
Abstract
Skin coloration and patterning play a key role in animal survival and reproduction. As a result, color phenotypes have generated intense research interest. In aposematic species, color phenotypes can be important in avoiding predation and in mate choice. However, we still know little about the underlying genetic mechanisms of color production, particularly outside of a few model organisms. Here we seek to understand the genetic mechanisms underlying the production of different colors and how these undergo shifting expression patterns throughout development. To answer this, we examine gene expression of two different color patches(yellow and green) in a developmental time series from young tadpoles through adults in the poison frog Oophaga pumilio. We identified six genes that were differentially expressed between color patches in every developmental stage (casq1, hand2, myh8, prva, tbx3, and zic1). Of these, hand2, myh8, tbx3, and zic1 have either been identified or implicated as important in coloration in other taxa. Casq1 and prva buffer Ca2+ and are a Ca2+ transporter, respectively, and may play a role in preventing autotoxicity to pumiliotoxins, which inhibit Ca2+-ATPase activity. We identify further candidate genes (e.g., adh, aldh1a2, asip, lef1, mc1r, tyr, tyrp1, xdh), and identify a suite of hub genes that likely play a key role in integumental reorganization during development (e.g., collagen type I–IV genes, lysyl oxidases) which may also affect coloration via structural organization of chromatophores that contribute to color and pattern. Overall, we identify the putative role of a suite of candidate genes in the production of different color types in a polytypic, aposematic species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Animal coloration has long been a focus of research in genetics, animal behavior, and evolutionary biology because it is a highly variable trait that is often shaped by natural and sexual selection (Endler and Mappes 2017). Colors are frequently used in communication (e.g. as sexual ornaments; (Hill et al. 2006; Caro and Allen 2017), predator deterrence (e.g. in aposematic signals; Ruxton et al. 2004), thermal regulation (Trullas et al. 2007), and various other ecological functions. In some species, two or more distinct, genetically-determined phenotypes exist in different geographic localities (i.e. color polytypism; (Huxley 1955; Endler 1980). Such polytypic species have the potential to shed light on fundamental processes including local adaptation, the maintenance of genetic variation, and speciation (Gray and McKinnon 2007; Bolton et al. 2015). To understand the role of animal coloration in organismal biology, we need to examine not only the selective forces acting on color traits, but also their genetic and developmental underpinnings. Unfortunately, our understanding of the genetics responsible for the diversity in animal coloration in non-model organisms is still in its infancy (Shawkey and D’Alba 2017). However, this is rapidly changing with the advent of newer sequencing technologies and computational approaches that make elucidating underlying genetic mechanisms much more tractable in non-model organisms (Price-Waldman and Stoddard 2021; Funk et al. 2018).
The poison frogs (family Dendrobatidae) comprise a taxon with high inter- and intra-specific variation in coloration (Brown et al. 2011). Adult coloration in poison frogs is shaped by several different evolutionary forces, including natural selection, sexual selection, and stochastic processes. Natural selection has produced several genera that display bright aposematic coloration that is used to warn predators of the presence of toxic alkaloids in their skin (Darst et al. 2006; Saporito et al. 2007; Noonan and Comeault 2009; Stuckert et al. 2014a, b). Sexual selection has also contributed to shaping coloration and the development of color polymorphism in some poison frogs. For example in O. pumilio, a species with several color morphs, females show color-based mate preferences for the most common local color morph (Reynolds and Fitzpatrick 2007; Maan and Cummings 2008, 2012; Yusan Yang et al. 2016) and males are also more aggressive in their territorial interactions toward other males bearing that color (Yang et al. 2018, 2019). Genetic drift has also likely played a role in shaping the color diversity of poison frogs. For example, evidence indicates drift is a primary driver of divergent color morphs of O. pumilio seen on different islands (Tazzyman and Iwasa 2010; Gehara et al. 2013). Additionally, work examining selection based on color between morphs of Dendrobates tinctorius has also revealed a pattern of neutral evolution consistent with drift (Wollenberg et al. 2008). Similarly, evidence indicates that variation in warning signals can persist even when warning signals are weak when there is limited gene flow (Lawrence et al. 2019). Thus, the evolutionary forces shaping poison frog coloration appear to be varied and complex, and we are just now beginning to understand the details of how color variation evolved in many species.
Recent genomic and transcriptomic studies have provided insight into the genetic mechanisms underlying color variation. Twomey et al. (2020a; b) examined a red/yellow polymorphism in the Sira poison frog, Ranitomeya sirensis, using pigment analysis and RNA sequencing of adult individuals. The authors attribute color differences between the two morphs to varying expression levels of carotenoid-modifying enzymes as well as a loss of function mutation in the gene Beta-Carotene Oxygenase 2 (bco2). Rodriguez et al. (2020) examined the genetic basis of coloration in O. pumilio by performing a differential gene expression analysis of red, green, and blue adult phenotypes. In that study, the authors observe differential expression across numerous functional gene categories, including carotenoid synthesis, melanin synthesis, pteridine synthesis, structural coloration, and pigment cell development. Most recently, several causative genes underlying color and pattern elements between color morphs were identified in the mimic poison frog Ranitomeya imitator. In this species, the color morphs appear to have evolved via selective sweeps on polygenic variation (Linderoth et al. 2023). Their findings highlight the complexity of the genetic mechanisms producing bright, aposematic coloration in amphibians and underscore the need for further coloration research in poison frogs.
A particularly understudied area of animal coloration is how integument color develops in animals with larval stages. There is a growing body of literature on the genetics of pattern formation in Danio fishes (Huang et al. 2021), made possible by decades of work and excellent genomic resources. However, while the morphogenesis, migration, and interactions among pigment cells during amphibian larval stages are described for a handful of anurans (Goldberg et al. 2019; Thibaudeau and Altig 2012), the genetics of these processes are almost entirely unknown. Stuckert et al. (2019) examined expression differences among color morphs in Dendrobates auratus using skin samples from final-stage larva (Gosner stages 41–43), providing candidate genes involved in melanin and iridophore pathways. Work examining gene expression across the development of mimetic Ranitomeya suggested similar pathways at play, but intriguingly suggested that even congeneric species which have evolved mimetic phenotypes may use different gene expression patterns throughout development to produce these convergent phenotypes (Stuckert et al. 2021). However, a limitation of each of these studies is that they used skin samples that included multiple colors which can muddle gene expression signals. Thus our expectations for how gene expression of color and pattern genes occur during development remains an unknown but important part of understanding color and pattern formation in amphibians.
Here we aim to fill a gap in our understanding of the development of coloration in amphibians by studying the development of coloration in the polytypic poison frog O. pumilio. Using whole-transcriptome sequences from pre- and post-metamorphic captive-bred individuals, we examine gene expression profiles across ontogenetic stages and compare green dorsal skin to yellow ventral skin. Frogs in this population exhibit a dark, forest green dorsum and bright yellow venter (Fig. 1C). In amphibians, yellow skin is likely produced by different pigments (e.g., pteridines, carotenoids) that are deposited within the xanthophores (Obika and Bagnara 1964). Green skin is primarily produced by (1) the orientation of structural elements in the iridophores, melanin deposited within melanophores absorbing some light, and possibly pigments in the xanthophores or (2) via solely the iridophores themselves (Bagnara et al. 1968). Green pigments have been identified from frogs (Taboada et al. 2020), but this is not a likely mechanism in poison frogs. Notably, in addition to possessing a green dorsum, frogs in this population display black spots on the dorsum. The size of these and how densely they are spread on the skin are both variable within the population. These are produced by the presence of melanophores and melanin deposited within them (Freeborn 2021).

