
Abstract
1H-detected solid-state NMR spectroscopy has been becoming increasingly popular for the characterization of protein structure, dynamics, and function. Recently, we showed that higher-dimensionality solid-state NMR spectroscopy can aid resonance assignments in large micro-crystalline protein targets to combat ambiguity (Klein et al., Proc. Natl. Acad. Sci. U.S.A. 2022). However, assignments represent both, a time-limiting factor and one of the major practical disadvantages within solid-state NMR studies compared to other structural-biology techniques from a very general perspective. Here, we show that 5D solid-state NMR spectroscopy is not only justified for high-molecular-weight targets but will also be a realistic and practicable method to streamline resonance assignment in small to medium-sized protein targets, which such methodology might not have been expected to be of advantage for. Using a combination of non-uniform sampling and the signal separating algorithm for spectral reconstruction on a deuterated and proton back-exchanged micro-crystalline protein at fast magic-angle spinning, direct amide-to-amide correlations in five dimensions are obtained with competitive sensitivity compatible with common hardware and measurement time commitments. The self-sufficient backbone walks enable efficient assignment with very high confidence and can be combined with higher-dimensionality sidechain-to-backbone correlations from protonated preparations into minimal sets of experiments to be acquired for simultaneous backbone and sidechain assignment. The strategies present themselves as potent alternatives for efficient assignment compared to the traditional assignment approaches in 3D, avoiding user misassignments derived from ambiguity or loss of overview and facilitating automation. This will ease future access to NMR-based characterization for the typical solid-state NMR targets at fast MAS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Owing to deuteration strategies (Akbey et al. 2009; Chevelkov et al. 2006; Linser et al. 2011b; Reif 2021; Vasa et al. 2018a) and facilitated by increasing Magic-Angle-Spinning (MAS) frequencies (Cala-De Paepe et al. 2017; Lewandowski et al. 2011; Schledorn et al. 2020), effectively reducing the influence of coherent contributions from proton-proton dipolar couplings (Böckmann et al. 2015; Malär et al. 2019; Penzel et al. 2019), proton detection has been facilitating effective resonance assignment (Andreas et al. 2015; Barbet-Massin et al. 2013; Klein et al. 2022a, b; Knight et al. 2011, 2012; Linser et al. 2010a, b; Orton et al. 2020; Schubeis et al. 2021; Stanek et al. 2016, 2020; Vasa et al. 2019; Xiang et al. 2016; Zhou et al. 2007c), assessment of protein dynamics with timescales overarching many orders of magnitudes (Chevelkov et al. 2007, 2009; Gauto et al. 2017; Grohe et al. 2020; Kurauskas et al. 2017; Ma et al. 2014; Rovó 2020; Rovó and Linser 2018; Rovó et al. 2019; Schanda and Ernst 2016; Singh et al. 2019; Vasa et al. 2018a), as well as protein structure elucidation (Andreas et al. 2016; Bertarello et al. 2020; Grohe et al. 2019; Huber et al. 2012; Jain et al. 2017; Klein et al. 2022b; Linser et al. 2011a, 2014; Mandala et al. 2018; Retel et al. 2017; Schubeis et al. 2020; Söldner et al. 2023; Vasa et al. 2019; Zhou et al. 2007b) based on proton-proton distances. Apart from micro-crystalline proteins, this has been including various sample types from membrane proteins (Lalli et al. 2017; Retel et al. 2017; Schubeis et al. 2020; Shi et al. 2019; Zhou et al. 2012) and protein amyloids (Becker et al. 2023; Stanek et al. 2016; Xiang et al. 2017) to supramolecular assemblies (Lu et al. 2020; Zinke et al. 2020) and others. Moreover, complex magnetization transfer pathways, correlating multiple nuclei similarly to solution NMR, are becoming increasingly established (Ahlawat et al. 2023; Barbet-Massin et al. 2014; Fraga et al. 2017; Klein et al. 2022a, b; Linser et al. 2010b; Orton et al. 2020; Penzel et al. 2015; Sharma et al. 2020; Stanek et al. 2020; Vasa et al. 2018b). Important established experiments for resonance assignment so far are those solution-NMR-derived experiments correlating inter- and intraresidual carbon chemical shifts with amide 15 N/1H shifts similar to solution HNCA, HNCO, HNCOCA, HNCACO, HNCACB, and HNCOCACB (Akbey et al. 2009; Barbet-Massin et al. 2013, 2014; Knight et al. 2011; Linser et al. 2010b, 2011b; Zhou et al. 2007a). In these experiments, proton magnetization is utilized both for excitation and detection, and either scalar or dipolar transfers or mixtures thereof are used to connect the involved spins, with several possibilities of invoking relay spins dependent on the level/sites of protonation. As an extension to HNCACB correlations, sidechain-to-backbone correlations (“S2B experiments”) help to assess the sidechain carbon (Kulminskaya et al. 2015, 2016; Linser 2011) and/or proton chemical shifts (Ahlawat et al. 2023; Klein et al. 2022b; Stanek et al. 2016), thus providing residue-type information, which is key for mapping stretches of sequential correlations to the known primary sequence or enable Hali-based distance restraints for structure calculation (Andreas et al. 2016; Jain et al. 2017; Klein et al. 2022b; Nimerovsky et al. 2022; Söldner et al. 2023; Vasa et al. 2019). In addition to these H/N/C correlations, HNCACONH-type experiments, concatenating sequential amide groups directly, have been suggested (Andreas et al. 2015; Klein et al. 2022a; Orton et al. 2020; Xiang et al. 2015) as more effective sequential correlations that avoid ambiguities associated with the match-making process based on 13 C spins (“amide-to-amide experiments”).
For solid samples, transfer efficiencies are irrespective of the target’s molecular weight. Hence, for a given rotor diameter, preparation scheme, and experiment, the signal intensity obtained only scales linearly with the number of molecules in the rotor or inversely with the molecular weight of the target. (In the case of homooligomeric complexes or assemblies, this refers to the asymmetric unit.) Even though innovations for more complex sequences, but also for accelerated spectral acquisition (Gallo et al. 2019; Sharma et al. 2020; Stanek et al. 2020) and automated assignment (Klukowski et al. 2022; Lee et al. 2019; Schmidt and Güntert 2012; Volk et al. 2008) are soaring, manual, 3D carbon matchmaking-based assignment still seems to represent the established state-of-the-art. The assignment relies on the consolidation of multiple, usually at least five or six (backbone), possibly plus three (including sidechain assignments) experiments to fight ambiguities. Modern software packages, such as CCPNmr 3 (Skinner et al. 2016) or Poky (Lee et al. 2021), aid the user through the assignment process, but the assignment process inevitably becomes increasingly challenging with a growing number of spectra to consider even for an expert. A reduction of the number of spectra used while simultaneously reducing the number of overlapping peaks has been achieved by concatenating two 3Ds to a 4D experiment (Fraga et al. 2017; Klein et al. 2022b; Vasa et al. 2018b; Xiang et al. 2014; Zinke et al. 2017). This was shown to be advantageous both, for broad resonance lines (as in the cases of low deuteration levels or heterogeneous sample preparations) as well as for proteins with an increasing number of residues. The strategy is particularly beneficial if the number of magnetization transfers remains unaltered by the concatenation and losses in signal-to-noise derived from additional phase increments is exactly compensated by making the second experiment unnecessary, which is the case, e.g., in 4D (Vasa et al. 2018b) or 5D amide-to-amide experiments (Klein et al. 2022a; Orton et al. 2020).
The pertinent hurdles of peak overlap and assignment ambiguity (the existence of multiple possible matches) of the process are excessively aggravated for an increasing number of residues, which entails an exponential growth of assignment possibilities. In the established approaches, a 2D H/X correlation (H/N or HA/CA) is used as a basis for identification of i ± 1 resonances of the same kind. Even when the problematic step of carbon-based match-making is overcome by said direct sequential linkages suggested by us and Pintacuda et al. (Andreas et al. 2015; Klein et al. 2022a; Vasa et al. 2018b; Xiang et al. 2015), overlap of residue i in the source H/X plane (Fig. 1A) will lead to ambiguities in the assignment process, further amplified by overlapping peaks at the position of the i + 1 signal and so on. 5D backbone experiments (Fig. 1) can partly levitate this limitation by a further increase of dispersion: In contrast to 4D HNNH amide-to-amide experiments, where only amide shifts are correlated with each other, for at least one of the two sequential amino acids they report on three nuclei, enabling more confident identification. This improves the concatenation of the sequential correlation with information from carbon-edited, in particular HNCACB-type and S2B-type, experiments, which allows for residue type information and hence for facilitated mapping of those correlations onto the primary sequence.
