
Abstract
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive, life-threatening disease characterized by deposition of insoluble amyloid fibrils in the myocardium, resulting in cardiac structural and functional abnormalities and ultimately heart failure. Disease frequency is reportedly lower in women than men, but sex-related differences have not been well established. We conducted a systematic literature review (SLR), based on PRISMA-P guidelines and registered with PROSPERO, to assess whether the epidemiology and clinical presentation of ATTR-CM differ between women and men. MEDLINE, Embase, and Cochrane databases and selected conference proceedings were searched (August 16, 2019) to identify observational and clinical studies reporting sex-specific data for patients with wild-type or hereditary ATTR-CM. Of 193 publications satisfying final eligibility criteria, 69 studies were included in our pooled analysis. Among the 4669 patients with ATTR-CM analyzed, 791 (17%) were women, including 174 (9%), 366 (29%), and 251 (18%) in studies of wild-type, hereditary, and undefined ATTR-CM, respectively. Data available on disease characteristics were limited and very heterogeneous, but trends suggested some cardiac structural/functional differences, i.e., lower interventricular septal and posterior wall thickness and left ventricular (LV) end diastolic diameter, and higher LV ejection fractions, in women versus men across ATTR-CM subtypes. Because LV wall thickness > 12 mm is generally the suggested threshold for ATTR-CM diagnosis in both sexes, smaller cardiac anatomy in women with the disease may lead to underdiagnosis. Additional research and studies are needed to elucidate potential disparities between sexes in ATTR-CM frequency, clinical characteristics, and underlying biological mechanisms. This study was registered within the International Prospective Register of Systematic Reviews (PROSPERO) database of the University of York (CRD42019146995).
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive, often underdiagnosed and misdiagnosed disease caused by the extracellular deposition of misfolded transthyretin protein (TTR) that forms insoluble amyloid fibrils in organs and tissues [1]. Infiltration of the myocardium by amyloid fibrils ultimately results in life-threatening cardiac structural and functional abnormalities and heart failure [1,2,3]. The condition may be inherited as an autosomal dominant trait associated with mutations of the transthyretin gene TTR (hereditary ATTR-CM) or caused by age-dependent, non-mutational mechanisms (wild-type ATTR-CM). The prevalence of hereditary ATTR-CM varies by region due to fluctuating geographic distribution of TTR mutations; the Val122lle mutation, the most common in the USA, has been reported in approximately 3–4% of African Americans [4]. Among hospitalized patients with heart failure and preserved ejection fraction, older than 60 years of age, with left ventricular wall thickness of 12 mm or greater on scintigraphy, the prevalence of wild-type ATTR-CM was 13% [5]. The frequency of ATTR-CM, particularly the wild-type form, is reportedly lower in women than men [6,7,8,9,10,11], and the female sex has been proposed as a protective factor [1]. However, the higher proportions (over 50%) of women with wild-type ATTR observed in some study populations [5, 12] provide theoretical support that ATTR-CM may be further underdiagnosed and/or misdiagnosed in women because of sex-related differences in clinical presentation or disease characteristics [7, 13].
Given the availability of definitive treatments for ATTR-CM, early diagnosis in women, as in all patients, is critical to slowing disease progression and improving outcomes before overt multi-organ dysfunction ensues [13,14,15,16,17]. Despite the multiple diagnostic imaging options currently available, including advanced techniques such as nuclear scintigraphy that allow non-invasive assessment in select populations, and expert consensus recommendations for their use in ATTR-CM [18, 19], diagnosis of the disease remains a challenge. Clinicians’ awareness of cardiac and non-cardiac clinical signs and symptoms, such as unexplained increases in left ventricular (LV) wall thickness on echocardiography, atrial fibrillation, bilateral carpal tunnel syndrome, and lumbar spinal stenosis, should raise suspicion of ATTR-CM and help facilitate prompt diagnosis [7]. However, the presumption of male predominance of wild-type and hereditary ATTR-CM may lower clinical suspicion of the disease when such “red flags” are observed in women.
We conducted a systematic literature review (SLR) to examine the epidemiology of ATTR-CM in women, to evaluate whether the clinical presentation and characteristics of the disease differ between women and men, and to explore potential reasons for these differences.
Methods
The SLR was designed to identify studies that reported sex-specific findings for patients with ATTR-CM, including wild-type and hereditary subtypes. The SLR was conceived and conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses Protocol (PRISMA-P) guidelines [20] and was registered within the International Prospective Register of Systematic Reviews (PROSPERO) database of the University of York (CRD42019146995) [21].
Search strategy and data sources
A comprehensive search strategy was developed to find studies reporting data on women with ATTR-CM in the published literature. Multiple search terms for transthyretin amyloidosis and variations of the wild-type and hereditary subtypes were used in combination with terms for cardiomyopathy and women using the Boolean operator “AND” (Supplementary Table 1). Searches were conducted for studies published in English through August 16, 2019, based on electronic searches of the following databases: MEDLINE®, Embase®, Cochrane Central Register of Controlled Trials, and Cochrane Database of Systematic Reviews. A manual cross-referencing search was also conducted of the PubMed® database. Reference lists of the published literature identified in the database searches, as well as relevant review articles, were manually searched to identify publications that may not have appeared in the original electronic searches. In addition, to capture the most recent and relevant studies on this topic that are not yet published, manual searches were conducted of proceedings from the following conferences: the European Society of Cardiology, Heart Failure Society of America, and International Society of Amyloidosis.