Visual representation of the Isla Popa phenotype. A Representative images of sampling timepoints throughout development (referred to by Gosner stages, Gosner 1960). B Spectral reflectance measurements. C Images of the dorsum and venter of an adult
In this study, we identify putative color genes differentially expressed between all sampled timepoints and other genes expressed during some, but not all, developmental stages. We also conduct differential expression analyses between dorsal and ventral skin throughout the developmental series, followed by network and GO enrichment analyses. We identify a priori coloration genes differentially expressed between the dorsum and venter at any age and reveal novel insights about the production of coloration across development.
Methods
IACUC statements
This work was done in compliance with Institutional Animal Care and Use Committee protocols at University of Pittsburgh (No. 18103943).
Data and code availability
All read data are available in the European Nucleotide Archive under accession number PRJEB41509. All code for analyses are available at https://github.com/AdamStuckert/Oophaga_pumilio and Stuckert 2023.
Husbandry and sample collection
Animals used in this experiment were sampled from a pure-breeding line established from wild-caught individuals collected from Isla Popa in the Bocas del Toro province of Panama. Within this population, adult males and females are qualitatively similar in coloration: green with black spots dorsally and bright yellow ventrally (Summers et al. 2003; Maan and Cummings 2009); Fig. 1).
All animals were housed in the same walk-in environmental chamber (Darwin Chambers Company, St. Louis, MO) at the University of Pittsburgh per Dugas et al. (Dugas et al. 2013) throughout development. Adult frogs were fed with springtails (Collembola) and fruit flies (wingless Drosophila melanogaster) dusted with powdered supplement (Repashy calcium supplement Plus ICB, Repashy Ventures Inc., San Marcos, CA). The fruit flies were raised on a growth medium supplemented with carotenoids (see Dugas et al. 2013 for details). Females regularly provide tadpoles with unfertilized eggs until close to metamorphosis (~ 45 days) and each tadpole is fed approximately five times (typically one trophic egg per feed) before metamorphosis (Pröhl and Hödl 1999).
We collected dorsal and ventral skin tissue from adults and tadpoles at four developmental stages (hereafter Gosner stages, or GS; (Gosner 1960)) that were easily distinguishable: GS26 (shortly after hatching), GS37 (hindlimb mostly developed), GS42 (forelimbs emerged), and GS46 (tail fully resorbed). Tadpoles of O. pumilio appear uniformly brown/black in color until late in development (~ GS42, Fig. 2) when they begin to take on their adult coloration. We collected samples from five tadpoles per Gosner stage (GS26, GS37, GS42, GS46), and 15 adults for this study.

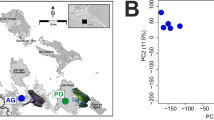
Principal components analysis of gene expression across samples. Age represents either adult frogs or is presented as the Gosner stage at which a tadpole was sampled. Location represents whether RNA was extracted from a small patch of skin on the dorsum (green on the frog) or venter (yellow on the frog). Axes are labeled with the proportion of the data that is explained by each principal component
Lab work
Adult frogs and tadpoles were euthanized by decapitation followed by double-pithing. The dorsal and ventral skins were dissected from the body and submerged immediately in vials containing ~ 1.5 ml of RNAlater (Ambion, Austin, Texas). The vials were then flash frozen in liquid nitrogen and stored at − 80 °C until shipment to the University of New Hampshire for RNA extraction. To extract RNA, skin patches were lysed in Trizol using a tissue homogenizer, treated with DNAse to remove any remaining genomic DNA, and total RNA was then extracted using a standardized Trizol protocol. mRNA libraries were prepared using standard poly-A tail purification, prepared using Illumina primers, and individually dual-barcoded using a New England Biolabs Ultra II Directional kit (E7760). Individually barcoded samples were pooled and sequenced on one lane of an Illumina HiSeq X at the Vincent J. Coates Genomics Sequencing Laboratory and Functional Genomics Laboratory at the University of California at Berkeley. Reads were paired-end and 150 base pairs in length.
Transcriptome assembly
We merged reads from dorsal and ventral samples from a single random individual per Gosner Stage to create a single paired-end dataset for transcriptome assembly. We then used seqtk’s “sample” function to subsample each species’ readset to 50 million read pairs (https://github.com/lh3/seqtk). We used this subsampled dataset to build a de novo transcriptome using the Oyster River Protocol version 2.2.7 (MacManes 2018) which automates the following processes. Initial error correction was done using RCorrector 1.01 (Song and Florea 2015), followed by quality trimming using Trimmomatic version 0.36 at a Phred score of ≤ 3 (Bolger et al. 2014) as aggressive quality trimming decreases assembly completeness (MacManes 2014). The Oyster River Protocol assembles a transcriptome by using a series of different transcriptome assemblers and also multiple kmer lengths, merging them into a single transcriptome. Assemblies were conducted using Trinity version 2.7.0 (Grabherr et al. 2011), Trans-ABySS (Robertson et al. 2010), and SPAdes assembler version 3.12.0 with a kmer length of 55 and a second time with a kmer length of 75 (Bankevich et al. 2012; Prjibelski et al. 2020). These individually built transcriptomes were then merged together using OrthoFuser (MacManes 2018). Unique contigs which were dropped in Orthofuser were recovered using a reciprocal blast search of the final assembly against the individual assemblies for unique contigs. We removed all contigs with expression lower than one Transcript Per Million (TPM) using the TPM = 1 flag in the ORP. Contigs that were dropped due to low expression but which likely represent expressed genes were recovered by blasting these against the Uniprot database (Bairoch and Apweiler 2000). Finally, transcriptome quality was assessed using BUSCO version 4 and the tetrapoda database “tetrapoda_odb10” (Simão et al. 2015) and TransRate 1.0.3 (Smith-Unna et al. 2016).
Gene expression analyses
We quantified sample libraries using salmon version 0.14.1 (Patro et al. 2017) to map reads back to our reference transcriptome. We then annotated our transcriptome using the SwissProt database (UniProt Consortium 2019) using Diamond version 0.9.10 (Buchfink et al. 2015) at an e-value cutoff of 0.001 and without the -max_target_seqs 1 flag. We imported quantification results into R version 4.0.2 (Team 2017) using the package “tximport” (Soneson, Love, and Robinson 2015), and conducted a gene level analysis using DESeq2 version 1.28.1 (Love et al. 2014). We shrunk log fold change estimates using the function “lfcShrink” in DESeq2, using the “normal” shrinkage estimate, which allows targeted comparisons. Since we were particularly interested in running comparisons between skin color patches (i.e., dorsum vs. venter), we then ran a Wald test comparing these two color patches at each time point from Gosner stage 26 through Gosner stage 46, as well as adult frogs. Additionally, we were interested in gene expression changes over time, so we ran a Likelihood Ratio Test to examine what genes change significantly throughout development (Gosner stages 26–46). We used a Benjamini-Hochberg (Benjamini and Hochberg 1995) correction for every analysis of gene expression, and considered a q-value of < 0.05 to be statistically significant. We then extracted data from our models for particular a priori color genes that play a role in color or pattern production in other taxa. This a priori list of candidate color genes is from (Stuckert et al. 2021) but was updated by searching for genes that have been implicated in coloration in genomics studies in the period 2019–2021 and includes 561 genes in total). Plots were produced using ggplot2 version 3.3.2 (Wickham 2011).
In this studywe use two approaches to our gene expression analysis. The first uses our de novo assembled transcriptome to analyze readsets. The second uses an O. pumilio assembly (Rogers et al. 2018) that was scaffolded with RNA seq data by Rodriguez et al. (Rodríguez et al. 2020) to analyze our data. Both approaches use the same overall methodological approach following this initial step, and our results are largely congruous between methods.
