
Abstract
Metastatic disease results from the dissemination of tumor cells beyond their organ of origin to grow in distant organs and is the primary cause of death in patients with advanced breast cancer. Preclinical murine models in which primary tumors spontaneously metastasize are valuable tools for studying metastatic progression and novel cancer treatment combinations. Here, we characterize a novel syngeneic murine breast tumor cell line that provides a model of spontaneously metastatic neu-expressing breast cancer with quicker onset of widespread metastases after orthotopic mammary implantation in immune-competent NeuN mice. The NT2.5-lung metastasis (-LM) cell line was derived from serial passaging of tumor cells that were macro-dissected from spontaneous lung metastases after orthotopic mammary implantation of parental NT2.5 cells. Within one week of NT2.5-LM implantation, metastases are observed in the lungs. Within four weeks, metastases are also observed in the bones, spleen, colon, and liver. We demonstrate that NT2.5-LM metastases are positive for NeuN—the murine equivalent of human epidermal growth factor 2 (HER2). We further demonstrate altered expression of markers of epithelial-to-mesenchymal transition (EMT), suggestive of their enhanced metastatic potential. Genomic analyses support these findings and reveal enrichment in EMT-regulating pathways. In addition, the metastases are rapidly growing, proliferative, and responsive to HER2-directed therapy. The new NT2.5-LM model provides certain advantages over the parental NT2/NT2.5 model, given its more rapid and spontaneous development of metastases. Besides investigating mechanisms of metastatic progression, this new model may be used for the rationalized development of novel therapeutic interventions and assessment of therapeutic responses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer remains one of the leading causes of cancer mortality among women worldwide, with metastatic burden as the major contributor of patient death [1, 2]. The development of murine models of breast cancer has provided researchers with the means to more intricately study tumor initiation, progression, metastasis, and response to therapies, leading to our current understanding of the complex physiological systems and molecular mechanisms underlying these processes [3, 4]. Various transgenic models of breast cancer that develop spontaneous mammary tumors and metastases exist [5,6,7,8,9,10]. However, only few of these models allow for efficient study of the metastatic tumor microenvironment (TME). Syngeneic models of breast cancer, which involve orthotopic implantation of tumor cells or tumor chunks, are widely utilized, but often times, these models are either slow-growing or do not develop clinically overt metastases. Experimental metastasis models, which involve tail vein injection of tumor cells, are also widely utilized, but these models are limited by lack of resolution in metastatic progression, and conclusions drawn from these models may be artificial. As such, development of appropriate mouse models of breast carcinoma that recapitulate metastatic progression in a pathophysiological and clinically relevant context is necessary.
The immunotolerant MMTV-HER2/Neu (ERBB2) transgenic murine model (NeuN) originally characterized by Guy et al. [9], in which FVB/N strain mice express the non-transforming rat Neu cDNA under control by a mammary tissue-specific promoter, gives rise to spontaneous mammary tumors between 125 and 300 days. This model yields spontaneously developing mammary tumors that closely mimic human epidermal growth factor 2-positive (HER2+) tumors [11]. One caveat of this model is its long latency for development of both primary and metastatic disease, as well as the lack of penetrance of metastatic disease. To circumvent these issues, previous efforts have focused on its improvement and have led to the development of a syngeneic tumor cell line derivative, known as NT2.5. The latter model has significantly shortened the time from tumor cell injection to tumor growth and is capable of establishing widespread distant metastases upon cardiac or tail vein injections [12]. Metastases in various organs can be observed within 3 weeks of NT2.5 tumor cell injection, but this model is also limited by its inability to recapitulate the process of spontaneous metastasis.
In this study, we report the serial passaging of the original NT2.5 cell line to generate a new subline called NT2.5-LM, which represents an orthotopic, immunotolerant model of HER2+ breast cancer capable of promoting development of spontaneous metastases. We also perform an in-depth characterization of the newly established NT2.5-LM cell line at both the genomic and proteomic levels to establish the foundations for its potential use in preclinical studies.
Methods
Cell lines
NT2.5-lung metastasis (-LM) cell line was derived from the parental NT2.5 cell line, which was originally derived from the NT2 cell line established by Guy et al.[9] 1 × 105 NT2.5 cells were injected intravenously by tail vein in five 8-week-old female NeuN mice. Three weeks after tail vein injection, lung metastases were macro-dissected from all mice, minced on ice, filtered using a 100 µm filter, and pooled. The pooled cells were used to repeat the process described above, starting with intravenous injection, and after the third round of lung metastasis harvest, pooled cells were injected into the mammary fat pad of five 8-week-old female NeuN mice for spontaneous lung metastasis formation. After confirmation of spontaneous lung metastasis formation by lung harvest and Hematoxylin and Eosin (H&E) stains, the cell line was propagated in cell culture and named NT2.5-LM. NT2.5 cells were derived from spontaneous mammary tumors growing in female NeuN mice and obtained from Jaffee Lab at Johns Hopkins University [14,15,, 15]. Culture conditions for NT2.5-LM and NT2.5 cells are as follows: 37 °C, 5% CO2 in RPMI 1640 (Gibco, cat. 11875-093) supplemented with 20% fetal bovine serum (Gemini, cat. 100-106), 1.2% HEPES (Gibco, cat. 15630-080), 1% L-glutamine (Gibco, cat. 25030-081), 1% MEM non-essential amino acids (Gibco, cat. 11140-050), 0.5% penicillin/streptomycin (Gibco, cat. 15140-122), 1% sodium pyruvate (Sigma, cat. S8636), 0.2% insulin (NovoLog, cat. U-100). Cell lines are tested for mycoplasma every 6 months.
Mice
A syngeneic mouse model of HER2+ breast cancer using the NT2.5 cell line was derived from the NeuN transgenic mouse developed by Guy et al. [9] NeuN transgenic mice overexpress non-transforming rat neu cDNA under the control of a mammary specific promoter and develop spontaneous focal mammary adenocarcinomas after a long latency of 125 days with the majority of mice developing tumors by 300 days. Injection of NT2.5 into NeuN mice leads to development of tumors 100% of the time, since mice are tolerized to Neu. Mice were kept in pathogen-free conditions and were treated in accordance with institutional and American Association of Laboratory Animal Committee policies. NeuN mice were originally from W. Muller McMaster University, Hamilton, Ontario, Canada and overexpress HER2 via the mouse mammary tumor virus (MMTV) promoter. Colonies are renewed yearly from Jackson labs and bred in-house by brother/sister mating.