In previous work (Klein et al. 2022a, b) we have demonstrated that 5D data can be beneficial for backbone assignment of large protein complexes and sidechain proton assignments in fully protonated samples studied by solid-state NMR. One would assume that such efforts are only justified when “everything else fails”. However, here we would like to convince the reader that higher-dimensionality approaches offer competitive alternatives for a reliable and simple assignment even for the usual fast-MAS ssNMR targets of lower molecular weight, manually or in combination with automated assignment – which would commonly be addressed using lower-dimensionality strategies. Given the high transfer efficiency of proton-detected solid-state NMR spectroscopy, in particular, 5D HNcaCONH and 5D HNcoCANH amide-to-amide experiments are practically well feasible and can leverage a simplified, high-fidelity assignment process from a minimal set of experiments and can be easily combined with S2B experiments for sidechain assignments.

H/N overlap as found even for well-behaved targets. (A) 2D hNH spectrum of the SH3 domain of chicken α-spectrin. Despite its small size (62 residues) and high β-sheet content, the HN correlation comprises multiple overlapping peaks, causing ambiguities for any HN-based experiments, e.g., the standard out-and-back experiments. (B) Overlap in 4D HNNH direct amide-to-amide correlations (top), resolved by the additional carbon dimension in a 5D HNcoCANH experiment (bottom row), leaving only unambiguous assignments here
Materials and methods
We used a perdeuterated sample of the SH3 domain of chicken α-spectrin, which was obtained by recombinant protein expression and purification in aqueous buffers as described previously (Linser et al. 2007). Micro-crystallization was achieved in aqueous buffer by pH-shift from 3.5 to 7.5, employing paramagnetic relaxation enhancement (PRE) with 75 mM Cu(EDTA) to accelerate data acquisition (Linser et al. 2007; Wickramasinghe et al. 2009). After precipitation overnight, the crystals were spun into a 1.3 mm rotor containing fluorinated rubber plugs, finally containing around 1 mg of protein. NMR experiments on the perdeuterated sample were carried out on a Bruker NEO spectrometer operating at a proton Larmor frequency of 700 MHz, using a triple-resonance HNC probe at 55.5 kHz MAS and approximately 25 °C. Detailed experimental conditions are listed in Tables S1 and S2. All 5D experiments were acquired using non-uniform sampling (NUS) with a Gaussian weighted, random schedule to minimize artifacts and interferences during reconstruction with the signal separating algorithm (SSA) as suggested by the authors (Kosiński et al. 2017). Exponential line broadening in the direct dimension was applied in all the spectra, while in some of the spectra, exponential apodization was applied in the indirect dimensions as well to maximize the signal-to-noise ratio. This ultimately results in a mixed window function along the indirect dimensions. 3D base experiments were recorded using either uniform or non-uniform sampling. NUS reconstruction was performed using SSA for 3D (Stanek and Koźmiński 2010) or 5D (Kosiński et al. 2017) experiments, respectively. Data analysis and manual assignments were done in CCPNmr V3 (Skinner et al. 2016), while FLYA (Schmidt and Güntert 2012) was used for automated resonance assignment. 5D experiments were recorded in blocks of 8 scans for ca. 2–2.5 days, after which a field reset, by tracking the shift of the water signal, was performed to account for the drift of the magnetic field in the absence of a lock signal. For the HNcoCANH experiment, three blocks of the same 2048 NUS points were recorded within a total time of ca. 6.5 days. The 5D HNcaCONH was acquired in two blocks, i.e., a total of 16 scans, and 1920 NUS points in a time of ca. 4 days. Addition of the individual blocks can be easily performed in TopSpin using addser as well as independent tools such as NMRPipe (Delaglio et al. 1995) or nmrglue (Helmus and Jaroniec 2013). The corresponding 3D base experiments were recorded as NUS experiments as well, using 16 scans and 1024 NUS points for the hCANH and the hCONH. The experimental time accounted for ca. 8 h in both cases. To obtain additional side-chain assignments, shifts from a 5D HCCNH were taken into account as recorded previously (Klein et al. 2022b). This 5D HCCNH experiment was recorded with a protonated sample on the same spectrometer using an HNC 0.7 mm probe at 100 kHz and approximately 20 °C.
Results
Expansion of amide-to-amide experiments to 5D
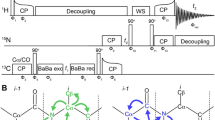
The pulse sequences for 5D HNcoCANH and 5D HNcaCONH (Fig. 2) here are obtained straightforwardly from their lower-dimensionality (HNNH) variants (Andreas et al. 2015; Xiang et al. 2015) and feature an additional carbon indirect dimension t1. Both 5D experiments were introduced as NUS or APSY previously (Klein et al. 2022a; Orton et al. 2020) using exclusively CP for heteronuclear transfers and either BSH-CP (Chevelkov et al. 2013; Shi et al. 2014) or INEPTs for homonuclear CC transfer. The two sequences are composed of the same number of transfer steps as their lower-dimensional counterparts. In the case of an INEPT transfer (Fig. 2B C), starting with in-phase CO magnetization, only the CO-Cα coupling is active during the INEPT transfer, which allows simple rectangular pulses. Even though during the refocusing period, the Cα-Cβ coupling is also active, hard pulses were again used here to avoid the complication of poor separation of Cα and Cβ resonances for Cα-selective pulses and losses during the rather lengthy selective pulses (Barbet-Massin et al. 2013).

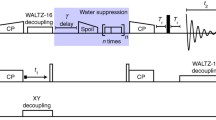
5D magnetization transfer scheme and pulse sequence for the HNcoCANH using carbon-carbon CP (i) or INEPT (ii). Both versions have been used previously (Andreas et al. 2015; Klein et al. 2022a; Orton et al. 2020; Stanek et al. 2020; Xiang et al. 2015) but are shown here again for completeness. For the INEPT transfer step, using a block similar to the one reported in Barbet-Massin et al. 2013 (Barbet-Massin et al. 2013), only hard pulses at a 13 C carrier position of 120 ppm were used here. Compare Orton et al., also involving selective pulses (Orton et al. 2020). Phase cycling for HNcoCANH: φ1 = x,-x; φ2 = x,x,-x,-x; φ3 = x,x,x,x,-x,-x,-x,-x; φrec = x,-x,-x,x,-x,x,x,-x. Carrier changes are marked by arrows. The Cα carrier position was set to 55 ppm, while 173 ppm was used for carbonyl carbons. Water suppression was achieved by the MISSISSIPPI scheme (Zhou and Rienstra 2008) without gradients, using a pulse length of 20 ms at 10 kHz rf field strength
In the following, using the SH3 domain of chicken α-spectrin, we demonstrate that the above strategies are a competitive approach for a virtually complete, highly effective, and user-friendly assignment process on small proteins that have been tackled with standard methodology. To identify a minimal set of higher-dimensional experiments required, we recorded different 5D backbone experiments on a perdeuterated and 100% proton back-exchanged sample and expanded the set by a sidechain-to-backbone (S2B) experiment acquired on a fully protonated, micro-crystalline sample. Figure 3 shows an exemplary set of 2D slices from the 5D HNcoCANH experiment (Fig. 3A) as well as from the reverse HNcaCONH experiment (Fig. 3B) in combination with a suitable 3D base experiment each. When Cβ information is sought, a 3D hcaCBCANH (using 1/(4 J)) experiment is often competitive to the 3D hcaCBcaNH with full J-transfer (over 1/(2 J)). This experiment includes both, Cα and Cβ shift information and can equally be used as a base experiment for reconstruction of the 5D directly instead of a 3D hCANH, which then becomes dispensable.