Weighted gene correlation network analysis
To identify networks of genes that interact in response to differences in tissue location or developmental stage, we used weighted gene co-expression network analysis (WGCNA) using the package WGCNA (Langfelder and Horvath 2008). For the WGCNA analysis, we used the variance stabilizing transformed data produced by DESeq2. WGCNA requires filtering out genes with low expression, which we did prior to differential expression analyses. We estimated a soft threshold power (β) that fits our data, by plotting this value against Mean Connectivity to determine the minimum value at which Mean Connectivity asymptotes, which represents scale free topology. For our data, we used β = 4, the recommended minimum module size of 30, and we merged modules with a dissimilarity threshold below 0.25. After module formation, we tested whether the eigen modules (conceptually equivalent to a first principal component of the modules) were correlated with sampling location (e.g., dorsum vs venter) at p < 0.05. To examine gene ontology of these modules we then took module membership for each gene within these modules and ranked them in decreasing order of module membership. We then supplied this single ranked list of module membership to the Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GOrilla), and ran it in fast mode using Homo sapiens set as the organism (Eden et al. 2009) to examine enrichment of Function. Gene Ontology (GO) analyses were conducted on 10 December, 2020. Finally, we extracted hub genes for WGCNA modules of interest, and plotted the interactions between these hub genes within a module using Cytoscape version 3.8.0, running on Java 11.0.6 (Su et al. 2014).
Results
Transcriptome assembly, annotation, and read mapping
We produced a de novo transcriptome assembly for the Isla Popa morph of O. pumilio. This transcriptome had a TransRate score of 0.371, and an optimal TransRate score of 0.447, indicating high read support for the assembled transcripts. Our BUSCO analysis indicated that this transcriptome had 70.7% of the complete tetrapod orthologs (62.5% complete, single copies; 8.2% duplicated copies), 7.1% of copies were present but fragmented, and 22.2% were missing. We annotated 37,164 assembled transcripts (24.3%) using the Swiss-Prot database. We mapped reads to our reference transcriptome using salmon, successfully mapping 65.5% (± 7.1% SD) of reads.
We also utilized an alternative reference transcriptome produced by Rodriguez et al. (Rodríguez et al. 2020). This transcriptome was built by scaffolding an existing O. pumilio reference genome using RNA seq data (Rogers et al. 2018). We annotated this transcriptome in the same manner with which we produced our de novo assembly and were able to annotate 40,383 contigs out of 55,640 total contigs (72.57%). When we mapped our reads to this assembly using Salmon, we were able to map 67.33% (± 6.42% SD).
Transcriptome guided differential expression analyses
We conducted six individual comparisons. One comparison was a likelihood ratio test between all tadpole timepoints to examine any change in expression patterns. This comparison yielded a total of 6460 differentially expressed genes after Benjamini–Hochberg correction (all numbers below represent significant genes after the same correction), 160 of which are putatively color genes as identified via our a priori list.
Five of the comparisons we ran were a Wald test of differential expression between the dorsal and ventral skin at each time point (i.e., tadpoles from Gosner stage 26 through Gosner stage 46, as well as adult frogs). Comparatively few genes were differentially expressed at each larval developmental stage relative to adulthood (Table 1). In total, there were 1,176 unique differentially expressed genes between the dorsum and venter at any age, 54 of which were in our a priori candidate list. These 1,176 unique genes that were differentially expressed at any age contained six genes which were differentially expressed between the dorsum and venter in every age class (casq1, hand2, myh8, prva, tbx3, and zic1). Of these, hand2, myh8, tbx3, and zic1 have been identified as being related to the development of coloration in other taxa and were in our a priori candidate list (Fig. 3). There were a number of additional genes that were differentially expressed between dorsum and venter at multiple time points, including gata3 (4 age classes), edn3 (3 age classes), and asip and oca2 (2 age classes). Of the 54 genes that we identified a priori which were differentially expressed, the vast majority of these were differentially expressed exclusively in adults (38 out of 43). Many of these genes are related to melanophore production or melanin synthesis, including asip, oca2, mc1r, kit, lef1, pmel, sox10, tyr and tyrp1 (Fig. 4). Fewer were related to pteridine synthesis (xdh, only at Gosner stage 26) or carotenoid metabolism (adh1, adh2, aldh1a2, rdh10; Fig. 5).

Genes that are differentially expressed between the dorsum (green) and venter (yellow) in every age class

Melanin genes that are differentially expressed between the dorsum (green) and venter (yellow)

Carotenoid (adh1, aldh1a2, rdh10) and pteridine (xdh) genes that are differentially expressed between the dorsum (green) and venter (yellow)
Genome guided differential expression analyses
Using the Rogers et al. (2018) genome that had been scaffolded by Rodriguez et al. (2020), we again conducted the same six individual comparisons. Results are qualitatively quite similar to our de novo transcriptome based approach. One comparison was a likelihood ratio test between all tadpole timepoints to examine any change in expression patterns. This comparison yielded a total of 5684 differentially expressed genes, 153 of which are putatively color genes as identified via our a priori list.
Again, comparatively few genes were differentially expressed at each developmental stage relative to adulthood (Table 1). In total, there were 1374 unique differentially expressed genes between the dorsum and venter at any age, 51 of which were in our a priori candidate list. These 1374 unique genes that were differentially expressed at any age contained nine genes that were differentially expressed between the dorsum and venter at each age (casq1, eno3, hand2, myom2, myoz1, pvalb, srl, tbx3, and zic1). Of these, eno3, hand2, myom2, myoz1, pvalb, tbx3, and zic1 have been identified as coloration in other taxa and found in our a priori candidate list. There were a number of additional genes that were differentially expressed between dorsum and venter at multiple time points, including myh8 (3 age classes, and differentially expressed at every time point in the transcriptome analysis) and asip, gata3, and oca2 (2 age classes). Of the 51 genes that we identified a priori which were differentially expressed, the vast majority of these were differentially expressed exclusively in adults (38 out of 43). Many of these genes are related to melanophore production of melanin synthesis, including asip, oca2, mc1r, kit, lef1, pmel, sox10, tyr and tyrp1. Fewer were related to pteridine synthesis (xdh, only at Gosner stage 26) or carotenoid metabolism (adh1, adh2, aldh1a2, rdh10).
Weighted gene correlation network analysis
A total of eight modules that were identified by WGCNA were significantly correlated with sample location (dorsum vs. venter) in adult samples (Table 2). Parsing the results of network analyses can be difficult, given the abundance of modules (identified herein by number, which is without biological significance) and GO terms that are identified as statistically significant, but may be unrelated to the phenotype of interest. The second module contained numerous GO terms related to actin binding or cytoskeletal binding, including actin binding (GO:0003779), actin filament binding (GO:0051015), and cytoskeletal protein binding (GO:0008092). These GO terms involve many myosin genes, including a number that were differentially expressed between the dorsum and venter in numerous age classes (e.g., myh8, myoz1). Further, four of the hub genes in the black module are those that were differentially expressed between the dorsum in every age class (casq1, myh8, myoz1, and prva; Fig. 6).