Survival, tumor growth, metastasis growth, necropsy
1 × 105 NT2.5 or NT2.5-LM cells were injected into the mammary fat pad. NT2.5-LM tumors were resected on day 12. Survival endpoint was determined to be mammary tumor volume exceeding 1500 mm3 (at time of tumor regrowth after surgical resection in NT2.5-LM) or morbidity symptoms due to lung metastatic tumor burden, such as breathing, coat condition, activity, and posture. Mammary tumor growth was measured by calipers (± 0.01 mm) three times a week, with weekly tumor growth determined by calculating the average of differences in tumor volumes per week for each mouse. Lung surface metastases were counted by visual inspection of collected lungs following euthanasia at survival endpoint and before fixation in formalin and paraffin-embedding. Lung sections were taken 40 µm apart, for a representative 3 sections per lung. H&E stained sections were scanned and analyzed using either HALO or NDPView.2 to quantify number and tumor area of lung metastases. For necropsy, various tissues were collected at survival endpoint, fixed in formalin, paraffin-embedded, sectioned, stained with H&E, and visualized by light microscopy. Necropsy tissues include heart, lymph nodes, lungs, kidney, adrenal gland, stomach, colon, spleen, skull, ear, body wall, and teeth.
Immunohistochemistry
Immunohistochemistry staining was performed at the Oncology Tissue Services Core of Johns Hopkins University. Immunolabeling for ErbB2, Ki67, CK5, CK6, AE1/3 and EGFR was performed on formalin‐fixed, paraffin-embedded sections. Briefly, following dewaxing and rehydration, slides were immersed in 1% Tween-20, then heat‐induced antigen retrieval was performed in a steamer using Antigen Unmasking Solution (catalog# H-3300, Vector Labs) for 25 min. Slides were rinsed in PBST, endogenous peroxidase and phosphatase were blocked (Dako, cat. S2003), and then incubated with the following primary antibodies for 45 min at room temperature: anti‐ErbB2 (1:400 dilution; ThermoFisher Scientific, cat. MA5-15050, SF23975824), anti‐Ki67 (1:200 dilution; Abcam, cat. Ab16667), anti-EGFR (1:50 dilution; LSBio, cat. LS-B2914-50), anti-CK5 (1:2000 dilution; BioLegend, cat. 905501), anti-CK6 (1:200 dilution; Novus Biologicals, cat. NBP2-34358), anti-AE-1/AE-3 (1:200 dilution; Novus Biologicals, cat. NBP2-29429). Slides were then incubated with HRP-conjugated anti-rabbit secondary antibody (Leica Microsystems, cat. PB6119) for 30 min at room temperature. Signal detection was conducted with 3,3′‐Diaminobenzidine (Sigma-Aldrich, cat. D4293). Counterstaining was conducted with Mayer’s hematoxylin.
Anti-HER2 treatment of mice
1 × 105 NT2.5-LM cells were injected into the mammary fat pad. Mammary tumors were resected on day 12, after which mice were treated with anti-HER2 antibody starting on day 23 to mimic standard therapy treatment with trastuzumab in patients with HER2+ breast cancer. Anti-HER2 monoclonal antibody (BioXCell, clone 7.16.4) and mouse IgG2a isotype vehicle antibody (BioXCell, clone C1.18.4) were dosed at 100 µg/mouse by intraperitoneal (i.p.) injection once a week for three weeks as described [16]. Following three weeks of treatment, either lung tissues were collected for tumor burden analysis, or maintenance dosing was continued once a week until survival endpoint. For tumor burden analysis, three different levels were taken from formalin-fixed and paraffin-embedded lungs sectioned 100 µm apart. Slides were H&E stained, scanned, and analyzed using HALO to obtain summed lung metastasis counts and percent tumor area.
Tumor dissociation
Following collection, mammary tumors were minced on ice and dissociated using a tumor dissociation kit (Miltenyi Biotec, cat. 130-096-730) and the 37C_m_TDK_2 program on the OctoDissociator (Miltenyi Biotec) per the manufacturer’s instructions. Cell suspensions were filtered using 70 µm cell strainers and red blood cells were lysed using ACK lysis buffer (Quality Biological, cat. 118-156-721). To submit for RNA sequencing, dead cells were removed using the MACS Dead Cell Removal Kit (Miltenyi Biotec).
Flow cytometry
NT2.5 and NT2.5-LM cells were cultured for at least two passages, washed with PBS, and stained with Live/Dead Fixable Aqua (ThermoFisher, cat. L10119) for 30 min at 4 °C, per the manufacturer’s instructions. Cells were fixed and permeabilized for 30 min at room temperature using the Foxp3 / Transcription Factor Staining Buffer Set (Life Technologies Corp., cat. 00-5523-00), followed by an Fc receptor block (BD Pharmingen, cat. 553142) for 10 min at room temperature. Cells were incubated with the following primary antibodies for 30 min at room temperature: anti-Vimentin (1:100 dilution; Cell Signaling Technology, cat. 5741). Cells were then incubated with FITC-conjugated anti-rabbit secondary antibody (1 µg/mL; BioLegend, cat. 406403) for 30 min at room temperature. Samples were run on the Attune NxT flow cytometer (Invitrogen) and analyzed using Kaluza software.
MenaINV immunofluorescence and image analysis
Immunofluorescence staining for MenaINV was performed on formalin‐fixed, paraffin-embedded (FFPE) sections. Briefly, slides were deparaffinized by melting for 5 min at 58 °C in an oven equipped with a fan, followed by two Xylene treatments for 20 min each. Slides were rehydrated and antigen retrieval was performed in 1 mM EDTA, pH 8.0 for 20 min at 97 °C in a conventional steamer. Slides were washed with 0.05% PBST and incubated in blocking solution (5% goat serum in 0.05% PBST) for 1 h at room temperature. Slides were then incubated with anti-MenaINV primary antibody (0.25 ug/mL; in-house developed in the lab of Dr. John S. Condeelis, AE1071, AP-4) overnight at 4 °C. After three washes in 0.05% PBST, slides were incubated with Alexa 488-conjugated goat anti-chicken secondary antibody at room temperature for 1 h. After three washes in 0.05% PBST, slides were incubated with spectral DAPI for 5 min and mounted with ProLong Gold Antifade Mountant (Life Technologies, cat. P36930). Slides were imaged using the Pannoramic 250 Flash II digital whole slide scanner. Up to 10 High-Power Field (HPF) images per mouse, depending on tumor and metastasis burden availability, were captured in TIFF format using Caseviewer v2.4 (3DHISTECH). Further image processing was performed in ImageJ. Single MenaINV channels were uploaded, converted to 8-bit, and binarized using intensity thresholding (default method). The DAPI channel confirmed that all HPFs chosen were within necrosis-free areas of the tumors and metastases. The MenaINV+ area in each HPF was then expressed as a fraction of the total tumor area, and the mean of all HPFs was calculated for each mouse. For visualization purposes only, images were enhanced in Caseviewer by exclusively using linear image modifications (i.e., brightness and contrast), and the signal was pseudo-colored for optimal representation of fields of interest.