Study screening, selection, and data extraction
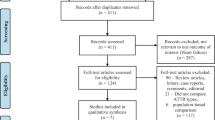
The flow of publications/studies through the search and screening process is summarized in Fig. 1. After removal of duplicate citations in the initial searches, publication titles and abstracts were assessed against inclusion/exclusion criteria in the first of 2 rounds of screening by 2 independent reviewers. In the second screening round, the reviewers examined complete texts of the publications for eligibility. If agreement on discrepancies between the 2 reviewers could not be reached, a third senior investigator reached a final decision. Data from the included publications were extracted by one reviewer and validated by the second reviewer for quality assurance, then reported in pre-specified data-extraction tables. Excluded studies were documented along with the reason for their exclusion.

Flow of publications/studies through the search and screening process
Based on the PICOS (Population, Intervention, Comparison, Outcome, Study design) framework [22], study inclusion criteria encompassed the following: (i) original studies of adult women with ATTR-CM, including subpopulations of women with wild-type and hereditary disease subtypes, (ii) interventional or non-interventional studies with or without comparator, (iii) studies reporting sex-specific data (e.g., demographic characteristics, ATTR genotype, clinical measures, biomarkers, imaging, incidence, frequency, mortality, comorbidities, and hormonal status), and (iv) controlled clinical trials (randomized and non-randomized) and observational studies (prospective and retrospective). Diagnostic criteria used to identify patients with ATTR-CM were based primarily on recommendations of the 2019 Multi-societal Expert Consensus Committee (Supplementary Table 2) [23]. The hereditary subtype of ATTR-CM was confirmed via TTR genopositivity established by DNA analysis or mass spectrometry.
Pooled analysis
Findings from selected included studies were pooled to analyze the proportions of women and men who had wild-type ATTR-CM, hereditary ATTR-CM, and undefined ATTR-CM. Studies that reported sample size were excluded from the pooled analysis if they were found to include data from convenient or pre-specified patient populations, or populations that overlapped with other included studies (e.g., publications from the same author/institution or registry/survey during a similar time frame). When studies overlapped, the publication with the largest reported patient population was retained.
Study quality assessment
The research quality of relevant case series and cohort studies identified in the searches (characteristic publications only) was critically appraised using the Joanna Briggs Institute Critical Appraisal Tool and the Newcastle–Ottawa scale, respectively (Supplementary Table 3). Congress abstracts were not assessed for quality. Studies that received a poor assessment on the risk of bias appraisal were excluded.
Results
Literature search/screening
Among 1170 total publications identified in the initial literature searches (Embase, n = 642; MEDLINE, n = 417; Cochrane, n = 111), 193 publications met the final eligibility criteria, including 185 full-text publications and 8 congress abstracts (Fig. 1). A total of 155 publications provided information only on sample sizes for women and men with ATTR-CM, and 38 provided a description of ATTR-CM characteristics in patients of both sexes, as well as diagnostic techniques. Among the publications reporting sample sizes, 8 (5%) were based on controlled studies (randomized, n = 5; non-randomized, n = 3), and 147 (95%) were based on observational studies (retrospective, n = 87; prospective, n = 60). The ATTR-CM subtype breakdown in these studies was wild-type ATTR-CM, 58 (36%); hereditary ATTR-CM, 62 (40%); and undefined ATTR-CM, 67 (43%). (Some studies included multiple ATTR-CM subtypes.) Among the publications reporting disease characteristics, 30 (79%) were based on case series and 8 (21%) were based on cohort studies. Of these, the ATTR-CM subtype breakdown was wild-type ATTR-CM, 15 (39%); hereditary ATTR-CM, 29 (76%); and undefined ATTR-CM, 5 (13%).
Findings from 69 selected studies (sample size studies, n = 52; disease characteristics studies, n = 17) were pooled to assess ATTR-CM epidemiology. Twenty-five of the pooled studies (36%) included patients with wild-type ATTR-CM, 25 (36%) included patients with hereditary ATTR-CM, and 30 (43%) included patients with undefined ATTR-CM. (As above, some studies included multiple ATTR-CM subtypes.) Studies of wild-type, hereditary, and undefined ATTR-CM included 2027, 1279, and 1363 individuals, respectively.
Frequency of wild-type and hereditary ATTR-CM
Across studies included in the pooled analysis, 791 of 4669 (17%) patients with ATTR-CM were women. In the studies of wild-type, hereditary, and undefined ATTR-CM, 174 (9%), 366 (29%), and 251 (18%) patients were women, respectively. When the proportions of women were analyzed in ranges (i.e., ≤ 10%, > 10–30%, and > 30%), some differences between studies were evident (Fig. 2). For studies of wild-type and undefined ATTR-CM, the majority—92% (23/25) and 77% (23/30), respectively—reported that less than 30% of patients were women. In contrast, for studies of hereditary ATTR-CM, the majority—56% (14/25)—reported that more than 30% of patients were women.