5D amide-to-amide backbone walks, illustrated for the HNcoCANH (A) and the HNcaCONH (B) experiment. Both experiments enable a straightforward backbone walk, as they provide direct, well-resolved i to i + 1 connectivities. If sidechain carbon information is desired anyways, an hcaCBcaNH with split CACB transfer can be used instead of an hCANH as the base experiment for 5D reconstruction. The HNcaCONH benefits from the higher resolution of the carbonyl carbons but lacks the possibility of connecting sequential with sidechain information
Data processing and spectral features
Recording NUS 5D experiments is straightforward using standard software. Processing of 5D data sets, on the other hand, can be leveraged by the SSA algorithm using sparse multi-dimensional Fourier transformation (SMFT), developed by Koźmiński and coworkers (Kazimierczuk et al. 2009; Kosiński et al. 2017; Stanek and Koźmiński 2010). In brief, this methodology does not reconstruct the entire 5D frequency domain spectrum but instead produces frequency-domain data only at predefined positions. These regions are preselected through a peak list obtained from picking a 3D spectrum that shares three dimensions with the 5D experiment (see Fig. 4A). The approach avoids the excessive amount of data and computation time required if 5D data sets were fully Fourier-transformed, using artifact reduction in all indirect dimensions at defined regions of the 5D space (Kosiński et al. 2017; Stanek and Koźmiński 2010). As undefined peaks, i.e., not peak-picked ones, will not be processed by SSA, high signal-to-noise ratio and resolution are desired in the 3D base experiments, which can again be leveraged via NUS. The experiments proposed in this work for the 3D base experiment are the 3D hCANH or 3D hCONH experiments, rather than the alternative hCAcoNH or hCOcaNH experiments, due to their better sensitivity.
The 5D experiments share an H/N/C shift triplet with the base experiment. Therefore, only the additional H and N dimensions, providing chemical shifts of the i + 1 residue (HNcoCANH) or the i-1 residue (HNcaCONH), are generated for each 3D source peak provided. The processed 5D spectrum eventually becomes a stack of 2D H/N planes at predefined H/N/C peak positions that can be superimposed onto ordinary 2D H/N correlation spectra. Evaluation can be done via common software packages, like NMRFAM-SPARKY (Lee et al. 2014), POKY (Lee et al. 2021), CCPNmr V3 (Skinner et al. 2016) etc. and does not require special data processing other than NUS reconstruction to make the spectra accessible. In addition to the gain in resolution, the absence of depth through a third dimension in 5D spectra processed this way strongly reduces the level of complexity upon analysis. In particular, the difficulty of finding and evaluating peaks and determining their maxima in three or four dimensions is replaced by mere 2D peak identification.
Selecting peak i of a sequential correlation via three coordinates effectively avoids peak overlap, which not only occurs for complex proteins or heterogeneous samples but even for small, micro-crystalline proteins (compare Fig. 1), and therefore facilitates identification of next- (or previous-) residue H/N shifts with high fidelity compared to HNNH 4Ds in a general sense. Not only the sparsity of peaks in 5D space, largely reducing overlapping correlations, and the direct residue linking using amide shifts dispensing the drawbacks of carbon-based matchmaking, but also the facilitated concatenation of sequential-walk information with residue-type information from hcaCBcaNH or S2B experiments (linked to the sequential correlations via the H/N/Cα triplet) are general advantages of the approach applicable for a wide variety of protein targets.
The i + 1 (HNcoCANH) or i-1 (HNcaCONH) peak, respectively, is the predominant signal found in the spectra and easily distinguished from remaining NUS artifacts or thermal noise in the regime of conditions described here. Interestingly, in the BSH-CP version, due to the possibility of magnetization transfer in CP steps to nuclei two bonds away, an out-and-back transfer to the H/N group of origin can be observed, leading to an additional (“diagonal”) peak in the processed 2D plane. This out-and-back pathway can likely be attributed to the upper-limit magnetization transfer of around 50% during the BSH-CP (Chevelkov et al. 2013; Shi et al. 2014). As similar CP conditions apply for NCA and NCO transfers, non-transferred CO magnetization after the BSH-CP may partially be transferred back to the amide nitrogen of origin. Whereas this diagonal peak is redundant and has to be taken into account upon judgement of bulk signal intensities (see below), it in fact practically facilitates identification/confirmation of the source resonances upon manual inspection. Rigorous analysis of the HNcoCANH spectrum shows that the ratio between the i + 1 and i peak (cross peak to diagonal peak) is constant at approximately 3:1 for all amino acids apart from glycine (Fig. 4B/C) which show a ratio of ca. 8:1. We ascribe these effects, which might be exploited for residue type identification, to higher BSH-CP transfer efficiency in the case that no Cβ is present (The latter may function as a sink of magnetization for rf fields close to or at the HORROR condition.) as well as possibly the NCO and NCA conditions being somewhat more distinct for Glycines.

Processing of 5D HNcoCANH experiment and information obtained. A) Using SSA for spectral reconstruction, a stack of 2D planes is returned (one for each previously defined 3D peak). B and C) In BSH-CP versions of the amide-to-amide correlations, incomplete magnetization transfers cause additional out-and-back-like correlations with a constant intensity ratio of around 3:1 for non-glycine residues but much higher for glycines (shown as a histogram in B) and as a function of sequence in the SH3 domain in C)
Sensitivity considerations
Figure 5 shows a comparison of bulk signal intensities for the same sample. HN bulk intensities of 12% and 6% are obtained in case of the HNcoCANH and the HNcaCONH, respectively, which is within the range of expected performance (Penzel et al. 2015). A strong difference between the CO-Cα and the Cα-CO BSH-CP is noted, which is in line with previous quantification of this pulse sequence element using DREAM transfer (Penzel et al. 2015) and presumably derives from competing Cα-Cβ transfer. For the perdeuterated and amide back-exchanged protein in a 1.3 mm rotor, the INEPT-based scheme (element ii) in Fig. 2) yields slightly reduced intensity of the HN bulk signal compared to the fully CP-based HNcoCANH version in our hands (Fig. 5C). However, almost all of this signal can be assumed to reflect the signal of interest, whereas in the dipolar sequence a quarter of the overall signal derives from back transfer (the diagonal peak, see above), making the INEPT version similarly sensitive in practice. Here, no diagonal peaks (no back-transfer) are/is evident as tested in 3D NNH-type experiments (hNcocaNH, recorded on a fully protonated sample), see Fig. S1. Given that for INEPTs, in contrast to BSH-CP, the Cα-CO transfer (in the other direction) performs with similar efficiency (Andreas et al. 2015; Penzel et al. 2015; Xiang et al. 2015) as the CO-Cα transfer, the INEPT version is a generally competitive alternative whenever heteronuclear T2 times are sufficiently long (i.e., using deuterated samples or MAS of around 100 kHz or faster).

Bulk signal intensities for the 5D HNcoCANH and 5D HNcaCONH compared to the hNH intensity. (A) The HNcoCANH maintains approximately 12% of the hNH when used with BSH-CP for homonuclear CC transfers. (B) Using the HNcaCONH with BSH-CP results in ca. 6% of the hNH bulk signal. (C) In case an INEPT block is used, the signal intensity is attenuated by an additional 30%, however, in contrast to the BSH version, the bulk signal represents only the peaks of interest. Also, the difference between CACO and COCA is not observed in the INEPT version
Naively, the increased number of transfer steps in the direct amide-to-amide experiments would suggest extremely poor sensitivity compared to assignment strategies based on complementary carbon matchmaking. In addition to the advantages in assignment fidelity, however, the sensitivity of the approach is in fact very competitive, as the set consisting of carbon matchmaking experiments relies on a greater number of individual experiments, including some with very low sensitivity: hCANH and hCONH schemes provide a bulk signal to noise of around 25%, however, their counterparts (hCAcoNH and hCOcaNH) only bear on the order of around 12%, due to losses during the carbon-carbon transfer step (Penzel et al. 2015; Shi et al. 2014). The pair of hcaCBcaNH and hcaCBcacoNH experiments only provide a bulk signal of around 8 and 3%, respectively. Even if, hypothetically, the full suite of six carbon-based 3D experiments did carry the same fidelity for unambiguous matchmaking as a 5D amide-to-amide correlation (which is not the case as explained above) and if an additional hcaCBcaNH is always desired even as a supplement for the latter case, the measurement time requirements would be higher for the set based on carbon-matchmaking-based 3Ds (see Table 1). (In this direct comparison with 3D experiments, an assumed 10% bulk signal of the 5D amide-to-amide correlation would have to be corrected by a √2 for each additional indirect dimension compared to a 3D (i.e., a factor of 2) as well as the ratio between intensity of the peak of interest and overall magnetization transfer derived from the bulk signal (0.75). This number is still higher than the bulk sensitivity of the weakest one (the hcaCBcacoNH, ca. 3%) of the six 3D experiments. It is further important to note that NUS spectra generally show equal or higher signal-to-noise ratios compared to time-equivalent US spectra when sampled with a decaying sampling function, e.g., exponential or gaussian weighting (Hyberts et al. 2013; Palmer et al. 2015; Paramasivam et al. 2012), the more indirect dimensions are present. 5D sensitivities therefore scale up favorably towards improved sensitivities. The actual scaling factor, however, is strongly depended on the bias of the sampling function as well as the recorded tmax in each indirect dimension, and 1.2-fold – 3-fold improvements per indirect dimension can be achieved (Palmer et al. 2015; Paramasivam et al. 2012; Suiter et al. 2014). For 5D and 4D experiments, the many indirect dimensions therefore tend to lead to substantial sensitivity gains. (To take this effect into account in Table 1, we also listed tentative numbers (in brackets) assuming an improvement of 1.2x per indirect dimension. (As also 3Ds can of course be recorded as NUS experiments, such number are also shown for those – again in brackets.) In addition to easing the biggest bottleneck of the set, fewer experiments have to be recorded for the 5D set, streamlining the actual assignment process for the user (see below for assignment success with different kinds of experiments taken into account within a computational framework). Even whenever hcaCBcaNH and hCONH are desired in any case (e.g., for complementing the 5D approach for residue-type information and secondary-structural analysis) and the hCANH is additionally recorded to serve as a basis for SMFT (As mentioned above, the hcaCBCANH can be used for this purpose, too.), the 3D hCAcoNH and 3D hCOcaNH become dispensable. The advantage of several-fold time saving stays even when the six 3Ds are all recorded under NUS (assumed 1.2x intensity gain per indirect dimension, numbers in brackets) and even when sampling-related sensitivity gain in NUS is ignored altogether, even for the 5D (numbers in squared brackets).