Whole exome sequencing (WES)
NT2.5 and NT2.5-LM cell lines were cultured as described above and sent for whole exome sequencing at the Johns Hopkins Genomics Core. One microgram or more of mouse genomic DNA from each sample was analyzed by whole exome sequencing using the SureSelectXT Mouse All Exon kit (Agilent), followed by next generation sequencing using the NovaSeq 6000 S4 flow cell (Illumina) with a 2 × 150 bp paired-end read configuration, per the manufacturer’s instructions. bcl2fastq v2.15.0 (Illumina) was used to convert BCL files to FASTQ files using default parameters. Running alignments against the mm10 genome was done by bwa v0.7.7 (mem) along with Piccard-tools1.119 to add read groups and remove duplicate reads. GATK v3.6.0 base call recalibration steps were used to create a final alignment file. MuTect2 v3.6.0 was used to call somatic variants against a panel of normal using default parameters. snpEFF (v4.1) was used to annotate the variant calls and to create a clean tab separated table of variants. IGV v2.13.2 was used to identify breast cancer specific mutations from MuTect2 files. SnapGene Viewer v.6.2 was used to visually align and determine the mutations between the two cell lines against the mRNA sequences of selected genes. Annotations were created to visualize mutational differences.
Single cell RNA sequencing (scRNA-seq)
For library preparation, 10 × Genomics Chromium Single Cell 3′ RNA-seq kits v3 were used. Gene expression libraries were prepared per the manufacturer’s instructions. 4 biological replicates totaling 8 processed tumors were sequenced in 2 batches: Run A—2 NT2.5 tumors, 2 NT2.5-LM tumors; Run B—2 NT2.5 tumors, 2 NT2.5-LM tumors. These tumors were taken as a subset from a larger batch of tumors that include various mouse treatments, with each batch having an equal assortment of samples from multiple treatment groups to reduce technical biases. Here, we restrict our analysis to replicates under the vehicle treatment condition. Illumina HiSeqX Ten or NovaSeq were used to generate total reads. Paired-end reads were processed using CellRanger v3.0.2 and mapped to the mm10 transcriptome with default settings. ScanPy v1.8.1 and Python v3 was used for quality control and basic filtering. For gene filtering, all genes expressed in less than 3 cells within a tumor (NT2.5 and NT2.5-LM) were removed. Cells expressing less than 200 genes or more than 8,000 genes or having more than 15% mitochondrial gene expression were also removed. Gene expression was total-count normalized to 10,000 reads per cell and log transformed. Highly variable genes were identified using default ScanPy parameters, and the total counts per cell and the percent mitochondrial genes expressed were regressed out. Finally, gene expression was scaled to unit variance and values exceeding 10 standard deviations were removed. Neighborhood graphs were constructed using 10 nearest neighbors and 30 principal components. Tumors were clustered together within cell lines using Louvain clustering (with resolution parameter 0.09) and cancer cells were identified as Lcn + , Wfd2c + , Cd24a + , Cd276 + , Col9a1 + , Erbb2 + [17,18,19,20,21,22,22]. All other cell clusters were removed. There were ~ 15,000 NT2.5 cancer cells and ~ 13,000 NT2.5-LM cancer cells. The 250 top differentially expressed genes in the cancer clusters from each cell line were identified using the Wilcoxon rank-sum test and compared for overlap with pathways from the ‘KEGG_2019_Mouse’ database using GSEAPY (Gene Set Enrichment Analysis in Python).
Statistics
For survival curves, Mantel-Cox log rank tests were used. For tumor growth rate, metastasis counts, and lung metastasis volumes, Mann Whitney tests were used. For quantification of immunohistochemistry staining, Welch’s T-tests were used. For flow cytometry, unpaired t-tests were used. For immunofluorescence staining of tumor and metastatic tissues, Mann Whitney U-tests were used. To aid in statistical choice, data were tested for normality using D’Agostino-Pearson omnibus normality tests, Anderson–Darling tests, Shapiro–Wilk normality tests, and Kolmogorov–Smirnov normality tests.
Results
Orthotopic implantation of NT2.5-LM leads to decreased survival, larger mammary tumors, and increased lung metastasis
In the NT2.5 syngeneic model, NT2.5 cells are implanted in the mammary fat pad of adult female NeuN mice, after which the maximum allowable volume of 1.5 cm3 is reached in 4–5 weeks [13, 16, 22], prior to the establishment of metastatic disease and preventing efficient study of metastatic tumor microenvironments (TMEs). To derive a highly metastatic cell line, lung metastases were macro-dissected from the lungs of NT2.5 mammary tumor-bearing NeuN mice, dissociated to single-cell suspensions, and intravenously injected into non-tumor-bearing NeuN mice, after which lung metastases could be harvested again and the process repeated. After the third round of harvest, spontaneous lung metastases could be observed 3 weeks following mammary fat pad injection of isolated cells, thus establishing the NT2.5-lung metastasis (-LM) cell line for use.
To characterize the phenotype of NT2.5-LM-derived tumors in vivo, we orthotopically injected NT2.5-LM cells into NeuN mice and measured survival, tumor burden, and metastatic burden. When compared to parental NT2.5 controls, mice orthotopically injected with NT2.5-LM cells experienced significantly decreased survival (Fig. 1a) and increased weekly mammary tumor growth rates (Fig. 1b). Despite surgical resection of NT2.5-LM mammary tumors at 12 days post-injection, tumors regrew at 24 days post-injection and reached endpoint criteria faster than NT2.5 mammary tumors (Fig. S1a–b). Necropsy analyses of mice with NT2.5-LM mammary tumors revealed widespread metastases in the heart, lymph nodes, lungs, kidneys, adrenal glands, stomach, colon, spleen, skull, ears, body walls, and teeth (Fig. S2), with high metastatic burden observed in the lungs. Due to the high metastatic burden, we focused on the lungs as a surrogate measure of total metastatic burden moving forward. When examining lungs of mice euthanized from 34 to 41 days post-injection, we found a significant increase in the number of lung metastases in the NT2.5-LM model, when compared to the NT2.5 control (Fig. 1c). NT2.5-LM lung micro-metastases could be observed by H&E staining as early as 7 days post-injection, with consistent growth observed at 10, 22, 28, and 35 days post-injection (Fig. 1d).