Relative frequency of studies in the pooled analysis that reported women by percentage range (n = 69). (Some studies included multiple ATTR-CM subtypes.) ATTR-CM transthyretin amyloid cardiomyopathy
Sex-related differences in demographic and clinical characteristics
Overall, no marked numerical differences between women and men with ATTR-CM in age or ethnicity were seen across studies that reported these characteristics (Table 1; Supplementary Tables 4–6) [24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. However, some anatomical differences in cardiac structure were identified (Fig. 3a–c); namely, women had numerically lower values for interventricular septal thickness, posterior wall thickness, and LV diastolic diameter compared with men in studies of wild-type [10, 31, 39], hereditary [29, 41,42,43], and undefined ATTR-CM [24, 26]. In addition, LV ejection fraction was numerically higher in women than men in several studies across ATTR-CM genotypes (Fig. 3d) [10, 31, 41, 44]. In studies that reported on survival and mortality [6, 10, 24, 35, 37, 38, 40, 41, 45,46,47], these outcomes did not appear to be different between women and men.

Screening/diagnostic techniques in ATTR-CM
A number of tests may be conducted in patients with suspected ATTR-CM to examine the risk that the condition is present. In the studies included in this review, echocardiography was the most frequently reported test, conducted in 14 of 38 (37%) studies [6, 10, 26, 31,32,33, 37, 39, 41, 42, 44, 48,49,50] (Fig. 4). Clinical symptoms (5 [13%]), magnetic resonance imaging (MRI; 4 [11%]), and Western blot assay (4 [11%]) were other features or techniques used to screen for ATTR-CM in similar proportions of studies.

Techniques used a to confirm a diagnosis of ATTR-CM or b to investigate suspected ATTR-CM (n = 38). ATTR-CM transthyretin amyloid cardiomyopathy, MRI magnetic resonance imaging
Multiple approaches can be taken to establish a diagnosis of ATTR-CM, including endomyocardial or extracardiac biopsy, mass spectrometry, and nuclear scintigraphy; in addition, DNA genotyping can be used to confirm the presence or absence of a TTR gene mutation after the diagnosis is established [18]. The number of published studies including these diagnostic techniques, alone or in combination, is shown in Fig. 4. The most common were cardiac biopsy [6, 10, 24,25,26, 28, 31,32,33,34,35,36,37,38,39, 41, 43, 44, 46, 47, 49,50,51,52,53,54], DNA genotyping [6, 10, 25, 27,28,29,30, 33, 37, 40,41,42,43,44,45, 47, 48, 51,52,53, 55,56,57,58], and extracardiac biopsy, performed in 26 (68%), 24 (63%), and 19 (50%) studies, respectively. Biopsy was not performed in 6 (16%) studies [27, 29, 30, 40, 55, 57], but DNA genotyping was conducted in all 6 of these studies and nuclear scintigraphy in 2 of the 6 studies. Cardiac and extracardiac biopsies were conducted together in 14 (37%) [6, 10, 26, 28, 33, 39, 41, 44, 46, 51,52,53,54] studies, and cardiac biopsy and DNA genotyping were conducted together in 13 (34%) studies [6, 10, 25, 28, 33, 37, 41, 43, 44, 47, 51,52,53].
Discussion
Based on a pooled analysis of 69 studies, among 4669 patients with ATTR-CM, we found that 17% were women and 83% were men. The lowest proportion of women (9%) was observed in studies of wild-type ATTR-CM, whereas the highest (29%) was observed in studies of hereditary ATTR-CM. These findings are consistent with previous reports that ATTR-CM is a condition predominantly identified in men [6, 39], especially for the wild-type versus the hereditary subtype [6,7,8].
The research reviewed in the current publication does not elucidate the potential underlying mechanisms for the predominance of this condition in men or the higher proportion of women presenting with hereditary versus wild-type ATTR-CM, nor does it capture potential bias in reporting. Sex hormones and sex chromosomes have been shown to account for sex differences in cardiovascular disease [59, 60], and findings from several studies suggest that these factors may also help explain discrepancies in the prevalence and onset of ATTR-CM between men and women [41, 54, 61,62,63]. In a study of differences in penetrance of hereditary amyloidosis (i.e., the Val30Met ATTR pathogenic variant), Hellman et al. observed that penetrance of the trait was significantly higher when inherited from the mother than from the father [62]. In an earlier Swedish genealogical study, the age of onset was shown to be significantly higher when the mutation was inherited from the father, and anticipation (i.e., higher penetrance in younger generations) was greater in descendants of mothers with the disease [63]. It is also possible that genetic counseling and mutations identified within families with a history of hereditary disease improve detection of this genotype in women, particularly as women may be more inclined to seek the help of healthcare providers [64,65,66]. These sex-biasing factors are complex, and many other factors, including age and ascertainment bias, are also expected to play a role.
