
Abstract
The ATP-dependent chromatin remodeling complex SWI/SNF (also called BAF) is critical for the regulation of gene expression. During the evolution from yeast to mammals, the BAF complex has evolved an enormous complexity that contains a high number of subunits encoded by various genes. Emerging studies highlight the frequent involvement of altered mammalian SWI/SNF chromatin-remodeling complexes in human cancers. Here, we discuss the recent advances in determining the structure of SWI/SNF complexes, highlight the mechanisms by which mutations affecting these complexes promote cancer, and describe the promising emerging opportunities for targeted therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Cancer is a genetic disease arising due to the various mutations in the DNA sequence that encodes key proteins essential for the functioning of the cell. The DNA sequence, therefore, receives a lot of attention, and rightly so. However, the DNA inside the cell is not naked. It is packaged with proteins in the form of chromatin. Hence, the cells need to have a mechanism by which the underlying DNA sequence can be made accessible to machinery such as those of transcription and repair. This is brought about largely by protein complexes known as chromatin remodelers—“remodelers” because they displace or evict the histone protein octamers around which the DNA is wrapped for packaging [1]. There are a variety of chromatin-remodeling protein complexes such as the SWI/SNF, CHD1, and RISC among others [1]. In this review, we will restrict our focus to the SWI/SNF complex because of its specific involvement in cancer.
SWI/SNF is an ATP-dependent, multi-subunit, chromatin remodeling complex. It uses the energy from ATP hydrolysis to perform its remodeling function. SWI/SNF subunits were first identified in yeast as genes essential for enabling mating-type switching and sucrose metabolism (Sucrose non-fermenting) [2, 3]. Many of the proteins encoded by these genes were later found to be part of a multi-subunit protein assembly that was named the SWI/SNF complex [4,5,6,7]. This 11-subunit complex (in yeast) has a DNA-stimulated ATP-dependent activity and it acts on the chromatin template [8]. It disrupts histone–DNA contacts, resulting in the sliding of the histone octamer on DNA or the eviction of histones making the DNA accessible for binding of various transcription factors [9, 10]. Perhaps the most essential component of this complex is the ATPase while the other subunits aid in either activating or recruitment of the complex on chromatin. Consistent with its important role, the SWI/SNF complex is conserved across species from yeast to humans, with homologous subunits.
Notably, 25% of cancers have mutations in at least one SWI/SNF subunit [11]. In this review, we will describe how various mutations in the SWI/SNF complex facilitate tumorigenesis and also examine the vulnerabilities of these cancers that can be targeted for treatment. However, to fully understand the implications of SWI/SNF mutations in cancer, a prologue is essential to understand what proteins constitute the SWI/SNF complex, what are their functions, and how the complex is assembled.
2 Composition of the SWI/SNF chromatin remodeling complex
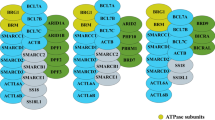
The mammalian SWI/SNF (mSWI/SNF) complex is also called the BAF complex. It consists of a total of 12–15 subunits encoded by 29 genes, including multiple paralogs [12]. BAF complexes typically contain one of two mutually exclusive catalytic subunits, SMARCA4 or SMARCA2, as well as several additional/accessory subunits [12] (Fig. 1). The ATPase subunits not only contain the catalytic domain but also a bromodomain, and an AT-hook which enables their interaction with the substrate–nucleosome core particle (NCP) [13], while the accessory subunits either play a role in maintaining the integrity and function of the complex and/or have certain essential domains, crucial for the complex targeting to chromatin.

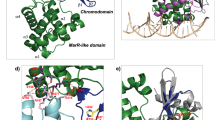
Mode of binding of SWI/SNF complex to nucleosome. Cartoon illustrating the binding of canonical BAF (cBAF) (a) and polybromo-associated BAF (PBAF) (b) with nucleosome core particle (NCP). The distinct modules are marked in distinct colors: blue for ATPase, green for ARP, and orange for base modules. Also, the unique subunits within the base modules are highlighted in darker orange color. PBAF complex uniquely contains a histone binding lobe which enables its interaction with modified histone tails in the NCP
SMARCC1/2 and SMARCD1/2/3 associate to form a structural matrix, on which various other proteins are assembled [14]. Subunits SMARCB1, SMARCE1, and ARID family proteins—ARID1A/1B and ARID2—contain regions that mediate their interaction with transcription factors or DNA [15,16,17]. For example, the HMG (high mobility group) and ARID (AT-rich interaction DNA) domains in SMARCE1 and ARID proteins, respectively, enable the complex interaction with DNA [16, 17]. Similarly, the N and C terminus of SMARCB1 enable its interaction with transcription factors and NCP, respectively [15]. The transactivation domain in SS18/L aids its interaction with transcription activators [18]. A few other subunits—BRD7, 9, PBRM1 (Polybromo 1), and DPF1/2/3 and PHF10—have bromodomains and PHD (plant homeodomain), respectively, which enables the recognition of acetylated/methylated histone tails [19]. Actin B (ACTB) and ACTL6A subunits form an ATP-binding cleft required for maximal ATPase activity of SMARCA4 [20], while few other subunits like B-cell CLL/lymphoma 7 (BCL7) protein family members a/b/c, BCL11, and Glioma tumor suppressor candidate region gene 1/L (GLTSCR1/L) do not have any unique structured or functional domains and their role in SWI/SNF is largely unknown.
Due to the presence of multiple subunits and paralogs, there could be hypothetically around 1500 different types of SWI/SNF complexes in a cell. However, there are three major biochemically distinct SWI/SNF complex assemblies—canonical BAF (cBAF), polybromo BAF (PBAF), and non-canonical BAF (ncBAF) [21, 22]. All three complexes share a few common subunits including the ATPase SMARCA4/2, but they also harbor a few exclusive protein components (Figs. 1, 2a). cBAF is the most abundant complex, around 1 MDa in size, and harbors two unique subunits—ARID1A/1B and DPF1/2/3 [21]. At the same time, PBAF is less abundant and contains ARID2, PBRM1, PHF10, and BRD7 as distinct subunits, whereas ncBAF (also called GBAF) is the smallest complex around 870 kDa in size, has GLTSCR1/L and BRD9 as two distinct protein components, and also does not contain cBAF subunits like SMARCB1, ARID, and SMARCE1 [21]. Interestingly, ncBAF complex subunit GLTSCR1 is present only in multicellular eukaryotes [23].

Mutational frequencies of SWI/SNF components in cancer. a Percentage of mutated samples for each SWI/SNF subunit was plotted by mining the data from two cancer datasets—Pan-Cancer Analysis of Whole Genomes and China pan-cancer dataset (N = 13,116) from The Cancer Genome Atlas (TCGA). The graph distinguishes various complex specific and shared subunits between cBAF, PBAF, and ncBAF complexes. b Heatmap of the frequency of a few frequently mutated subunits across various cancer types. The TCGA data set (N = 13,116) was used to generate the heatmap. The map also depicts that ARID1A is frequently mutated in various cancers, while PBRM1 is most mutated in renal cell carcinoma
3 Biochemical variations enable functional complexity in higher eukaryotes
Heterogeneity in SWI/SNF subunit composition enables heterogeneity in its function allowing SWI/SNF complexes to contribute to transcriptional regulation across cell types and stages of development. For example, SWI/SNF complexes contribute to the development of T cells, hepatocytes, oligodendrocytes, and the maintenance of embryonic stem cell self-renewal and pluripotency [24,25,26,27].
Specificity in the control of these developmental programs is achieved in part through restricted expression and combinatorial assembly of various SWI/SNF subunits and their paralogs. For instance, the SMARCD3 subunit is expressed specifically in the embryonic heart, where it regulates the heart-specific enhancers essential for the control of cardiac development [28]. Similarly, a switch from the PHF10 and ACTL6A subunits, expressed in neural stem cells, to DPF1, DPF3, and ACTL6B subunits are essential for the transition of proliferating neural progenitors into differentiation to post-mitotic mature neurons [29]. Subunit switching can modulate interaction with specific transcription factors and facilitates differential activation of transcriptional pathways. Furthermore, the careful balance between gene activation and repression is also achieved in part through BAF complexes opposing polycomb-mediated repression [30,31,32,33]. BAF complex recruitment leads to the rapid eviction of polycomb repressive complexes (PRCs) and their associated histone modification marks in the absence of Pol II occupancy, transcription, and replication [34]. The recruitment of SWI/SNF complexes to target genes largely happens (1) by the direct protein–protein interactions between various subunits and transcription activators [35,36,37], (2) by the direct interaction of SWI/SNF with modified histones due to the numerous interaction domains of the SWI/SNF subunits [38], and (3) indirectly by interacting with other transcriptional apparatus like histone acetyltransferases [36]. The recruitment then ultimately governs gene expression in various cellular pathways.
4 SWI/SNF complex has distinct structural and functional modules
BAF complexes contain many subunits, which function together to bring changes in gene expression, but how these subunits work together to achieve this common goal has been a question of great interest. Recent biochemical and structural studies highlight that the mSWI/SNF subunits are assembled into distinct modules essential for the functioning of the complex [7, 21, 39, 40]. The complex is arranged in a “C shape clamp” around the nucleosome core particle (NCP) (Fig. 1). Their organization and interaction with the NCP suggest that there are three functional modules in the cBAF complexes. The ATPase module consists of the SMARCA4 subunit, which interacts with the topmost part of the NCP. The bulk of the base module is made up of SMARCB1, SMARCC, SMARCD, SMARCE1, and DPF subunits, where SMARCB1 interacts with the acidic patch of the NCP at the bottom. The subunits ACTB and ACTL6A form the regulatory ARP (actin-related protein) module connecting the ATPase and the base modules. This connection is further strengthened by the association of ARID1, which also stabilizes the base module binding to the NCP (Fig. 1a). The bilateral nucleosome engagement by SMARCB1 and SMARCA4/2 is essential as any deletion in the sequences interacting with NCP disrupts the maximum chromatin-remodeling activity of the cBAF complex [40, 41]. Interestingly, the mode of interaction and function of the PBAF complex with NCP is also similar with few changes; the base module in PBAF additionally has PBRM1, PHF10, BRD7, and ARID2 proteins. The role of ARID2 is highly similar to ARID1, although the rest of the subunits form a histone-binding lobe unique to PBAF and this probably enables its genome recruitment via sensing the chromatin environment (Fig. 1b). The three distinct complexes also show a varying degree of remodeling activity toward various modified nucleosomes [38]. Poly-acetylation of H3 tails activates all three complexes, while H4 acetylation selectively promotes the binding and activity of ncBAF, while H3K4me3 selectively inhibits cBAF activity while having minimal impact on PBAF and ncBAF remodeling activities [38]. The differential remodeling activities also may be the reason behind the distinct genome enrichment of the three complexes at specific chromatin signatures. cBAF complexes occupy mostly the active enhancer sites marked by the presence of H3K27ac and H3K4me1 suggesting their role in enhancer regulation [42, 43]. In contrast, PBAF is predominantly localized to active promoters harboring H3K27ac and H3K4me3 marks. Finally, ncBAF complexes are mostly enriched at CTCF sites co-localized with H3K4me1 [43].
5 Cancer and SWI/SNF complexes
Advances in cancer genome sequencing revealed that SWI/SNF complex subunits are highly mutated in various cancers with a cumulative frequency approaching 25%, even higher than that seen for the tumor suppressor p53 [44]. In addition to the mutations within the complex subunits, their expression changes, and any mutations in the proteins enabling their interaction with chromatin can also have a direct or indirect regulatory effect on the functioning of the complex.
Large-scale pan-cancer genome-sequencing studies have reported that mutations occur across most of the genes encoding SWI/SNF subunits (Fig. 2a) [45, 46]. These mutations are widespread across various cancer types (Fig. 2b). They include nonsense, missense, frameshift, and deletion, which occur across the entire length of the genes. However, the most common type of mutation is missense mutations which are frequently located in the conserved domains of the SWI/SNF subunits [47, 48]. These mutations usually can lead to the degradation of the complex and, thus, loss from its target sites and/or formation of aberrant complexes that have either gain- or loss-of-function phenotypes. Interestingly, certain rare cancers like synovial sarcoma (SS), malignant rhabdoid tumor (MRT), and clear-cell meningioma are known to contain very few other genetic mutations apart from in SWI/SNF subunits [49,50,51]. This suggests that in these cancers SWI/SNF alterations have the potential to be driver mutations by giving a significant advantage for tumor initiation or growth [11]. Of the many subunits of the SWI/SNF complex, mainly five—SMARCA4/2, SMARCB1, ARID1A/B, PBRM1, and ARID2—are significantly mutated compared to the others (Fig. 2a) [52].