NT2.5-LM leads to decreased survival, larger mammary tumors, and increased lung metastasis. a 1 × 105 NT2.5 or NT2.5-LM cells were injected into a mammary fat pad of NeuN mice (NT2.5, n = 10; NT2.5-LM, n = 7). After surgical resection of NT2.5-LM tumor-bearing mice at 12 days post-injection (dpi), mice were allowed to reach human survival endpoint with tumor volume exceeding 1500 mm3. b Mammary tumor sizes of mice in (a) were measured at least 3 × a week by calipers, averaged, and used to calculate differences in average weekly tumor growth rate. c At survival endpoint of mice in (a), the number of surface metastases was counted by visual inspection. d H&E staining of lungs in NT2.5-LM tumor-bearing mice collected at 7, 10, 22, 28, and 35 days post-injection (dpi). Black arrows point to lung metastases. Scale bars as shown. e Immunohistochemistry (IHC) staining of Erbb2 and f Ki67 in NT2.5 mammary tumors (top) and NT2.5-LM lung metastases (bottom) collected at 35 days post-injection. Scale bars as shown. g Percentage of Ki67 + cells from 10 regions of interest (ROIs) were counted from Ki67 IHC staining in (F). Statistics used: Mantel-Cox Log-rank test for (a), Mann–Whitney U-test for (b-d), Welch’s T-test for (g), *p < 0.05, **p < 0.01, ****p < 0.0001
To further illuminate on the phenotypic characteristics of NT2.5-LM metastases, we performed immunohistochemical staining for ERBB2, Ki67, CK5, CK6, AE1/3, and EGFR. NT2.5-LM lung metastases are ERBB2-positive (Fig. 1e), express similarly low levels of AE1/3 and EGFR, and are similarly negative for CK5 and CK6, when compared to NT2.5 mammary tumors (Fig. S3). Finally, NT2.5-LM lung metastases are more proliferative, as observed by increased numbers of Ki67 + cells (Fig. 1f–g).
NT2.5-LM does not exhibit altered mutational landscape compared to parental NT2.5
With the increased number of lung metastases in NT2.5-LM model, we hypothesized that there might be differences in the genomic landscape and pathogenic mutational burden between the NT2.5 and NT2.5-LM tumors. First, we performed whole exome sequencing on the NT2.5 and NT2.5-LM cell lines to identify potential variations in genes with known pathogenic mutations and in genes known to affect proliferation and metastasis. Many pathogenic gene mutations common to breast cancer [23], such as Pten, Brca2, Atm, Cdh1, Chek2, Nf1, Arid1a, Pik3ca, and Esr1, revealed no alterations between NT2.5 and NT2.5-LM (Fig. 2a). Of note, NT2.5-LM contained mutations in Brca1 and NT2.5 contained mutations in Rad51c, but both were found within intron regions, thus not affecting protein sequence. Since NT2.5-LM is a HER2+ cell line, we examined the Erbb2 transcript sequence across both cell lines more thoroughly and found six mutations within the protein coding sequence. However, all six mutations were silent (Fig. 2b). Lastly, we assessed tumor mutational burden, given that it represents another factor that could affect response to therapy. We found 11.45 mutations per megabase in the NT2.5 and 13.45 mutations per megabase in the NT2.5-LM models, with similar distributions of high missense mutations, single nucleotide polymorphisms (SNPs), and tyrosine-to-cytosine and cytosine-to-tyrosine mutations (Fig. 2c–d). Collectively, these data suggest that phenotypic differences between the NT2.5 and NT2.5-LM models are not the result of diversified mutational burden in NT2.5-LM.

NT2.5-LM does not exhibit altered mutational landscape compared to parental NT2.5. a Alignment of NT2.5 and NT2.5-LM whole exome sequencing reads to the mm10 genome reveal cell line-specific and –overlapping mutations common in breast cancer. b Erbb2 transcript sequence with identified mutation sites in NT2.5 and NT2.5-LM. All mutations were identified to be silent mutations. Nucleotide numbering is based on DNA reference sequence NM_001003817.1. Note that the version number of this reference sequence may be frequently updated. c Distributions of mutation classifications, variant types, single nucleotide variant (SNV) classes, and top 10 mutated genes for NT2.5 and (d) NT2.5-LM are shown
NT2.5-LM exhibits altered signaling indicative of epithelial-to-mesenchymal transition (EMT)
Given the non-significant alterations in mutational burden, we sought to explain the differences in pro-metastatic phenotypes by comparing gene expression profiles between NT2.5 and NT2.5-LM. Four NT2.5 tumors and four NT2.5-LM tumors were collected from NeuN mice and subjected to unsorted single-cell RNA sequencing (scRNAseq), yielding approximately 9.6 × 108 total reads. From Louvain clustering, approximately 15,000 NT2.5 and 13,000 NT2.5-LM cancer cells were identified as Lcn + , Wfd2c + , Cd24a + , Cd276 + , Col9a1 + , Erbb2 + , [17,18,19,20,21,22,22] subsetted out, and visualized by Uniform Manifold Approximation and Projection (UMAP) (Fig. 3a). An analysis of the top 25 differentially expressed genes between the two cancer cell clusters revealed an upregulation of genes associated with increased cellular proliferation (Pdgfa, Sox9) [25,26,, 26], invasion and migration (Lrp1, Cd9, Cxcl1, Anxa1, Areg, Ifitm3), [27,28,29,30,31,32,33,33] epithelial-to-mesenchymal transition (EMT) (Vim, Inhba], [34, 35] and stemness and metastatic potential (S100A4, Nrp2, Aldh2, JunB) [36,37,38,39,40,41,41] in NT2.5-LM. Concurrently, there was a downregulation of genes associated with decreased cellular proliferation (Crip1), [42] decreased invasion (Cldn7), [43, 44] and decreased epithelial phenotype and polarization (Epcam) [45] in NT2.5-LM (Fig. 3b–c). We validated the increased gene expression of Vim and decreased gene expression of Epcam in NT2.5-LM at the protein level by flow cytometry, demonstrating a significant increase in the percentage of Vimentin-positive cells and significant decrease in the percentage of Epcam-positive cells. (Fig. 3d–e).