The large degree of heterogeneity between studies reporting characteristics of ATTR-CM prevented meta-analysis of the data, but we observed trends suggestive of potential sex-based differences. Across studies, lower interventricular septal and posterior wall thickness and higher LV ejection fractions were reported in women compared with men. These trends are consistent with reported measurements for normal and abnormal hearts [67]. A suggested cut-off criterion for the diagnosis of ATTR-CM is an LV wall thickness > 12 mm in both sexes [23]. Given that both normal and abnormal cardiac anatomy is smaller in women than men, which our findings suggest is also the case for women and men with ATTR-CM, women with ATTR-CM could be at risk for potential underdiagnosis because they are less likely to meet the diagnostic wall-thickness threshold. Further research is warranted to determine whether use of lower cut-off values or cardiac dimensions indexed by body size for women will affect rates of suspicion and ultimately diagnosis of ATTR-CM in women.
Various approaches may be used to help raise suspicion of ATTR-CM and facilitate earlier identification of the disease, including echocardiogram (ECHO), electrocardiogram (ECG), cardiac magnetic resonance imaging (cMRI), and biomarker clues [18, 23, 68]. ECHO was the most frequently reported test conducted in the studies in this review, reflecting the prominent role this test has traditionally played in the evaluation of cardiac structure and function in patients with troubling heart symptoms. However, the limited sensitivity and specificity of ECHO for ATTR-CM may contribute to missed or late diagnoses of amyloidosis. Although ECG may be useful in identifying ATTR-CM, none of the studies reported use of this diagnostic technique. Similarly, no studies used the cardiac biomarkers N-terminal pro-B-type natriuretic peptide or troponin to confirm the diagnosis of ATTR-CM, although these markers reportedly predict mortality in patients with wild-type ATTR-CM and are thus used for risk stratification [69]. Therefore, our findings suggest that additional guidance on the assessment of patients with suspected ATTR-CM is needed to help raise awareness among clinicians and encourage earlier diagnosis.
In recently published multi-societal expert consensus recommendations, several diagnostic criteria were proposed for ATTR-CM, primarily based on endomyocardial biopsy, extracardiac biopsy, or scintigraphy, as were guidelines for the appropriate use of multimodality imaging [18]. The majority of studies included in the current systematic review were designed and conducted prior to publication of these recommendations/guidelines (in 2019), but the study findings related to diagnostic testing are nonetheless of interest. The use of non-invasive scintigraphy was not commonly reported in this SLR, but substantial research supports its utilization. In a large, multicenter study in > 1000 patients with biopsy-proven cardiac amyloidosis, Gillmore et al. demonstrated that positive nuclear cardiac scintigraphy was 100% specific for ATTR-CM when light-chain amyloidosis (AL) was concurrently ruled out in patients with a visual myocardial uptake grading of 2 and 3 [19]. The range of diagnostic tests reported in the studies included in our review (i.e., cardiac and extracardiac biopsy, mass spectrometry, DNA genotyping, and scintigraphy) does not reveal a breach between the consensus ATTR-CM diagnostic recommendations and practice in clinical research. However, we found some evidence of inconsistencies in the approach: for example, cardiac and extracardiac biopsies were conducted together in 14 (37%) studies, and cardiac biopsy and DNA genotyping were conducted together in 13 (34%) studies. These findings indicate that broader education on the consensus recommendations is needed to encourage more uniform application of assessment and diagnostic criteria.
A number of limitations should be considered when assessing the findings of this SLR, including the small sample sizes of the studies, particularly for women, and the limited data available, particularly in cohort studies. The majority of these data were derived from case series rather than cohort studies, impeding direct comparison of data between women and men. Other limitations include heterogeneity in the stage at which patients were recruited in the studies (i.e., previously diagnosed patients, patients with suspected ATTR-CM, and patients pre-selected for another condition), heterogeneity in the statistical reporting of data (e.g., means vs. medians), and heterogeneity in time frames and duration of follow-up (i.e., from onset or time of biopsy or pre- and post-liver transplant to variable follow-up visit or death). Finally, the criteria used to confirm the presence of ATTR-CM (e.g., interventricular septal thickness > 12 mm, positive biopsy or scintigraphy) may have biased patient selection and may have been inaccurate for identifying women with ATTR-CM.
The results of this literature review indicate that ATTR-CM is a condition predominantly reported in men compared with women. The disparity between sexes in ATTR-CM frequency may be explained by several factors, including potential cardioprotective effects of estrogen, smaller heart structures in women that do not meet diagnostic thresholds for ATTR-CM, and a lack of awareness of red flags for ATTR-CM in women compared with men. Interpretation of the literature was limited by the heterogeneous nature of the studies included and the small patient populations involved, but our findings raise important questions and reinforce the need for additional investigation into sex-related differences in ATTR-CM.