6 Mutations in SWI/SNF subunits
6.1 SMARCA4/2
SMARCA4 the ATPase is a common subunit with all three BAF complexes, and it is also frequently mutated in various cancers including breast, lung, and colorectal cancers [53,54,55]. It has been identified as a major tumor suppressor in various pan-cancer studies [56]. Interestingly, many of these mutations are missense and heterozygous in nature [48]. Furthermore, half of them occur within the conserved domains and, hence, potentially affect the function of the complex [48]. Therefore, these mutations can be subcategorized as loss- and gain-of-function mutations [57]. The former impairs DNA translocation, thereby hindering chromatin remodeling (inactivates) whereas the latter increases DNA translocation efficiency, nucleosome remodeling, and hence, chromatin accessibility (hyperactivates) [57].
Accordingly, heterozygous expression of SMARCA4 also mimics a dominant negative phenotype, as DNA accessibility at various enhancers is lost inducing pro-oncogenic expression changes via the MYC signaling pathway [48]. Another way these mutations bring about their effect is by increasing the chromatin retention of Polycomb repressive complex (PRC) complexes, leading to H3K27me3 changes at CpG-island promoters and contributing indirectly to tumor development [58]. SMARCA4 mutations are also found in genetically complex tumors, presumably adding to their genetic burden. These mutations can act as progression events and usually correlate with a poorer prognosis as observed with non-small cell lung cancer [54]. Elevated SMARCA4 expression has also been reported in many cancers including colorectal, gastric, prostate, and intestinal cancers [59,60,61,62,63].
Unlike SMARCA4, its paralog SMARCA2 is not frequently mutated in cancers but rather is epigenetically silenced across numerous tumor types and cancer cell lines [64]. The exact mechanism of epigenetic silencing varies but can arise through promoter methylation, polymorphisms, or HDAC/EZH2-driven mechanisms [65,66,67,68]. Furthermore, its transcriptional reactivation by knockdown or use of HDAC inhibitors prevents cell proliferation [69]. In a certain subset of tumors like small cell carcinoma of the ovary, hypercalcemic type (SCCOHT), both ATPases are not expressed [70, 71]. Loss of both the catalytic subunits leads to a formation of a residual SWI/SNF complex which still can bind to chromatin but with less affinity; however, there is no formation of distinct cBAF and PBAF complexes [72]. The reintroduction of ATPase-deficient SMARCA4 in SCCOHT cell lines restores complex localization to a few enhancers and promoters, but does not promote tumor-suppressive gene expression programs, suggesting that the catalytic activity is essential for mediating these functions [72]. In SMARCA4-deficient cancers that retain SMARCA2 expression, several reports have shown that SMARCA2 acts as a synthetic lethal target, making it a potential therapeutic vulnerability [48, 49].
6.2 SMARCB1
SMARCB1 is present in both cBAF and PBAF complexes. The discovery of mutations in the SMARCB1 gene in rhabdoid tumors in 1998 was the first evidence linking the SWI/SNF complex to cancer [73]. Beyond the mutations in SMARCB1 which lead to the loss of its expression, these tumors are genetically simple, bearing no other frequent driver mutations [74]. Studies in mouse models demonstrate that homozygous inactivation of SMARCB1 is embryonically lethal, although induced somatic homozygous loss results in the rapid onset of cancer in 100% of mice at 11 weeks [75,76,77]. This remarkably takes half the time required for tumor formation following P53 inactivation [78].
Heterozygous mutations cause rhabdoid tumor formation in 10–30% of mice, suggesting that SMARCB1 is a bonafide tumor suppressor [75,76,77]. Mechanistically, the loss of SMARCB1 protein affects the integrity of the SWI/SNF complex and also dissociates the SWI/SNF complex from chromatin [79, 80]. This probably is due to the direct interaction of SMARCB1 with the NCP, necessary for the proper functioning of the BAF and PBAF complexes as discussed above. However, it is the residual BAF complexes deficient in SMARCB1 which drive tumor formation. These aberrant complexes are unable to evict Polycomb, resulting in an elevated H3K27me3 mark, especially at the p16Ink4a tumor suppressor locus thus driving oncogenesis in malignant rhabdoid tumors [81, 82].
Not only mutations but expression changes have also been reported for SMARCB1 in cancer. It is highly upregulated in hepatocellular carcinoma (HCC), leading to the expression of Nucleoporin 210 (NUP210), important for xenobiotic metabolism. This, in turn, promotes tumor formation and is therefore associated with a poor prognosis of the disease [83]. A reduced expression of SMARCB1 has been reported for synovial sarcoma (SS). The translocation of 78 amino acids of SSX at a locus of SS18, an SWI/SNF subunit, leads to the expression of SS18–SSX fusion mutant protein [84, 85]. The fusion protein has been shown to integrate into the BAF complex and acts in a dominant negative manner, displacing not only the wild-type SS18 but also SMARCB1 [86, 87]. The dislocated SMARCB1 is immediately degraded, mimicking reduced SMARCB1 expression, a molecular signature associated with SS. This change in biochemical composition causes cBAF complex degradation and an increase in the prevalence of PBAF and ncBAF complexes [88]. These changes ultimately redirect the BAF complexes from enhancers to broad polycomb domains, activating bivalent genes and leading to oncogenesis [87].
6.3 ARID1A/1B
ARID1A is a cBAF-specific subunit and the most frequently mutated SWI/SNF subunit across cancer types [11]. It was first noted for its mutation status in ovarian clear cell carcinoma, where it is mutated in nearly 60% of the cases [89, 90]. Most of the mutations are loss of function, with nonsense or frameshift mutations occurring all through the gene length. Furthermore, the knockdown of ARID1A is enough for the malignant transformation of immortalized endometrial cells, suggestive of its role as a tumor suppressor [91]. However, studies using mice models suggest that the role of ARID1A in cancer is complex, and often context dependent, and it has both tumor-suppressive and oncogenic roles [42, 92]. Homozygous or heterozygous loss of ARID1A is tumor suppressive due to the decreased chromatin accessibility at enhancers and also decreased expression of genes linked to migration, invasion, and metastasis [42, 93]. Interestingly, elevated ARID1A levels promote tumor initiation by increasing oxidative stress through Cytochrome P450 pathways [93]. Although not that abundant, mutations in ARID1B are also identified in neuroblastoma and pancreatic cancer [94, 95]. Interestingly, mutations in the nuclear localization signal of ARID1B are observed in pancreatic cancer [96]. This leads to the cytoplasm localization of ARID1B which has been shown to promote oncogenesis by stimulating the ERK signaling pathway [96].
Dual mutations in the ARID1 paralogs have been reported in gastric, endometrial, and liver cancers [97]. Furthermore, more than 30% of the reported ARID1A mutant cell lines also harbor loss of ARID1B protein [98]. Mechanistically, dual loss leads to the formation of residual complexes, which associate with PBAF and disrupt its oligomerization [97]. This in turn affects PBAF chromatin distribution contributing to aggressive carcinogenesis in skin and liver mice models [42, 97].
6.4 PBRM1 and ARID2
PBRM1 is a PBAF-specific subunit that contains six bromodomains (BD) [99]. It is mutated in 40% of clear-cell renal carcinoma tumors (ccRCCs) [100]. After VHL, PBRM1 is the second most frequently mutated gene in ccRCC. The combined loss of VHL and PBRM1 is necessary and sufficient for renal malignancy as demonstrated by genetic mouse models [101]. Together, they govern hypoxia gene expression and relieve cells of replication stress [100, 102]. The mutations in PBRM1 often cause loss of protein expression, although few missense mutations are concentrated in the bromodomains [99]. Mutations in the BD2 and BD4 disrupt the chromatin binding of PBRM1 in vitro, negatively affecting gene expression pathways necessary for cell proliferation in vivo [99, 103]. These mutations probably influence the histone-binding lobe of the PBAF complex, rendering it incapable to bind to acetylated chromatin.
ARID2 is also a PBAF-specific subunit and acts similarly to ARID1. It is frequently mutated in HCC, melanoma, ovarian, breast, and lung cancers [104,105,106]. In liver cells, ARID2 enables the transcription of interferon (IFN)-γ [107]. Therefore, the loss of ARID2 leads to disruption in IFN-γ signaling pathways essential for the maintenance of a tumor-suppressive environment in hepatocytes [107].
Studies across various SWI/SNF subunit mutations in many cancers reveal a common theme where these mutations inactivate or destabilize the complex. This ultimately affects the gene regulatory pathways in place to prevent oncogenesis. However, the superimposition of various common mutations on the structure of the BAF complex suggests that only 44% account for their role in complex formation and stability [108]. This suggests that there could be additional mechanisms concerning how these mutations affect the complex function. These mechanisms might include various mutations on the subunits, influencing their binding to a transcription factor, or changes in post-translational modifications manipulating the binding activity of the complex. For example, mutations in BD4 of PBRM1 promote tumor progression by disrupting P53 transcriptional activity and by failing to recruit acetylated P53 at its target promoters [109]. Furthermore, not only mutations within the SWI/SNF complex subunits but any mutations/alteration in its interacting partners essential for the complex recruitment to chromatin also can lead to disruption in its genome distribution promoting tumor conducive environment.
7 Mutations in the interaction partners of SWI/SNF subunits
Studies done in yeast have identified that the binding of SWI/SNF to chromatin is largely dependent on its association with various transcription factors (TFs) [110, 111]. Genetic alterations in these TFs may also play an important role in many tumor types in which SWI/SNF subunits are not genetically altered, thereby expanding the already wide-spanning role of these complexes in human cancer.
Many tumor suppressors proteins like TP53, MYC, Retinoblastoma (Rb), and BRCA1 interact with various SWI/SNF subunits [112,113,114,115,116,117,118]. Furthermore, more than 40% of P53 mutants bring about their effect by modulating SWI/SNF activity or recruitment to chromatin targets [117, 119, 120]. Similarly, MYC binds to various components of SWI/SNF and also regulates the expression of SWI/SNF subunits themselves [112, 113]. In addition, as SMARCB1 has a binding preference to the acidic patch of the nucleosome, any mutations in the histones also affect the binding and remodeling activities of SWI/SNF [121,122,123].
SWI/SNF complexes also display a gain-of-function activity in cancers harboring chromosomal translocations leading to the expression of oncofusion TFs. Particularly, FET (EWSR1::FLI1, FUS::DDIT3) and ETS (TMPRSS2::ERG) oncofusions’ role in perturbing SWI/SNF complex genome binding has been noted. EWSR1::FLI1, found in Ewing sarcoma (EWS), gains the interaction with the SWI/SNF complex and guides it to genes having GGAA microsatellite repeats, enabling oncogenic gene transcription [124, 125]. Similarly, TMPRSS2::ERG, occurring in approximately 50% of prostate cancer cases, gains interaction with the SWI/SNF complex and retargets it from AR to ETS sites [126,127,128]. This ultimately facilitates basal to the luminal transition of cells which is essential for prostate cancer progression [128, 129]. Unlike these two fusion proteins that direct the targeting of SWI/SNF complexes to loci that support oncogenic gene expression and proliferation, FUS::DDIT3 binding uniquely acts as a loss-of-function mutation. FUS::DDIT3 is found in 95% of myxoid liposarcoma (MLS), binds to SWI/SNF and prevents its binding at adipogenic enhancers, and upregulates tumorigenic pathways [130,131,132].
Collectively, the mutations and aberrant expression in SWI/SNF subunits or their interactors may contribute to disease progression in even more than 25% of the cancers than initially anticipated.
8 Therapeutic modalities for targeting SWI/SNF-altered cancers
Systemic investigation on SWI/SNF mutant cancer lines using shRNA and CRISPR libraries has identified several promising candidates for targeted therapy of SWI/SNF-altered cancers. These genes not only include other SWI/SNF subunits but also their interactors. Furthermore, several of these vulnerabilities are being pursued in their therapeutic translation, and a few of these approaches are being tested in ongoing clinical trials.