Even when the most insensitive experiment of the traditional set (the hcaCBcacoNH) is omitted (Set 2,3,4,6,7, with a relative time to 2D hNH of 710 – which can work for very well-behaved proteins as the SH3 domain in the following but further reduces the fidelity of sequential connections to the dispersion of the CO/Cα 2D plane), the time required for the 5D set with its direct connections (relative time to 2D hNH also being 710) is still on par. With the possibility of sensitivity enhancement for all or most of the indirect dimensions (Blahut et al. 2022), further strong increase of the competitiveness of higher-dimensionality approaches vs. standard approaches can be expected.
A direct, purely experimental signal-to-noise comparison for a given peak would be preferred over the above theoretical assessment but is compromised by the necessity of NUS (and reconstruction) for the 5Ds and the associated difficulties of measuring noise there. (The acquisition of four indirect dimensions in a uniform manner is practically infeasible.) However, the propagation from bulk signal-to-noise is straightforward and rigorous when considering all relevant factors. In either way, one has to keep a sample- and setup-dependent variability of transfer efficiencies and NUS reconstruction in mind.
Application to the SH3 domain of chicken α-spectrin
In a manual backbone walk based on the 5D HNcoCANH experiment recorded on the SH3 domain with 8 scans and 1805 NUS points (36 h on a 700 MHz spectrometer), 46 out of 48 expected resonances can be assigned. Concatenated with a 3D hcaCBCANH (2d), complete backbone assignment can be obtained from this minimal set of experiments. In order to see whether assignments improve with an increased signal to noise ratio, we recorded the HNcoCANH step-wise longer in blocks of 2 days and with slightly more NUS points (up to in total three blocks of 8 scans, 2048 points) and again performed a manual assignment. Naturally, the best spectrum was obtained after the longest (6 days of) measurement time, but the two weakest resonances were unambiguously reconstructed after 4 days (or two blocks). Assuming experimental times of approx. 2 days for a uniformly sampled 3D hcaCBcaNH and 12 h for an hCONH experiment (compare Table 1), a complete backbone assignment can therefore be realized within 6.5 days. This time is on par or faster compared to established 3D-based strategies, but warrants reduced ambiguity and a more intuitive and straightforward assignment procedure. Alternatively, the backbone walk can also be followed using the HNcaCONH experiment. However, when recorded with 16 scans and 1920 NUS points, only 41 out of 48 expected resonances can be unambiguously assigned.
Maybe more interestingly, to assess the minimal required measurement time for a given performance, we artificially truncated the sparse FID recorded with a minimal phase cycle of 8 scans (2048 points, 52 h experimental time), leaving either 1536, 1024, 512, 256, 192, or only 64 time-domain complex points (Fig. 6), which is commensurate to 39 h, 26 h, 13 h, 6.5 h, 5 h, and 1.5 h experimental time, respectively (Fig. 6A). As the signal-to-artifact ratio improves with the square root of recorded NUS points (Kazimierczuk et al. 2009) and the thermal signal-to-noise ratio (SNR) generally improves with an increasing number of FIDs (assuming tmax < 1.3 T2), maximizing the number of NUS points over the number of scans in a time-equivalent manner will usually lead to equivalent or better spectra. In case of the SH3 domain, > 75%, i.e., 36 out of 48 possible resonances, can be identified starting from around 256 NUS points with 8 scans (i.e., around 6 h of measurement time). For this condition, an average SNR of ca. 15 is achieved, whereas for shorter times the spectral quality breaks down (Fig. 6). Considering the quadratic dependency of the SNR and the molecular weight, i.e., copies of the molecule in the rotor, a protein of 35 kDa or 5-times the size of the SH3 domain would require at least 25-times more scans, i.e., 6400 NUS points (when the minimal phase cycle of 8 is employed). It should, however, be emphasized here that this is merely a qualitative approximation as spectral crowding may require an even higher number of NUS points. Besides the molecular weight, the SNR of (any) NMR experiment heavily depends on other factors, such as sample packing, homogeneity, etc. and the above recommendations will only be partially translatable, which in turn would require constant processing and evaluation of the 5D until convergence is reached. To introduce another qualitative guideline, the SNR of an hCANH experiment recorded for the same sample over 12 h (the one that we used as a source spectrum for the SMFT reconstruction of the 5D HNcoCANH) is depicted as a function of residue in Fig. S2. This comparison might be used to qualitatively estimate the required measurement time for a 5D to reach the different levels of performance shown in Fig. 6 for other samples that an hCANH has previously been recorded for. Here, the hCANH signals show a SNR of around 20–40 after 12 h of measurement time with similar trends for the individual residues as the 5D (Fig. S2). If for future samples the 3D will take n-times longer to reach a similar average SNR, the measurement times, i.e., minimum number of NUS points, for the 5D would accordingly have to be increased by n. (Mind the general inaccuracies upon quantifying the SNR of NUS data sets.)