Hub genes for the second module associated with sampling location, which WGCNA identified as correlated with coloration in adult frogs. The four genes highlighted in red (casq1, myh8, myoz1, and prva) are those that were differentially expressed between the dorsum and venter in every age class (tadpoles of Gosner stage 26 through adults)
Additionally, a total of eight gene modules were identified by WGCNA to be significantly associated with developmental stage (Table 3). As mentioned above, parsing these results and their significance can be difficult. Many of the enriched gene ontology terms in these modules relate to the production of the extracellular matrix. For example, the five most enriched GO terms in the first module are protein-lysine 6-oxidase activity (GO:0004720), chondroitin sulfate binding (GO:0035374), extracellular matrix structural constituent conferring compression resistance (GO:0030021), extracellular matrix constituent conferring elasticity (GO:0030023), and procollagen-lysine 5-dioxygenase activity (GO:0097493). Of these five most enriched gene ontology terms, all are related to collagen or the extracellular matrix. Further, a number of the hub genes in this module were collagen type 1–6 genes (10 out of 36 hub genes) and two were in the lysyl oxidase gene family (LOXL1, LOXL2; Fig. 7). Similarly, the fourth module has a variety of enriched GO terms related to keratinization (GO:0031424), cornification (GO:0070268), or intermediate filament organization (GO:0045109). Most of these modules are likely to be strictly related to development, and not coloration related per se. However, certain modules (e.g., the second module) are associated with an abundance of GO terms related to G proteins: GDP binding (GO:0019003), GTPase activity (GO:0003924), GTP binding (GO:0005525), purine ribonucleoside binding (GO:0032550), purine nucleoside binding (GO:0001883), ribonucleoside binding (GO:0032549), guanyl ribonucleotide binding (GO:0032561), guanyl nucleotide binding (GO:0019001), and nucleoside binding (GO:0001882). These modules are driven by a number of genes in the ras oncogene family. While these ras oncogenes are best known for cancer phenotypes in mutants, many of them have been implicated in coloration in other taxa (Houben et al. 2004; McLean et al. 2017).

Hub genes for the first gene module associated with development, which WGCNA identified as correlated with age class. Genes in the lysyl oxidase gene family (LOXL1, LOXL2) are highlighted in yellow, while collagen type 1–6 genes are highlighted in red
Discussion
In this paper, we examined gene expression in the dorsum and venter throughout development in the strawberry poison frog, O. pumilio, to better understand both differential color production and ontogenetic changes in the integument throughout development. These data allow us to look at the differences in color production between green skin (dorsum) and yellow skin (venter), while also examining how changes in gene expression contribute to external development prior to, and after metamorphosis. Here we highlight genes that are differentially expressed between the dorsum and venter at multiple developmental stages (e.g., casq1, hand2, myh8, prva, tbx3, and zic1), key pathways varying across developmental stages, and finally candidate color genes for producing the difference between green and yellow integument.
Genes differentially expressed between the dorsum and venter at multiple developmental stages
A total of six genes were differentially expressed between the dorsum and venter at each time point: casq1, hand2, myh8, prva, tbx3, and zic1. Their differential expression throughout the developmental stages we tracked (recently hatched tadpoles through froglets) make these excellent candidate genes for the production of different structures in the integument between the dorsum and venter. This includes both structures related to color (chromatophores) as well as structures related to toxicity (granular glands). Hand2, myh8, tbx3, and zic1 have been putatively identified as players in coloration in other taxa. Heart- and neural crest derivatives-expressed protein 2 (hand2) encodes a transcription factor that plays a role in a variety of developmental processes, including heart morphogenesis (Shen et al. 2010) and limb development (Yelon et al. 2000). In dark-eyed juncos (Junco hyemalis), expression of hand2 is correlated with feather coloration, with higher expression in gray feathers than black feathers (Abolins-Abols et al. 2018). Similarly, hand2 has higher expression in the lighter-colored egg spots in certain cichlids (Santos et al. 2016). We see expression patterns that recapitulate the findings in both of these species: higher expression in lighter colored regions of the body (the venter in our study system).
Hand2 plays a role in maintaining expression of T-Box Transcription Factor 5 (Yelon et al. 2000). While we did not see the same pattern of differential expression in hand2 and tbx5, we did see the same expression between hand2 and the T-Box Transcription Factor 3 gene (tbx3). This potentially indicates that hand2 expression is also related to tbx3 expression in a manner similar to tbx5, which is plausible given they are both in the tbx2 subfamily. Tbx3 has been shown to inhibit pigment synthesis and produce lighter colored hair in horses (Imsland et al. 2016). We see a similar pattern in our data with much higher expression in the lighter ventral integument, potentially indicating that tbx3 is inducing lighter phenotypes in O. pumilio as it does in other taxa.
While hand2 and tbx3 seem to play a role in coloration specifically, other genes differentially expressed between the dorsum and venter seem to be related to early differentiation during development and not coloration. Myosin Heavy Chain 8 (myh8) is associated with multiple human diseases associated with variety of developmental defects, as well as abnormal spotted pigmentation such as Trismus‐pseudocamptodactyly syndrome (Toydemir et al. 2006) and Cushing’s syndrome (Majumder et al. 2020). This gene is clearly important to a variety of developmental processes, and it is likely that its effect on pigmentation is due to abnormal organization in the integument. Zic Family Member 1 (zic1) is critically important to basic neural crest development and signaling (Milet et al. 2013; Plouhinec et al. 2014). Further, zic1 is involved in dorsoventral patterning in zebrafish, and is affected by both BMP and hedgehog signaling (Rohr et al. 1999). Zic1 is known to influence the location of iridophores and melanophores in medaka (Oryzias latipes) and thus impact color and pattern phenotypes. However, this seems to largely be a result of how zic1 mutants cause mirroring effects between the dorsum and venter, seemingly indicating that this gene largely drives differences in dorsoventral patterning in general, and not coloration per se(Ohtsuka et al. 2004). Despite this, zic1 is a good candidate gene for coloration in this taxon, and plausibly others.
We also discovered differential expression between the dorsum and venter at every time point in the genes Calsequestrin 1 (casq1) and Parvalbumin (pvalb). These genes buffer Ca2+ and are a Ca2+ transporter respectively. Alkaloids are known to affect voltage gated channels. Pumiliotoxins in particular appear to be fairly specific inhibitors of Ca2+-ATPase activity (Tamburini et al. 1981). O. pumilio is an obligate egg-feeding species (Weygoldt et al. 1980). In addition to providing key nutrients to their young, these mothers also provision trophic eggs with alkaloids. Tadpoles first exhibit very low levels of toxicity around Gosner stage 34, and show an increase in both toxin quantity and diversity after this developmental stage (Stynoski et al. 2014). In O. pumilio, granular glands in this species do not begin developing until around Gosner stage 32, and are visible by Gosner stage 40 (Stynoski and O’Connell 2017). This is perhaps surprising, as O. pumilio tadpoles feed on a diet of trophic eggs provisioned with alkaloids through development and before they have the physiology to sequester them in the skin. We expect a greater number and possibly overall larger size of granular glands in the dorsum, as this is where potential predators are most likely to attack and in many toxic dendrobatids, including O. pumilio, the dorsal skin is visibly more granular than the ventral skin (CR-Z and YY pers. obs.). In this study, we found differential expression with higher expression in the venter in every time point in the genes casq1 and pvalb, but that the difference between the dorsum and venter dramatically decreased around Gosner stage 37–42, which corresponds with the onset and maturation of granular gland development in O. pumilio. Although this expression pattern is in the opposite direction than what we would have predicted, these results still lead to us proposing casq1 and pvalb as candidate genes. This finding, coupled with evidence that pumiliotoxins (and other alkaloids) impact Ca2+ channels and the developmental data lead us to hypothesize that casq1 and pvalb are associated with granular glands and play a role in preventing autotoxicity in O. pumilio.
Key pathways in developmental stages
Amphibian metamorphosis involves vast reorganization of the body. The process of metamorphosis is initiated via Thyroid Hormone (TH), which causes cascading effects throughout the organism, including organogenesis, reorganization of the intestinal tract, reorganization/ossification of the skull, and the initiation of apoptosis in the tail (Mukhi et al. 2010). Most work focuses on one of those processes, with less work focusing on reorganization of the integument. Our gene network analyses indicate a number of gene pathways and key hub genes that are putatively driving reorganization of the body and integument in frogs throughout development.