8.1 Targeting intracomplex vulnerabilities
One vulnerability that emerges in cancers is the mutations in a few SWI/SNF subunits that lead to specific dependency on other SWI/SNF genes. For example, the loss of ARID1A in ovarian and colorectal cancers creates a dependency on its paralog, ARID1B [98, 133]. Similarly, SMARCA4 mutant cells show an enriched dependency on SMARCA2 [134, 135]. This is due to the compensatory mechanism inside the cells. Nevertheless, there are cancers having mutations in both the paralogous subunits of SWI/SNF, but these are rare [71]. Therefore, the development of specific chemical degraders such as PROTACs makes this approach tractable [136,137,138]. Proteolysis targeting chimeras (PROTACs) use structure-based design to direct E3 ubiquitin ligases to the specific protein of interest for their degradation [136]. ACBI1 is a bifunctional degrader developed specifically against the bromodomain of SMARCA4 and SMARCA2, inducing their degradation [139]. Treatment with this degrader led to cell death in SMARCA4 mutant cell lines [139]. PROTACs are highly specific, and accordingly, ACBI1 selectively degrades only SMARCA2 and SMARCA4 and does not show any effect on other bromodomain-containing proteins [139]. Furthermore, recently developed ACBI2 preferentially degrades SMARCA2 and induces lung cancer tumor growth inhibition [140].
Small molecule inhibitor PFI-3 has been developed against the bromodomains of SMARCA2, SMARCA4, and PBRM1 [141, 142]. However, it was an ineffective treatment for SMARCA4 mutant cell lines [141]. Furthermore, cDNA complementation studies suggest that the inhibition of SMARCA2 ATPase activity has a negative influence on the growth of SMARCA4 mutant cells [141]. Accordingly, dual ATPase inhibitors for SMARCA2 and SMARCA4 have been developed and tested for their antitumor activity in cells deficient in SMARCA4 [143]. In addition, such dependencies are also reported between SMARCC1/SMARCC2, SMARCA4/ACTB, and SMARCA4/ARID2, although they are yet to be functionally tested before developing targeted strategies [144].
8.2 Targeting inter-complex vulnerabilities
Screening for genetic and/or pharmacological vulnerabilities in various SWI/SNF-altered cancer cell lines has not only yielded new mechanistic insights into the functioning of the complex but also revealed a new group of targets, which should be further explored for their therapeutic potential.
PRC2 and mSWI/SNF complexes have opposing effects on gene expression [81]. Furthermore, mutations in SMARCB1 and SMARCA4 lead to widespread changes in the H3K27Me3 distribution, changing gene expression patterns, and suggesting that PRC2 can be targeted in these cancers [58, 81, 82]. Furthermore, SWI/SNF mutant cell lines and xenograft models are particularly dependent on PRC2 activity, and its inhibition suppresses the oncogenic signaling [145]. The use of EZH2 (PRC2 catalytic subunit)-specific chemical inhibitor tazemetostat has been approved for the treatment of MRT and epithelioid sarcomas [146, 147]. The efficacy of this treatment is being currently tested in other cancers having SMARCB1 or SMARCA4 mutations [148, 149]. In addition, alternative strategies are also being tested to inhibit PRC2 with newly developed inhibitors targeting other components of the PRC2 complex [149, 150]. Inhibition of EZH2 has also been shown to be lethal for ARID1A mutant ovarian cancer cells due to the activation of the PI3K/AKT signaling pathway [151]. On the other hand, the ATPase switch from SMARCA4 to SMARCA2 during the EZH2 treatment can lead to acquired resistance, which also accompanies BCL2 upregulation [152]. BCL2 is an anti-apoptotic gene, which can be inactivated by the use of its inhibitor ABT263 [153]. Therefore, a combination treatment of inhibiting BCL2 and EZH2 is a better therapeutic strategy for ARID1A mutated cancers.
Treatment with pan-HDAC inhibitor panobinostat or SAHA induces cellular senescence in SMARCB1 mutant rhabdoid and ovarian tumor cells [154, 155], while ARID1A-deficient tumors rely on HDAC6 activity for the deacetylation of P53, required for repression of pro-apoptotic genes [156]. The use of ricolinostat, an HDAC6-specific inhibitor, increases P53 acetylation, promotes apoptotic response, and improves the survival of mice harboring ARID1A mutant cancer [156].
SMARCA4 inactivating mutations in lung cancers increase their sensitivity to CDK4/6 and Aurora Kinase inhibition [157, 158]. Interestingly, ARID1A mutations co-occur with PIK3CA mutations [159]. Loss of ARID1A in breast cancer cells activates AKT; furthermore, treating these cells with MK-2206 (AKT inhibitor) and buparlisib (PI3K inhibitor) increased apoptosis [160]. A similar effect was also observed in various ARID1A mutant ovarian cancer cell lines and mice [159]. Therefore, ARID1A mutant cancers are highly susceptible to the inhibition of PI3K and AKT kinases.
The role of ARID1A in regulating genome stability by recruiting DNA repair proteins like MSH2 at damage sites is known [161, 162]. Accordingly, the loss of ARID1A makes cells sensitive to DNA-damaging agents like radiation, ATR, and PARP inhibitors [163, 164]. Xenograft mice having ARID1A tumors show a drastic decrease in tumor burden upon combined radiation and PARP inhibitor treatment [164]. Therefore, several clinical trials are underway in patients with ARID1A mutant cancers with inhibitors of ATR and PARP (ClinicalTrials.gov Identifiers NCT03207347, NCT04042831, NCT02576444, NCT04065269). Similarly, SMARCA4- and PBRM1-deficient tumors also show a sensitivity to ATR and PARP inhibition [165,166,167].
Large-scale CRISPR screening, aimed at understanding the genetic elements enabling T-cell-mediated tumor killing of melanoma cells, revealed the role of the PBAF complex in immune cell signaling pathways [168]. Depletion of PBRM1, BRD7, ARID2, or any of the PBAF-specific components enhances T-cell response against cancer cells in mouse xenograft models [168]. Accordingly, treatment of ccRCC patients with anti-programmed cell death 1 (PD-1), an immune checkpoint inhibitor, positively correlates with reduced tumor burden and therefore is clinically beneficial [169, 170]. A similar observation has also been made in mice bearing ARID1A-deficient ovarian and gastric cancer [162, 171].
Metabolic vulnerabilities have also been reported for BAF mutant cancers. Especially, ARID1A is shown essential for maintaining glutathione homeostasis by promoting the expression of cystine transporter, SLC7A11 [172]. Hence, ARID1A mutant cells are particularly sensitive to inhibition of the GSH metabolic pathway [173, 174]. Treatment with APR-246 (which targets GSH) and buthionine sulfoximine (for glutamate–cysteine ligase synthetase catalytic subunit) leads to apoptotic cell death due to elevated reactive oxygen species [172]. SMARCB1-deficient tumors show sensitivity to inhibition of the proteasome machinery and autophagy pathways [175], while SMARCA4 mutated lung cancer cells have increased oxygen consumption and enhanced respiratory capacity. Therefore, they are sensitive to IACS-010759, an oxidative phosphorylation (OXPHOS) inhibitor [176].
These studies demonstrate clearly that there are multiple prospects available for targeting SWI/SNF mutant tumors. However, a detailed investigation in both pre-clinical models, as well as large-scale patient trials, is essential for understanding which options are tractable, and this should be the focus in the future.
9 Conclusion
In yeast, SW/SNF complex regulates the expression of around 5% of the genome. In humans, nonetheless, they seem to have a much more enormous impact on chromatin structure and therefore regulate almost all the cellular pathways. Recent discoveries on how the complex assembles, its 3D structure, TF interactions, and genome-wide distribution in wild-type and mutant cells have revealed intricate mechanistic details of the working of the complex. This has also revealed the basis of oncogenesis in SWI/SNF mutated cancers.
SWI/SNF subunits are mutated at a collective frequency of 25% in all cancers. However, considering the broad role of the complex and its interactors in various physiological pathways, by both direct and indirect mechanisms, SWI/SNF may be affected at much higher frequency in cancers. Many mutations in various subunits display cell line– or tissue-specific dependency; therefore, there is a need for the development of tailor-made therapies for individual cancer types. However, elucidating the distinct functions and networks of SWI/SNF complex members across various cancer types as well as developmental stages will enable the development of more holistic therapies targeting a wide variety of cancers.
References
Becker, P. B., & Workman, J. L. (2013). Nucleosome remodeling and epigenetics. Cold Spring Harbor Perspectives in Biology, 5(9), a017905. https://doi.org/10.1101/cshperspect.a017905
Carlson, M., Osmond, B. C., & Botstein, D. (1981). Mutants of yeast defective in sucrose utilization. Genetics, 98(1), 25–40. https://doi.org/10.1093/genetics/98.1.25
Stern, M., Jensen, R., & Herskowitz, I. (1984). Five SWI genes are required for expression of the HO gene in yeast. Journal of Molecular Biology, 178(4), 853–868. https://doi.org/10.1016/0022-2836(84)90315-2
Peterson, C. L., Dingwall, A., & Scott, M. P. (1994). Five SWI/SNF gene products are components of a large multisubunit complex required for transcriptional enhancement. Proc Natl Acad Sci U S A, 91(8), 2905–2908. https://doi.org/10.1073/pnas.91.8.2905
Côté, J., Quinn, J., Workman, J. L., & Peterson, C. L. (1994). Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science, 265(5168), 53–60. https://doi.org/10.1126/science.8016655
Cairns, B. R., Kim, Y. J., Sayre, M. H., Laurent, B. C., & Kornberg, R. D. (1994). A multisubunit complex containing the SWI1/ADR6, SWI2/SNF2, SWI3, SNF5, and SNF6 gene products isolated from yeast. Proceedings of the National Academy of Sciences of the United States of America, 91(5), 1950–1954. https://doi.org/10.1073/pnas.91.5.1950
Mashtalir, N., Suzuki, H., Farrell, D. P., Sankar, A., Luo, J., Filipovski, M., D’Avino, A. R., St. Pierre, R., Valencia, A. M., Onikubo, T., Roeder, R. G., Han, Y., He, Y., Ranish, J. A., DiMaio, F., Walz, T., & Kadoch, C. (2020). A structural model of the endogenous human BAF complex informs disease mechanisms. Cell, 183(3), 802-817.e824. https://doi.org/10.1016/j.cell.2020.09.051
Côté, J., Quinn, J., Workman Jerry, L., & Peterson Craig, L. (1994). Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science, 265(5168), 53–60. https://doi.org/10.1126/science.8016655
Vignali, M., Hassan, A. H., Neely, K. E., & Workman, J. L. (2000). ATP-dependent chromatin-remodeling complexes. Molecular and Cellular Biology, 20(6), 1899–1910. https://doi.org/10.1128/mcb.20.6.1899-1910.2000
Workman, J. L., & Kingston, R. E. (1998). Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annual Review of Biochemistry, 67, 545–579. https://doi.org/10.1146/annurev.biochem.67.1.545
Kadoch, C., Hargreaves, D. C., Hodges, C., Elias, L., Ho, L., Ranish, J., & Crabtree, G. R. (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature genetics, 45(6), 592–601. https://doi.org/10.1038/ng.2628