NT2.5-LM exhibits altered signaling indicative of increased EMT. a Four NT2.5 and four NT2.5-LM mammary tumors were collected from NeuN mice, dissociated to single cell suspensions, and sent for unsorted single-cell RNA sequencing. Cancer cell clusters were annotated as Lcn + , Wfd2c + , Cd24a + , Cd276 + , Col9a1 + , Erbb2 + , and subsetted out for UMAP visualization. b Top 25 significantly up- and down-regulated genes in NT2.5-LM. c Violin plots of key metastasis-related genes identified in (b). d Flow cytometry staining of epithelial-to-mesenchymal transition (EMT) gene identified in (c) in NT2.5 and NT2.5-LM cell lines for Vimentin and e Epcam. Statistics used: Unpaired T-test for (d–e), ****p < 0.0001
Further investigation into differential pathway regulation was performed by comparing the top 250 differentially expressed genes for overlap with pathways from the ‘KEGG_2019_Mouse’ database using Gene Set Enrichment Analysis. The most significantly up- and down-regulated pathway in NT2.5-LM was the ribosome pathway, suggestive of distinct ribosomal profiles between cell lines. NT2.5-LM exhibited significant upregulation of the glycolysis pathway and downregulation of oxidative phosphorylation, ECM-receptor interaction, focal adhesion, protein digestion and absorption, and adherens junction pathways (p-adj < 0.05) (Fig. S4, Table S1). Dissolution of adherens junctions and alterations in cell–cell interactions is a hallmark of EMT [46, 47], and these data offer increased EMT as an explanation for the increased metastatic phenotype of NT2.5-LM.
Proof-of-utility: NT2.5-LM responds to HER2 directed therapy
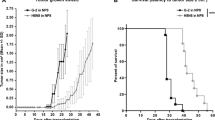
Patients with HER2+ breast cancer demonstrate a response rate of over 35% when treated with HER2-directed monoclonal antibody therapy [48]. To demonstrate how this model could be used to study novel HER2 directed therapies and to characterize the sensitivity of the NT2.5-LM model, NT2.5-LM metastasis-bearing mice were treated with anti-HER2 antibody by intraperitoneal (i.p.) injection once a week and assessed for survival (Fig. S5). Anti-HER2-treated mice showed improved survival when compared to isotype-treated mice, with a ~ 35% response rate to therapy (Fig. 4a), similar to that observed in patients treated with single agent therapy [48]. When assessing the anti-HER2 treatment effects on lung metastases, we found that treatment did not change the number of lung metastases (Fig. 4b), but it significantly decreased the surface area of metastases within the lung (Fig. 4c). Together, these data suggest that the new NT2.5-LM model demonstrates clinical relevance with regards to its therapeutic response to anti-HER treatments.

NT2.5-LM responds to HER2-directed therapy. a 1 × 105 NT2.5-LM cells were injected into a mammary fat pad of NeuN mice. After surgical resection of NT2.5-LM tumor-bearing mice at 12 days post-injection (dpi), treatment with isotype or anti-HER2 monoclonal antibody (100 µg/mouse, 1x/week, intraperitoneal injection) began at 23 dpi (n = 12 per treatment group) and continued until survival endpoint at 70 dpi. b 1 × 105 NT2.5-LM cells were injected into a mammary fat pad of NeuN mice, tumors were surgically resected at 12 dpi, and anti-HER2 treatment (100 µg/mouse, 1x/week, intraperitoneal injection) began at 23 dpi (n = 10 per treatment group). Lungs were collected at 38 dpi. Three different levels were taken from formalin-fixed and paraffin-embedded lungs sectioned 100 µm apart. Slides were H&E stained, scanned, and analyzed using HALO to obtain summed lung metastasis counts and c percent tumor area over normal lung tissue. Two mice in the vehicle group were removed due to inconsistencies between HALO results and physical examination of H&E slides. Statistics used: Mantel-Cox Log-rank test for (a), Mann–Whitney U-test for (b–c), ns = not-significant, **p < 0.01
Proof-of-utility: NT2.5-LM expresses increased levels of MenaINV—a marker of metastatic potential
Another proof-of-utility of the NT2.5-LM model is to investigate mechanisms of enhanced invasive potential with translatable outcomes. Our group has performed extensive work on mechanisms of metastatic dissemination and has previously reported that pro-migratory/pro-invasive tumor cells primed for the metastatic journey tend to upregulate the expression of MenaINV, a spliced isoform of the actin-regulatory protein mammalian enabled (Mena) that conveys increased metastatic potential. Specifically, previous studies have collectively shown that MenaINV is correlated with increased breast cancer cell migration, invasion, and metastasis [49,50,51,52,53,53], and is significantly upregulated in response to cytotoxic treatments in patients [54]. In view of observed alterations in various ECM and cell–cell adhesion interaction pathways, (Fig. S4, Table S1), we expected an enrichment of MenaINV-positive tumor cells in NT2.5-LM metastatic tumors. Indeed, immunofluorescence analysis of MenaINV revealed significantly increased expression in the metastatic NT2.5-LM tumors, when compared to the NT2.5 mammary tumors (Fig. 5a–b). The enhanced expression of MenaINV in the NT2.5-LM model enables use of this model for future studies investigating mechanisms of enhanced metastatic potential.

NT2.5-LM expresses increased levels of MenaINV—a marker of metastatic potential. a Representative immunofluorescence images of MenaINV (red) and DAPI (blue) staining in NT2.5 mammary tumor (top), and NT2.5-LM lung metastases (bottom) collected 34–41 days post-injection (dpi). Middle column and right column panels correspond to dotted square in left column panels. Scale bars as shown. b Quantification of MenaINV staining from NT2.5 mammary tumor (n = 6) and NT2.5-LM lung metastases (n = 6) by averaging signal intensity from up to 10 regions of interest (ROIs) in each sample. Statistics used: Mann–Whitney U-test for (b), **p < 0.01
Discussion
Spontaneously metastatic breast cancer cell lines are valuable tools for studying how metastatic tumors differ from primary tissue tumors in mice, but the time for spontaneous lung metastases to develop after injection of cancer cells into the breast tissue site is prolonged and inconsistent. In this study, we generated a more aggressively metastatic breast cancer cell line, NT2.5-LM, that spontaneously metastasizes to distant organs as early as one week post-injection. This not only allows us to study the effects of treatment interventions on metastatic progression in the most biologically accurate setting, but also utilizes surgical removal of the primary tumor early on to ensure that we are not limited by humane endpoints of primary tumor growth.
NT2.5-LM exhibited poorer survival, faster primary tumor growth, and more widespread metastases. Because the NT2.5-LM cell line was derived from NT2.5, we sought to understand the differences that would cause it to be more widely metastatic and proliferative compared to the parent cell line. We hypothesized that increased expression of HER2 or a novel mutation in the ErbB2 gene could be driving increased proliferation. NT2.5-LM did not exhibit new pathogenic mutations in ErbB2, and increased expression of HER2 was not observed by immunohistochemistry. Furthermore, pathways analyses conducted on scRNAseq data demonstrated no significant difference in expression of genes within the ErbB pathway. Thus, change in HER2 signaling is not a likely mechanism driving the increased metastatic and proliferative phenotype observed in NT2.5-LM.