References
Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, Ciliberti P, Biagini E, Salvi F, Branzi A (2010) Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol 7(7):398–408. https://doi.org/10.1038/nrcardio.2010.67
Donnelly JP, Hanna M (2017) Cardiac amyloidosis: an update on diagnosis and treatment. Cleve Clin J Med 84(12 Suppl 3):12–26. https://doi.org/10.3949/ccjm.84.s3.02
Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez-Naharro A, Quarta CC, Rezk T, Whelan CJ, Gonzalez-Lopez E, Lane T, Gilbertson JA, Rowczenio D, Petrie A, Hawkins PN (2018) A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 39(30):2799–2806
Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN (2015) Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid 22(3):171–174. https://doi.org/10.3109/13506129.2015.1051219
González-López E, Gallego-Delgado M, Guzzo-Merello G, de Haro-del Moral FJ, Cobo-Marcos M, Robles C, Bornstein B, Salas C, Lara-Pezzi E, Alonso-Pulpon L, Garcia-Pavia P (2015) Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 36(38):2585–2594. https://doi.org/10.1093/eurheartj/ehv338
Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, Rowczenio DM, Gilbertson JA, Hutt DF, Rezk T, Strehina SG, Caringal-Galima J, Manwani R, Sharpley FA, Wechalekar AD, Lachmann HJ, Mahmood S, Sachchithanantham S, Drage EPS, Jenner HD, McDonald R, Bertolli O, Calleja A, Hawkins PN, Gillmore JD (2019) Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 140(1):16–26
Yamamoto H, Yokochi T (2019) Transthyretin cardiac amyloidosis: an update on diagnosis and treatment. ESC Heart Fail 6(6):1128–1139. https://doi.org/10.1002/ehf2.12518
Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen AV, Damy T, Plante-Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C (2016) Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol 68(2):161–172. https://doi.org/10.1016/j.jacc.2016.03.596
Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SDJ, Venner CP, Wassef N, McCarthy CA, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ (2013) Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2(2):e000098
Gonzalez-Lopez E, Gagliardi C, Dominguez F, Quarta CC, De Haro-Del Moral FJ, Milandri A, Salas C, Cinelli M, Cobo-Marcos M, Lorenzini M, Lara-Pezzi E, Foffi S, Alonso-Pulpon L, Rapezzi C, Garcia-Pavia P (2017) Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J 38(24):1895–1904
Connors LH, Sam F, Skinner M, Salinaro F, Sun F, Ruberg FL, Berk JL, Seldin DC (2016) Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation 133(3):282–290. https://doi.org/10.1161/circulationaha.115.018852
Winburn I, Ishii T, Sumikawa T, Togo K, Yasunaga H (2019) Estimating the prevalence of transthyretin amyloid cardiomyopathy in a large in-hospital database in Japan. Cardiol Ther 8:297–316. https://doi.org/10.1007/s40119-019-0142-5
Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, Gospodinova M, Obici L, Rapezzi C, Garcia-Pavia P (2019) Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail 7(8):709–716. https://doi.org/10.1016/j.jchf.2019.04.010
Alnylam (2019) APOLLO-B: a study to evaluate patisiran in participants with transthyretin amyloidosis with cardiomyopathy (ATTR amyloidosis with cardiomyopathy) (NCT03997383) (2019) https://clinicaltrials.gov/ct2/show/NCT03997383. Accessed 31 October 2019
Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, Grogan M, Selby VN, Jacoby D, Hanna M, Nativi-Nicolau J, Patel J, Rao S, Sinha U, Turtle CW, Fox JC (2019) Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol 74(3):285–295. https://doi.org/10.1016/j.jacc.2019.03.012
Falk RH (2018) 24 month open label study of the tolerability and efficacy of inotersen in TTR amyloid cardiomyopathy patients (NCT03702829). https://clinicaltrials.gov/ct2/show/NCT03702829. Accessed 31 October 2019
Pfizer (2019) US FDA approves Vyndaqel® and Vyndamax™ for use in patients with transthyretin amyloid cardiomyopathy, a rare and fatal disease (press release). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_vyndaqel_and_vyndamax_for_use_in_patients_with_transthyretin_amyloid_cardiomyopathy_a_rare_and_fatal_disease. Accessed 30 June 2019
Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans A, Hanna MA, Hazenberg BPC, Kristen AV, Kwong RY, Maurer MS, Merlini G, Miller EJ, Moon JC, Murthy VL, Quarta CC, Rapezzi C, Ruberg FL, Shah SJ, Slart R, Verberne HJ, Bourque JM (2019) ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. J Card Fail 25(11):e1–e39. https://doi.org/10.1016/j.cardfail.2019.08.001
Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, Lachmann HJ, Bokhari S, Castano A, Dorbala S, Johnson GB, Glaudemans AW, Rezk T, Fontana M, Palladini G, Milani P, Guidalotti PL, Flatman K, Lane T, Vonberg FW, Whelan CJ, Moon JC, Ruberg FL, Miller EJ, Hutt DF, Hazenberg BP, Rapezzi C, Hawkins PN (2016) Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24):2404–2412. https://doi.org/10.1161/circulationaha.116.021612
Shamseer L, Moher D, Clarke M, Ghersi D, Liberati A, Petticrew M, Shekelle P, Stewart LA (2015) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ 350:g7647. https://doi.org/10.1136/bmj.g7647
National Institute for Health Research, PROSPERO International Prospective Register of Systematic Reviews (2019) A systematic review to identify and understand gender characteristics in men and women with transthyretin amyloidosis with cardiomyopathy (ATTR-CM). https://www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=146995]. Accessed June 22 2020
Schardt C, Adams MB, Owens T, Keitz S, Fontelo P (2007) Utilization of the PICO framework to improve searching PubMed for clinical questions. BMC Med Inform Decis Mak 7:16. https://doi.org/10.1186/1472-6947-7-16
Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans A, Hanna MA, Hazenberg BPC, Kristen AV, Kwong RY, Maurer MS, Merlini G, Miller EJ, Moon JC, Murthy VL, Quarta CC, Rapezzi C, Ruberg FL, Shah SJ, Slart R, Verberne HJ, Bourque JM (2019) ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 2 of 2-diagnostic criteria and appropriate utilization. J Card Fail 25(11):854–865. https://doi.org/10.1016/j.cardfail.2019.08.002
Galat A, Guellich A, Bodez D, Slama M, Dijos M, Zeitoun DM, Milleron O, Attias D, Dubois-Rande JL, Mohty D, Audureau E, Teiger E, Rosso J, Monin JL, Damy T (2016) Aortic stenosis and transthyretin cardiac amyloidosis: the chicken or the egg? Eur Heart J 37(47):3525–3531
Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN (1997) Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med 336(7):466–473
Wizenberg TA, Muz J, Sohn YH, Samlowski W, Weissler AM (1982) Value of positive myocardial technetium-99m-pyrophosphate scintigraphy in the noninvasive diagnosis of cardiac amyloidosis. Am Heart J 103(4 Pt 1):468–473. https://doi.org/10.1016/0002-8703(82)90331-3
Choi K, Seok JM, Kim BJ, Choi YC, Shin HY, Sunwoo IN, Kim DS, Sung JJ, Lee GY, Jeon ES, Kim NH, Min JH, Oh J (2018) Characteristics of South Korean patients with hereditary transthyretin amyloidosis. J Clin Neurol (Korea) 14(4):537–541
Damy T, Costes B, Hagege AA, Donal E, Eicher JC, Slama M, Guellich A, Rappeneau S, Gueffet JP, Logeart D, Planté-Bordeneuve V, Bouvaist H, Huttin O, Mulak G, Dubois-Rand JL, Goossens M, Canoui-Poitrine F, Buxbaum JN (2016) Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J 37(23):1826–1834
Morner S, Hellman U, Suhr OB, Kazzam E, Waldenstrom A (2005) Amyloid heart disease mimicking hypertrophic cardiomyopathy. J Intern Med 258(3):225–230
Vermeer AMC, Janssen A, Boorsma PC, Mannens M, Wilde AAM, Christiaans I (2017) Transthyretin amyloidosis: a phenocopy of hypertrophic cardiomyopathy. Amyloid 24(2):87–91. https://doi.org/10.1080/13506129.2017.1322573
aus dem Siepen F, Bauer R, Voss A, Hein S, Aurich M, Riffel J, Mereles D, Rocken C, Buss SJ, Katus HA, Kristen AV (2018) Predictors of survival stratification in patients with wild-type cardiac amyloidosis. Clin Res Cardiol 107(2):158–169
Kim D, Lee GY, Choi JO, Kim K, Kim SJ, Jeon ES (2019) Associations of electrocardiographic parameters with left ventricular longitudinal strain and prognosis in cardiac light chain amyloidosis. Sci Rep 9(1):7746
Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RM, Bacchi-Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S (2009) Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 120(13):1203–1212
Helder MRK, Schaff HV, Nishimura RA, Gersh BJ, Dearani JA, Ommen SR, Mereuta OM, Theis JD, Dogan A, Edwards WD (2014) Impact of incidental amyloidosis on the prognosis of patients with hypertrophic cardiomyopathy undergoing septal myectomy for left ventricular outflow tract obstruction. Am J Cardiol 114(9):1396–1399
Satoskar AA, Efebera Y, Hasan A, Brodsky S, Nadasdy G, Dogan A, Nadasdy T (2011) Strong transthyretin immunostaining: potential pitfall in cardiac amyloid typing. Am J Surg Pathol 35(11):1685–1690. https://doi.org/10.1097/PAS.0b013e3182263d74
Sharma PP, Payvar S, Litovsky SH (2008) Histomorphometric analysis of intramyocardial vessels in primary and senile amyloidosis: epicardium versus endocardium. Cardiovasc Pathol 17(2):65–71
Treibel TA, Fontana M, Gilbertson JA, Castelletti S, White SK, Scully PR, Roberts N, Hutt DF, Rowczenio DM, Whelan CJ, Ashworth MA, Gillmore JD, Hawkins PN, Moon JC (2016) Occult transthyretin cardiac amyloid in severe calcific aortic stenosis. Circ Cardiovasc Imaging 9(8):e005066