Kadoch, C., & Crabtree, G. R. (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv, 1(5), e1500447. https://doi.org/10.1126/sciadv.1500447
Morrison, E. A., Sanchez, J. C., Ronan, J. L., Farrell, D. P., Varzavand, K., Johnson, J. K., Gu, B. X., Crabtree, G. R., & Musselman, C. A. (2017). DNA binding drives the association of BRG1/hBRM bromodomains with nucleosomes. Nature Communications, 8(1), 16080. https://doi.org/10.1038/ncomms16080
Dong, C., Zhang, R., Xu, L., Liu, B., & Chu, X. (2022). Assembly and interaction of core subunits of BAF complexes and crystal study of the SMARCC1/SMARCE1 binary complex. Biochemical and Biophysical Research Communications, 599, 9–16. https://doi.org/10.1016/j.bbrc.2022.02.007
Weissmiller, A. M., Wang, J., Lorey, S. L., Howard, G. C., Martinez, E., Liu, Q., & Tansey, W. P. (2019). Inhibition of MYC by the SMARCB1 tumor suppressor. Nature Communications, 10(1), 2014. https://doi.org/10.1038/s41467-019-10022-5
Heo, Y., Park, J. H., Kim, J., Han, J., Yun, J. H., & Lee, W. (2020). Crystal structure of the HMG domain of human BAF57 and its interaction with four-way junction DNA. Biochemical and Biophysical Research Communications, 533(4), 919–924. https://doi.org/10.1016/j.bbrc.2020.09.094
Patsialou, A., Wilsker, D., & Moran, E. (2005). DNA-binding properties of ARID family proteins. Nucleic Acids Research, 33(1), 66–80. https://doi.org/10.1093/nar/gki145
de Bruijn, D. R., van Dijk, A. H., Willemse, M. P., & van Kessel, A. G. (2008). The C terminus of the synovial sarcoma-associated SSX proteins interacts with the LIM homeobox protein LHX4. Oncogene, 27(5), 653–662. https://doi.org/10.1038/sj.onc.1210688
Ho, P. J., Lloyd, S. M., & Bao, X. (2019). Unwinding chromatin at the right places: how BAF is targeted to specific genomic locations during development. Development, 146(19), dev178780. https://doi.org/10.1242/dev.178780
Schubert, H. L., Wittmeyer, J., Kasten, M. M., Hinata, K., Rawling, D. C., Héroux, A., Cairns, B. R., & Hill, C. P. (2013). Structure of an actin-related subcomplex of the SWI/SNF chromatin remodeler. Proceedings of the National Academy of Sciences of the United States of America, 110(9), 3345–3350. https://doi.org/10.1073/pnas.1215379110
Mashtalir, N., D’Avino, A. R., Michel, B. C., Luo, J., Pan, J., Otto, J. E., Zullow, H. J., McKenzie, Z. M., Kubiak, R. L., St. Pierre, R., Valencia, A. M., Poynter, S. J., Cassel, S. H., Ranish, J. A., & Kadoch, C. (2018). Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell, 175(5), 1272-1288.e1220. https://doi.org/10.1016/j.cell.2018.09.032
Alpsoy, A., & Dykhuizen, E. C. (2018). Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. Journal of Biological Chemistry, 293(11), 3892–3903. https://doi.org/10.1074/jbc.RA117.001065
Innis, S. M., & Cabot, B. (2020). GBAF, a small BAF sub-complex with big implications: A systematic review. Epigenetics & Chromatin, 13(1), 48. https://doi.org/10.1186/s13072-020-00370-8
Chi, T. H., Wan, M., Zhao, K., Taniuchi, I., Chen, L., Littman, D. R., & Crabtree, G. R. (2002). Reciprocal regulation of CD4/CD8 expression by SWI/SNF-like BAF complexes. Nature, 418(6894), 195–199. https://doi.org/10.1038/nature00876
Gresh, L., Bourachot, B., Reimann, A., Guigas, B., Fiette, L., Garbay, S., Muchardt, C., Hue, L., Pontoglio, M., Yaniv, M., & Klochendler-Yeivin, A. (2005). The SWI/SNF chromatin-remodeling complex subunit SNF5 is essential for hepatocyte differentiation. The EMBO Journal, 24(18), 3313–3324. https://doi.org/10.1038/sj.emboj.7600802
Ho, L., Jothi, R., Ronan, J. L., Cui, K., Zhao, K., & Crabtree, G. R. (2009). An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proceedings of the National Academy of Sciences of the United States of America, 106(13), 5187–5191. https://doi.org/10.1073/pnas.0812888106
Yu, Y., Chen, Y., Kim, B., Wang, H., Zhao, C., He, X., Liu, L., Liu, W., Wu, L. M., Mao, M., Chan, J. R., Wu, J., & Lu, Q. R. (2013). Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell, 152(1–2), 248–261. https://doi.org/10.1016/j.cell.2012.12.006
Lickert, H., Takeuchi, J. K., Von Both, I., Walls, J. R., McAuliffe, F., Adamson, S. L., Henkelman, R. M., Wrana, J. L., Rossant, J., & Bruneau, B. G. (2004). Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature, 432(7013), 107–112. https://doi.org/10.1038/nature03071
Lessard, J., Wu, J. I., Ranish, J. A., Wan, M., Winslow, M. M., Staahl, B. T., Wu, H., Aebersold, R., Graef, I. A., & Crabtree, G. R. (2007). An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron, 55(2), 201–215. https://doi.org/10.1016/j.neuron.2007.06.019
Ho, L., Miller, E. L., Ronan, J. L., Ho, W. Q., Jothi, R., & Crabtree, G. R. (2011). esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nature Cell Biology, 13(8), 903–913. https://doi.org/10.1038/ncb2285
Kia, S. K., Gorski, M. M., Giannakopoulos, S., & Verrijzer, C. P. (2008). SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Molecular and Cellular Biology, 28(10), 3457–3464. https://doi.org/10.1128/mcb.02019-07
Weber, C. M., Hafner, A., Kirkland, J. G., Braun, S. M. G., Stanton, B. Z., Boettiger, A. N., & Crabtree, G. R. (2021). mSWI/SNF promotes Polycomb repression both directly and through genome-wide redistribution. Nature Structural & Molecular Biology, 28(6), 501–511. https://doi.org/10.1038/s41594-021-00604-7
Drosos, Y., Myers, J. A., Xu, B., Mathias, K. M., Beane, E. C., Radko-Juettner, S., Mobley, R. J., Larsen, M. E., Piccioni, F., Ma, X., Low, J., Hansen, B. S., Peters, S. T., Bhanu, N. V., Dhanda, S. K., Chen, T., Upadhyaya, S. A., Pruett-Miller, S. M., Root, D. E., … Roberts, C. W. M. (2022). NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition. Molecular Cell, 82(13), 2472-2489.e2478. https://doi.org/10.1016/j.molcel.2022.04.015
Kadoch, C., Williams, R. T., Calarco, J. P., Miller, E. L., Weber, C. M., Braun, S. M., Pulice, J. L., Chory, E. J., & Crabtree, G. R. (2017). Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nature Genetics, 49(2), 213–222. https://doi.org/10.1038/ng.3734
Neely, K. E., Hassan, A. H., Brown, C. E., Howe, L., & Workman, J. L. (2002). Transcription activator interactions with multiple SWI/SNF subunits. Molecular and Cellular Biology, 22(6), 1615–1625. https://doi.org/10.1128/mcb.22.6.1615-1625.2002
Alver, B. H., Kim, K. H., Lu, P., Wang, X., Manchester, H. E., Wang, W., Haswell, J. R., Park, P. J., & Roberts, C. W. M. (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nature Communications, 8(1), 14648. https://doi.org/10.1038/ncomms14648
Yudkovsky, N., Logie, C., Hahn, S., & Peterson, C. L. (1999). Recruitment of the SWI/SNF chromatin remodeling complex by transcriptional activators. Genes & Development, 13(18), 2369–2374. https://doi.org/10.1101/gad.13.18.2369
Mashtalir, N., Dao, H. T., Sankar, A., Liu, H., Corin, A. J., Bagert, J. D., Ge, E. J., D’Avino, A. R., Filipovski, M., Michel, B. C., Dann, G. P., Muir, T. W., & Kadoch, C. (2021). Chromatin landscape signals differentially dictate the activities of mSWI/SNF family complexes. Science, 373(6552), 306–315. https://doi.org/10.1126/science.abf8705
Kadoch, C., & Crabtree, G. R. (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Science Advances, 1(5), e1500447. https://doi.org/10.1126/sciadv.1500447
He, S., Wu, Z., Tian, Y., Yu, Z., Yu, J., Wang, X., Li, J., Liu, B., & Xu, Y. (2020). Structure of nucleosome-bound human BAF complex. Science, 367(6480), 875–881. https://doi.org/10.1126/science.aaz9761
Mashtalir, N., Suzuki, H., Farrell, D. P., Sankar, A., Luo, J., Filipovski, M., D’Avino, A. R., St Pierre, R., Valencia, A. M., Onikubo, T., Roeder, R. G., Han, Y., He, Y., Ranish, J. A., DiMaio, F., Walz, T., & Kadoch, C. (2020). A structural model of the endogenous human BAF complex informs disease mechanisms. Cell, 183(3), 802-817.e24. https://doi.org/10.1016/j.cell.2020.09.051
Mathur, R., Alver, B. H., San Roman, A. K., Wilson, B. G., Wang, X., Agoston, A. T., Park, P. J., Shivdasani, R. A., & Roberts, C. W. M. (2017). ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nature Genetics, 49(2), 296–302. https://doi.org/10.1038/ng.3744
Michel, B. C., D’Avino, A. R., Cassel, S. H., Mashtalir, N., McKenzie, Z. M., McBride, M. J., Valencia, A. M., Zhou, Q., Bocker, M., Soares, L. M. M., Pan, J., Remillard, D. I., Lareau, C. A., Zullow, H. J., Fortoul, N., Gray, N. S., Bradner, J. E., Chan, H. M., & Kadoch, C. (2018). A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nature Cell Biology, 20(12), 1410–1420. https://doi.org/10.1038/s41556-018-0221-1
Mittal, P., & Roberts, C. W. M. (2020). The SWI/SNF complex in cancer — biology, biomarkers and therapy. Nature Reviews Clinical Oncology, 17(7), 435–448. https://doi.org/10.1038/s41571-020-0357-3
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. (2020). Pan-cancer analysis of whole genomes. Nature, 578(7793), 82–93. https://doi.org/10.1038/s41586-020-1969-6
Wu, L., Yao, H., Chen, H., Wang, A., Guo, K., Gou, W., Yu, Y., Li, X., Yao, M., Yuan, S., Pang, F., Hu, J., Chen, L., Liu, W., Yao, J., Zhang, S., Dong, X., Wang, W., Hu, J., … Wang, M. (2022). Landscape of somatic alterations in large-scale solid tumors from an Asian population. Nature Communications, 13(1), 4264. https://doi.org/10.1038/s41467-022-31780-9
Shain, A. H., & Pollack, J. R. (2013). The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One, 8(1), e55119. https://doi.org/10.1371/journal.pone.0055119
Hodges, H. C., Stanton, B. Z., Cermakova, K., Chang, C.-Y., Miller, E. L., Kirkland, J. G., Ku, W. L., Veverka, V., Zhao, K., & Crabtree, G. R. (2018). Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nature Structural & Molecular Biology, 25(1), 61–72. https://doi.org/10.1038/s41594-017-0007-3
Chun, H. E., Lim, E. L., Heravi-Moussavi, A., Saberi, S., Mungall, K. L., Bilenky, M., Carles, A., Tse, K., Shlafman, I., Zhu, K., Qian, J. Q., Palmquist, D. L., He, A., Long, W., Goya, R., Ng, M., LeBlanc, V. G., Pleasance, E., Thiessen, N., … Marra, M. A. (2016). Genome-wide profiles of extra-cranial malignant rhabdoid tumors reveal heterogeneity and dysregulated developmental pathways. Cancer Cell, 29(3), 394–406. https://doi.org/10.1016/j.ccell.2016.02.009
Kadoch, C., & Crabtree, G. R. (2013). Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell, 153(1), 71–85. https://doi.org/10.1016/j.cell.2013.02.036
St Pierre, R., Collings, C. K., Samé Guerra, D. D., Widmer, C. J., Bolonduro, O., Mashtalir, N., Sankar, A., Liang, Y., Bi, W. L., Gerkes, E. H., Ramesh, V., Qi, J., Smith, M. J., Meredith, D. M., & Kadoch, C. (2022). SMARCE1 deficiency generates a targetable mSWI/SNF dependency in clear cell meningioma. Nature genetics, 54(6), 861–873. https://doi.org/10.1038/s41588-022-01077-0
Centore, R. C., Sandoval, G. J., Soares, L. M. M., Kadoch, C., & Chan, H. M. (2020). Mammalian SWI/SNF chromatin remodeling complexes: Emerging mechanisms and therapeutic strategies. Trends in Genetics, 36(12), 936–950. https://doi.org/10.1016/j.tig.2020.07.011
Bai, J., Mei, P., Zhang, C., Chen, F., Li, C., Pan, Z., Liu, H., & Zheng, J. (2013). BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PLOS ONE, 8(3), e59772. https://doi.org/10.1371/journal.pone.0059772
Reisman, D. N., Sciarrotta, J., Wang, W., Funkhouser, W. K., & Weissman, B. E. (2003). Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: Correlation with poor prognosis. Cancer Research, 63(3), 560–566.