Prominent among these genes are those related to the extracellular matrix, collagen, and keratin. Collagen is a large family of extracellular matrix proteins, and are extremely abundant molecules in vertebrates (Myllyharju and Kivirikko 2004). We identified many collagen type 1–6 genes as hub genes in gene networks associated with age, indicating their potential importance in the reorganization of the integument which tadpoles undergo during metamorphosis. Not only are these genes likely candidates for reorganizing the integument during metamorphosis, but collagen genes may influence coloration as well. Coloration is at least in part related to physical structures in the skin. Recent work has found that the orientation and thickness of guanine platelets in iridophores plays a key role in light reflectance and thus the color of the skin (Twomey et al. 2020a; b). Genes which play a role in integumental structure (e.g., keratin, collagen, etc.) may change the orientation of these elements in subtle ways, and are thus plausible candidates for producing different colors. Several collagen genes (col1a1, col5a1, and col14a1) have already been implicated in the production of different colors in another study of O. pumilio or in the poison frog D. auratus (Stuckert et al. 2019; Rodríguez et al. 2020).
Candidate color genes
We also examined specific a priori candidate genes for coloration within our data. Of particular note are genes influencing melanin synthesis or carotenoid metabolism within our data, as those are genes we would predict to be playing a role in the differences between green and yellow skin. In amphibians, yellow and green skin is likely to derive from either pteridines or carotenoids deposited in xanthophores, the outermost chromatophore layer (Bagnara et al. 1968). We hypothesize that there are different quantities of carotenoids between yellow and green skin, although which carotenoids are at play and in which color skin carotenoids are in a greater abundance in this morph of O. pumilio are outstanding questions.Therefore it is difficult to identify a priori expectations for carotenoid gene expression because it is unclear which skin has more carotenoids. We identified a number of carotenoid genes that are differentially expressed between green skin (dorsum) and yellow skin (venter), including adh1, adh2, aldh1a2, and rdh10. All of these are genes in the retinoic acid pathway, and together they catalyze the first two steps in the transition from cellular retinol into retinoic acid (Parés et al. 2008; Thompson et al. 2019; Sandell et al. 2007; Niederreither and Dollé 2008). Genes in this pathway have been repeatedly linked to coloration in other taxa (eg (McLean et al. 2017, 2019; Stuckert et al. 2021)). In fact, adh1, rdh10 and aldh1a1 (note we found differential expression in aldh1a2, but they are likely playing similar roles here) were differentially expressed between red, blue, and green O. pumilio in another study (Rodríguez et al. 2020). Furthermore, the retinoic acid pathway is known to activate tbx3 (Ballim et al. 2012), one of the genes differentially expressed between the dorsum and venter at every developmental stage, and one that impacts color phenotypes in other taxa. Although these genes have been implicated in coloration and are correlated with coloration in this study, whether these genes are producing color or are merely associated with color due to downstream processes related to cleaved carotenoids remains unclear and requires functional testing.
Greens are likely to be derived from a combination of certain wavelengths of light being transmitted through pigments in the xanthophores (again, either pteridines or carotenoids), reflection from iridophores, and light absorption via melanins in the melanophores (although it is possible to produce greens via other mechanisms, see Taboada et al. 2020). Darker greens are putatively derived from higher quantities of melanin (Bagnara et al. 1968). However, the dorsum of these frogs also exhibits small black spots, and these black spots are likely contributing to some expression patterns related to melanin production. We identified a number of melanin genes that are differentially expressed between the and the lighter venter in our samples. We found a number of genes in the melanin synthesis pathway which are differentially expressed between the dorsum and venter, many of which have been identified as important in frogs or other vertebrates. Prominent amongst these are agouti signaling protein (asip) and melanocortin receptor 1 (mc1r). Mc1r is found in the plasma membrane of melanocytes, and activation of this gene leads to melanogenesis. We find higher expression of mc1r in darker skin, concordant with other examples in a variety of taxa (e.g., (Mandal et al. 2020; Espinasa et al. 2018; Mundy 2005; Suzuki et al. 2001). Asip is an agonist of mc1r, and when it binds to mc1r it can cause a switch from the production of eumelanin (brown/black pigment) to pheomelanin (red to reddish-brown pigment). Note that while pheomelanin has been implicated as a primary pigment in one frog species to date (Wolnicka-Glubisz et al. 2012), its presence and role in amphibians largely remains unexplored. We found that asip has the highest expression in the light venter of adults, a result that tracks with other taxa in which asip is correlated with lighter phenotypes.
Similarly, tyrosinase related protein 1 (tyrp1) is capable of producing a lighter colored phenotype via changing the ratios of eumelanin and pheomelanin (Videira et al. 2013). While tyrp1’s role in melanin synthesis is not entirely clear, it is possible that it is important for maintaining the stability of the tyrosinase protein (Sarangarajan and Boissy 2001). We also saw differential expression of the gene tyrosinase (tyr). Tyrosinase is the rate limiting enzyme in the production of melanin, and as a result is important to the ultimate color phenotype. Tyrp1 is also differentially expressed between colors between morphs in O. pumilio and D. auratus (Rodríguez et al. 2020; Stuckert et al. 2019). Counter to predictions, we see higher expression of tyrp1 and lower expression of tyr in the dark green skin. Indeed, our results for tyrp1 expression are counter to what was seen in Dendrobates auratus, in which lighter morphs had higher expression of tyrp1 (Stuckert et al. 2019). We also found differential expression in Proto-oncogene c-KIT (kit), which exhibited a parabolic expression pattern, peaking around Gosner stage 42. Expression was consistently higher in the green dorsal skin, consistent with evidence that kit is required for migration and survival of melanocyte precursors during development (Wehrle-Haller 2003). Mutations in this gene are responsible for piebaldism in humans, in which there are unpigmented areas in the body, typically the forehead and belly (Giebel and Spritz 1991). Finally, we discovered a similar pattern of expression in Lymphoid enhancer-binding factor 1 (lef1) to kit, with a parabolic expression pattern peaking around Gosner stage 42. Lef1 mediates Wnt signaling and ultimately impacts melanogenesis. In minks and the green-and-black poison frog, lef1 expression is correlated with lighter phenotypes (Song et al. 2017; Stuckert et al. 2019). Given their roles in development, and the somewhat surprising expression patterns between the dorsum and venter in our data, it is possible that the expression patterns we observed in lef1, tyr, and tyrp1 are primarily a result of development rather than coloration itself. Despite this, we believe a number of the genes we have discussed related to melanin synthesis are playing a role in coloration differences between the dorsum and venter in O. pumilio, particularly asip and mc1r.
Conclusion
In this paper, we examined gene expression in the dorsum and venter throughout development in the strawberry poison frog, O. pumilio. We used these data to examine the differences in color production between green skin (dorsum) and yellow skin (venter), while also examining how changes in gene expression contribute to external development. We have highlighted key genes that are likely related to coloration. These include melanin synthesis genes such as asip and mc1r, genes in the retinoic acid pathway (e.g., adh1, adh2, aldh1a2, and rdh10), and candidate color genes that were differentially expressed between the dorsum and venter at every developmental stage (hand2, myh8, tbx3, and zic1). Further, our data indicate that a number of genes related to Ca2+ buffering and transport (casq1 and pvalb) may be critically important to preventing autotoxicity, and are especially good candidates for allowing the sequestration and maintenance of pumiliotoxins. Finally, we discuss a number of enriched pathways associated with development that are likely key in the transition from aquatic tadpole to terrestrial froglet, such as those associated with oxidoreductase activity, the extracellular matrix, keratinization, and cornification.
Data availability
All read data are available in the European Nucleotide Archive under accession number PRJEB41509.
Code availability
All code for analyses are available at https://github.com/AdamStuckert/Oophaga_pumilio and on Zenodo (link upon acceptance).