Xu B, Godoy Rivas C, Rodriguez ER, Tan C, Gillinov AM, Harb S, Jellis C, Griffin B (2019) Unrecognized cardiac amyloidosis at the time of mitral valve surgery: incidence and outcomes. Cardiology 142(4):253–258. https://doi.org/10.1159/000499933
Yamamoto Y, Onoguchi M, Haramoto M, Kodani N, Komatsu A, Kitagaki H, Tanabe K (2012) Novel method for quantitative evaluation of cardiac amyloidosis using 201Tl-Cl and 99mTc-PYP SPECT. Ann Nucl Med 26(8):634–643
Zegri-Reiriz I, de Haro-Del Moral FJ, Dominguez F, Salas C, de la Cuadra P, Plaza A, Krsnik I, Gonzalez-Lopez E, Garcia-Pavia P (2019) Prevalence of cardiac amyloidosis in patients with carpal tunnel syndrome. J Cardiovasc Transl Res 12(6):507–513. https://doi.org/10.1007/s12265-019-09895-0
Rapezzi C, Riva L, Quarta CC, Perugini E, Salvi F, Longhi S, Ciliberti P, Pastorelli F, Biagini E, Leone O, Cooke RM, Bacchi-Reggiani L, Ferlini A, Cavo M, Merlini G, Perlini S, Pasquali S, Branzi A (2008) Gender-related risk of myocardial involvement in systemic amyloidosis. Amyloid 15(1):40–48. https://doi.org/10.1080/13506120701815373
Arvidsson S, Pilebro B, Westermark P, Lindqvist P, Suhr OB (2015) Amyloid cardiomyopathy in hereditary transthyretin V30M amyloidosis - impact of sex and amyloid fibril composition. PLoS One 10(11):e0143456. https://doi.org/10.1371/journal.pone.0143456
Nelson WW, Choi JC, Vanderpoel J, Damaraju CV, Wildgoose P, Fields LE, Schein JR (2013) Impact of co-morbidities and patient characteristics on international normalized ratio control over time in patients with nonvalvular atrial fibrillation. Am J Cardiol 112(4):509–512
Dungu JN, Valencia O, Pinney JH, Gibbs SD, Rowczenio D, Gilbertson JA, Lachmann HJ, Wechalekar A, Gillmore JD, Whelan CJ, Hawkins PN, Anderson LJ (2014) CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging 7(2):133–142. https://doi.org/10.1016/j.jcmg.2013.08.015
Gustafsson S, Ihse E, Henein MY, Westermark P, Lindqvist P, Suhr OB (2012) Amyloid fibril composition as a predictor of development of cardiomyopathy after liver transplantation for hereditary transthyretin amyloidosis. Transplantation 93(10):1017–1023
Oshima T, Kawahara S, Ueda M, Kawakami Y, Tanaka R, Okazaki T, Misumi Y, Obayashi K, Yamashita T, Ohya Y, Ihse E, Shinriki S, Tasaki M, Jono H, Asonuma K, Inomata Y, Westermark P, Ando Y (2014) Changes in pathological and biochemical findings of systemic tissue sites in familial amyloid polyneuropathy more than 10 years after liver transplantation. J Neurol Neurosurg Psychiatry 85(7):740–746
Yazaki M, Tokuda T, Nakamura A, Higashikata T, Koyama J, Higuchi K, Harihara Y, Baba S, Kametani F, Ikeda S (2000) Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild-type transthyretin. Biochem Biophys Res Commun 274(3):702–706. https://doi.org/10.1006/bbrc.2000.3203
Haagsma EB, Van Gameren II, Bijzet J, Posthumus MD, Hazenberg BPC (2007) Familial amyloidotic polyneuropathy: long-term follow-up of abdominal fat tissue aspirate in patients with and without liver transplantation. Amyloid 14(3):221–226
Kero T, Sorensen J, Antoni G, Wilking H, Carlson K, Vedin O, Rosengren S, Wikstrom G, Lubberink M (2018) Quantification of (11)C-PIB kinetics in cardiac amyloidosis. J Nucl Cardiol 27:774–784. https://doi.org/10.1007/s12350-018-1349-x
Nelson LM, Gustafsson F, Gimsing P (2015) Characteristics and long-term outcome of patients with systemic immunoglobulin light-chain amyloidosis. Acta Haematol 133(4):336–346
Abulizi M, Cottereau AS, Guellich A, Vandeventer S, Galat A, Van Der Gucht A, Planté-Bordeneuve V, Dubois-Rande JL, Bodez D, Rosso J, Damy T, Itti E (2018) Early-phase myocardial uptake intensity of99mTc-HMDP vs 99mTc-DPD in patients with hereditary transthyretin-related cardiac amyloidosis. J Nucl Cardiol 25(1):217–222
Ihse E, Rapezzi C, Merlini G, Benson MD, Ando Y, Suhr OB, Ikeda SI, Lavatelli F, Obici L, Quarta CC, Leone O, Jono H, Ueda M, Lorenzini M, Liepnieks J, Ohshima T, Tasaki M, Yamashita T, Westermark P (2013) Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid 20(3):142–150
Pilebro B, Arvidsson S, Lindqvist P, Sundstrom T, Westermark P, Antoni G, Suhr O, Sorensen J (2018) Positron emission tomography (PET) utilizing Pittsburgh compound B (PIB) for detection of amyloid heart deposits in hereditary transthyretin amyloidosis (ATTR). J Nucl Cardiol 25(1):240–248
Tasaki M, Ueda M, Obayashi K, Koike H, Kitagawa K, Ogi Y, Jono H, Su Y, Suenaga G, Oshima T, Misumi Y, Yoshida M, Yamashita T, Sobue G, Ando Y (2013) Effect of age and sex differences on wild-type transthyretin amyloid formation in familial amyloidotic polyneuropathy: a proteomic approach. Int J Cardiol 170(1):69–74