Influence of the number of NUS points (complex time points) and hence overall experimental time invested on the number of signals, i.e., residues found in the spectrum (A/C) and the signal-to-noise ratio (SNR) (B/C) after reconstruction of the FID recorded with 8 scans (minimal phase cycle). The dashed lines in (A) mark the threshold at which 75% of all 48 possible signals are found, arbitrarily chosen as the limit for a minimal acceptable performance. In (B) the averaged SNR over all identified signals is shown with blue crosses, while the red curve marks the calculated SNR extrapolated down from the SNR measured for 2048 NUS points. The dashed lines mark the SNR reached for the 256 points, for which 75% (36 out of 48) of all signals are found. In (C) the signal-to-noise ratio for the individual residues is shown in dependence of the number of NUS points included for reconstruction. Prolines 21 and 54 and CP inaccessible residues 47 and 48 are marked in gray (problematic both in the roles of residue i or i-1). R21 and N38 are mobile residue always difficult to detect. (Linser et al. 2010a) The data reduction (i.e., reduction of measurement time effectively included in the sparse FID) was implemented by truncating to only the desired subset of time-domain points. As the NUS schedule was randomized before acquisition, the maximum resolution and sampling density weighting is maintained for all subsets. 2048, 1536, 1024, 512, 256, 192, and 64 time-domain complex points correspond to effective experimental times of 52 h, 39 h, 26 h, 13 h, 6.5 h, 5 h, and 1.5 h, respectively
Automated assignment approaches, which have been becoming increasingly powerful throughout the last years, are becoming an indispensable support for leveraging resonance assignment of large proteins. Nevertheless, they also streamline the assignment of smaller proteins to the point of fully automated peak picking, assignment, and structure calculation (Klukowski et al. 2022) and can be generally used as a tool to validate manual assignments. Figure 7 shows the application of the FLYA algorithm (Schmidt and Güntert 2012) for the peak list obtained from different combinations of 5D experiments and different other experiments for automated assignment of the deuterated sample of the SH3 domain. The caption of Fig. 7 explains the commonly used color coding of FLYA, which reports (dis-)agreement with reference assignments as well as reliability of the automated assignments. These results are validated by comparison with the manual assignments obtained from 3D experiments for the residues visible in CP-based experiments, i.e., excluding residues 1–6 and 47–48. (These residues escape CP-based schemes due to their extensive fast dynamics.) The classical set of 3D experiments, however leaving out the hcaCBcacoNH as the weakest and most time-consuming one yields 99% strong assignments that are correct (Fig. 7A; Table 2). (For the SH3 domain no carbon-matchmaking ambiguities are present in the Cα/CO 2D plane, hence retaining hCANH, hCAcoNH, hCONH, hCOcaNH, and hcaCBcaNH is sufficient. In other words, for this set of five 3D experiments FLYA is in agreement with the manual assignments; exceptions are residues 44–46 at the beginning of the distal loop. A similar outcome is obtained for each of the combinations including 5D amide-to-amide correlations (Fig. 7B-F; Table 2). The weaker HNcaCONH suffers from an overall reduced number of visible peaks and reduced chemical shift redundancy (Fig. 7C; Table 2). The combination of both 5Ds and their base experiments, on the other hand (here, the hcaCBCANH was always used as a base), returns complete and 100% correct assignments (Fig. 7B; Table 1) but requires increased measurement time investments. However, nearly perfect assignment is obtained also for a minimal set of one (HNcoCANH, here with 24 scans) 5D amide-to-amide experiment in conjunction with the hcaCBCANH (Fig. 7D; Table 2, no errors). When the short 5D HNcoCANH is employed (i.e., the 36 h version), FLYA yields remarkably complete and correct assignments (Fig. 7E; Tables 2 and 98% correct, i.e., 2% mismatch, in total approx. 3.5 days of measurement time). Only two resonances are erroneous, which are next to two residues that are not always correctly assigned by FLYA. Interestingly, the two residues directly missing in the 5D (R21 and T37), can be indirectly assigned by FLYA, probably in the same way the expert is able to assign those in the manual assignment process. Importantly, in both of these cases (Fig. 7D and E, and Table 2), backbone assignments are obtained from only two experiments, avoiding lots of the error-prone peak-picking and minimizing combinatorial efforts of standard assignment tasks. (A reduced set of experiments also simplifies chemical-shift referencing, a common problem for solid-state NMR as a lock signal is absent.) For comparison, a FLYA run using a 4D HNNH instead of the 5D HNcoCANH yields similar results to the run with the quick (36 h, lower-s/n) 5D (Fig. 7F; Tables 2 and 97% correct, 3% mismatch). This 4D spectrum was recorded for approx. 30 h (581 NUS points and 16 scans), i.e., it would have a higher signal to noise than the combination in Fig. 7D. As such, as expected, the vacancies in the assignment are a result of H/N overlap rather than signal to noise. Missing CO resonances can in either case be filled in a straightforward way either by FLYA (see Fig. S3, no strong violations after 36 h) or the user. The other residues missed out even for excessive sets of experiments (Fig. 7B/D and Table 2) are invisible in any CP-based experiments and have been assigned previously only via fully INEPT-based experiments (Linser et al. 2010a).

FLYA assignments using different levels of input data. The classical set of 3D experiments for backbone assignment was used as input for FLYA in (A) and can be seen as a point of orientation for the performance of FLYA. For (B) to E), different combinations of 5D and 3D experiments were used (see the text for more details, HNcoCANH with 24 scans in B) and (C) and with 8 scans in E). F) The 5D HNcoCANH was replaced with a 4D HNcocaNH experiment, yielding slightly worse but similar results. For all runs, the manual assignments were used as a reference. 100 independent runs were performed, using tolerances of 0.1 ppm for 1 H and 0.5 ppm for 13 C and 15 N, respectively, and ultimately combined into a consensus chemical shift assignment. (A 2D hNH is usually submitted as well, but implies negligible costs, relatively speaking.) Color scheme: Green: In agreement with reference assignment (here the manual assignment). Red: Disagreement with reference assignment. Blue: Additional assignments, not in reference. Black: In reference but not assigned by FLYA. Dark colors generally indicate “strong”/reliable assignments that are found in at least 80% of the independent FLYA runs, lighter colors indicate they have been found less than 80% of the time
Simultaneous backbone and sidechain assignment
As highlighted above, a particular advantage of 5D over 4D amide-to-amide experiments is the use of a shift triplet as a source for assignment, facilitating matchmaking with side-chain resonances. This concerns Cβ resonances, obtained via hcaCBcaNH type experiments, or the set of sidechain carbons, obtained via S2B experiments. In addition to sidechain carbons, S2B experiments can be used for the assignment of protons in sidechains of protonated samples. This is highly sought for structure calculation, chemical-shift perturbations outside the set of backbone shifts, and other analyses of the protein (Andreas et al. 2016; Stanek et al. 2016; Vasa et al. 2019). Recently we demonstrated that sidechain protons can be reliably assigned using a 5D S2B, in particular the combined 5D HCCNH/4D HCCH experiment (Klein et al. 2022b). Whereas a similar approach could be used for partially protonated sidechains (for example from RAP (Asami and Reif 2013) or iFD labeling (Medeiros-Silva et al. 2016), we demonstrated this for non-deuterated samples using high spinning frequencies (> 100 kHz) for narrow proton linewidth. The 5D HCCNH experiment presents essentially the same HN bulk sensitivity as the 3D hcaCBcaNH, but one HN signal detected in the bulk carries cumulative magnetization stemming from a multitude of individual H/C moieties. In congruence with the above thought of streamlining backbone assignments in small proteins by a minimal set of higher-dimensionality experiments, here we tested whether a full (backbone and sidechain) assignment can also be leveraged by a minimal number of (accordingly higher-complexity) experiments. For expansion of resonance assignment to the sidechain H/C moieties we replaced the hcaCBcaNH experiment in the sets described above with the 5D HCCNH. This demands more measurement time (The S2B experiment was recorded in a 0.7 mm rotor for 6 days.), but the set allows coverage of all but the carbonyl resonances from only two experiments (Fig. 8A) within cumulative measurement time of 8 days at 700 MHz (or 8.5 d when an hCONH is included for added CO assignments). These minimal sets of experiments combine data from different preparations, namely the deuterated/amide-back-exchanged sample spun at 55 kHz for the backbone experiment(s) and the fully protonated sample spun at 110 kHz for the S2B. The assignments are successful even though we did not take specific care of adjusting the temperature to a common value (25 °C in the 1.3 mm rotor vs. 20 °C in the 0.7 mm rotor), which entails slight chemical-shift deviations between the experiments. For comparison, the set of established 3D experiments of an equivalently complete strategy would at least incorporate hCANH/hCAcoNH, hCONH/hCOcaNH, hcaCBcaNH, HNHA, hCCH, and HCcH, i.e., eight experiments and a larger extent of manual handling, including referencing the individual experiments and substantial efforts of picking the many peaks in crowded 3D side-chain spectra. Following a manual assignment strategy with exclusively the two 5D experiments (recording the 5D HCCNH with a simultaneous 4D HCCH, as shown in Klein et al. 2022b), 88% of all resonances, excluding the side-chain protons of aromatic residues, can be assigned from two 5Ds. (Mixing between aliphatic and aromatic carbons still represents a hurdle and has only recently been overcome in solid-state NMR for uniformly labelled proteins (Ahlawat et al. 2023).) In this case, an additional 3D hCANH experiment served as a base for reconstruction but could in principle be replaced by the H/N/CA information (the strongest peaks) already intrinsically included in 5D S2B experiment.