References
Abolins-Abols M, Kornobis E, Ribeca P, Wakamatsu K, Peterson MP, Ketterson ED, Milá B (2018) Differential gene regulation underlies variation in melanic plumage coloration in the dark-eyed junco (Junco Hyemalis). Mol Ecol 27(22):4501–4515
Bagnara JT, Taylor JD, Hadley ME (1968) The dermal chromatophore unit. J Cell Biol 38(1):67–79
Bairoch A, Apweiler R (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucl Acids Res 28(1):45–48
Ballim RD, Mendelsohn C, Papaioannou VE, Prince S (2012) The ulnar-mammary syndrome gene, Tbx 3, is a direct target of the retinoic acid signaling pathway, which regulates its expression during mouse limb development. Mol Biol Cell 23(12):2362–2372
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol J Comput Mol Cell Biol 19(5):455–477
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B Methodol. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30(15):2114–2120
Bolton PE, Rollins LA, Griffith SC (2015) The danger within: the role of genetic, behavioural and ecological factors in population persistence of colour polymorphic species. Mol Ecol 24(12):2907–2915
Brown JL, Twomey E, Amezquita A, DeSouza MB, Caldwell J, Lötters S, Von May R et al (2011) A taxonomic revision of the neotropical poison frog genus Ranitomeya (Amphibia: Dendrobatidae). Zootaxa 3083:1–120
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12(1):59–60
Caro T, Allen WL (2017) Interspecific visual signalling in animals and plants: a functional classification. Philos Trans R Soc London Ser B Biol Sci. https://doi.org/10.1098/rstb.2016.0344
Darst CR, Cummings ME, Cannatella DC (2006) A Mechanism for diversity in warning signals: conspicuousness versus toxicity in poison frogs. Proc Natl Acad Sci USA 103(15):5852–5857
Dugas MB, Yeager J, Richards-Zawacki CL (2013) Carotenoid supplementation enhances reproductive success in captive strawberry poison frogs (Oophaga Pumilio). Zoo Biol 32(6):655–658
Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinf 10(February):48
Endler JA (1980) Natural selection on color patterns in Poecilia Reticulata. Evol Int J Org Evol 34(1):76–91
Endler JA, Mappes J (2017) The current and future state of animal coloration research. Philos Trans R Soc London Ser B Biol Sci. https://doi.org/10.1098/rstb.2016.0352
Espinasa L, Robinson J, Espinasa M (2018) Mc1r gene in Astroblepus Pholeter and Astyanax Mexicanus: convergent regressive evolution of pigmentation across cavefish species. Dev Biol 441(2):305–310
Freeborn LR (2021) The genetic, cellular, and evolutionary basis of skin coloration in the highly polymorphic poison frog, Oophaga pumilio. Doctoral Dissertation, University of Pittsburgh
Funk WC, Zamudio KR, Crawford AJ (2018) Advancing understanding of amphibian evolution, ecology, behavior, and conservation with massively parallel sequencing. Popul Genom. https://doi.org/10.1007/13836_2018_61
Garrone R, Lethias C, Le Guellec D (1997) Distribution of minor collagens during skin development. Microsc Res Tech 38(4):407–412
Gehara M, Summers K, Brown JL (2013) Population expansion, isolation and selection: novel insights on the evolution of color diversity in the strawberry poison frog. Evol Ecol 27(4):797–824
Giebel LB, Spritz RA (1991) Mutation of the KIT (mast/stem cell growth factor receptor) protooncogene in human piebaldism. Proc Natl Acad Sci USA 88(19):8696–8699
Godwin JW, Debuque R, Salimova E, Rosenthal NA (2017) Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regener Med. https://doi.org/10.1038/s41536-017-0027-y
Goldberg J, Quinzio SI, Cruz JC, Fabrezi M (2019) Intraspecific developmental variation in the life cycle of the andean treefrog (Boana Riojana): a temporal analysis. J Morphol 280(4):480–493
Gosner KL (1960) A simplified table for staging anuran embryos and larvae with notes on identification. Herpetologica 16(3):183–190
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X et al (2011) Full-LENGTH TRANSCRIPTOME ASSEMBLY FROM RNA-Seq data without a reference genome. Nat Biotechnol 29(7):644–652
Gray SM, McKinnon JS (2007) Linking color polymorphism maintenance and speciation. Trends Ecol Evol 22(2):71–79
Hill GE, Hill GE, McGraw KJ, McGraw KJ (2006) Bird coloration. Harvard University Press
Houben R, Becker JC, Kappel A, Terheyden P, Bröcker E-B, Goetz R, Rapp UR (2004) Constitutive activation of the ras-raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog 3(March):6
Huang D, Lewis VM, Foster TN, Toomey MB, Corbo JC, Parichy DM (2021) Development and genetics of red coloration in the zebrafish relative Danio Albolineatus. Elife 10:e70253. https://doi.org/10.7554/eLife.70253
Huxley J (1955) Morphism and evolution. Heredity 9(1):1–52
Imsland F, McGowan K, Rubin C-J, Henegar C, Sundström E, Berglund J, Schwochow D et al (2016) Regulatory mutations in TBX3 disrupt asymmetric hair pigmentation that underlies dun camouflage color in horses. Nat Genet 48(2):152–158
Langfelder P, Horvath S (2008) WGCNA: an r package for weighted correlation network analysis. BMC Bioinf 9(December):559
Langton AK, Sherratt MJ, Griffiths CEM, Watson REB (2012) Differential expression of elastic fibre components in intrinsically aged skin. Biogerontology 13(1):37–48
Lawrence JP, Rojas B, Fouquet A, Mappes J, Blanchette A, Saporito RA, Bosque RJ, Courtois EA, Noonan BP (2019) Weak warning signals can persist in the absence of gene flow. Proc Natl Acad Sci USA 116(38):19037–19045
Linderoth T, Aguilar-Gómez D, White E, Twomey E, Stuckert A, Bi K, Ko A, et al. (2023) Genetic basis of aposematic coloration in a mimetic radiation of poison frogs. bioRxiv https://doi.org/10.1101/2023.04.20.537757.