Di Bella G, Minutoli F, Mazzeo A, Vita G, Oreto G, Carerj S, Anfuso C, Russo M, Gaeta M (2010) MRI of cardiac involvement in transthyretin familial amyloid polyneuropathy. AJR Am J Roentgenol 195(6):W394–W399. https://doi.org/10.2214/AJR.09.3721
Hattori T, Takei Y, Koyama J, Nakazato M, Ikeda S (2003) Clinical and pathological studies of cardiac amyloidosis in transthyretin type familial amyloid polyneuropathy. Amyloid 10(4):229–239
Koike H, Hashimoto R, Tomita M, Kawagashira Y, Iijima M, Tanaka F, Sobue G (2011) Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: a practical analysis. Amyloid 18(2):53–62
Okamoto S, Yamashita T, Ando Y, Ueda M, Misumi Y, Obayashi K, Horibata Y, Uchino M (2008) Evaluation of myocardial changes in familial amyloid polyneuropathy after liver transplantation. Intern Med 47(24):2133–2137. https://doi.org/10.2169/internalmedicine.47.1399
Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M (2017) The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ 8(1):33. https://doi.org/10.1186/s13293-017-0152-8
Arnold AP, Cassis LA, Eghbali M, Reue K, Sandberg K (2017) Sex hormones and sex chromosomes cause sex differences in the development of cardiovascular diseases. Arterioscler Thromb Vasc Biol 37(5):746–756. https://doi.org/10.1161/ATVBAHA.116.307301
Ton V-K, Mukherjee M, Judge DP (2015) Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol 8(Suppl 1):39–44. https://doi.org/10.4137/CMC.S15719
Hellman U, Alarcon F, Lundgren H-E, Suhr OB, Bonaiti-Pellié C, Planté-Bordeneuve V (2008) Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid 15(3):181–186. https://doi.org/10.1080/13506120802193720
Drugge U, Andersson R, Chizari F, Danielsson M, Holmgren G, Sandgren O, Sousa A (1993) Familial amyloidotic polyneuropathy in Sweden: a pedigree analysis. J Med Genet 30(5):388–392. https://doi.org/10.1136/jmg.30.5.388
Thompson AE, Anisimowicz Y, Miedema B, Hogg W, Wodchis WP, Aubrey-Bassler K (2016) The influence of gender and other patient characteristics on health care-seeking behaviour: a QUALICOPC study. BMC Fam Pract 17:38. https://doi.org/10.1186/s12875-016-0440-0
Mackenzie CS, Gekoski WL, Knox VJ (2006) Age, gender, and the underutilization of mental health services: the influence of help-seeking attitudes. Aging Ment Health 10(6):574–582. https://doi.org/10.1080/13607860600641200
Matheson FI, Smith KLW, Fazli GS, Moineddin R, Dunn JR, Glazier RH (2014) Physical health and gender as risk factors for usage of services for mental illness. J Epidemiol Community Health 68(10):971–978. https://doi.org/10.1136/jech-2014-203844
Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt J-U (2015) Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 28(1):1–39.e14. https://doi.org/10.1016/j.echo.2014.10.003
Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C (2017) Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 135(14):1357–1377. https://doi.org/10.1161/circulationaha.116.024438
Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, Klarich KW, Miller WL, Maleszewski JJ, Dispenzieri A (2016) Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 68(10):1014–1020. https://doi.org/10.1016/j.jacc.2016.06.033
Acknowledgments
The authors would like to thank Selma Mohammed, MD, of the Division of Cardiology, Creighton University, Omaha, NE, for her contributions to the development of the systematic literature review and the valuable comments on drafts of this manuscript.
Funding
The systematic literature review to support this manuscript was sponsored by Pfizer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Drs. Bruno, Castaño, and Burton are full-time employees of Pfizer and may hold Pfizer stock and/or stock options. Dr. Grodin has received funding as a consultant for Pfizer and Eidos.
Medical writing support for this article was provided by Donna McGuire of Engage Scientific Solutions and was funded by Pfizer. Sara Lucas, PhD, and Catherine Rolland, PhD, are employees of Envision Pharma Group and paid consultants to Pfizer in connection with the development of the SLR report that forms the basis of this manuscript. They were not compensated for their role in the review of this manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors provided consent for the publication of this article.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 171 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bruno, M., Castaño, A., Burton, A. et al. Transthyretin amyloid cardiomyopathy in women: frequency, characteristics, and diagnostic challenges. Heart Fail Rev 26, 35–45 (2021). https://doi.org/10.1007/s10741-020-10010-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-020-10010-8