
Abstract
Although family history is a major risk factor for colorectal cancer (CRC) a genetic diagnosis cannot be obtained in over 50 % of familial cases when screened for known CRC cancer susceptibility genes. The genetics of undefined-familial CRC is complex and recent studies have implied additional clinically actionable mutations for CRC in susceptibility genes for other cancers. To clarify the contribution of non-CRC susceptibility genes to undefined-familial CRC we conducted a mutational screen of 114 cancer susceptibility genes in 847 patients with early-onset undefined-familial CRC and 1609 controls by analysing high-coverage exome sequencing data. We implemented American College of Medical Genetics and Genomics standards and guidelines for assigning pathogenicity to variants. Globally across all 114 cancer susceptibility genes no statistically significant enrichment of likely pathogenic variants was shown (6.7 % cases 57/847, 5.3 % controls 85/1609; P = 0.15). Moreover there was no significant enrichment of mutations in genes such as TP53 or BRCA2 which have been proposed for clinical testing in CRC. In conclusion, while we identified genes that may be considered interesting candidates as determinants of CRC risk warranting further research, there is currently scant evidence to support a role for genes other than those responsible for established CRC syndromes in the clinical management of familial CRC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Family history is a major risk factor for colorectal cancer (CRC) with around 15 % of patients reporting having a first-degree relative affected with CRC [1]. Understanding the genetic basis of familial CRC risk is clinically relevant for discriminating between high- and low-risk groups; important not only in defining screening requirements and genetic counselling but increasingly for optimising chemotherapy [2].
Although Lynch syndrome and familial adenomatous polyposis (FAP), caused by inherited germline mismatch repair (MMR) and APC gene mutations respectively, contribute significantly to CRC a genetic diagnosis cannot be obtained in over 50 % of familial cases [3]. While Amsterdam positive families negative for MMR mutations have been labelled as Familial CRC Type X [4] this is merely a descriptive definition and the genetic basis of all forms of undefined-familial CRC is likely to be complex.
Many cancer susceptibility genes are pleiotropic (i.e. influence multiple types of malignancies) [5] and epidemiological studies have reported moderate increases in risk of CRC associated with a number of cancer susceptibility genes. It has been recently suggested that mutations in cancer susceptibility genes not normally considered primarily as determinants of CRC risk such as BRCA2 [6] and TP53 [7] contribute significantly to CRC and are of clinical utility.
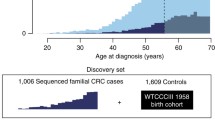
To clarify the contribution of non-CRC cancer susceptibility genes to undefined-familial CRC we report a comprehensive mutational screen of 114 such genes in 847 patients systematically ascertained with early-onset undefined-familial CRC.
Materials and methods
Cancer susceptibility genes evaluated
We evaluated a set of 114 well established cancer susceptibility genes in which rare mutations have been documented to confer high or moderate risk of cancer [5].
Subjects, sequencing and quality control
We report data on 857 unrelated cases of undefined-familial CRC that were negative for a mutation in a known cancer susceptibility genes for CRC. Specifically, there was no evidence of a likely pathogenic mutation in one of the known CRC genes—APC, MLH1, SMAD4, BMPR1A, MUTYH, MSH2, MSH6, PMS2, POLE or POLD1. The cases were derived from a previous whole exome sequencing (WES) gene discovery project based on 1028 familial cases (≥1 first-degree relative) with early-onset CRC (≤55 years) ascertained through the UK National Study of Colorectal Cancer Genetics [8, 9]. To determine the population prevalence of cancer susceptibility gene mutations we analysed WES data on 1644 healthy individuals (with no personal history of malignancy) from the UK 1958 Birth cohort (58BC [10]—974 from the ICR1000 dataset (EGAD00001001021) [11] and an additional 670 individuals all sequenced at The Institute of Cancer Research as per cases.
