
Abstract
Germline pathogenic variants in the DNA mismatch repair (MMR) genes (Lynch syndrome) predispose to colorectal (CRC) and endometrial (EC) cancer. However, mosaic variants in the MMR genes have been rarely described. We identified a likely de novo mosaic MSH6:c.1135_1139del p.Arg379* pathogenic variant in a patient diagnosed with suspected Lynch syndrome/Lynch-like syndrome. The patient developed MSH6-deficient EC and CRC at 54 and 58 years of age, respectively, without a detectable germline MMR pathogenic variant. Multigene panel sequencing of tumor and blood-derived DNA identified an MSH6 somatic mutation (MSH6:c.1135_1139del p.Arg379*) common to both the EC and CRC, raising suspicion of mosaicism. A droplet digital polymerase chain reaction (ddPCR) assay detected the MSH6 variant at 5.34% frequency in normal colonic tissue, 3.49% in saliva and 1.64% in blood DNA, demonstrating the presence of the MSH6 variant in all three germ layers. This study highlights the utility of tumor sequencing to guide sensitive ddPCR testing to detect low-level mosaicism in the MMR genes. Further investigation of the prevalence of MMR mosaicism is needed to inform routine diagnostic approaches and genetic counselling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lynch syndrome is caused by germline pathogenic variants in one of the DNA mismatch repair (MMR) genes where carriers have an increased risk of developing colorectal (CRC) and endometrial (EC) cancer, among other cancers. Mosaicism of hereditary CRC genes is not uncommon [1], but mosaicism in the MMR genes is thought to be rare with only a few cases reported to date (Table 1) [2,3,4,5]. Here, we report the first case of a mosaic MSH6 gene pathogenic variant in an EC- and CRC-affected individual diagnosed with suspected Lynch syndrome.
Case Presentation
The patient (ID_151-1) developed EC at the age of 54 and underwent a total hysterectomy and bilateral salpingo-oophorectomy. The cancer was a well-differentiated (FIGO grade 1) endometrioid adenocarcinoma showing superficial myometrial invasion. There was no evidence of cervical or adnexal involvement. MMR immunohistochemical staining of the tumor showed solitary loss of MSH6 protein expression (Fig. 1a). In December 2016, the patient was referred to a family cancer clinic where germline MMR gene testing was completed by next-generation sequencing (NGS) and multiplex ligation-dependent probe amplification. No clinically actionable germline pathogenic variants in MSH6 or the other MMR genes were identified. Patients with tumor MMR-deficiency without evidence of a germline pathogenic variant or tumor MLH1 methylation are given a diagnosis of suspected Lynch syndrome/Lynch-like syndrome, also referred to as unexplained tumor MMR-deficiency, therefore, the patient (ID_151-1) was diagnosed with suspected Lynch syndrome.
In September 2017, the patient developed a second primary cancer within the caecum at the age of 58. A right hemicolectomy was performed to remove a stage IIA high-grade mucinous carcinoma that demonstrated solitary loss of MSH6 protein expression by MMR immunohistochemistry. The personal and family cancer histories are shown in Fig. 1b. The patient had no children. She was referred from the clinic to the ANGELS study (Applying Novel Genomic approaches to Early-onset and suspected Lynch Syndrome colorectal and endometrial cancers) for tumor sequencing [6]. The study was approved by the University of Melbourne human research ethics committee (HREC#1750748) and the institutional review boards at the Austin Health Clinical Genetics Service. All participants in this study signed an ethics-approved consent form.

a DNA mismatch repair immunohistochemical staining of the endometrial tumor showing loss of MSH6 protein expression and retained expression of MLH1, MSH2 and PMS2 proteins. b Display of the family pedigree. The proband (ID_151-1) is indicated by the black arrow. The carrier of the MSH6:c.1135_1139del p.Arg379* pathogenic variant is indicated with a red plus symbol and the two additional family members who were tested were non-carriers indicated with a black minus symbol
Investigations
The patient’s (ID_151-1) EC and CRC tumor tissue DNA and matched blood-derived DNA were tested on a custom-designed multigene panel. Details of the panel sequencing assay and bioinformatic pipeline have been published previously [6]. The mean on-target coverage for the EC, CRC and blood-derived DNA were 489x, 927x and 69x, respectively. The genomic calculated tumor cellularity for the EC and CRC were 21% and 37%, respectively. MANTIS determined both the EC and CRC to be MSI-H, with scores of 0.22 and 0.49 respectively (> 0.16 = MSI-H) [7]. Panel sequencing identified a single MSH6 somatic mutation (NM_000179.2: c.1135_1139del p.Arg379*) at a variant allele frequency (VAF) of 10.1% in the EC and two MSH6 somatic mutations (c.3261del p.Phe1088Serfs*2 and c.1135_1139del p.Arg379*) in the CRC at VAFs of 23% and 18.6%, respectively. The MSH6:c.1135_1139del p.Arg379* mutation, common to both tumors, had a VAF of 2.3% in the matched blood-derived DNA (Table 2; Fig. 2a). No other variants were in common between the EC and CRC. These results suggested the MSH6:c.1135_1139del p.Arg379* mutation was potentially mosaic in at least two germ layers. To exclude the possibility of a sequencing artefact, Sanger sequencing confirmed the presence of the MSH6 mutation in the CRC tumor but did not detect the variant in the normal non-adjacent colonic mucosa or blood DNA samples (Fig. 2b; Table 2).