Pyo, J. S., Son, B. K., Oh, D., & Kim, E. K. (2018). BRG1 is correlated with poor prognosis in colorectal cancer. Human Pathology, 73, 66–73. https://doi.org/10.1016/j.humpath.2017.12.013
Mardinian, K., Adashek, J. J., Botta, G. P., Kato, S., & Kurzrock, R. (2021). SMARCA4: Implications of an altered chromatin-remodeling gene for cancer development and therapy. Molecular Cancer Therapeutics, 20(12), 2341–2351. https://doi.org/10.1158/1535-7163.Mct-21-0433
Clapier, C. R., Verma, N., Parnell, T. J., & Cairns, B. R. (2020). Cancer-associated gain-of-function mutations activate a SWI/SNF-family regulatory hub. Molecular Cell, 80(4), 712-725.e715. https://doi.org/10.1016/j.molcel.2020.09.024
Stanton, B. Z., Hodges, C., Calarco, J. P., Braun, S. M., Ku, W. L., Kadoch, C., Zhao, K., & Crabtree, G. R. (2017). Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nature Genetics, 49(2), 282–288. https://doi.org/10.1038/ng.3735
Dal Molin, M., Hong, S.-M., Hebbar, S., Sharma, R., Scrimieri, F., De Wilde, R. F., Mayo, S. C., Goggins, M., Wolfgang, C. L., & Schulick, R. D. (2012). Loss of expression of the SWI/SNF chromatin remodeling subunit BRG1/SMARCA4 is frequently observed in intraductal papillary mucinous neoplasms of the pancreas. Human pathology, 43(4), 585–591. https://doi.org/10.1016/j.humpath.2011.06.009
Lin, S., Jiang, T., Ye, L., Han, Z., Liu, Y., Liu, C., Yuan, C., Zhao, S., Chen, J., & Wang, J. (2016). The chromatin-remodeling enzyme BRG1 promotes colon cancer progression via positive regulation of WNT3A. Oncotarget, 7(52), 86051. https://doi.org/10.18632/oncotarget.13326
Roy, N., Malik, S., Villanueva, K. E., Urano, A., Lu, X., Von Figura, G., Seeley, E. S., Dawson, D. W., Collisson, E. A., & Hebrok, M. (2015). Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes & development, 29(6), 658–671. https://doi.org/10.1101/gad.256628.114
Sentani, K., Oue, N., Kondo, H., Kuraoka, K., Motoshita, J., Ito, R., Yokozaki, H., & Yasui, W. (2001). Increased expression but not genetic alteration of BRG1, a component of the SWI/SNF complex, is associated with the advanced stage of human gastric carcinomas. Pathobiology, 69(6), 315–320. https://doi.org/10.1159/000064638
Sun, A., Tawfik, O., Gayed, B., Thrasher, J. B., Hoestje, S., Li, C., & Li, B. (2007). Aberrant expression of SWI/SNF catalytic subunits BRG1/BRM is associated with tumor development and increased invasiveness in prostate cancers. The Prostate, 67(2), 203–213. https://doi.org/10.1002/pros.20521
Glaros, S., Cirrincione, G. M., Muchardt, C., Kleer, C. G., Michael, C. W., & Reisman, D. (2007). The reversible epigenetic silencing of BRM: Implications for clinical targeted therapy. Oncogene, 26(49), 7058–7066. https://doi.org/10.1038/sj.onc.1210514
Wu, J., He, K., Zhang, Y., Song, J., Shi, Z., Chen, W., & Shao, Y. (2019). Inactivation of SMARCA2 by promoter hypermethylation drives lung cancer development. Gene, 687, 193–199. https://doi.org/10.1016/j.gene.2018.11.032
Wang, J. R., Gramling, S. J., Goldstein, D. P., Cheng, D., Chen, D., Azad, A. K., Tse, A., Hon, H., Chen, Z., Mirshams, M., Simpson, C., Huang, S. H., Marquez, S., O’Sullivan, B., Liu, F. F., Roberts, H., Xu, W., Brown, D. H., Gilbert, R. W., … Liu, G. (2013). Association of two BRM promoter polymorphisms with head and neck squamous cell carcinoma risk. Carcinogenesis, 34(5), 1012–1017. https://doi.org/10.1093/carcin/bgt008
Chan-Penebre, E., Armstrong, K., Drew, A., Grassian, A. R., Feldman, I., Knutson, S. K., Kuplast-Barr, K., Roche, M., Campbell, J., Ho, P., Copeland, R. A., Chesworth, R., Smith, J. J., Keilhack, H., & Ribich, S. A. (2017). Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: In vitro and in vivo preclinical models. Molecular Cancer Therapeutics, 16(5), 850–860. https://doi.org/10.1158/1535-7163.Mct-16-0678
Yamamichi, N., Yamamichi-Nishina, M., Mizutani, T., Watanabe, H., Minoguchi, S., Kobayashi, N., Kimura, S., Ito, T., Yahagi, N., Ichinose, M., Omata, M., & Iba, H. (2005). The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene, 24(35), 5471–5481. https://doi.org/10.1038/sj.onc.1208716
Kahali, B., Yu, J., Marquez, S. B., Thompson, K. W., Liang, S. Y., Lu, L., & Reisman, D. (2014). The silencing of the SWI/SNF subunit and anticancer gene BRM in rhabdoid tumors. Oncotarget, 5(10), 3316–3332. https://doi.org/10.18632/oncotarget.1945
Witkowski, L., Donini, N., Byler-Dann, R., Knost, J. A., Albrecht, S., Berchuck, A., McCluggage, W. G., Hasselblatt, M., & Foulkes, W. D. (2017). The hereditary nature of small cell carcinoma of the ovary, hypercalcemic type: Two new familial cases. Familial Cancer, 16(3), 395–399. https://doi.org/10.1007/s10689-016-9957-6
Karnezis, A. N., Wang, Y., Ramos, P., Hendricks, W. P., Oliva, E., D’Angelo, E., Prat, J., Nucci, M. R., Nielsen, T. O., Chow, C., Leung, S., Kommoss, F., Kommoss, S., Silva, A., Ronnett, B. M., Rabban, J. T., Bowtell, D. D., Weissman, B. E., Trent, J. M., … Huntsman, D. G. (2016). Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. The Journal of Pathology, 238(3), 389–400. https://doi.org/10.1002/path.4633
Pan, J., McKenzie, Z. M., D’Avino, A. R., Mashtalir, N., Lareau, C. A., St Pierre, R., Wang, L., Shilatifard, A., & Kadoch, C. (2019). The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity-independent genomic targeting. Nature Genetics, 51(4), 618–626. https://doi.org/10.1038/s41588-019-0363-5
Versteege, I., Sévenet, N., Lange, J., Rousseau-Merck, M. F., Ambros, P., Handgretinger, R., Aurias, A., & Delattre, O. (1998). Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature, 394(6689), 203–206. https://doi.org/10.1038/28212
Lee, R. S., Stewart, C., Carter, S. L., Ambrogio, L., Cibulskis, K., Sougnez, C., Lawrence, M. S., Auclair, D., Mora, J., Golub, T. R., Biegel, J. A., Getz, G., & Roberts, C. W. (2012). A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. The Journal of Clinical Investigation, 122(8), 2983–2988. https://doi.org/10.1172/jci64400
Roberts, C. W., Galusha, S. A., McMenamin, M. E., Fletcher, C. D., & Orkin, S. H. (2000). Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proceedings of the National Academy of Sciences of the United States of America, 97(25), 13796–13800. https://doi.org/10.1073/pnas.250492697
Guidi, C. J., Sands, A. T., Zambrowicz, B. P., Turner, T. K., Demers, D. A., Webster, W., Smith, T. W., Imbalzano, A. N., & Jones, S. N. (2001). Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Molecular and Cellular Biology, 21(10), 3598–3603. https://doi.org/10.1128/mcb.21.10.3598-3603.2001
Klochendler-Yeivin, A., Fiette, L., Barra, J., Muchardt, C., Babinet, C., & Yaniv, M. (2000). The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Reports, 1(6), 500–506. https://doi.org/10.1093/embo-reports/kvd129
Roberts, C. W., Leroux, M. M., Fleming, M. D., & Orkin, S. H. (2002). Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell, 2(5), 415–425. https://doi.org/10.1016/s1535-6108(02)00185-x
Wang, X., Lee, R. S., Alver, B. H., Haswell, J. R., Wang, S., Mieczkowski, J., Drier, Y., Gillespie, S. M., Archer, T. C., Wu, J. N., Tzvetkov, E. P., Troisi, E. C., Pomeroy, S. L., Biegel, J. A., Tolstorukov, M. Y., Bernstein, B. E., Park, P. J., & Roberts, C. W. M. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nature Genetics, 49(2), 289–295. https://doi.org/10.1038/ng.3746
Nakayama, R. T., Pulice, J. L., Valencia, A. M., McBride, M. J., McKenzie, Z. M., Gillespie, M. A., Ku, W. L., Teng, M., Cui, K., Williams, R. T., Cassel, S. H., Qing, H., Widmer, C. J., Demetri, G. D., Irizarry, R. A., Zhao, K., Ranish, J. A., & Kadoch, C. (2017). SMARCB1 is required for widespread BAF complex–mediated activation of enhancers and bivalent promoters. Nature Genetics, 49(11), 1613–1623. https://doi.org/10.1038/ng.3958
Wilson, B. G., Wang, X., Shen, X., McKenna, E. S., Lemieux, M. E., Cho, Y. J., Koellhoffer, E. C., Pomeroy, S. L., Orkin, S. H., & Roberts, C. W. (2010). Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell, 18(4), 316–328. https://doi.org/10.1016/j.ccr.2010.09.006
Oruetxebarria, I., Venturini, F., Kekarainen, T., Houweling, A., Zuijderduijn, L. M., Mohd-Sarip, A., Vries, R. G., Hoeben, R. C., & Verrijzer, C. P. (2004). P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. Journal of Biological Chemistry, 279(5), 3807–3816. https://doi.org/10.1074/jbc.M309333200
Hong, S. H., Son, K. H., Ha, S. Y., Wee, T. I., Choi, S. K., Won, J. E., Han, H. D., Ro, Y., Park, Y. M., Eun, J. W., Nam, S. W., Han, J. W., Kang, K., & You, J. S. (2021). Nucleoporin 210 serves a key scaffold for SMARCB1 in liver cancer. Cancer Research, 81(2), 356–370. https://doi.org/10.1158/0008-5472.Can-20-0568
dos Santos, N. R., de Bruijn, D. R., Balemans, M., Janssen, B., Gärtner, F., Lopes, J. M., de Leeuw, B., & Geurts van Kessel, A. (1997). Nuclear localization of SYT, SSX and the synovial sarcoma-associated SYT-SSX fusion proteins. Human Molecular Genetics, 6(9), 1549–1558. https://doi.org/10.1093/hmg/6.9.1549
Kato, H., Tjernberg, A., Zhang, W., Krutchinsky, A. N., An, W., Takeuchi, T., Ohtsuki, Y., Sugano, S., de Bruijn, D. R., Chait, B. T., & Roeder, R. G. (2002). SYT associates with human SNF/SWI complexes and the C-terminal region of its fusion partner SSX1 targets histones. Journal of Biological Chemistry, 277(7), 5498–5505. https://doi.org/10.1074/jbc.M108702200
Middeljans, E., Wan, X., Jansen, P. W., Sharma, V., Stunnenberg, H. G., & Logie, C. (2012). SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS ONE, 7(3), e33834–e33834. https://doi.org/10.1371/journal.pone.0033834
McBride, M. J., Pulice, J. L., Beird, H. C., Ingram, D. R., D’Avino, A. R., Shern, J. F., Charville, G. W., Hornick, J. L., Nakayama, R. T., Garcia-Rivera, E. M., Araujo, D. M., Wang, W.-L., Tsai, J.-W., Yeagley, M., Wagner, A. J., Futreal, P. A., Khan, J., Lazar, A. J., & Kadoch, C. (2018). The SS18-SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell, 33(6), 1128-1141.e1127. https://doi.org/10.1016/j.ccell.2018.05.002
Li, J., Mulvihill, T. S., Li, L., Barrott, J. J., Nelson, M. L., Wagner, L., Lock, I. C., Pozner, A., Lambert, S. L., Ozenberger, B. B., Ward, M. B., Grossmann, A. H., Liu, T., Banito, A., Cairns, B. R., & Jones, K. B. (2021). A role for SMARCB1 in synovial sarcomagenesis reveals that SS18-SSX induces canonical BAF destruction. Cancer Discovery, 11(10), 2620–2637. https://doi.org/10.1158/2159-8290.Cd-20-1219
Wiegand, K. C., Shah, S. P., Al-Agha, O. M., Zhao, Y., Tse, K., Zeng, T., Senz, J., McConechy, M. K., Anglesio, M. S., Kalloger, S. E., Yang, W., Heravi-Moussavi, A., Giuliany, R., Chow, C., Fee, J., Zayed, A., Prentice, L., Melnyk, N., Turashvili, G., … Huntsman, D. G. (2010). ARID1A mutations in endometriosis-associated ovarian carcinomas. New England Journal of Medicine, 363(16), 1532–1543. https://doi.org/10.1056/NEJMoa1008433
Jones, S., Wang, T. L., Shih Ie, M., Mao, T. L., Nakayama, K., Roden, R., Glas, R., Slamon, D., Diaz, L. A., Jr., Vogelstein, B., Kinzler, K. W., Velculescu, V. E., & Papadopoulos, N. (2010). Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science, 330(6001), 228–231. https://doi.org/10.1126/science.1196333
Lakshminarasimhan, R., Andreu-Vieyra, C., Lawrenson, K., Duymich, C. E., Gayther, S. A., Liang, G., & Jones, P. A. (2017). Down-regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non-tumorigenic endometriotic cells. Cancer Letters, 401, 11–19. https://doi.org/10.1016/j.canlet.2017.04.040