Full details of sample ascertainment, sequencing pipeline for these samples have been reported [9]. Briefly, samples with non-northern European ancestry, high levels of heterozygosity, sex discrepancy, poor call rate and contamination were excluded. We considered only canonical transcripts and for each variant, assumed the most deleterious predicted effect for each Ensembl transcript according to Variant Effect Predictor [12]. To identify false positives we adopted an automated approach imposing: GQ ≥ 30, for a heterozygous call an alternate depth ≥3 and χ2 < 10.83 (i.e. P > 0.0001) for the observed versus expected distribution of alternate/reference alleles (alt-ref-ratio), UCSC alignability (100 bp window size) = 1, not in simple repeat, Hardy–Weinberg Equilibrium (HWE) test (P > 1.0 × 10−8) in cases and controls and an overall call rate ≥75 % in both cases and controls. We evaluated the fidelity of sequencing in 1332 samples which had also been genotyped using Illumina HumanExome-12v1_A Beadchip arrays (Illumina, San Diego, CA, USA). Specificity and sensitivity of across all alleles with MAF < 0.05 was >99.99 % and 78.4 % respectively for filtered variants.
Interpretation of variant pathogenicity
We implemented the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for determining the pathogenicity of variants [13]. ACMG definitions are contingent on the population frequencies of variants. Here we utilised frequency information from the non-Finnish European Exome Aggregation Consortium (NFE-ExAC), excluding The Cancer Genome Atlas (TCGA) samples when appropriate (nonTCGA-NFE-ExAC). For assigning pathogenicity of novel missense mutations ACMG requires the rate of benign missense variation to be determined in genes and functional domains (PM1 and PP2). ACMG defines mutations as benign if seen in >5 % of the population (BA1). We exploited this definition, counting the frequency of missense mutations in our dataset with NFE-ExAC frequency >5 %, to quantify the rate of benign missense variation. When normalised for protein length, none of the cancer susceptibility genes evaluated in this study fell outside of two standard deviations from the average mutational rate of 1.2 mutations per 1000 amino acids (<6.6). ACMG also requires novel mutations to conform to established mechanisms of action for each gene, therefore established mechanisms were determined by mining pathogenic/likely pathogenic (P/LP) mutations for any disease in ClinVar (NFE-ExAC < 1 %) and combined with annotations from the expertly curated Cosmic Gene Census [14] and where available relevant literature.
Definition of pathogenic/likely pathogenic (P/LP) variants: We required all variants to be rare with a nonTCGA-NFE-ExAC frequency <0.01 % for dominantly acting genes or <0.5 % for genes with evidence of recessive action. We automatically included variants marked as “pathogenic” or “likely pathogenic” in ClinVar which met the above frequency conditions and where ClinVar annotations were not conflicted. Novel loss of function (LOF) and splice site variants met the criteria for likely pathogenic only when the mechanism of action (e.g. splice site) was established for that gene. Only canonical transcripts were considered. LOF variants at the extreme 3′ end of the gene were excluded from analysis (final coding 5 % [15]). Missense mutations were included where the same amino acid change was observed as a previously established P/LP variant regardless of nucleotide change.
Definition of novel likely pathogenic (LP) missense set: We also defined a set of novel (not documented in Clinvar) likely pathogenic missense mutations requiring, in addition to the criteria outlined above: (1) a consensus that the mutation is deleterious in a minimum of 6/8 computational tools calculated via ANNOVAR [16] (satisfying PP3 criteria; SIFT, Polyphen-pp2hvar, LRT, MutationTaster, MutationAssessor, FATHMM, RadialSVM and LR) (2) that the variant is in a gene with a low rate of benign missense variation (PP2) (3) located in PFAM domain with documented P/LP variants with no benign variation (PM1) and (4) for dominantly acting variants only: absent from the nonTCGA-NFE-ExAC population (PM2).
P values, where reported, were calculated using a two-sided Fisher’s exact test in R [17].
Results and discussion
Overall 6.7 % of the undefined-familial CRC cases (57/847) and 5.3 % of the controls (85/1609) were identified as being a carrier of a P/LP mutation in one of the 114 cancer susceptibility genes surveyed (Table 1; Supplementary Table 2). Globally the difference is not statistically significant implying that pleiotropic effects across cancer susceptibility genes are not widespread with respect to CRC and certainly rare and/or not highly penetrant. Furthermore, after correcting for multiple testing, no individual gene was significantly mutated in undefined-familial CRC cases. This does not preclude the possibility that some of the mutations we have identified are causal, but does place bounds on their prevalence and clinical utility.