Results of FLYA assignments via an experimental pair of an amide-to-amide experiment and an S2B experiment in comparison to the manual assignments. (A) Sketch of magnetization transfers of the experiments in the set. The orange HNcoCANH was recorded on a 1.3 mm sample, the 4D HCCH (cyan) is an additional pathway available for free while recording the 5D HCCNH (blue); both were simultaneously recorded on a 0.7 mm sample. (B) to E) FLYA results from different combinations of input data as denoted on the right. Although the assignment success is very similar for the different combinations, the best agreement with the manual assignments is achieved through the combination of two 5Ds, likely due to the higher chemical shift redundancy. The tolerances used are 0.1 ppm for 1 H, 0.8 ppm for 13 C, and 0.5 ppm for 15 N, respectively. Compare Fig. S4 for additional experimental sets. Color scheme as in Fig. 7
While the manual sidechain assignment strategy remains straightforward when using the 5D HCCNH experiment (The complete set of sidechain H/C peaks is obtained in a 2D hCH type spectrum per residue, assuming that no overlap in the hCANH 3D space is present.), the application of FLYA can accelerate the process further, with no or little compromise for the assignment quality, based on the results for the backbone assignments. From FLYA assignments, based on the same minimal set as for the manual assignments (the 5D HNcoCANH, 5D HCCNH, and the hCANH as a base for SMFT), 95% of the strongly assigned resonances are identical with the manual assignments (dark green) and 4% are erroneous (Fig. 8B; Table 3). An output with this level of quality is sufficiently good for structure calculation without further manual corrections (Klein et al. 2022b). When using the 4D HNNH version for backbone sequential connectivities, only slightly lower assignment success is achieved (Fig. 8C; Table 3). This is due to the fact that the S2B experiment is the bottleneck and a slightly lower backbone assignment fidelity does not lead to a further decrease of the success rate. If sufficient resolution in the H/N correlation is warranted, the S2B and amide-to-amide experiments combined here can also both be acquired as 4Ds, i.e., 4D HNcocaNH and 4D HCcNH (Fig. 8D; Table 3). Despite the low molecular weight and high β-sheet content of the SH3-domain, the hNH spectrum does comprise regions with signal overlap (Fig. 1) and hence ambiguities in the backbone assignment are obtained. However, given the intrinsic gain in sensitivity by \(\surd 2\), the detection of the weakest resonances within the large dynamic range is facilitated. As such, even under the conditions chosen here (4.25 days and 6 days in the case of the 4D HCcNH and 5D HCCNH experiment, respectively), around 13% more resonances are found using the 4D HCcNH than in the 5D HCCNH (when used on its own, ignoring the interleaved 4D HCCH), upon manual assignment. The additional assignments generally refer to weak resonances, emphasizing sensitivity as the critical aspect here. However, when processed in conjunction with the simultaneous 4D HCCH experiment from orphaned magnetization, in the framework of manual assignment, the 5D HCCNH approach facilitates even slightly more assignments than the 4D HCcNH version. Conversely, as reported previously (Klein et al. 2022b), the poor chemical shift redundancy in the 4D HCCH and the associated many assignment possibilities lead to no further improvements in the automated assignment process with FLYA, such that the FLYA results shown in Fig. 8 and S4 are always obtained without the 4D HCCH experiment.
When the 5D HCCNH S2B (without the 4D HCCH peak list) is combined with a 4D backbone experiment, only 89% of all strong assignments are correct (Fig. 8C; Table 3), which rate is further decreased to 83% when both experiments are taken into account as 4Ds (Fig. 8D). Even when supplementing the FLYA runs with an additional hCANH peak list, the trends are maintained (see Fig. S4, 88% for 4D/4D and 91% for 4D/5D). Additionally, we assessed the assignment success of the minimal set involving two 5D spectra when the HNcoCANH is recorded for only 36 h (8 scans, Fig. 8E). The assignment success (90% correct assignments, 8% erroneous) lies in between what is obtained when acquiring this backbone experiment longer (Fig. 8B) and slightly above the FLYA assignment involving the 4D HNNH experiment instead (Fig. 8C). Despite the increase in mismatching assignments, a remarkable extent of backbone assignments is still obtained through automated assignment from these two data sets. Even though in this case, the 5D versions do not outcompete the computational use of 4D sets by much, the results still show that not only manual assignments can benefit from 5D data, but even in automated routines for combined backbone and sidechain assignments, the fifth dimension can be of advantage. The FLYA results in total highlight that high degrees of assignments (backbone and sidechains) of proteins can be obtained with minimal manual expert user interference from only two (highly complementary, higher-dimensionality) experiments.
Discussion
The above data suggests that even for small to mid-range molecular-weight targets, the traditional assignment process, based on pairs of complementary carbon match-making experiments, can realistically be alleviated by replacing the numerous spectra of lower information content by fewer, higher-information-content experiments. The compressed information content addresses multiple hurdles associated with the assignment process, in particular upon manual expert user assignments. The number of spectra that the user has to oversee simultaneously, display and maintain their orientation within, is much lower. Finding the matching resonance of the next/previous neighbor by combined processing of multiple strips by traditional means is usually not straightforward. In particular, when a complementary pair of 3Ds derives from partially overlapping H/N peaks, consolidating 6 spectra to validate each one of the numerous trials necessary with the usual degrees of carbon shift similarity easily becomes confusing. Instead, from fewer, higher-dimensionality experiments, the user benefits from a direct identification of the next/previous amide coordinates from a simple 2D plane out of a single, preassembled stack directly linked to residue type information. Similarly, referencing of spectra to each other is facilitated. Even though sensitivities of the higher-dimensionality experiments are still lower than the average 3D experiment, the measurement times for a set of experiments are highly competitive, as very insensitive experiments like the hcaCBcacoNH are unnecessary. Current developments, e.g., in the field of optimal-control NMR, hold promise that the sensitivity of higher-dimensionality experiments can be further (drastically) increased in the future (Blahut et al. 2022) and are currently investigated by us. Within the boundaries of the above assessments, automated assignments seem to be performing at least similarly well, comparing carbon-match-making and higher-dimensionality direct-concatenation approaches, but (for this sample) also demonstrating the performance of 4D experiments as maintained alternatives to the 5Ds. We realize that a first implementation of the processing routines is associated at current with an activation barrier, as the way of data handling feels different to long-established procedures, which combats the simplification of the assignment process itself. We hope that a more broadband implementation and standardization of 4D and 5D processing into increasingly user-friendly software packages will overcome this hurdle for future users and allow an easy access to the benefits of higher-dimensionality data for the next generation of solid-state NMR spectroscopists.
Conclusion
Here we have demonstrated the very general value of 5D amide-to-amide experiments for facilitated assignment of micro-crystalline or quasi-crystalline proteins including low to mid-range molecular weight. Due to their low level of ambiguity upon sequential correlations and high-fidelity match-making between sequential information and information on residue type, the higher-dimensionality approaches effectively reduce ambiguity upon peak assignment with less or at least comparable measurement times relative to traditional triple-resonance experiments. In addition, the low number of complementary experiments with compressed information content each minimize the combinatorial and organizational effort on the user side and further facilitate the assignment process by eradication of human errors in practice. As such, the experiments used here provide competitive performance for backbone and side-chain assignments over traditional techniques and can be effectively combined with automated assignment algorithms. Once the pipelines for setup and processing of higher-dimensionality spectra have been established as routine tools in a given research environment, these approaches can streamline manual and automated assignment of future solid-state NMR targets and hence leverage one of the currently most time-consuming and cumbersome steps of bio-ssNMR studies for new target systems.