Other potential mechanisms driving observed differences in NT2.5-LM include the differential regulation of proliferation- and metastasis-promoting pathways. We observed a shift in metabolic pathways with an upregulation of glycolysis and a downregulation of oxidative phosphorylation KEGG pathways, which have been previously implicated in more metastatic cancers [55, 56], supporting our observations that NT2.5-LM is more widely metastatic. We observed a downregulation of ECM receptor interaction, focal junction, and adheres junction pathways, which are interactors in the intravasation and extravasation processes of metastasis [57]. We also identified differential expression of key genes involved in EMT that favored a more mesenchymal phenotype in NT2.5-LM, which could explain the increased number of metastases in lung and other distant organs. Given our finding that the ribosome pathway was both significantly up- and down-regulated in NT2.5-LM, it is likely that different sets of ribosome genes may be correlated with other respectively up- and down-regulated pathways, prompting further examinations. Our observed alterations in expression of epithelial markers, mesenchymal markers, cell adhesion and extracellular matrix pathways, and metabolic pathways are characteristic of EMT [58, 59].
One interesting alteration associated with the loss of epithelial cell–cell contacts is the increased expression of invasive actin regulatory protein isoform MenaINV [60]. MenaINV-expressing breast cancer cells participate in a paracrine loop with intratumoral macrophages, which facilitates their translocation to the perivascular niche. Once they reach the vasculature, MenaINV-expressing tumor cells associate with perivascular macrophages to intravasate into the blood vessel. These tripartite microanatomical structures composed of endothelial cells, perivascular macrophages, and MenaINV-expressing tumor cells are key prerequisites of metastatic dissemination and have been previously called Tumor Microenvironment of Metastasis (TMEM) doorways [51,52,52, 54, 61, 62]. Of note, NT2.5-LM tumors exhibit increased expression of MenaINV, which could explain its highly metastatic nature. As such, this model may be efficiently used in the future to study mechanisms of breast cancer cell dissemination associated with TMEM doorways and MenaINV-dependent pathways.
In summary, our findings distinguish NT2.5-LM as a more proliferative and metastatic model of breast cancer for experimental use that also preserves the spontaneous metastatic process within a shorter timeline. Various genetic and epigenetic changes can occur in a cancer cell as it accumulates mutations, proceeds through EMT, interacts with the TME, and forms distant metastases. Our group and others have shown that the addition of epigenetic modulators to various therapies in multiple cancer models has decreased tumor growth and improved response [16, 22, 63, 64]. Moving forward, we envision the use of this NT2.5-LM model to facilitate efficient future studies of novel treatment combinations for metastatic disease and evaluation of different metastatic TME contributions to therapeutic response.
Data availability
All WES and scRNAseq raw and processed data files are available on GEO NCBI under accession numbers GSE261252 and GSE261253.
References
Sung H et al (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71(3):209–249. https://doi.org/10.3322/CAAC.21660
Riggio AI, Varley KE, Welm AL (2020) The lingering mysteries of metastatic recurrence in breast cancer. Br J Cancer 124(1):13–26. https://doi.org/10.1038/s41416-020-01161-4
Park MK, Lee CH, Lee H (2018) Mouse models of breast cancer in preclinical research. Lab Anim Res 34(4):160. https://doi.org/10.5625/LAR.2018.34.4.160
Kim IS, Baek SH (2010) Mouse models for breast cancer metastasis. Biochem Biophys Res Commun 394(3):443–447. https://doi.org/10.1016/J.BBRC.2010.03.070
Macleod KF, Jacks T (1999) Insights into cancer from transgenic mouse models. J Pathol 187:43–60. https://doi.org/10.1002/(SICI)1096-9896(199901)187:1
Green JE et al (2000) The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene 19(8):1020–1027. https://doi.org/10.1038/SJ.ONC.1203280
Siegel PM, Shu W, Cardiff RD, Muller WJ, Massagué J (2003) Transforming growth factor β signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A 100(14):8430. https://doi.org/10.1073/PNAS.0932636100
Lin SCJ et al (2004) Somatic mutation of p53 leads to estrogen receptor alpha-positive and -negative mouse mammary tumors with high frequency of metastasis. Cancer Res 64(10):3525–3532. https://doi.org/10.1158/0008-5472.CAN-03-3524
Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ (1992) Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci 89(22):10578–10582. https://doi.org/10.1073/PNAS.89.22.10578
Guy CT, Cardiff RD, Muller WJ (1992) Induction of mammary tumors by expression of polyomavirus middle T oncogene a transgenic mouse model for metastatic disease. Mol Cell Biol 12(3):954. https://doi.org/10.1128/MCB.12.3.954
Fry EA, Taneja P, Inoue K (2017) Oncogenic and tumor-suppressive mouse models for breast cancer engaging HER2/neu. Int J Cancer 140(3):495–503. https://doi.org/10.1002/IJC.30399
Song H et al (2008) An immunotolerant HER-2/neu transgenic mouse model of metastatic breast cancer. Clin Cancer Res 14(19):6116. https://doi.org/10.1158/1078-0432.CCR-07-4672
Reilly RT et al (2000) HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res 60(13):3569–3576
Machiels JP et al (2001) Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res 61(9):3689–3697
Jaffee EM et al (1998) Development and characterization of a cytokine-secreting pancreatic adenocarcinoma vaccine from primary tumors for use in clinical trials. Cancer J Sci Am 4(3):194–203
Christmas BJ et al (2018) Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res 6(12):1561–1577. https://doi.org/10.1158/2326-6066.CIR-18-0070
Gündüz UR, Gunaldi M, Isiksacan N, Gündüz S, Okuturlar Y, Kocoglu H (2016) A new marker for breast cancer diagnosis, human epididymis protein 4: A preliminary study. Mol Clin Oncol 5(2):355. https://doi.org/10.3892/MCO.2016.919