Three cases (0.3 %) were identified as being carriers of P/LP mutations in the folliculin gene FLCN: a stop gain (p.Ser386Ter), splice donor (c.1432 + 1G>A) and a frameshift variant (p.Glu297AlafsTer25) catalogued by ClinVar as pathogenic (Table 2). These mutations are extremely rare with only the frameshift variant present in a single sample in nonTCGA-NFE-ExAC. FLCN is a highly conserved gene and recent computational methods have predicted FLCN to be intolerant to LOF variants (analysis of protein-coding genetic variation in 60,706 humans, Lek et al. preprint). While mutations in FLCN cause Birt-Hogg-Dube syndrome (BHD) [18], and are seen in 5 % of familial renal cell cancer patients suggesting a role in cancer predisposition [19], none of the gene carriers we identified had a personal or family history of renal cancer. Although recent data has been conflicted as to whether there is an increased incidence of CRC associated with BHD, Nahorski et al. [20] have reported FLCN deactivation contributes to colorectal tumourigenesis with somatic frameshift mutations being identifiable in 23 % of microsatellite instable CRC.
A further three cases had P/LP frameshift mutations in the nucleotide excision repair (NER) gene ERCC3; two cases with p.Gln586ArgfsTer25 and a single sample with p.Asp474GlufsTer2 (Table 2). We observe a LP mutation in a single control: p.Arg109Ter. ERCC3 forms a subunit of the basal transcription factor 2 (TFIIH, Table 1) and is associated with Xeroderma pigmentosum B [21], Cockayne’s syndrome [22] and trichothiodystrophy [23]. A variant in the related NER protein ERCC6 was recently suggested as a candidate for familial CRC following exome sequencing with functional data supporting a reduction in capacity for repairing DNA double strand breaks [24]. In transcription coupled (TC)-NER, blockage of transcribing RNA Polymerase II (RNA-Pol II) on the damaged DNA template is thought to initiate the repair reaction in a process that requires ERCC6 in combination with ERCC2 (TFIIH subunit), ERCC3 (TFIIH subunit), ERCC1-ERCC4 (XPF), ERCC5 (XPG), ERCC8 (CSA) and XPA. We observed P/LP mutations in six of these genes in total identifying 13 in cases and eight in controls (Table 2, 1.5 vs 0.5 %, P = 0.011; 15 vs 9 including LP missense, P = 0.008). We identify two patients with first degree relatives with malignant melanoma of the skin (ERCC4 p.Arg468AspfsTer25 and ERCC6 p.Thr699HisfsTer61). With the exception of ERCC6 thus far mutations of the NER genes have not been implicated as risk factors for CRC. It is however noteworthy that ERRC1 expression has been shown to be reduced by 84–100 % in CRC [25, 26].
Three cases and one control were found to harbour rare novel LOF mutations in the recessively acting Bloom syndrome gene BLM (Table 2): one case with the frameshift mutation p.Cys361Ter, another with the stop gain mutation p.Gln645Ter and an additional case and control both having the stop gain mutation p.Arg899Ter catalogued as pathogenic and likely pathogenic in ClinVar for hereditary cancer predisposition syndrome and Bloom’s respectively. An additional rare missense mutation meeting ACMG guidelines for likely pathogenic variants was identified in the cases (p.Arg808Cys, no such variants in controls). While the family histories of these samples are varied, only one Bloom’s related malignancy (myeloma) was reported in the mother of an affected individual where the father was diagnosed with CRC. While our results could be considered to support a possible role for BLM in CRC risk, we do not observe as high a frequency of P/LP mutations as a recent study which found enrichment in early-onset CRC patients with deleterious BLM mutations (1.6 % of patients and 0.02 % controls [27]).
With respect to BRCA1/2 mutations we observed a range of mutation types including ten frameshift (6 cases, 5 controls) three stop gained (1 case, 2 controls) and two missense (1 case, 1 control) with the majority of variants (12/15) documented as pathogenic by ClinVar (0.9 % vs 0.5 %, P = 0.20, Table 2). Whilst 5/8 of cases had a family history of breast and/or ovarian cancer, in three cases CRC was observed in the father with the BRCA associated cancer observed in the mother. Of these three patients, one carried BRCA1:p.Gly1077AlafsTer8 and BRCA2:p.Ser1230LeufsTer9, both catalogued as pathogenic in ClinVar and absent in nonTCGA-NFE-ExAC.