References
Ahlawat S, Mopidevi SMV, Taware PP, Raran-Kurussi S, Mote KR, Agarwal V (2023) Assignment of aromatic side-chain spins and characterization of their distance restraints at fast MAS. J Struct Biology: X 7:100082
Akbey Ü et al (2009) Optimum levels of exchangeable protons in perdeuterated proteins for proton detection in MAS solid-state NMR spectroscopy. J Biomol NMR 46:67–73
Andreas LB et al (2015) Protein residue linking in a single spectrum for magic-angle spinning NMR assignment. J Biomol NMR 62:253–261
Andreas LB et al (2016) Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Proc Natl Acad Sci USA 113:9187–9192
Asami S, Reif B (2013) Proton-detected solid-state NMR spectroscopy at aliphatic sites: application to crystalline systems. Acc Chem Res 46:2089–2097
Barbet-Massin E et al (2013) Out-and-back 13 C-13 C scalar transfers in protein resonance assignment by proton-detected solid-state NMR under ultra-fast MAS. J Biomol NMR 56:379–386
Barbet-Massin E et al (2014) Rapid Proton-Detected NMR assignment for proteins with fast Magic Angle Spinning. J Am Chem Soc 136:11
Becker LM et al (2023) The rigid core and flexible surface of ayloid fibrils probed by magic-angel-spinning NMR spectroscopy of aromatic residues. Angew Chem Int Ed e202219314
Bertarello A et al (2020) Picometer resolution structure of the coordination sphere in the metal-binding site in a metalloprotein by NMR. J Am Chem Soc 142:16757–16765
Blahut J, Brandl MJ, Pradhan T, Reif B, Tosner Z (2022) Sensitivity-enhanced Multidimensional solid-state NMR spectroscopy by optimal-control-based transverse mixing sequences. J Am Chem Soc 144:17336–17340
Böckmann A, Ernst M, Meier BH (2015) Spinning proteins, the faster, the better? J Magn Reson 253:71–79
Cala-De Paepe D, Stanek J, Jaudzems K, Tars K, Andreas LB, Pintacuda G (2017) Is protein deuteration beneficial for proton detected solid-state NMR at and above 100 kHz magic-angle spinning? Solid State Nucl Magn Reson 87:126–136
Chevelkov V, Rehbein K, Diehl A, Reif B (2006) Ultrahigh Resolution in Proton Solid-State NMR spectroscopy at high levels of Deuteration. Angew Chem Int Ed 45:3878–3881
Chevelkov V, Faelber K, Schrey A, Rehbein K, Diehl A, Reif B (2007) Differential line broadening in MAS solid-state NMR due to dynamic interference. J Am Chem Soc 129:10195–10200
Chevelkov V, Fink U, Reif B (2009) Accurate determination of Order parameters from 1H,15 N Dipolar Couplings in MAS solid-state NMR experiments. J Am Chem Soc 131:14018–14022
Chevelkov V, Giller K, Becker S, Lange A (2013) Efficient CO-CA transfer in highly deuterated proteins by band-selective homonuclear cross-polarization. J Magn Reson 230:205–211
Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. Journal of Biomolecular NMR 6
Fraga H et al (2017) Solid-state NMR H–N–(C)–H and H–N–C–C 3D/4D correlation experiments for Resonance assignment of large proteins. ChemPhysChem 18:2697–2703
Gallo A, Franks WT, Lewandowski JR (2019) A suite of solid-state NMR experiments to utilize orphaned magnetization for assignment of proteins using parallel high and low gamma detection. J Magn Reson
Gauto DF, Hessel A, Rovó P, Kurauskas V, Linser R, Schanda P (2017) Protein conformational dynamics studied by 15 N and 1H R1ρ relaxation dispersion: application to wild-type and G53A ubiquitin crystals. Solid State Nucl Magn Reson 87:86–95
Grohe K et al (2019) Exact distance measurements for structure and dynamics in solid proteins by fast-magic-angle-spinning NMR. Chem Commun 55:7899–7902
Grohe K et al (2020) Protein Motional Details revealed by complementary Structural Biology techniques. Structure 28:1–11
Helmus JJ, Jaroniec CP (2013) Nmrglue: an open source Python package for the analysis of multidimensional NMR data. J Biomol NMR 55:355–367
Huber M, Böckmann A, Hiller S, Meier BH (2012) 4D solid-state NMR for protein structure determination. Phys Chem Chem Phys 14:5239–5246
Hyberts SG, Robson SA, Wagner G (2013) Exploring signal-to-noise ratio and sensitivity in non-uniformly sampled multi-dimensional NMR spectra. J Biomol NMR 55:167–178
Jain MG et al (2017) Selective 1H-1H Distance Restraints in fully protonated proteins by very fast Magic-Angle spinning solid-state NMR. J Phys Chem Lett 8
Kazimierczuk K, Zawadzka A, Koźmiński W (2009) Narrow peaks and high dimensionalities: exploiting the advantages of random sampling. J Magn Reson 197:219–228
Klein A et al (2022a) Atomic-resolution Chemical Characterization of (2x)72 kDa Tryptophan Synthase via Four- and Five-Dimensional 1H-Detected Solid-State NMR. Proc Natl Acad Sci USA 119:e2114690119
Klein A, Vasa SK, Söldner B, Grohe K, Linser R (2022b) Unambiguous side-chain assignments for solid-state NMR structure elucidation of Nondeuterated Proteins via a combined 5D/4D side-chain-to-backbone experiment. J Phys Chem Lett 13:1644–1651
Klukowski P, Riek R, Guntert P (2022) Rapid protein assignments and structures from raw NMR spectra with the deep learning technique ARTINA. Nat Commun 13:6151
Knight MJ et al (2011) Fast resonance assignment and fold determination of human superoxide dismutase by high-resolution proton-detected solid-state MAS NMR spectroscopy. Angew Chem Int Ed 50:11697–11701
Knight MJ et al (2012) Structure and backbone dynamics of a microcrystalline metalloprotein by solid-state NMR. Proc Natl Acad Sci USA 109:11095–11100
Kosiński K, Stanek J, Górka MJ, Żerko S, Koźmiński W (2017) Reconstruction of non-uniformly sampled five-dimensional NMR spectra by signal separation algorithm. J Biomol NMR 68:129–138
Kulminskaya N, Vasa SK, Giller K, Becker S, Linser R (2015) Asynchronous through-bond homonuclear isotropic mixing: application to carbon-carbon transfer in perdeuterated proteins under MAS. J Biomol NMR 63:245–253
Kulminskaya N, Vasa SK, Giller K, Becker S, Kwan A, Sunde M, Linser R (2016) Access to side-chain Carbon Information in Deuterated solids under fast MAS through Non-Rotor-Synchronized Mixing. Chem Commun 52:268–271
Kurauskas V et al (2017) Slow conformational exchange and overall rocking motion in ubiquitin protein crystals. Nat Commun 8:1–11
Lalli D et al (2017) Proton-Based structural analysis of a Heptahelical transmembrane protein in lipid bilayers. J Am Chem Soc 139:13006–13012
Lee W, Tonelli M, Markley JL (2014) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31:1325–1327
Lee W, Bahrami A, Dashti HT, Eghbalnia HR, Tonelli M, Westler WM, Markley JL (2019) I-PINE web server: an integrative probabilistic NMR assignment system for proteins. J Biomol NMR 73:213–222
Lee W, Rahimi M, Lee Y, Chiu A (2021) POKY: a software suite for multidimensional NMR and 3D structure calculation of biomolecules. Bioinformatics 37:3041–3042
Lewandowski JR, Dumez JN, Akbey Ü, Lange S, Emsley L, Oschkinat H (2011) Enhanced resolution and coherence lifetimes in the solid-state NMR spectroscopy of perdeuterated proteins under ultrafast magic-angle spinning. J Phys Chem Lett 2:2205–2211
Linser R (2011) Side-chain to backbone correlations from solid-state NMR of Perdeuterated Proteins through combined excitation and long-range magnetization transfers. J Biomol NMR 51:221–226
Linser R, Chevelkov V, Diehl A, Reif B (2007) Sensitivity enhancement using paramagnetic relaxation in MAS solid-state NMR of Perdeuterated Proteins. J Magn Reson 189:209–216
Linser R, Fink U, Reif B (2010a) Assignment of dynamic regions in biological solids enabled by spin-state selective NMR experiments. J Am Chem Soc 132:8891–8893
Linser R, Fink U, Reif B (2010b) Narrow carbonyl resonances in Proton-Diluted Proteins facilitate NMR assignments in the solid state. J Biomol NMR 47:1–6
Linser R, Bardiaux B, Higman V, Fink U, Reif B (2011a) Structure calculation from unambiguous long-range Amide and Methyl 1H – 1H Distance Restraints for a microcrystalline protein with MAS solid-state NMR spectroscopy. J Am Chem Soc 133:5905–5912
Linser R et al (2011b) Proton-Detected solid-state NMR spectroscopy of fibrillar and membrane proteins. Angew Chem Int Ed 50:4508–4512
Linser R et al (2014) Solid-state NMR structure determination from Diagonal-Compensated, sparsely nonuniform-sampled 4D Proton-Proton Restraints. J Am Chem Soc 136:11002–11010
Lu M et al (2020) Atomic-resolution structure of HIV-1 capsid tubes by magic-angle spinning NMR. Nat Struct Mol Biol 27:863–869