The MSH6:c.1135_1139del p.Arg379* variant, confirmed as pathogenic in ClinVar and InSiGHT databases, was tested across different germ layer DNA samples from the proband (ID_151-1), father (ID_151-3), sister (ID_151-9) and unrelated controls using a customized ultra-sensitive droplet digital polymerase chain reaction (ddPCR) assay (Table 2). The MSH6 variant was detected at low levels in the normal colonic mucosa (5.3% VAF), saliva (3.5% VAF) and blood (1.6% VAF) DNA from the patient but in none of the controls (#1-#8) (Table 2; Fig. 2c), confirming mosaicism in all three germ layers thus suggesting an early embryonic event post zygosis. The MMR-proficient CRC tissue and non-adjacent normal colonic tissue from the father (ID_151-3) and blood and saliva DNA from the sister (ID_151-9) did not show evidence of the MSH6 variant by ddPCR (Table 2). The mother was deceased prior to study recruitment and could not be screened for the MSH6 variant.

a Integrative Genomics Viewer display of the MSH6:c.1135_1139del p.Arg379* pathogenic variant and its read depth in the patient’s blood, endometrial cancer and colorectal cancer DNA from targeted multigene panel sequencing. b Sanger sequencing analysis of the probands (ID_151-1) colorectal cancer tumor tissue, colonic normal tissue and blood, showing the presence of the MSH6:c.1135_1139del p.Arg379* variant in the tumor tissue but not detectable in the colonic normal or blood-derived DNA. c Results from the droplet digital polymerase chain reaction (ddPCR) assay displaying the abundance of the detected MSH6:c.1135_1139del p.Arg379* variant in different tissue DNA samples from the patient (ID_151-1), but not in the father (ID_151-3), sister (ID_151-9) or controls. The purple line indicates a manually placed threshold. *Only the colorectal tumor tissue was tested
Discussion
Currently, MMR mosaicism appears to be rare with only a handful of cases reported to date [2–5] (Table 1). The MSH6:c.1135_1139del p.Arg379* variant is the first report of a mosaic pathogenic variant in the MSH6 gene and was identified as a somatic mutation in both the EC and CRC following panel testing in a woman presenting with MSH6-deficiency in both her tumors. ddPCR of the MSH6 variant enabled confirmation of mosaicism demonstrating the variant at low-levels in multiple tissue samples encompassing the endoderm (colon), ectoderm (saliva) and mesoderm (blood). This suggests the variant occurred early in embryogenic development and is potentially present in the primordial germ cells. The MSH6 pathogenic variant may therefore be heritable. Since the patient did not have and can no longer have biological offspring, we did not test for gonadal mosaicism but preimplantation genetic testing may be recommended where a patient is planning to have children, since detection of a mosaic pathogenic MMR variant in gonadal cells would increase the risk of cancer for all carrier children. The patient (ID_151-1), now diagnosed with mosaic Lynch syndrome, can undergo risk-appropriate clinical management while the father and sister, who had no evidence of the MSH6 variant in their DNA samples, can now be confirmed as non-carriers and are released from intensive screening surveillance.
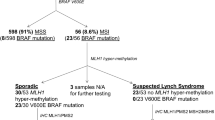
This case of an MSH6 mosaic variant was identified in a person with a diagnosis of suspected Lynch syndrome/Lynch-like syndrome. Tumor testing of suspected Lynch syndrome cases has shown that the predominant etiology is two somatic MMR mutations causing biallelic MMR gene inactivation [5]. When considering who to screen for MMR mosaic variants, cases with somatic MMR mutations in the absence of a germline MMR pathogenic variant are the ideal candidates. Of the MMR mosaic cases identified to date, 3/5 developed multiple Lynch syndrome spectrum cancers (Table 1) raising the suspicion of an undetected germline pathogenic variant. It is likely that the presentation of these MMR mosaicism cases with multiple cancers is a bias of cases selected for mosaicism testing. The ability to test multiple tumors for somatic MMR mutations, both with loss of MSH6 expression, enabled us to target the ddPCR screening to a single variant shared between the tumors. A common MMR mutation in multiple tumors from the same patient may also be indicative of a primary and metastatic lesion, although more than one somatic mutation in common would support this rather than mosaicism. Testing of multiple adenomas to identify a common somatic mutation via the “adenoma first” approach, has been successfully used to identify APC mosaic variants in adenomatous polyposis [8]. This approach has shown APC mosaicism to be a more common mechanism than previously thought in unexplained adenomatous polyposis [1]. Guillerm et al. [5] and Lucia Jansen et al. [1] have both proposed decision tree models for triaging individuals diagnosed with suspected Lynch syndrome for identifying MMR gene mosaicism in patients and first-degree relatives, albeit for the research setting.
