynamic assay. British Journal of Haematology, 173, 884–895.
de Groot, K. A., Tsang, A., Sjoe, M., Niewerth, D., et al. (2015). Pharmacodynamic monitoring of (immuno)proteasome inhibition during bortezomib treatment of a critically ill patient with lupus nephritis and myocarditis. Lupus Science & Medicine, 2, e000121.
Acknowledgements
This research was supported by KIKA (children cancer free) and the Egbers Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cloos, J., Roeten, M.S., Franke, N.E. et al. (Immuno)proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev 36, 599–615 (2017). https://doi.org/10.1007/s10555-017-9699-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-017-9699-4