Love M, Anders S, Huber W (2014) Differential analysis of count data–the DESeq2 package. Genome Biol 15(550):10–1186
Maan ME, Cummings ME (2008) Female preferences for aposematic signal components in a polymorphic poison frog. Evolution 62(9):2334–2345
Maan ME, Cummings ME (2009) Sexual dimorphism and directional sexual selection on aposematic signals in a poison frog. Proc Natl Acad Sci 106(45):19072–19077
Maan ME, Cummings ME (2012) Poison frog colors are honest signals of toxicity, particularly for bird predators. Am Nat 179(1):E1–E14
MacManes MD (2014) On the optimal trimming of high-throughput mRNA Sequence Data. Front Genet 5:1–7
MacManes MD (2018) The oyster river protocol: a multi-assembler and kmer approach for de novo transcriptome assembly. PeerJ 6:e5428
Majumder S, Chakraborty PP, Ghosh PC, Bera M (2020) Cushing’s syndrome in early infancy due to isolated sporadic bilateral micronodular adrenocortical disease associated with myosin heavy chain 8 mutation: diagnostic challenges, too many! BMJ Case Rep. https://doi.org/10.1136/bcr-2020-236850
Mandal BK, Chen H, Si Z, Hou X, Yang He, Xidan Xu, Wang J, Wang C (2020) Shrunk and scattered black spots turn out due to MC1R knockout in a white-black oujiang color common carp (Cyprinus Carpio Var. Color). Aquaculture 518(March):734822
McEwan J, Lynch J, Beck CW (2011) Expression of key retinoic acid modulating genes suggests active regulation during development and regeneration of the amphibian limb. Dev Dyn 240(5):1259–1270
McLean CA, Lutz A, Rankin KJ, Stuart-Fox D, Moussalli A (2017) Revealing the biochemical and genetic basis of color variation in a polymorphic lizard. Mol Biol Evol 34(8):1924–1935
McLean CA, Lutz A, Rankin KJ, Elliott A, Moussalli A, Stuart-Fox D (2019) Red Carotenoids and associated gene expression explain colour variation in frillneck lizards. Proc Biol Sci R Soc 286(1907):20191172
Milet C, Maczkowiak F, Roche DD, Monsoro-Burq AH (2013) Pax3 and Zic1 drive induction and differentiation of multipotent, migratory, and functional neural crest in Xenopus embryos. Proc Natl Acad Sci USA 110(14):5528–5533
Mukhi S, Cai L, Brown DD (2010) Gene switching at Xenopus Laevis metamorphosis. Dev Biol 338(2):117–126
Mundy NI (2005) A window on the genetics of evolution: MC1R and plumage colouration in birds. Proc Biol Sci R Soc 272(1573):1633–1640
Myllyharju J, Kivirikko KI (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet TIG 20(1):33–43
Niederreither K, Dollé P (2008) Retinoic acid in development: towards an integrated view. Nat Rev Genet 9(7):541–553
Noonan BP, Comeault AA (2009) The role of predator selection on polymorphic aposematic poison frogs. Biol Lett 5(1):51–54
Obika M, Bagnara JT (1964) Pteridines as pigments in amphibians. Science 143(3605):485–487
Ohtsuka M, Kikuchi N, Yokoi H, Kinoshita M, Wakamatsu Y, Ozato K, Takeda H, Inoko H, Kimura M (2004) Possible roles of zic1 and zic4, identified within the medaka double anal fin (da) locus, in dorsoventral patterning of the trunk-tail region (related to phenotypes of the da mutant). Mech Dev 121(7–8):873–882
Parés X, Farrés J, Kedishvili N, Duester G (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families. Cell Mol Life Sci CMLS 65(24):3936
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14(4):417–419
Plouhinec J-L, Roche DD, Pegoraro C, Figueiredo AL, Maczkowiak F, Brunet LJ, Milet C et al (2014) Pax3 and Zic1 trigger the early neural crest gene regulatory network by the direct activation of multiple key neural crest specifiers. Dev Biol 386(2):461–472
Price-Waldman R, Stoddard MC (2021) Avian coloration genetics: recent advances and emerging questions. J Hered 112(5):395–416
Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A (2020) Using SPAdes de novo assembler. Curr Protoc Bioinf 70(1):e102
Pröhl H, Hödl W (1999) Parental investment, potential reproductive rates, and mating system in the strawberry dart-poison frog, Dendrobates Pumilio. Behav Ecol Sociobiol 46(4):215–220
Reilly DS, Tomassini N, Zasloff M (1994) Expression of magainin antimicrobial peptide genes in the developing granular glands of Xenopus skin and induction by thyroid hormone. Dev Biol 162(1):123–133
Reynolds RG, Fitzpatrick BM (2007) Assortative mating in poison-dart frogs based on an ecologically important trait. Evolution 61(9):2253–2259
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K et al (2010) De novo assembly and analysis of RNA-Seq data. Nat Methods 7(11):909–912
Rodríguez A, Mundy NI, Ibáñez R, Pröhl H (2020) Being red, blue and green: the genetic basis of coloration differences in the strawberry poison frog (Oophaga Pumilio). BMC Genomics 21(1):301
Rogers RL, Zhou L, Chu C, Marquez R, Corl A, Linderoth T, Freeborn L et al (2018) Genomic takeover by transposable elements in the strawberry poison frog. Mol Biol Evol 35(12):2913–2927
Rohr KB, Schulte-Merker S, Tautz D (1999) Zebrafish zic1 expression in brain and somites is affected by BMP and hedgehog signalling. Mech Dev 85(1–2):147–159
Ruxton GD, Sherratt TN, Speed MP (2004) Avoiding attack: the evolutionary ecology of crypsis, warning signals and mimicry, vol 17. Oxford University Press, Oxford
Sandell LL, Sanderson BW, Moiseyev G, Johnson T, Mushegian A, Young K, Rey J-P, Ma J-X, Staehling-Hampton K, Trainor PA (2007) RDH10 Is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev 21(9):1113–1124
Santos ME, Baldo L, Langyu Gu, Boileau N, Musilova Z, Salzburger W (2016) Comparative transcriptomics of anal fin pigmentation patterns in cichlid fishes. BMC Genomics 17(September):712
Saporito RA, Zuercher R, Roberts M, Gerow KG, Donnelly MA (2007) Experimental evidence for aposematism in the dendrobatid poison frog Oophaga Pumilio. Copeia 4:1006–1011
Sarangarajan R, Boissy RE (2001) Tyrp1 and oculocutaneous albinism type 3. Pigment Cell Res Spons Eur Soc Pigment Cell Res Int Pigment Cell Soc 14(6):437–444
Satoh A, Kashimoto R, Ohashi A, Furukawa S, Yamamoto S, Inoue T, Hayashi T, Agata K (2022) An approach for elucidating dermal fibroblast dedifferentiation in amphibian limb regeneration. Zool Lett 8(1):6
Shawkey MD, D’Alba L (2017) Interactions between colour-producing mechanisms and their effects on the integumentary colour palette. Philos Trans R Soc London Ser B Biol Sci. https://doi.org/10.1098/rstb.2016.0536
Shen L, Li XF, Shen AD, Wang Q, Liu CX, Guo YJ, Song ZJ, Li ZZ (2010) Transcription factor HAND2 mutations in sporadic chinese patients with congenital heart disease. Chin Med J 123(13):1623–1627
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212
Smith-Unna R, Boursnell C, Patro R, Hibberd JM, Kelly S (2016) TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res 26(8):1134–1144
Soneson C, Love MI, Robinson MD (2015) Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4:1521
Song Li, Florea L (2015) Rcorrector: efficient and accurate error correction for illumina RNA-Seq reads. GigaScience 4(1):48
Song X, Chao Xu, Liu Z, Yue Z, Liu L, Yang T, Cong Bo, Yang F (2017) Comparative transcriptome analysis of mink (Neovison Vison) skin reveals the key genes involved in the melanogenesis of black and white coat colour. Sci Rep 7(1):1–11
Stuckert AMM, Saporito RA, Venegas PJ, Summers K (2014a) Alkaloid defenses of co-mimics in a putative müllerian mimetic radiation. BMC Evol Biol 14:1–8
Stuckert AMM, Venegas PJ, Summers K (2014b) Experimental evidence for predator learning and müllerian mimicry in peruvian poison frogs (Ranitomeya, Dendrobatidae). Evol Ecol 28(3):413–426
Stuckert AMM, Moore E, Coyle KP, Davison I, MacManes MD, Roberts R, Summers K (2019) Variation in pigmentation gene expression is associated with distinct aposematic color morphs in the poison frog, Dendrobates Auratus. BMC Evol Biol 19(85):1–15
Stuckert AMM, Chouteau M, McClure M, LaPolice TM, Linderoth T, Nielsen R, Summers K, MacManes MD (2021) The genomics of mimicry: gene expression throughout development provides insights into convergent and divergent phenotypes in a müllerian mimicry system. Mol Ecol. https://doi.org/10.1111/mec.16024
Stuckert A (2023) AdamStuckert/Oophaga_pumilio: v1.1 (v_1.1). Zenodo. https://doi.org/10.5281/zenodo.8346530
Stynoski JL, O’Connell LA (2017) Developmental morphology of granular skin glands in pre-metamorphic egg-eating poison frogs. Zoomorphology 136(2):219–224
Stynoski JL, Torres-Mendoza Y, Sasa-Marin M, Saporito RA (2014) Evidence of maternal provisioning of alkaloid-based chemical defenses in the strawberry poison frog Oophaga Pumilio. Ecology 95(3):587–593
Su G, Morris JH, Demchak B, Bader GD (2014) Biological network exploration with cytoscape 3. Curr Protoc Bioinf 47(1):8–13
Summers K, Cronin TW, Kennedy T (2003) Variation in spectral reflectance among populations of Dendrobates Pumilio, the strawberry poison frog, in the Bocas Del Toro archipelago, panama. J Biogeogr 30:35–53
Suzuki T, Li W, Zhang Q, Novak EK, Sviderskaya EV, Wilson A, Bennett DC, Roe BA, Swank RT, Spritz RA (2001) The gene mutated in cocoa mice, carrying a defect of organelle biogenesis, is a homologue of the human hermansky-pudlak syndrom-3 gene. Genomics 78(November):30–37
Suzuki K-I, Utoh R, Kotani K, Obara M, Yoshizato K (2002) Lineage of anuran epidermal basal cells and their differentiation potential in relation to metamorphic skin remodeling. Dev Growth Differ 44(3):225–238
Taboada C, Brunetti AE, Lyra ML, Fitak RR, Soverna AF, Ron SR, Lagorio MG et al (2020) Multiple origins of green coloration in frogs mediated by a novel biliverdin-binding serpin. Proc Natl Acad Sci USA 117(31):18574–18581
Tamburini R, Albuquerque EX, Daly JW, Kauffman FC (1981) Inhibition of calcium-dependent atpase from sarcoplasmic reticulum by a new class of indolizidine alkaloids, pumiliotoxins A, B, and 251D. J Neurochem 37(3):775–780
Tazzyman SJ, Iwasa Y (2010) Sexual selection can increase the effect of random genetic drift-a quantitative genetic model of polymorphism in Oophaga Pumilio, the strawberry poison-dart frog. Evol Int J Org Evol 64(6):1719–1728
Team R Core (2017) R Development Core Team. http://www.mendeley.com/research/r-language-environment-statistical-computing-96/%5Cnpapers2://publication/uuid/A1207DAB-22D3-4A04-82FB-D4DD5AD57C28.