This study highlights the importance of screening for mosaicism in patients with a diagnosis of suspected Lynch syndrome and somatic MMR mutations in their tumors. Consistent with literature, this study has shown that Sanger sequencing is not sensitive enough to reliably detect low level variants [9] and alternate, more sensitive methods are required to screen for mosaic variants. The stepwise approach of MMR gene sequencing using NGS methodology in MMR-deficient tumors followed by sensitive ddPCR testing of a specific variant in DNA from multiple tissue sources from different germ layers is a recommended approach moving forward. As tumor screening and sensitive methods such as ddPCR become more widely adopted, the prevalence of MMR mosaicism may also be shown to be higher. As the true prevalence of MMR mosaicism becomes known, improvements to the diagnostic workflow can enable efficient and cost-effective screening approaches to detect all cases of Lynch syndrome, including those with mosaicism.
Data availability
The anonymized data analyzed during the current study is available from the corresponding author on reasonable request.
References
Lucia Jansen AM, Goel A (2020) Mosaicism in patients with Colorectal Cancer or Polyposis Syndromes: a systematic review. Clin Gastroenterol Hepatol 18:1949–1960. https://doi.org/10.1016/j.cgh.2020.02.049
Sourrouille I, Coulet F, Lefevre JH et al (2013) Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer 12:27–33. https://doi.org/10.1007/s10689-012-9568-9
Pastrello C, Fornasarig M, Pin E et al (2009) Somatic mosaicism in a patient with Lynch syndrome. Am J Med Genet 149:212–215. https://doi.org/10.1002/ajmg.a.32620
Geurts-Giele WR, Rosenberg EH, van Rens A et al (2019) Somatic mosaicism by a de novo MLH1 mutation as a cause of Lynch syndrome. Mol Genet Genomic Med 7:e00699. https://doi.org/10.1002/mgg3.699
Guillerm E, Svrcek M, Bardier-Dupas A et al (2020) Molecular tumor testing in patients with Lynch-like syndrome reveals a de novo mosaic variant of a mismatch repair gene transmitted to offspring. Eur J Hum Genet 28:1624–1628. https://doi.org/10.1038/s41431-020-0689-6
Walker R, Mahmood K, Joo JE et al (2023) A tumor focused approach to resolving the etiology of DNA mismatch repair deficient tumors classified as suspected Lynch syndrome. medRxiv. https://doi.org/10.1101/2023.02.27.23285541
Kautto EA, Bonneville R, Miya J et al (2017) Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 8:7452–7463. https://doi.org/10.18632/oncotarget.13918
Jansen AML, Crobach S, Geurts-Giele WRR et al (2017) Distinct patterns of somatic mosaicism in the APC gene in Neoplasms from patients with unexplained adenomatous polyposis. Gastroenterology 152:546–549e3. https://doi.org/10.1053/j.gastro.2016.10.040
Chin EL, da Silva C, Hegde M (2013) Assessment of clinical analytical sensitivity and specificity of next-generation sequencing for detection of simple and complex mutations. BMC Genet 14:6. https://doi.org/10.1186/1471-2156-14-6
Acknowledgements
The authors thank the patient and their family for their support and permission to publish this case report. We further thank members of the Colorectal Oncogenomics Group and members from Austin Health Family Cancer Clinic for their support of this manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. Funding by a National Health and Medical Research Council of Australia (NHMRC) project grant 1125269 (PI- Daniel Buchanan) supported the design, analysis, and interpretation of data. DDB is supported by an NHMRC Investigator grant (GNT1194896), and the University of Melbourne Dame Kate Campbell Fellowship. MAJ is supported by an NHMRC Investigator grant (GNT1195099). RW is supported by the Margaret and Irene Stewardson Fund Scholarship and by the Melbourne Research Scholarship. PG is supported by the University of Melbourne Research Scholarship.
Author information
Authors and Affiliations
Contributions
RW and DDB designed the concept of the study. SJ, MAJ, FAM, IMW, SDP and AC recruited the patient and family members. SJ, MC and SGP collected clinical and biological data. MC, JX, JEJ, JC, SGP and RW performed the molecular analysis. KM and PG ran the bioinformatic pipeline. CR and JC performed and oversaw the histological analysis. JMC provided the initial Python code to generate the pedigree. MC, JX, JEJ, RW, PG, KM and DDB analyzed the data. RW and DDB drafted the manuscript. All authors have approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the Ethics Committee of the University of Melbourne (Melbourne, Australia) and a written informed consent was obtained from all individual participants included in the study.
Consent to participate
A written informed consent was obtained from all individual participants included in the study.
Consent for publication
The participants have consented to the submission of an article about their family to a scientific journal
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Walker, R., Clendenning, M., Joo, J.E. et al. A mosaic pathogenic variant in MSH6 causes MSH6-deficient colorectal and endometrial cancer in a patient classified as suspected Lynch syndrome: a case report. Familial Cancer 22, 423–428 (2023). https://doi.org/10.1007/s10689-023-00337-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-023-00337-0