Of the other cancer susceptibility genes recently implicated in CRC, including WRN [24], SMARCA4 [28], AXIN2 [29, 30] and TP53 [7], we only identified a single case with a mutation: TP53 p.Glu68Ter. This case, a male aged 48 at diagnosis, had no personal history of other cancers or a family history of any Li-Fraumeni associated malignancy. Our results do not support the recent assertion of a clinically important role for TP53 in CRC [7]. It is however noteworthy that the TP53 mutations reported by Yurgelun et al. [7] were all (bar one) predicted benign missense changes and no reference to gene burden in a comparison with controls was performed.
Conclusion
In a large number of patients with familial CRC no alteration in any known CRC susceptibility gene can be identified. An explanation of their susceptibility is a priority in order to offer accurate genetic counselling and determine appropriate screening and/or treatment. Many cancer susceptibility genes have pleiotropic effects increasing the risk of a spectrum of cancers to varying degrees [5]. Hence the suggestion that non-CRC cancer susceptibility genes contribute to familial CRC is an attractive proposition.
However the risks for the minor type of cancer are in general modest with, for example, studies suggesting a 20–60 % increase in risk associated with BRCA2 mutations and cancers outside of breast and ovarian [6]. This is in contrast to mutations in genes such as TP53 that are typified by a constellation of cancers in the same family. In addition to the phenotypic variability associated with the classical dominantly acting cancer susceptibility genes there is evidence of increased cancer risk in carriers of recessive cancer syndrome mutations; exemplified by heterozygous ATM mutations associated with a two-fold elevated breast cancer risk [31]. The magnitude of these effects are therefore insufficient to result in families segregating only the minor tumour. While this means cancer susceptibility gene pleiotropism will not significantly account for undefined-familial CRC families per se such effects have the potential to impact on the overall burden of CRC.
Even accepting the potential inflation introduced through using cancer free controls, when considering all 114 cancer susceptibility genes, we did not observe a significant difference in frequency of pathogenic mutations between cases and controls. Although only nominally significant we did identify P/LP mutations in a number of interesting candidate genes including FLCN, BLM, ERCC-genes and BRCA1/2 as possible determinants of CRC risk.
Accurately ascribing pathogenicity to variants is a key challenge in interpreting sequencing data and we must be cognisant of variation that is disregarded or missing such as splice region or copy number variation. While our estimated frequency of P/LP mutations broadly fits with epidemiological estimates, it is likely that some of the cancer susceptibility genes included in this analysis are able to tolerate apparent LOF variants. Indeed we observed some over-representation of BRIP1 P/LP mutations amongst cases (6 cases, 5 controls) and a study by Seal et al. [32] had reported rare truncating mutations in BRIP1 with increased breast cancer risk. However a large replication effort of the most common truncating variant found no evidence to support an association between BRIP1 with breast cancer [33].
While we identified genes that may be considered interesting candidates for further research our inability to replicate other recent studies highlights the caution required when interpreting such research. Additional much larger data sets, familial studies and/or functional follow up would be required to confirm the role and scope of cancer susceptibility genes outside of those already clearly established with heritable CRC syndromes. However, compared to contemporaneous research efforts, a major strength of our study is its size enabling us to explore the maximum likely impact of non-CRC cancer susceptibility genes to undefined-familial CRC. In conclusion there is currently scant evidence to support a role for genes other than those responsible for established CRC syndromes in the clinical management of CRC patients. While testing for such genes has no immediate clinical utility, the accumulation of such data, in combination with functional studies and familial segregation, has the potential to robustly determine the role of these genes in CRC aetiology. Furthermore, as the cost of high throughput sequencing continues to reduce, such efforts may become economically justifiable.