Seaman S et al (2017) Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3 Positive Tumor Cells and Tumor Vasculature. Cancer Cell 31(4):501. https://doi.org/10.1016/J.CCELL.2017.03.005
Yang J et al (2009) Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci USA 106(10):3913–3918. https://doi.org/10.1073/PNAS.0810617106/SUPPL_FILE/0810617106SI.PDF
Berger T, Cheung CC, Elia AJ, Mak TW (2010) Disruption of the Lcn2 gene in mice suppresses primary mammary tumor formation but does not decrease lung metastasis. Proc Natl Acad Sci USA 107(7):2995–3000. https://doi.org/10.1073/PNAS.1000101107/SUPPL_FILE/PNAS.201000101SI.PDF
Yeo SK et al (2020) Single-cell RNA-sequencing reveals distinct patterns of cell state heterogeneity in mouse models of breast cancer. Elife 9:1–24. https://doi.org/10.7554/ELIFE.58810
Sidiropoulos DN et al (2022) Entinostat decreases immune suppression to promote anti-tumor responses in a HER2+ breast tumor microenvironment. Cancer Immunol Res 10(5):565–669. https://doi.org/10.1158/2326-6066.CIR-21-0170
Gil Del Alcazar CR et al (2022) Insights into Immune Escape During Tumor Evolution and Response to Immunotherapy Using a Rat Model of Breast Cancer. Cancer Immunol Res 10(6):680. https://doi.org/10.1158/2326-6066.CIR-21-0804
Pinto MP, Dye WW, Jacobsen BM, Horwitz KB (2014) Malignant stroma increases luminal breast cancer cell proliferation and angiogenesis through platelet-derived growth factor signaling. BMC Cancer 14(1):735. https://doi.org/10.1186/1471-2407-14-735
Jansson S et al (2018) The PDGF pathway in breast cancer is linked to tumour aggressiveness, triple-negative subtype and early recurrence. Breast Cancer Res Treat 169(2):231. https://doi.org/10.1007/S10549-018-4664-7
Ma Y et al (2020) SOX9 Is essential for triple-negative breast cancer cell survival and metastasis. Mol Cancer Res 18(12):1825–1838. https://doi.org/10.1158/1541-7786.MCR-19-0311
Xing P et al (2016) Roles of low-density lipoprotein receptor-related protein 1 in tumors. Chin J Cancer 35(1):6. https://doi.org/10.1186/S40880-015-0064-0
Fayard B et al (2009) The serine protease inhibitor protease nexin-1 controls mammary cancer metastasis through LRP-1-mediated MMP-9 expression. Cancer Res 69(14):5690–5698. https://doi.org/10.1158/0008-5472.CAN-08-4573
Rappa G, Green TM, Karbanová J, Corbeil D, Lorico A (2015) Tetraspanin CD9 determines invasiveness and tumorigenicity of human breast cancer cells. Oncotarget 6(10):7970. https://doi.org/10.18632/ONCOTARGET.3419
Yang C et al (2019) CXCL1 stimulates migration and invasion in ER-negative breast cancer cells via activation of the ERK/MMP2/9 signaling axis. Int J Oncol 55(3):684–696. https://doi.org/10.3892/IJO.2019.4840
Moraes LA, Ampomah PB, Lim LHK (2018) Annexin A1 in inflammation and breast cancer: a new axis in the tumor microenvironment. Cell Adh Migr 12(5):417. https://doi.org/10.1080/19336918.2018.1486143
Baillo A, Giroux C, Ethier SP (2011) Knock-down of amphiregulin inhibits cellular invasion in inflammatory breast cancer. J Cell Physiol 226(10):2691–2701. https://doi.org/10.1002/JCP.22620
Yang M, Gao H, Chen P, Jia J, Wu S (2013) Knockdown of interferon-induced transmembrane protein 3 expression suppresses breast cancer cell growth and colony formation and affects the cell cycle. Oncol Rep 30(1):171–178. https://doi.org/10.3892/OR.2013.2428
Paulin D, Lilienbaum A, Kardjian S, Agbulut O, Li Z (2022) Vimentin: Regulation and pathogenesis. Biochimie 197:96–112. https://doi.org/10.1016/J.BIOCHI.2022.02.003
Yu Y, Wang W, Lu W, Chen W, Shang A (2021) Inhibin β-A (INHBA) induces epithelial–mesenchymal transition and accelerates the motility of breast cancer cells by activating the TGF-β signaling pathway. Bioengineered 12(1):4681. https://doi.org/10.1080/21655979.2021.1957754
Helfman DM, Kim EJ, Lukanidin E, Grigorian M (2005) The metastasis associated protein S100A4: role in tumour progression and metastasis. Br J Cancer 92(11):1955–1958. https://doi.org/10.1038/sj.bjc.6602613
Elaimy AL et al (2018) VEGF-neuropilin-2 signaling promotes stem-like traits in breast cancer cells by TAZ-mediated repression of the Rac GAP β2-chimaerin. Sci Signal 11(528):eaao6897. https://doi.org/10.1126/SCISIGNAL.AAO6897/SUPPL_FILE/AAO6897_SM.PDF
Yasuoka H et al (2009) Neuropilin-2 expression in breast cancer: correlation with lymph node metastasis, poor prognosis, and regulation of CXCR4 expression. BMC Cancer 9(1):220. https://doi.org/10.1186/1471-2407-9-220/FIGURES/4
Zhang H, Fu L (2021) The role of ALDH2 in tumorigenesis and tumor progression: Targeting ALDH2 as a potential cancer treatment. Acta Pharm Sin B 11(6):1400. https://doi.org/10.1016/J.APSB.2021.02.008
Sundqvist A et al (2018) JUNB governs a feed-forward network of TGFβ signaling that aggravates breast cancer invasion. Nucleic Acids Res 46(3):1180. https://doi.org/10.1093/NAR/GKX1190
Qiao Y et al (2015) AP-1-mediated chromatin looping regulates ZEB2 transcription: new insights into TNFα-induced epithelial-mesenchymal transition in triple-negative breast cancer. Oncotarget 6(10):7804–7814. https://doi.org/10.18632/ONCOTARGET.3158
Ludyga N et al (2013) The impact of cysteine-rich intestinal protein 1 (CRIP1) in human breast cancer. Mol Cancer 12(1):28. https://doi.org/10.1186/1476-4598-12-28
Kominsky SL et al (2003) Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 22(13):2021–2033. https://doi.org/10.1038/SJ.ONC.1206199
Martin TA, Jiang WG (2009) Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta (BBA)—Biomembranes 1788(4):872–891. https://doi.org/10.1016/J.BBAMEM.2008.11.005
Hyun K-A et al (2016) Epithelial-to-mesenchymal transition leads to loss of EpCAM and different physical properties in circulating tumor cells from metastatic breast cancer. Oncotarget 7(17):24677. https://doi.org/10.18632/ONCOTARGET.8250
Liu F, Gu LN, Shan BE, Geng CZ, Sang MX (2016) Biomarkers for EMT and MET in breast cancer: an update. Oncol Lett 12(6):4869. https://doi.org/10.3892/OL.2016.5369
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119(6):1420. https://doi.org/10.1172/JCI39104
Vogel CL et al (2002) Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 20(3):719–726. https://doi.org/10.1200/JCO.20.3.719