Zhai, Y., Kuick, R., Tipton, C., Wu, R., Sessine, M., Wang, Z., Baker, S. J., Fearon, E. R., & Cho, K. R. (2016). Arid1a inactivation in an Apc- and Pten-defective mouse ovarian cancer model enhances epithelial differentiation and prolongs survival. The Journal of Pathology, 238(1), 21–30. https://doi.org/10.1002/path.4599
Sun, X., Wang, S. C., Wei, Y., Luo, X., Jia, Y., Li, L., Gopal, P., Zhu, M., Nassour, I., Chuang, J. C., Maples, T., Celen, C., Nguyen, L. H., Wu, L., Fu, S., Li, W., Hui, L., Tian, F., Ji, Y., … Zhu, H. (2017). Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell, 32(5), 574-589.e576. https://doi.org/10.1016/j.ccell.2017.10.007
Sausen, M., Leary, R. J., Jones, S., Wu, J., Reynolds, C. P., Liu, X., Blackford, A., Parmigiani, G., Diaz, L. A., Papadopoulos, N., Vogelstein, B., Kinzler, K. W., Velculescu, V. E., & Hogarty, M. D. (2013). Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nature Genetics, 45(1), 12–17. https://doi.org/10.1038/ng.2493
Shain, A. H., Giacomini, C. P., Matsukuma, K., Karikari, C. A., Bashyam, M. D., Hidalgo, M., Maitra, A., & Pollack, J. R. (2012). Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America, 109(5), E252-259. https://doi.org/10.1073/pnas.1114817109
Animireddy, S., Kavadipula, P., Kotapalli, V., Gowrishankar, S., Rao, S., & Bashyam, M. D. (2021). Aberrant cytoplasmic localization of ARID1B activates ERK signaling and promotes oncogenesis. J Cell Sci, 134(4), jcs251637. https://doi.org/10.1242/jcs.251637
Wang, Z., Chen, K., Jia, Y., Chuang, J.-C., Sun, X., Lin, Y.-H., Celen, C., Li, L., Huang, F., Liu, X., Castrillon, D. H., Wang, T., & Zhu, H. (2020). Dual ARID1A/ARID1B loss leads to rapid carcinogenesis and disruptive redistribution of BAF complexes. Nature Cancer, 1(9), 909–922. https://doi.org/10.1038/s43018-020-00109-0
Helming, K. C., Wang, X., Wilson, B. G., Vazquez, F., Haswell, J. R., Manchester, H. E., Kim, Y., Kryukov, G. V., Ghandi, M., Aguirre, A. J., Jagani, Z., Wang, Z., Garraway, L. A., Hahn, W. C., & Roberts, C. W. (2014). ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nature Medicine, 20(3), 251–254. https://doi.org/10.1038/nm.3480
Porter, E. G., & Dykhuizen, E. C. (2017). Individual bromodomains of polybromo-1 contribute to chromatin association and tumor suppression in clear cell renal carcinoma. Journal of Biological Chemistry, 292(7), 2601–2610. https://doi.org/10.1074/jbc.M116.746875
Espana-Agusti, J., Warren, A., Chew, S. K., Adams, D. J., & Matakidou, A. (2017). Loss of PBRM1 rescues VHL dependent replication stress to promote renal carcinogenesis. Nature Communications, 8(1), 2026. https://doi.org/10.1038/s41467-017-02245-1
Nargund, A. M., Pham, C. G., Dong, Y., Wang, P. I., Osmangeyoglu, H. U., Xie, Y., Aras, O., Han, S., Oyama, T., Takeda, S., Ray, C. E., Dong, Z., Berge, M., Hakimi, A. A., Monette, S., Lekaye, C. L., Koutcher, J. A., Leslie, C. S., Creighton, C. J., … Hsieh, J. J. (2017). The SWI/SNF protein PBRM1 restrains VHL-loss-driven clear cell renal cell carcinoma. Cell Reports, 18(12), 2893–2906. https://doi.org/10.1016/j.celrep.2017.02.074
Slaughter, M. J., Shanle, E. K., McFadden, A. W., Hollis, E. S., Suttle, L. E., Strahl, B. D., & Davis, I. J. (2018). PBRM1 bromodomains variably influence nucleosome interactions and cellular function. Journal of Biological Chemistry, 293(35), 13592–13603. https://doi.org/10.1074/jbc.RA118.003381
Li, M., Zhao, H., Zhang, X., Wood, L. D., Anders, R. A., Choti, M. A., Pawlik, T. M., Daniel, H. D., Kannangai, R., Offerhaus, G. J., Velculescu, V. E., Wang, L., Zhou, S., Vogelstein, B., Hruban, R. H., Papadopoulos, N., Cai, J., Torbenson, M. S., & Kinzler, K. W. (2011). Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nature Genetics, 43(9), 828–829. https://doi.org/10.1038/ng.903
Moreno, T., Monterde, B., González-Silva, L., Betancor-Fernández, I., Revilla, C., Agraz-Doblas, A., Freire, J., Isidro, P., Quevedo, L., Blanco, R., Montes-Moreno, S., Cereceda, L., Astudillo, A., Casar, B., Crespo, P., Morales Torres, C., Scaffidi, P., Gómez-Román, J., Salido, E., & Varela, I. (2021). ARID2 deficiency promotes tumor progression and is associated with higher sensitivity to chemotherapy in lung cancer. Oncogene, 40(16), 2923–2935. https://doi.org/10.1038/s41388-021-01748-y
Lawrence, M. S., Stojanov, P., Mermel, C. H., Robinson, J. T., Garraway, L. A., Golub, T. R., Meyerson, M., Gabriel, S. B., Lander, E. S., & Getz, G. (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature, 505(7484), 495–501. https://doi.org/10.1038/nature12912
Zhou, C., Zhang, Y., Yan, R., Huang, L., Mellor, A., Yang, Y., Chen, X., Wei, W., Wu, X., Yu, L., Liang, L., Zhang, D., Wu, S., & Wang, W. (2020). Exosome-derived miR-142–5p remodels lymphatic vessels and induces IDO to promote immune privilege in the tumour microenvironment. Cell death and differentiation, 28, 715–729. https://doi.org/10.1038/s41418-020-00618-6
Mashtalir, N., Suzuki, H., Farrell, D. P., Sankar, A., Luo, J., Filipovski, M., D’Avino, A. R., St Pierre, R., Valencia, A. M., Onikubo, T., Roeder, R. G., Han, Y., He, Y., Ranish, J. A., DiMaio, F., Walz, T., & Kadoch, C. (2020). A structural model of the endogenous human BAF complex informs disease mechanisms. Cell, 183(3), 802-817.e824. https://doi.org/10.1016/j.cell.2020.09.051
Cai, W., Su, L., Liao, L., Liu, Z. Z., Langbein, L., Dulaimi, E., Testa, J. R., Uzzo, R. G., Zhong, Z., Jiang, W., Yan, Q., Zhang, Q., & Yang, H. (2019). PBRM1 acts as a p53 lysine-acetylation reader to suppress renal tumor growth. Nature Communications, 10(1), 5800. https://doi.org/10.1038/s41467-019-13608-1
Prochasson, P., Neely, K. E., Hassan, A. H., Li, B., & Workman, J. L. (2003). Targeting activity is required for SWI/SNF function in vivo and is accomplished through two partially redundant activator-interaction domains. Molecular Cell, 12(4), 983–990. https://doi.org/10.1016/s1097-2765(03)00366-6
Woodley, C. M., Romer, A. S., Wang, J., Guarnaccia, A. D., Elion, D. L., Maxwell, J. N., Guerrazzi, K., McCann, T. S., Popay, T. M., Matlock, B. K., Flaherty, D. K., Lorey, S. L., Liu, Q., Tansey, W. P., & Weissmiller, A. M. (2021). Multiple interactions of the oncoprotein transcription factor MYC with the SWI/SNF chromatin remodeler. Oncogene, 40(20), 3593–3609. https://doi.org/10.1038/s41388-021-01804-7
Srikanth, S., Ramachandran, S., & Mohan, S. S. (2020). Construction of the gene regulatory network identifies MYC as a transcriptional regulator of SWI/SNF complex. Scientific Reports, 10(1), 158. https://doi.org/10.1038/s41598-019-56844-7
Dunaief, J. L., Strober, B. E., Guha, S., Khavari, P. A., Alin, K., Luban, J., Begemann, M., Crabtree, G. R., & Goff, S. P. (1994). The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell, 79(1), 119–130. https://doi.org/10.1016/0092-8674(94)90405-7
Trouche, D., Le Chalony, C., Muchardt, C., Yaniv, M., & Kouzarides, T. (1997). RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci U S A, 94(21), 11268–11273. https://doi.org/10.1073/pnas.94.21.11268
Zhang, H. S., Gavin, M., Dahiya, A., Postigo, A. A., Ma, D., Luo, R. X., Harbour, J. W., & Dean, D. C. (2000). Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell, 101(1), 79–89. https://doi.org/10.1016/s0092-8674(00)80625-x
Lee, D., Kim, J. W., Seo, T., Hwang, S. G., Choi, E.-J., & Choe, J. (2002). SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription*. Journal of Biological Chemistry, 277(25), 22330–22337. https://doi.org/10.1074/jbc.M111987200
Bochar, D. A., Wang, L., Beniya, H., Kinev, A., Xue, Y., Lane, W. S., Wang, W., Kashanchi, F., & Shiekhattar, R. (2000). BRCA1 is associated with a human SWI/SNF-related complex: Linking chromatin remodeling to breast cancer. Cell, 102(2), 257–265. https://doi.org/10.1016/s0092-8674(00)00030-1
Oh, J., Sohn, D. H., Ko, M., Chung, H., Jeon, S. H., & Seong, R. H. (2008). BAF60a interacts with p53 to recruit the SWI/SNF complex*. Journal of Biological Chemistry, 283(18), 11924–11934. https://doi.org/10.1074/jbc.M705401200
Valencia, A. M., Collings, C. K., Dao, H. T., St Pierre, R., Cheng, Y. C., Huang, J., Sun, Z. Y., Seo, H. S., Mashtalir, N., Comstock, D. E., Bolonduro, O., Vangos, N. E., Yeoh, Z. C., Dornon, M. K., Hermawan, C., Barrett, L., Dhe-Paganon, S., Woolf, C. J., Muir, T. W., & Kadoch, C. (2019). Recurrent SMARCB1 mutations reveal a nucleosome acidic patch interaction site that potentiates mSWI/SNF complex chromatin remodeling. Cell, 179(6), 1342-1356.e1323. https://doi.org/10.1016/j.cell.2019.10.044
McBride, M. J., Mashtalir, N., Winter, E. B., Dao, H. T., Filipovski, M., D’Avino, A. R., Seo, H.-S., Umbreit, N. T., St. Pierre, R., Valencia, A. M., Qian, K., Zullow, H. J., Jaffe, J. D., Dhe-Paganon, S., Muir, T. W., & Kadoch, C. (2020). The nucleosome acidic patch and H2A ubiquitination underlie mSWI/SNF recruitment in synovial sarcoma. Nature Structural & Molecular Biology, 27(9), 836–845. https://doi.org/10.1038/s41594-020-0466-9
Dao, H. T., & Pham, L. T. D. (2022). Acidic patch histone mutations and their effects on nucleosome remodeling. Biochemical Society Transactions, 50(2), 907–919. https://doi.org/10.1042/bst20210773
Guillon, N., Tirode, F., Boeva, V., Zynovyev, A., Barillot, E., & Delattre, O. (2009). The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS One, 4(3), e4932. https://doi.org/10.1371/journal.pone.0004932
Tomlins, S. A., Laxman, B., Dhanasekaran, S. M., Helgeson, B. E., Cao, X., Morris, D. S., Menon, A., Jing, X., Cao, Q., Han, B., Yu, J., Wang, L., Montie, J. E., Rubin, M. A., Pienta, K. J., Roulston, D., Shah, R. B., Varambally, S., Mehra, R., & Chinnaiyan, A. M. (2007). Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature, 448(7153), 595–599. https://doi.org/10.1038/nature06024
Tomlins, S. A., Mehra, R., Rhodes, D. R., Smith, L. R., Roulston, D., Helgeson, B. E., Cao, X., Wei, J. T., Rubin, M. A., Shah, R. B., & Chinnaiyan, A. M. (2006). TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Research, 66(7), 3396–3400. https://doi.org/10.1158/0008-5472.Can-06-0168
Sandoval, G. J., Pulice, J. L., Pakula, H., Schenone, M., Takeda, D. Y., Pop, M., Boulay, G., Williamson, K. E., McBride, M. J., Pan, J., St Pierre, R., Hartman, E., Garraway, L. A., Carr, S. A., Rivera, M. N., Li, Z., Ronco, L., Hahn, W. C., & Kadoch, C. (2018). Binding of TMPRSS2-ERG to BAF chromatin remodeling complexes mediates prostate oncogenesis. Molecular Cell, 71(4), 554-566.e557. https://doi.org/10.1016/j.molcel.2018.06.040
Zhang, D., Zhao, S., Li, X., Kirk, J. S., & Tang, D. G. (2018). Prostate luminal progenitor cells in development and cancer. Trends in cancer, 4(11), 769–783. https://doi.org/10.1016/j.trecan.2018.09.003
Zullow, H. J., Sankar, A., Ingram, D. R., Samé Guerra, D. D., D’Avino, A. R., Collings, C. K., Lazcano, R., Wang, W.-L., Liang, Y., Qi, J., Lazar, A. J., & Kadoch, C. (2022). The FUS::DDIT3 fusion oncoprotein inhibits BAF complex targeting and activity in myxoid liposarcoma. Molecular Cell, 82(9), 1737-1750.e8. https://doi.org/10.1016/j.molcel.2022.03.019
Conyers, R., Young, S., & Thomas, D. M. (2011). Liposarcoma: molecular genetics and therapeutics. Sarcoma, 2011, 483154. https://doi.org/10.1155/2011/483154
Jain, S., Xu, R., Prieto, V. G., & Lee, P. (2010). Molecular classification of soft tissue sarcomas and its clinical applications. International Journal of Clinical and Experimental Pathology, 3(4), 416–429. https://www.scopus.com/inward/record.uri?eid=2-s2.0-77955880551&partnerID=40&md5=15b403479578d7635be30eb1993e03a2.