Ma P et al (2014) Probing transient Conformational States of Proteins by solid-state R1ρ relaxation-dispersion NMR spectroscopy. Angew Chem Int Ed 53:4312–4317
Malär AA et al (2019) Quantifying proton NMR coherent linewidth in proteins under fast MAS conditions: a second moment approach. PhysChemChemPhys 21:18850–18865
Mandala VS, Williams JK, Hong M (2018) Structure and Dynamics of membrane proteins from solid-state NMR. Annu Rev Biophys 47:201–222
Medeiros-Silva J, Mance D, Daniëls M, Jekhmane S, Houben K, Baldus M, Weingarth M (2016) 1H-Detected solid-state NMR studies of Water-Inaccessible Proteins in Vitro and in situ. Angew Chem. Int Ed 55
Nimerovsky E, Najbauer EE, Movellan KT, Xue K, Becker S, Andreas LB (2022) Modest offset difference internuclear selective transfer via Homonuclear Dipolar Coupling. J Phys Chem Lett 13:1540–1546
Orton HW et al (2020) Protein NMR resonance assignment without spectral analysis: 5D SOlid-state automated projection SpectroscopY (SO-APSY). Angew Chem Int Ed 59:2380–2384
Palmer MR, Suiter CL, Henry GE, Rovnyak J, Hoch JC, Polenova T, Rovnyak D (2015) Sensitivity of nonuniform sampling NMR. J Phys Chem B 119:6502–6515
Paramasivam S et al (2012) Enhanced sensitivity by nonuniform sampling enables multidimensional MAS NMR spectroscopy of protein assemblies. J Phys Chem B 116:7416–7427
Penzel S et al (2015) Protein resonance assignment at MAS Frequencies approaching 100 kHz: a quantitative comparison of J-Coupling and Dipolar-Coupling-Based transfer methods. J Biomol NMR 63:165–186
Penzel S, Oss A, Org ML, Samoson A, Böckmann A, Ernst M, Meier BH (2019) Spinning faster: protein NMR at MAS frequencies up to 126 kHz. J Biomol NMR 73
Reif B (2021) Deuteration for high-resolution detection of Protons in Protein Magic Angle Spinning (MAS) solid-state NMR. Chemical Reviews
Retel JS et al (2017) Structure of outer membrane protein G in lipid bilayers. Nat Commun 8:1–10
Rovó P (2020) Recent advances in solid-state relaxation dispersion techniques. Solid State Nucl Magn Reson 00:101665
Rovó P, Linser R (2018) Microsecond Timescale Protein Dynamics: a combined solid-state NMR Approach. ChemPhysChem 19:34–39
Rovó P, Smith CA, Gauto D, De Groot BL, Schanda P, Linser R (2019) Mechanistic insights into Microsecond Time-Scale motion of solid proteins using complementary 15 N and 1H relaxation dispersion techniques. J Am Chem Soc 141:858–869
Schanda P, Ernst M (2016) Studying dynamics by magic-angle spinning solid-state NMR spectroscopy: principles and applications to biomolecules. Prog Nucl Magn Reson Spectrosc
Schledorn M et al (2020) Protein NMR spectroscopy at 150 kHz Magic-Angle Spinning continues to improve Resolution and Mass Sensitivity. ChemBioChem 21:2540–2548
Schmidt E, Güntert P (2012) A New Algorithm for Reliable and General NMR resonance assignment. J Am Chem Soc 134:12817–12829
Schubeis T et al (2020) A β-barrel for oil transport through lipid membranes: dynamic NMR structures of AlkL. Proc Natl Acad Sci USA 117:21014–21021
Schubeis T, Stanek J, Pintacuda G (2021) Backbone assignment of crystalline E. coli maltose binding protein. Biomol NMR Assignments 15:317–322
Sharma K, Madhu PK, Agarwal V, Mote KR (2020) Simultaneous recording of intra- and inter-residue linking experiments for backbone assignments in proteins at MAS frequencies higher than 60 kHz. J Biomol NMR 74:229–237
Shi C et al (2014) BSH-CP based 3D solid-state NMR experiments for protein resonance assignment. J Biomol NMR 59:15–22
Shi C, Öster C, Bohg C, Li L, Lange S, Chevelkov V, Lange A (2019) Structure and Dynamics of the rhomboid protease GlpG in Liposomes studied by solid-state NMR. J Am Chem Soc 141:17314–17321
Singh H et al (2019) Fast Microsecond Dynamics of the Protein–Water Network in the active site of human carbonic anhydrase II studied by solid-state NMR spectroscopy. J Am Chem Soc 141:19276–19288
Skinner SP, Fogh RH, Boucher W, Ragan TJ, Mureddu LG, Vuister GW (2016) CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis. J Biomol NMR 66:111–124
Söldner B et al (2023) Integrated Assessment of structure and Dynamics of Solid Proteins. J Phys Chem Lett 14:1725–1731
Stanek J, Koźmiński W (2010) Iterative algorithm of discrete Fourier transform for processing randomly sampled NMR data sets. J Biomol NMR 47:65–77
Stanek J et al (2016) NMR spectroscopic assignment of backbone and side-chain Protons in fully protonated proteins: Microcrystals, Sedimented Assemblies, and amyloid fibrils. Angew Chem Int Ed 55:15504–15509
Stanek J, Schubeis T, Paluch P, Güntert P, Andreas LB, Pintacuda G (2020) Automated backbone NMR resonance assignment of large proteins using redundant linking from a single simultaneous acquisition. J Am Chem Soc 142:5793–5799
Suiter CL et al (2014) Sensitivity gains, linearity, and spectral reproducibility in nonuniformly sampled multidimensional MAS NMR spectra of high dynamic range. J Biomol NMR 59:57–73
Vasa SK, Rovó P, Linser R (2018a) Protons as versatile reporters in solid-state NMR spectroscopy. Acc Chem Res 51:1386–1395
Vasa SK, Singh H, Rovó P, Linser R (2018b) Dynamics and interactions of a 29-kDa human enzyme studied by solid-state NMR. J Phys Chem Lett 9:1307–1311
Vasa SK, Singh H, Grohe K, Linser R (2019) Assessment of a large enzyme–drug Complex by Proton-Detected solid‐state NMR spectroscopy without Deuteration. Angew Chem Int Ed 58:5758–5762
Volk J, Herrmann T, Wüthrich K (2008) Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J Biomol NMR 41:127–138
Wickramasinghe NP et al (2009) Nanomole-scale protein solid-state NMR by breaking intrinsic 1 H T1 boundaries. Nat Methods 6:215–218
Xiang S, Chevelkov V, Becker S, Lange A (2014) Towards Automatic protein backbone assignment using Proton-Detected 4D solid-state NMR data. J Biomol NMR 60:85–90
Xiang S, Grohe K, Rovó P, Vasa S, Giller K, Becker S, Linser R (2015) Sequential backbone assignment based on Dipolar Amide-to-Amide correlation experiments. J Biomol NMR 62:303–311
Xiang SQ, Biernat J, Mandelkow E, Becker S, Linser R (2016) Backbone assignment for minimal protein amounts of low structural homogeneity in the absence of deuteration. Chem Commun 52:4002–4005
Xiang S et al (2017) A two-component Adhesive: tau fibrils arise from a combination of a well-defined motif and conformationally flexible. Interact J Am Chem Soc 139:2639–2646
Zhou DH, Rienstra CM (2008) High-performance solvent suppression for proton detected solid-state NMR. J Magn Reson 192:167–172
Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, Rienstra CM (2007a) Proton-Detected solid-state NMR spectroscopy of fully protonated proteins at 40 kHz Magic-Angle Spinning. J Am Chem Soc 129:11791–11801
Zhou DH et al (2007b) Solid-state protein structure determination with Proton-Detected triple resonance 3D Magic-Angle Spinning NMR spectroscopy. Angew Chem Int Ed 46:8380–8383
Zhou DH et al (2007c) Solid-state protein-structure determination with Proton-Detected triple-resonance 3D Magic-Angle-Spinning NMR spectroscopy. Angew Chem Int Ed 46:8380–8383
Zhou DH et al (2012) Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy. J Biomol NMR 54:291–305
Zinke M et al (2017) Bacteriophage tail-tube Assembly studied by Proton-Detected 4D solid-state NMR. Angew Chem Int Ed 56:9497–9501
Zinke M et al (2020) Architecture of the flexible tail tube of bacteriophage SPP1. Nat Commun 11:5759
Acknowledgements
Financial support is acknowledged from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) in the context of SFB 749, TP A13 (project number 27112786), SFB 1309, TP 03 (project number 325871075), and the Emmy Noether program. This work was funded under Germany’s Excellence Strategy – EXC 2033 – 390677874 – RESOLV and EXC 114 – 24286268. The authors gratefully acknowledge the computing time provided on the Linux HPC cluster at Technical University Dortmund (LiDO3), partially funded in the course of the Large-Scale Equipment Initiative by the German Research Foundation (DFG) as project 271512359.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.K. and S.K.V. performed research, A.K. and R.L. wrote the main manuscript text and prepared figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klein, A., Vasa, S.K. & Linser, R. 5D solid-state NMR spectroscopy for facilitated resonance assignment. J Biomol NMR 77, 229–245 (2023). https://doi.org/10.1007/s10858-023-00424-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-023-00424-5