References
Fuchs CS, Giovannucci EL, Colditz GA, Hunter DJ, Speizer FE, Willett WC (1994) A prospective study of family history and the risk of colorectal cancer. N Engl J Med 331(25):1669–1674. doi:10.1056/NEJM199412223312501
Lee GH, Payne SJ, Melville A, Clark SK (2014) Genetic testing in inherited polyposis syndromes—how and why? Colorectal Dis 16(8):595–602. doi:10.1111/codi.12600
Chubb D, Broderick P, Frampton M, Kinnersley B, Sherborne A, Penegar S, Lloyd A, Ma YP, Dobbins SE, Houlston RS (2015) Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J Clin Oncol 33(5):426–432. doi:10.1200/JCO.2014.56.5689
Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, Gallinger S, Bapat B, Aronson M, Hopper J, Jass J, LeMarchand L, Grove J, Potter J, Newcomb P, Terdiman JP, Conrad P, Moslein G, Goldberg R, Ziogas A, Anton-Culver H, de Andrade M, Siegmund K, Thibodeau SN, Boardman LA, Seminara D (2005) Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 293(16):1979–1985. doi:10.1001/jama.293.16.1979
Rahman N (2014) Realizing the promise of cancer predisposition genes. Nature 505(7483):302–308. doi:10.1038/nature12981
Friedenson B (2005) BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. Med Gen Med 7(2):60
Yurgelun MB, Masciari S, Joshi VA, Mercado RC, Lindor NM, Gallinger S, Hopper JL, Jenkins MA, Buchanan DD, Newcomb PA, Potter JD, Haile RW, Kucherlapati R, Syngal S, Colon Cancer Family R (2015) Germline TP53 Mutations in Patients with early-onset colorectal cancer in the colon cancer family registry. JAMA Oncol 1(2):214–221. doi:10.1001/jamaoncol.2015.0197
Penegar S, Wood W, Lubbe S, Chandler I, Broderick P, Papaemmanuil E, Sellick G, Gray R, Peto J, Houlston R (2007) National study of colorectal cancer genetics. Br J Cancer 97(9):1305–1309. doi:10.1038/sj.bjc.6603997
Chubb D, Broderick P, Dobbins SE, Frampton M, Kinnersley B, Penegar S, Price A, Ma YP, Sherborne AL, Palles C, Timofeeva MN, Bishop DT, Dunlop MG, Tomlinson I, Houlston RS (2016) Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun. doi:10.1038/ncomms11883
Power C, Elliott J (2006) Cohort profile: 1958 British birth cohort (National Child Development Study). Int J Epidemiol 35(1):34–41. doi:10.1093/ije/dyi183
Ruark E, Münz M, Renwick A, Clarke M, Ramsay E, Hanks S, Mahamdallie S, Elliott A, Seal S, Strydom A, Gerton L, Rahman N (2015) The ICR1000 UK exome series: a resource of gene variation in an outbred population. F1000Res 4:883. doi:10.12688/f1000research.7049.1
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F (2010) Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26(16):2069–2070. doi:10.1093/bioinformatics/btq330
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. doi:10.1038/gim.2015.30
Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR (2004) A census of human cancer genes. Nat Rev Cancer 4(3):177–183. doi:10.1038/nrc1299
MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZD, Conrad DF, Lunter G, Zheng H, Ayub Q, DePristo MA, Banks E, Hu M, Handsaker RE, Rosenfeld JA, Fromer M, Jin M, Mu XJ, Khurana E, Ye K, Kay M, Saunders GI, Suner MM, Hunt T, Barnes IH, Amid C, Carvalho-Silva DR, Bignell AH, Snow C, Yngvadottir B, Bumpstead S, Cooper DN, Xue Y, Romero IG, Genomes Project C, Wang J, Li Y, Gibbs RA, McCarroll SA, Dermitzakis ET, Pritchard JK, Barrett JC, Harrow J, Hurles ME, Gerstein MB, Tyler-Smith C (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science 335(6070):823–828. doi:10.1126/science.1215040
Yang H, Wang K (2015) Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 10(10):1556–1566. doi:10.1038/nprot.2015.105
Team RC (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing. https://www.R-project.org
Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P, Pavlovich CP, Sharma N, Walther M, Munroe D, Hill R, Maher E, Greenberg C, Lerman MI, Linehan WM, Zbar B, Schmidt LS (2002) Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt–Hogg–Dube syndrome. Cancer Cell 2(2):157–164
Woodward ER, Ricketts C, Killick P, Gad S, Morris MR, Kavalier F, Hodgson SV, Giraud S, Bressac-de Paillerets B, Chapman C, Escudier B, Latif F, Richard S, Maher ER (2008) Familial non-VHL clear cell (conventional) renal cell carcinoma: clinical features, segregation analysis, and mutation analysis of FLCN. Clin Cancer Res 14(18):5925–5930. doi:10.1158/1078-0432.CCR-08-0608
Nahorski MS, Lim DH, Martin L, Gille JJ, McKay K, Rehal PK, Ploeger HM, van Steensel M, Tomlinson IP, Latif F, Menko FH, Maher ER (2010) Investigation of the Birt–Hogg–Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J Med Genet 47(6):385–390. doi:10.1136/jmg.2009.073304
Weeda G, van Ham RC, Vermeulen W, Bootsma D, van der Eb AJ, Hoeijmakers JH (1990) A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne’s syndrome. Cell 62(4):777–791
Vermeulen W, Scott RJ, Rodgers S, Muller HJ, Cole J, Arlett CF, Kleijer WJ, Bootsma D, Hoeijmakers JH, Weeda G (1994) Clinical heterogeneity within xeroderma pigmentosum associated with mutations in the DNA repair and transcription gene ERCC3. Am J Hum Genet 54(2):191–200
Cleaver JE, Thompson LH, Richardson AS, States JC (1999) A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat 14(1):9–22. doi:10.1002/(SICI)1098-1004(1999)14:1<9::AID-HUMU2>3.0.CO;2-6
Arora S, Yan H, Cho I, Fan HY, Luo B, Gai X, Bodian DL, Vockley JG, Zhou Y, Handorf EA, Egleston BL, Andrake MD, Nicolas E, Serebriiskii IG, Yen TJ, Hall MJ, Golemis EA, Enders GH (2015) Genetic variants that predispose to DNA double-strand breaks in lymphocytes from a subset of patients with familial colorectal carcinomas. Gastroenterology 149(7):1872–1883 e1879. doi:10.1053/j.gastro.2015.08.052
Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM, Stern S, Oatman N, Banerjee B, Bernstein C (2012) Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer. Genome Integr 3(1):3. doi:10.1186/2041-9414-3-3
Smith DH, Fiehn AM, Fogh L, Christensen IJ, Hansen TP, Stenvang J, Nielsen HJ, Nielsen KV, Hasselby JP, Brunner N, Jensen SS (2014) Measuring ERCC1 protein expression in cancer specimens: validation of a novel antibody. Sci Rep 4:4313. doi:10.1038/srep04313
de Voer RM, Hahn MM, Mensenkamp AR, Hoischen A, Gilissen C, Henkes A, Spruijt L, van Zelst-Stams WA, Kets CM, Verwiel ET, Nagtegaal ID, Schackert HK, van Kessel AG, Hoogerbrugge N, Ligtenberg MJ, Kuiper RP (2015) Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci Rep 5:14060. doi:10.1038/srep14060
Esteban-Jurado C, Vila-Casadesus M, Garre P, Lozano JJ, Pristoupilova A, Beltran S, Munoz J, Ocana T, Balaguer F, Lopez-Ceron M, Cuatrecasas M, Franch-Exposito S, Pique JM, Castells A, Carracedo A, Ruiz-Ponte C, Abuli A, Bessa X, Andreu M, Bujanda L, Caldes T, Castellvi-Bel S (2015) Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet Med 17(2):131–142. doi:10.1038/gim.2014.89
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P (2004) Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 74(5):1043–1050. doi:10.1086/386293
Lejeune S, Guillemot F, Triboulet JP, Cattan S, Mouton C, Porchet N, Manouvrier S, Buisine MP (2006) Low frequency of AXIN2 mutations and high frequency of MUTYH mutations in patients with multiple polyposis. Hum Mutat 27(10):1064. doi:10.1002/humu.9460
Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Breast Cancer Susceptibility C, Easton DF, Stratton MR, Rahman N (2006) ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 38(8):873–875. doi:10.1038/ng1837
Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Breast Cancer Susceptibility C, Easton DF, Stratton MR, Rahman N (2006) Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 38(11):1239–1241. doi:10.1038/ng1902
Easton DF, Lesueur F, Decker B, Michailidou K, Li J, Allen J, Luccarini C, Pooley KA, Shah M, Bolla MK, Wang Q, Dennis J, Ahmad J, Thompson ER, Damiola F, Pertesi M, Voegele C, Mebirouk N, Robinot N, Durand G, Forey N, Luben RN, Ahmed S, Aittomaki K, Anton-Culver H, Arndt V, Australian Ovarian Cancer Study G, Baynes C, Beckman MW, Benitez J, Van Den Berg D, Blot WJ, Bogdanova NV, Bojesen SE, Brenner H, Chang-Claude J, Chia KS, Choi JY, Conroy DM, Cox A, Cross SS, Czene K, Darabi H, Devilee P, Eriksson M, Fasching PA, Figueroa J, Flyger H, Fostira F, Garcia-Closas M, Giles GG, Glendon G, Gonzalez-Neira A, Guenel P, Haiman CA, Hall P, Hart SN, Hartman M, Hooning MJ, Hsiung CN, Ito H, Jakubowska A, James PA, John EM, Johnson N, Jones M, Kabisch M, Kang D, kConFab I, Kosma VM, Kristensen V, Lambrechts D, Li N, Lifepool I, Lindblom A, Long J, Lophatananon A, Lubinski J, Mannermaa A, Manoukian S, Margolin S, Matsuo K, Meindl A, Mitchell G, Muir K, Investigators N, Nevelsteen I, van den Ouweland A, Peterlongo P, Phuah SY, Pylkas K, Rowley SM, Sangrajrang S, Schmutzler RK, Shen CY, Shu XO, Southey MC, Surowy H, Swerdlow A, Teo SH, Tollenaar RA, Tomlinson I, Torres D, Truong T, Vachon C, Verhoef S, Wong-Brown M, Zheng W, Zheng Y, Nevanlinna H, Scott RJ, Andrulis IL, Wu AH, Hopper JL, Couch FJ, Winqvist R, Burwinkel B, Sawyer EJ, Schmidt MK, Rudolph A, Dork T, Brauch H, Hamann U, Neuhausen SL, Milne RL, Fletcher O, Pharoah PD, Campbell IG, Dunning AM, Le Calvez-Kelm F, Goldgar DE, Tavtigian SV, Chenevix-Trench G (2016) No evidence that protein truncating variants in BRIP1 are associated with breast cancer risk: implications for gene panel testing. J Med Genet. doi:10.1136/jmedgenet-2015-103529
Acknowledgments
This work was supported by Cancer Research UK Research (C1298/A8362, Bobby Moore Fund for Cancer Research UK) and the European Union (FP7/207-2013) under Grant No. 258236, FP7 collaborative project. Systems Biology of Colorectal Cancer provided principal funding for this study. D.C. was funded by a grant from Bloodwise. Additional support was provided by the National Cancer Research Network and the National Health Service (NHS). This study makes use of the ICR1000 UK exome series data generated by Professor Nazneen Rahman’s team at The Institute of Cancer Research, London. This work made use of samples generated by the 1958 Birth Cohort. Access to these resources was enabled via the 58READIE Project funded by Wellcome Trust and Medical Research Council (Grant Numbers WT095219MA and G1001799).
Author contributations
Conception and design: Sara E. Dobbins, Peter Broderick, Daniel Chubb, Richard S. Houlston, Collection and assembly of data: Daniel Chubb, Peter Broderick, Amy Sherborne, Ben Kinnersley, Sara E. Dobbins. Data analysis and interpretation: Daniel Chubb, Ben Kinnersley, Sara E. Dobbins, Richard S. Houlston. Manuscript writing: All authors. Final approval of manuscript: All authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Sara E. Dobbins, Peter Broderick and Daniel Chubb have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dobbins, S.E., Broderick, P., Chubb, D. et al. Undefined familial colorectal cancer and the role of pleiotropism in cancer susceptibility genes. Familial Cancer 15, 593–599 (2016). https://doi.org/10.1007/s10689-016-9914-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-016-9914-4