Thibaudeau G, Altig R (2012) Coloration of Anuran Tadpoles (amphibia): development, dynamics, function, and hypotheses. ISRN Zool 2012(November):1–16
Thompson B, Katsanis N, Apostolopoulos N, Thompson DC, Nebert DW, Vasiliou V (2019) Genetics and functions of the retinoic acid pathway, with special emphasis on the eye. Hum Genomics 13(1):61
Toydemir RM, Chen H, Proud VK, Martin R, van Bokhoven H, Hamel BCJ, Tuerlings JH, Stratakis CA, Jorde LB, Bamshad MJ (2006) Trismus-pseudocamptodactyly syndrome is caused by recurrent mutation of MYH8. Am J Med Genet A 140(22):2387–2393
Trullas SC, van Wyk JH, Spotila JR (2007) Thermal melanism in ectotherms. J Therm Biol 32(5):235–245
Twomey E, Johnson JD, Castroviejo-Fisher S, Van Bocxlaer I (2020a) A ketocarotenoid-based color polymorphism in the sira poison frog Ranitomeya Sirensis indicates novel gene interactions underlying aposematic signal variation. Mol Ecol 29(11):2004–2015
Twomey E, Kain M, Claeys M, Summers K, Castroviejo-Fisher S, Van Bocxlaer I (2020b) Mechanisms for color convergence in a mimetic radiation of poison frogs. Am Nat Jan. https://doi.org/10.1086/708157
UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47(D1):D506–D515
Utoh R, Asahina K, Suzuki K, Kotani K, Obara M, Yoshizato K (2000) Developmentally and regionally regulated participation of epidermal cells in the formation of collagen lamella of anuran tadpole skin. Dev Growth Differ 42(6):571–580
Vallet SD, Ricard-Blum S (2019) Lysyl oxidases: from enzyme activity to extracellular matrix cross-links. Essays Biochem 63(3):349–364
Videira IF, Santos D, Moura DFL, Magina S (2013) Mechanisms regulating melanogenesis. An Bras Dermatol 88(1):76–83
Watkins T, Schmitt H, Li G, Stamer D (2020) Genetic background impacts phenotypic severity in LOXL1 null mice. Invest Ophthalmol vis Sci 61(7):4630–4630
Wehrle-Haller B (2003) The Role of Kit-Ligand in Melanocyte Development and Epidermal Homeostasis. Pigment Cell Res Spons Eur Soc Pigment Cell Res Int Pigment Cell Soc 16(3):287–296
Weygoldt P, Ecology SB, Schmidt D (1980) Complex brood care and reproductive behavior in captive poison-arrow frogs, Dendrobates Pumilio O. schmidt. Behav Ecol Sociobiol 7(4):329–332
Wickham H (2011) ggplot2. Wiley Interdiscip Rev Comput Stat 3(2):180–185
Wollenberg KC, Lötters S, Mora-Ferrer C, Veith M (2008) Disentangling composite colour patterns in a poison frog species. Biol J Linn Soc 93(3):433–444
Wolnicka-Glubisz A, Pecio A, Podkowa D, Kolodziejczyk LM, Plonka PM (2012) Pheomelanin in the skin of Hymenochirus Boettgeri (Amphibia: Anura: Pipidae). Exp Dermatol 21(7):537–540
Yamauchi M, Shiiba M (2008) Lysine hydroxylation and cross-linking of collagen. Methods Mol Biol 446:95–108
Yang Y, Richards-Zawacki CL, Devar A, Dugas MB (2016) Poison frog color morphs express assortative mate preferences in allopatry but not sympatry. Evol Int J Org Evol 70(12):2778–2788
Yang Y, Dugas MB, Sudekum HJ, Murphy SN, Richards-Zawacki CL (2018) Male-male aggression is unlikely to stabilize a poison frog polymorphism. J Evol Biol 31(3):457–468
Yang Y, Servedio MR, Richards-Zawacki CL (2019) Imprinting sets the stage for speciation. Nature 574(7776):99–102
Yelon D, Ticho B, Halpern ME, Ruvinsky I, Ho RK, Silver LM, Stainier DY (2000) The bHLH transcription factor hand2 Plays parallel roles in zebrafish heart and pectoral fin development. Development 127(12):2573–2582
Zahn N, James-Zorn C, Ponferrada VG, Adams DS, Grzymkowski J, Buchholz DR, Nascone-Yoder NM et al (2022) Normal table of Xenopus development: a new graphical resource. Development. https://doi.org/10.1242/dev.200356
Zhao L, Liu L, Wang S, Wang H, Jiang J (2016) Transcriptome profiles of metamorphosis in the ornamented pygmy frog Microhyla Fissipes clarify the functions of thyroid hormone receptors in metamorphosis. Sci Rep 6(February):1–11
Acknowledgements
This work was supported by the US National Science Foundation to CLR-Z (1146370), US National Science Foundation to MDM (DEB 1655585), XSEDE XRAC to MDM and AMMS (MCB110134), and startup funds from the University of Houston to AMMS. We are grateful to Anna Tigano for comments on an earlier draft of this manuscript.
Funding
The authors have not disclosed any funding.
Author information
Authors and Affiliations
Contributions
Conceived of the project: AMMS, RN, CR-Z, MDM, Animal work: LF, KAH, YY, CR-Z, Wet lab work: AMMS, Bioinformatics and analyses: AMMS, Initial writing: AMMS, Manuscript revision: AMMS, LF, KAH, YY, RN, CR-Z, MDM.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Consent to participate
Not applicable
Consent for publication
Consenting.
Ethics approval
This work was done in compliance with Institutional Animal Care and Use Committee protocols at University of Pittsburgh (No. 18103943).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stuckert, A.M.M., Freeborn, L., Howell, K.A. et al. Transcriptomic analyses during development reveal mechanisms of integument structuring and color production. Evol Ecol (2023). https://doi.org/10.1007/s10682-023-10256-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10682-023-10256-2