Roussos ET et al (2011) Mena invasive (MenaINV) and Mena11a isoforms play distinct roles in breast cancer cell cohesion and association with TMEM. Clin Exp Metastasis 28(6):515–527. https://doi.org/10.1007/S10585-011-9388-6/FIGURES/7
Philippar U et al (2008) A mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev Cell 15(6):813. https://doi.org/10.1016/J.DEVCEL.2008.09.003
Sharma VP et al (2021) Live tumor imaging shows macrophage induction and TMEM-mediated enrichment of cancer stem cells during metastatic dissemination. Nat Commun 12(1):1–24. https://doi.org/10.1038/s41467-021-27308-2
Borriello L et al (2022) Primary tumor associated macrophages activate programs of invasion and dormancy in disseminating tumor cells. Nat Commun 13(1):1–19. https://doi.org/10.1038/s41467-022-28076-3
Karagiannis GS, Goswami S, Jones JG, Oktay MH, Condeelis JS (2016) Signatures of breast cancer metastasis at a glance. J Cell Sci 129(9):1751–1758. https://doi.org/10.1242/JCS.183129/-/DC2
Karagiannis GS et al (2017) Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci Transl Med 9(397):1–15. https://doi.org/10.1126/SCITRANSLMED.AAN0026
Ashton TM, Gillies McKenna W, Kunz-Schughart LA, Higgins GS (2018) Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res 24(11):2482–2490. https://doi.org/10.1158/1078-0432.CCR-17-3070
Gaude E, Frezza C (2016) Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun 7:13041. https://doi.org/10.1038/NCOMMS13041
Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y (2020) Molecular principles of metastasis: a hallmark of cancer revisited. Sig Transduct Target Ther 5(1):1–17. https://doi.org/10.1038/s41392-020-0134-x
Pal AK et al (2022) Metabolomics and EMT markers of breast cancer a crosstalk and future perspective. Pathophysiology 29(2):200–222. https://doi.org/10.3390/PATHOPHYSIOLOGY29020017
Le Bras GF, Taubenslag KJ, Andl CD (2012) The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh Migr 6(4):365. https://doi.org/10.4161/CAM.21326
Goswami S et al (2009) Identification of invasion specific splice variants of the cytoskeletal protein Mena present in mammary tumor cells during invasion in vivo. Clin Exp Metastasis 26(2):153. https://doi.org/10.1007/S10585-008-9225-8
Roussos ET et al (2011) Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J Cell Sci 124(13):2120. https://doi.org/10.1242/JCS.086231
Robinson BD et al (2009) Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res 15(7):2433. https://doi.org/10.1158/1078-0432.CCR-08-2179
Orillion A et al (2017) Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res 23(17):5187–5201. https://doi.org/10.1158/1078-0432.CCR-17-0741
Kim K et al (2014) Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci USA 111(32):11774–11779. https://doi.org/10.1073/PNAS.1410626111/-/DCSUPPLEMENTAL
Acknowledgements
We would like to thank all members of the Elizabeth Jaffee and Elana Fertig lab for help throughout the course of these experiments. Additionally, we would like to thank the Molecular Genomics Core at USC, the Flow Cytometry Core at USC, the Translational Pathology Adult Tissue Core at USC, the SKCCC Experimental and Computational Genomics Core at Johns Hopkins, and the Oncology Tissue Services Core at Johns Hopkins for help with sequencing experiments, specimen processing, and data processing. We would like to thank the Analytical Imaging Facility at the Albert Einstein College of Medicine for immunofluorescence and tissue slide scanning, and the Integrated Imaging Program for Cancer Research IIPCR for discussion of data.
Funding
Open access funding provided by SCELC, Statewide California Electronic Library Consortium. This work was supported through funding from: Tower Cancer Research Foundation Career Development Award (ETRT); P30CA014089 from the National Cancer Institute (ETRT); NIH NCI P30 CA014089 (ETRT); MacMillan Pathway to Independence Fellowship (ETRT); Concern Foundation Conquer Cancer Now Award (ETRT); USC NCCC Core Voucher Program, NIH (NCI R01CA184926 for EMJ; P50CA062924 for EMJ, and LTK; NCI R01CA177669 for LTK); the Broccoli Foundation (EMJ and ETRT); The Bloomberg-Kimmel Institute for Cancer Immunotherapy; The Skip Viragh Center for Pancreas Cancer Clinical Research and Patient Care; The Commonwealth Foundation for Cancer Research (ETRT, LTK); The Allegheny Foundation (LTK); The Emerson Foundation (EMJ); Cancer Center Support Grant (P30CA013330 for GSK); Share Instrumentation Grant (1S10OD026852-01A1 for GSK); NIH-NCI K99/R00 Transition to Independence Award (R00CA237851 for GSK); The Integrated Imaging Program for Cancer Research IIPCR (GSK, ETRT); The Evelyn-Lipper Charitable Foundation (GSK); The Montefiore-Einstein Comprehensive Cancer Center (MECCC) start-up fund (GSK); The EGL Charitable Foundation (GSK, ETRT).
Author information
Authors and Affiliations
Contributions
ETRT, AGB, EG, VHN prepared manuscript. ETRT, AGB, EG, VHN, SC, CR, JJ, LTK, TA, JL conducted murine experimentation. AGB, VHN, CR performed pathology experiments. AGB and VHN performed flow cytometry experiments. JJ, SC, ETRT, LTK, IC performed scRNA-seq laboratory experiments and study design. AGB, JK, YL, AM analyzed scRNAseq data. VHN performed analysis of whole exome sequencing data. DPA and GSK performed immunofluorescence staining and analysis. EMJ provided funding for creation of original NT2.5-LM cell line and initial murine experimentation. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
EMJ is a paid consultant for Adaptive Biotech, CSTONE, Achilles, DragonFly, and Genocea. She receives funding from Lustgarten Foundation and Bristol Myer Squibb. She is the Chief Medical Advisor for Lustgarten and SAB advisor to the Parker Institute for Cancer Immunotherapy (PICI) and for the C3 Cancer Institute.
Ethical approval
All animal studies were approved by the Institutional Review Board of USC and Johns Hopkins University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baugh, A.G., Gonzalez, E., Narumi, V.H. et al. A new Neu—a syngeneic model of spontaneously metastatic HER2-positive breast cancer. Clin Exp Metastasis (2024). https://doi.org/10.1007/s10585-024-10289-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10585-024-10289-z