Hoffman, G. R., Rahal, R., Buxton, F., Xiang, K., McAllister, G., Frias, E., Bagdasarian, L., Huber, J., Lindeman, A., Chen, D., Romero, R., Ramadan, N., Phadke, T., Haas, K., Jaskelioff, M., Wilson, B. G., Meyer, M. J., Saenz-Vash, V., Zhai, H., … Jagani, Z. (2014). Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proceedings of the National Academy of Sciences of the United States of America, 111(8), 3128–3133. https://doi.org/10.1073/pnas.1316793111
Oike, T., Ogiwara, H., Tominaga, Y., Ito, K., Ando, O., Tsuta, K., Mizukami, T., Shimada, Y., Isomura, H., Komachi, M., Furuta, K., Watanabe, S., Nakano, T., Yokota, J., & Kohno, T. (2013). A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Research, 73(17), 5508–5518. https://doi.org/10.1158/0008-5472.Can-12-4593
Winter, G. E., Buckley, D. L., Paulk, J., Roberts, J. M., Souza, A., Dhe-Paganon, S., & Bradner, J. E. (2015). DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science, 348(6241), 1376–1381. https://doi.org/10.1126/science.aab1433
Zengerle, M., Chan, K. H., & Ciulli, A. (2015). Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chemical Biology, 10(8), 1770–1777. https://doi.org/10.1021/acschembio.5b00216
Lu, J., Qian, Y., Altieri, M., Dong, H., Wang, J., Raina, K., Hines, J., Winkler, J. D., Crew, A. P., Coleman, K., & Crews, C. M. (2015). Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chemistry & Biology, 22(6), 755–763. https://doi.org/10.1016/j.chembiol.2015.05.009
Farnaby, W., Koegl, M., Roy, M. J., Whitworth, C., Diers, E., Trainor, N., Zollman, D., Steurer, S., Karolyi-Oezguer, J., Riedmueller, C., Gmaschitz, T., Wachter, J., Dank, C., Galant, M., Sharps, B., Rumpel, K., Traxler, E., Gerstberger, T., Schnitzer, R., … Ciulli, A. (2019). BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nature Chemical Biology, 15(7), 672–680. https://doi.org/10.1038/s41589-019-0294-6
Kofink, C., Trainor, N., Mair, B., Wöhrle, S., Wurm, M., Mischerikow, N., Roy, M. J., Bader, G., Greb, P., Garavel, G., Diers, E., McLennan, R., Whitworth, C., Vetma, V., Rumpel, K., Scharnweber, M., Fuchs, J. E., Gerstberger, T., Cui, Y., … Farnaby, W. (2022). A selective and orally bioavailable VHL-recruiting PROTAC achieves SMARCA2 degradation in vivo. Nature Communications, 13(1), 5969. https://doi.org/10.1038/s41467-022-33430-6
Gerstenberger, B. S., Trzupek, J. D., Tallant, C., Fedorov, O., Filippakopoulos, P., Brennan, P. E., Fedele, V., Martin, S., Picaud, S., Rogers, C., Parikh, M., Taylor, A., Samas, B., O’Mahony, A., Berg, E., Pallares, G., Torrey, A. D., Treiber, D. K., Samardjiev, I. J., … Owen, D. R. (2016). Identification of a chemical probe for family VIII bromodomains through optimization of a fragment hit. Journal of Medicinal Chemistry, 59(10), 4800–4811. https://doi.org/10.1021/acs.jmedchem.6b00012
Papillon, J. P. N., Nakajima, K., Adair, C. D., Hempel, J., Jouk, A. O., Karki, R. G., Mathieu, S., Möbitz, H., Ntaganda, R., Smith, T., Visser, M., Hill, S. E., Hurtado, F. K., Chenail, G., Bhang, H.-E.C., Bric, A., Xiang, K., Bushold, G., Gilbert, T., … Jagani, Z. (2018). Discovery of orally active inhibitors of Brahma homolog (BRM)/SMARCA2 ATPase activity for the treatment of Brahma related gene 1 (BRG1)/SMARCA4-mutant cancers. Journal of Medicinal Chemistry, 61(22), 10155–10172. https://doi.org/10.1021/acs.jmedchem.8b01318
Vangamudi, B., Paul, T. A., Shah, P. K., Kost-Alimova, M., Nottebaum, L., Shi, X., Zhan, Y., Leo, E., Mahadeshwar, H. S., Protopopov, A., Futreal, A., Tieu, T. N., Peoples, M., Heffernan, T. P., Marszalek, J. R., Toniatti, C., Petrocchi, A., Verhelle, D., Owen, D. R., … Andersen, J. N. (2015). The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: Insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Research, 75(18), 3865–3878. https://doi.org/10.1158/0008-5472.Can-14-3798
Schick, S., Rendeiro, A. F., Runggatscher, K., Ringler, A., Boidol, B., Hinkel, M., Májek, P., Vulliard, L., Penz, T., Parapatics, K., Schmidl, C., Menche, J., Boehmelt, G., Petronczki, M., Müller, A. C., Bock, C., & Kubicek, S. (2019). Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nature Genetics, 51(9), 1399–1410. https://doi.org/10.1038/s41588-019-0477-9
Kim, K. H., Kim, W., Howard, T. P., Vazquez, F., Tsherniak, A., Wu, J. N., Wang, W., Haswell, J. R., Walensky, L. D., Hahn, W. C., Orkin, S. H., & Roberts, C. W. (2015). SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nature Medicine, 21(12), 1491–1496. https://doi.org/10.1038/nm.3968
Kurmasheva, R. T., Sammons, M., Favours, E., Wu, J., Kurmashev, D., Cosmopoulos, K., Keilhack, H., Klaus, C. R., Houghton, P. J., & Smith, M. A. (2017). Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer, 64(3), 10.1002/pbc.26218. https://doi.org/10.1002/pbc.26218
Knutson, S. K., Warholic, N. M., Wigle, T. J., Klaus, C. R., Allain, C. J., Raimondi, A., Porter Scott, M., Chesworth, R., Moyer, M. P., Copeland, R. A., Richon, V. M., Pollock, R. M., Kuntz, K. W., & Keilhack, H. (2013). Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proceedings of the National Academy of Sciences of the United States of America, 110(19), 7922–7927. https://doi.org/10.1073/pnas.1303800110
Maruyama, D., Tobinai, K., Makita, S., Ishida, T., Kusumoto, S., Ishitsuka, K., Yoshimitsu, M., Imaizumi, Y., Sawayama, Y., Takeuchi, S., Utsunomiya, A., Tsukasaki, K., Fujitani, S., & Araki, K. (2017). First-in-human study of the EZH1/2 dual inhibitor DS-3201b in patients with relapsed or refractory non-Hodgkin lymphomas — preliminary results. Blood, 130, 4070. https://doi.org/10.1182/blood.V130.Suppl_1.4070.4070
Dong, H., Liu, S., Zhang, X., Chen, S., Kang, L., Chen, Y., Ma, S., Fu, X., Liu, Y., Zhang, H., & Zou, B. (2019). An allosteric PRC2 inhibitor targeting EED suppresses tumor progression by modulating the immune response. Cancer Research, 79(21), 5587–5596. https://doi.org/10.1158/0008-5472.Can-19-0428
Bitler, B. G., Aird, K. M., Garipov, A., Li, H., Amatangelo, M., Kossenkov, A. V., Schultz, D. C., Liu, Q., Shih, I.-M., Conejo-Garcia, J. R., Speicher, D. W., & Zhang, R. (2015). Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nature Medicine, 21(3), 231–238. https://doi.org/10.1038/nm.3799
Wu, S., Fatkhutdinov, N., Fukumoto, T., Bitler, B. G., Park, P. H., Kossenkov, A. V., Trizzino, M., Tang, H.-Y., Zhang, L., Gardini, A., Speicher, D. W., & Zhang, R. (2018). SWI/SNF catalytic subunits’ switch drives resistance to EZH2 inhibitors in ARID1A-mutated cells. Nature Communications, 9(1), 4116. https://doi.org/10.1038/s41467-018-06656-6
Tse, C., Shoemaker, A. R., Adickes, J., Anderson, M. G., Chen, J., Jin, S., Johnson, E. F., Marsh, K. C., Mitten, M. J., Nimmer, P., Roberts, L., Tahir, S. K., Xiao, Y., Yang, X., Zhang, H., Fesik, S., Rosenberg, S. H., & Elmore, S. W. (2008). ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Research, 68(9), 3421–3428. https://doi.org/10.1158/0008-5472.Can-07-5836
Fukumoto, T., Park, P. H., Wu, S., Fatkhutdinov, N., Karakashev, S., Nacarelli, T., Kossenkov, A. V., Speicher, D. W., Jean, S., Zhang, L., Wang, T. L., Shih, I. M., Conejo-Garcia, J. R., Bitler, B. G., & Zhang, R. (2018). Repurposing pan-HDAC inhibitors for ARID1A-mutated ovarian cancer. Cell Reports, 22(13), 3393–3400. https://doi.org/10.1016/j.celrep.2018.03.019
Altucci, L. (2017). A key HDAC6 dependency of ARID1A-mutated ovarian cancer. Nature Cell Biology, 19(8), 889–890. https://doi.org/10.1038/ncb3588
Xue, Y., Meehan, B., Fu, Z., Wang, X. Q. D., Fiset, P. O., Rieker, R., Levins, C., Kong, T., Zhu, X., Morin, G., Skerritt, L., Herpel, E., Venneti, S., Martinez, D., Judkins, A. R., Jung, S., Camilleri-Broet, S., Gonzalez, A. V., Guiot, M. C., … Huang, S. (2019). SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nature Communications, 10(1), 557. https://doi.org/10.1038/s41467-019-08380-1
Chandler, R. L., Damrauer, J. S., Raab, J. R., Schisler, J. C., Wilkerson, M. D., Didion, J. P., Starmer, J., Serber, D., Yee, D., Xiong, J., Darr, D. B., Pardo-Manuel de Villena, F., Kim, W. Y., & Magnuson, T. (2015). Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nature Communications, 6, 6118. https://doi.org/10.1038/ncomms7118
Samartzis, E. P., Gutsche, K., Dedes, K. J., Fink, D., Stucki, M., & Imesch, P. (2014). Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget, 5(14), 5295–5303. https://doi.org/10.18632/oncotarget.2092
Shen, J., Ju, Z., Zhao, W., Wang, L., Peng, Y., Ge, Z., Nagel, Z. D., Zou, J., Wang, C., Kapoor, P., Ma, X., Ma, D., Liang, J., Song, S., Liu, J., Samson, L. D., Ajani, J. A., Li, G.-M., Liang, H., … Peng, G. (2018). ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nature Medicine, 24(5), 556–562. https://doi.org/10.1038/s41591-018-0012-z
Shen, J., Peng, Y., Wei, L., Zhang, W., Yang, L., Lan, L., Kapoor, P., Ju, Z., Mo, Q., Shih, I.-M., Uray, I. P., Wu, X., Brown, P. H., Shen, X., Mills, G. B., & Peng, G. (2015). ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discovery, 5(7), 752–767. https://doi.org/10.1158/2159-8290.CD-14-0849
Park, Y., Chui, M. H., Suryo Rahmanto, Y., Yu, Z.-C., Shamanna, R. A., Bellani, M. A., Gaillard, S., Ayhan, A., Viswanathan, A., Seidman, M. M., Franco, S., Leung, A. K. L., Bohr, V. A., Shih, I.-M., & Wang, T.-L. (2019). Loss of ARID1A in tumor cells renders selective vulnerability to combined ionizing radiation and PARP inhibitor therapy. Clinical Cancer Research, 25(18), 5584–5594. https://doi.org/10.1158/1078-0432.Ccr-18-4222
Gupta, M., Concepcion, C. P., Fahey, C. G., Keshishian, H., Bhutkar, A., Brainson, C. F., Sanchez-Rivera, F. J., Pessina, P., Kim, J. Y., Simoneau, A., Paschini, M., Beytagh, M. C., Stanclift, C. R., Schenone, M., Mani, D. R., Li, C., Oh, A., Li, F., Hu, H., … Kim, C. F. (2020). BRG1 loss predisposes lung cancers to replicative stress and ATR dependency. Cancer Research, 80(18), 3841–3854. https://doi.org/10.1158/0008-5472.Can-20-1744
Chabanon, R. M., Morel, D., Eychenne, T., Colmet-Daage, L., Bajrami, I., Dorvault, N., Garrido, M., Meisenberg, C., Lamb, A., Ngo, C., Hopkins, S. R., Roumeliotis, T. I., Jouny, S., Hénon, C., Kawai-Kawachi, A., Astier, C., Konde, A., Del Nery, E., Massard, C., … Postel-Vinay, S. (2021). PBRM1 deficiency confers synthetic lethality to DNA repair inhibitors in cancer. Cancer Research, 81(11), 2888–2902. https://doi.org/10.1158/0008-5472.Can-21-0628
Guo, T., Li, B., & Xu, C. (2019). PARP inhibition shows anticancer activity in small cell carcinoma of the ovary hypercalcemic type (SCCOHT) [40E]. Obstetrics & Gynecology, 133, 62S. https://journals.lww.com/greenjournal/Fulltext/2019/05001/PARP_Inhibition_Shows_Anticancer_Activity_in_Small.213.aspx.
Pan, D., Kobayashi, A., Jiang, P., Ferrari de Andrade, L., Tay, R. E., Luoma, A. M., Tsoucas, D., Qiu, X., Lim, K., Rao, P., Long, H. W., Yuan, G.-C., Doench, J., Brown, M., Liu, X. S., & Wucherpfennig, K. W. (2018). A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science, 359(6377), 770–775. https://doi.org/10.1126/science.aao1710
McDermott, D. F., Huseni, M. A., Atkins, M. B., Motzer, R. J., Rini, B. I., Escudier, B., Fong, L., Joseph, R. W., Pal, S. K., Reeves, J. A., Sznol, M., Hainsworth, J., Rathmell, W. K., Stadler, W. M., Hutson, T., Gore, M. E., Ravaud, A., Bracarda, S., Suárez, C., … Powles, T. (2018). Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nature Medicine, 24(6), 749–757. https://doi.org/10.1038/s41591-018-0053-3
Buglioni, S., Melucci, E., Sperati, F., Pallocca, M., Terrenato, I., De Nicola, F., Goeman, F., Casini, B., Amoreo, C. A., Gallo, E., Diodoro, M. G., Pescarmona, E., Vici, P., Sergi, D., Pizzuti, L., Di Lauro, L., Mazzotta, M., Barba, M., Fanciulli, M., … Maugeri-Saccà, M. (2018). The clinical significance of PD-L1 in advanced gastric cancer is dependent on ARID1A mutations and ATM expression. Oncoimmunology, 7(8), e1457602. https://doi.org/10.1080/2162402x.2018.1457602
Ogiwara, H., Takahashi, K., Sasaki, M., Kuroda, T., Yoshida, H., Watanabe, R., Maruyama, A., Makinoshima, H., Chiwaki, F., Sasaki, H., Kato, T., Okamoto, A., & Kohno, T. (2019). Targeting the vulnerability of glutathione metabolism in ARID1A-deficient cancers. Cancer Cell, 35(2), 177-190.e178. https://doi.org/10.1016/j.ccell.2018.12.009
Sasaki, M., Chiwaki, F., Kuroda, T., Komatsu, M., Matsusaki, K., Kohno, T., Sasaki, H., & Ogiwara, H. (2020). Efficacy of glutathione inhibitors for the treatment of ARID1A-deficient diffuse-type gastric cancers. Biochemical and Biophysical Research Communications, 522(2), 342–347. https://doi.org/10.1016/j.bbrc.2019.11.078
Carugo, A., Minelli, R., Sapio, L., Soeung, M., Carbone, F., Robinson, F. S., Tepper, J., Chen, Z., Lovisa, S., Svelto, M., Amin, S., Srinivasan, S., Del Poggetto, E., Loponte, S., Puca, F., Dey, P., Malouf, G. G., Su, X., Li, L., … Genovese, G. (2019). p53 is a master regulator of proteostasis in SMARCB1-deficient malignant rhabdoid tumors. Cancer Cell, 35(2), 204-220.e209. https://doi.org/10.1016/j.ccell.2019.01.006
Lissanu Deribe, Y., Sun, Y., Terranova, C., Khan, F., Martinez-Ledesma, J., Gay, J., Gao, G., Mullinax, R. A., Khor, T., Feng, N., Lin, Y. H., Wu, C. C., Reyes, C., Peng, Q., Robinson, F., Inoue, A., Kochat, V., Liu, C. G., Asara, J. M., … Futreal, P. A. (2018). Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nature Medicine, 24(7), 1047–1057. https://doi.org/10.1038/s41591-018-0019-5
Gao, W., Li, W., Xiao, T., Liu, X. S., & Kaelin, W. G., Jr. (2017). Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL−/− clear cell renal carcinoma. Proceedings of the National Academy of Sciences of the United States of America, 114(5), 1027–1032. https://doi.org/10.1073/pnas.1619726114
Neely, K. E., Hassan, A. H., Wallberg, A. E., Steger, D. J., Cairns, B. R., Wright, A. P. H., & Workman, J. L. (1999). Activation domain–mediated targeting of the SWI/SNF complex to promoters stimulates transcription from nucleosome arrays. Molecular Cell, 4(4), 649–655. https://doi.org/10.1016/S1097-2765(00)80216-6
Pfister, N. T., Fomin, V., Regunath, K., Zhou, J. Y., Zhou, W., Silwal-Pandit, L., Freed-Pastor, W. A., Laptenko, O., Neo, S. P., Bargonetti, J., Hoque, M., Tian, B., Gunaratne, J., Engebraaten, O., Manley, J. L., Børresen-Dale, A.-L., Neilsen, P. M., & Prives, C. (2015). Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes & development, 29(12), 1298–1315. https://doi.org/10.1101/gad.263202.115
Lindén, M., Thomsen, C., Grundevik, P., Jonasson, E., Andersson, D., Runnberg, R., Dolatabadi, S., Vannas, C., Luna Santamarίa, M., Fagman, H., Ståhlberg, A., & Åman, P. (2019). FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex. EMBO Reports, 20(5), 6. https://doi.org/10.15252/embr.201845766
Kelso, T. W. R., Porter, D. K., Amaral, M. L., Shokhirev, M. N., Benner, C., & Hargreaves, D. C. (2017). Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife, 6, e30506. https://doi.org/10.7554/eLife.30506
Simeone, N., Frezza, A. M., Zaffaroni, N., & Stacchiotti, S. (2021). Tazemetostat for advanced epithelioid sarcoma: Current status and future perspectives. Future Oncology, 17(10), 1253–1263. https://doi.org/10.2217/fon-2020-0781
Muscat, A., Popovski, D., Jayasekara, W. S. N., Rossello, F. J., Ferguson, M., Marini, K. D., Alamgeer, M., Algar, E. M., Downie, P., Watkins, D. N., Cain, J. E., & Ashley, D. M. (2016). Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of malignant rhabdoid tumors. Clinical Cancer Research, 22(14), 3560–3570. https://doi.org/10.1158/1078-0432.Ccr-15-2260
Tagal, V., Wei, S., Zhang, W., Brekken, R. A., Posner, B. A., Peyton, M., Girard, L., Hwang, T., Wheeler, D. A., Minna, J. D., White, M. A., Gazdar, A. F., & Roth, M. G. (2017). SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nature Communications, 8, 14098. https://doi.org/10.1038/ncomms14098
Brownlee, P. M., Meisenberg, C., & Downs, J. A. (2015). The SWI/SNF chromatin remodelling complex: Its role in maintaining genome stability and preventing tumourigenesis. DNA Repair, 32, 127–133. https://doi.org/10.1016/j.dnarep.2015.04.023
Miao, D., Margolis, C. A., Gao, W., Voss, M. H., Li, W., Martini, D. J., Norton, C., Bossé, D., Wankowicz, S. M., Cullen, D., Horak, C., Wind-Rotolo, M., Tracy, A., Giannakis, M., Hodi, F. S., Drake, C. G., Ball, M. W., Allaf, M. E., Snyder, A., … Van Allen, E. M. (2018). Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science, 359(6377), 801–806. https://doi.org/10.1126/science.aan5951
Gorrini, C., & Mak, T. W. (2019). Glutathione metabolism: An Achilles’ heel of ARID1A-deficient tumors. Cancer Cell, 35(2), 161–163. https://doi.org/10.1016/j.ccell.2019.01.017
Funding
This work was supported by funding from the National Institute of General Medical Sciences (grant no. R35GM118068) and the Stowers Institute for Medical Research to JLW.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reddy, D., Bhattacharya, S. & Workman, J.L. (mis)-Targeting of SWI/SNF complex(es) in cancer. Cancer Metastasis Rev 42, 455–470 (2023). https://doi.org/10.1007/s10555-023-10102-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-023-10102-5