
Abstract
High mammographic density (MD) is associated with a 4–6 times increase in breast cancer risk. For post-menopausal women, MD often decreases over time, but little is known about the underlying biological mechanisms. MD reflects breast tissue composition, and may be associated with microenvironment subtypes previously identified in tumor-adjacent normal tissue. Currently, these subtypes have not been explored in normal breast tissue. We obtained biopsies from breasts of healthy women at two different time points several years apart and performed microarray gene expression analysis. At time point 1, 65 samples with both MD and gene expression were available. At time point 2, gene expression and MD data were available from 17 women, of which 11 also had gene expression data available from the first time point. We validated findings from our previous study; negative correlation between RBL1 and MD in post-menopausal women, indicating involvement of the TGFβ pathway. We also found that breast tissue samples from women with a large decrease in MD sustained higher expression of genes in the histone family H4. In addition, we explored the previously defined active and inactive microenvironment subtypes and demonstrated that normal breast samples of the active subtype had characteristics similar to the claudin-low breast cancer subtype. Breast biopsies from healthy women are challenging to obtain, but despite a limited sample size, we have identified possible mechanisms relevant for changes in breast biology and MD over time that may be of importance for breast cancer risk and tumor initiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Breast cancer cells are extensively influenced by their non-cancerous surroundings, the microenvironment. The microenvironment consists of cells (such as fibroblasts, immune cells, endothelial cells and normal epithelial cells) and extracellular matrix (ECM) including collagen, which all may influence initiation and progression of cancer [1, 2]. Mammographic density (MD) is a measure of radiologic density of the breast [3]. It varies extensively between individuals and may be seen as a radiologic reflection of breast tissue composition; epithelial and non-epithelial cells as well as collagen increase MD whereas fatty tissue reduces MD [4]. High MD is a strong independent risk factor for breast cancer, but the underlying mechanisms are still unclear [5,6,7,8]. Reduction in MD has been linked to a reduction in breast cancer incidence for women using Tamoxifen as primary prevention [9] and for patients receiving adjuvant hormonal therapy [10].
Normal breast tissue changes throughout life and is influenced by different hormonal events such as menarche, pregnancy, lactation and menopause [11]. The composition of breast tissue is also influenced by heritability [12, 13], use of hormonal therapy [14], nutrition [15, 16] and changes in Body Mass Index (BMI) [17]. MD decreases with age [18] and continues to decrease after menopause [19, 20]. The paradox of decreasing MD in parallel with increasing breast cancer incidence with age, can be explained by the model proposed by Pike et al. [11] which states that biological “breast tissue age” is determined by the cumulative exposure of damaging events to the breast tissue. High MD can reflect such damaging exposure, and thus contribute to increased breast cancer risk. It is important to note that MD is not a single biological state by itself, but recapitulates complex physiological and pathological conditions [2, 21].
Breast tissue from healthy women not undergoing surgery is extremely hard to obtain. The women in this study had previously donated tissue to research when they were examined at breast diagnostic centers. In order to allow a longitudinal study, these women agreed to undergo a second invasive procedure, which allowed us to present the first data on gene expression changes in normal breast tissue over time.
In our previous studies of normal breast tissue [22, 23], we identified a group of normal breast tissue samples exhibiting upregulation of mesenchymal and stem cell genes and downregulation of epithelial markers and adhesion genes, a trait identified in tumors of the claudin-low breast tumor subtype. Furthermore, we identified 24 genes that were negatively correlated to MD, including RBL1 (Retinoblastoma-like protein 1, p107) and three uridine 5′-diphospho-glucuronosyltransferase (UGT) genes whose protein products are known to inactivate estrogen metabolites. RBL1 is expressed at high levels in normal breast epithelium [24], and is thought to have similar tumor suppressive effects as its cousin gene RB1. In addition to acting as gatekeepers of the G1-S transition, the RB proteins may play roles in preservation of chromosomal stability, induction and maintenance of senescence, and regulation of apoptosis, cellular differentiation and angiogenesis [25].
The microenvironment is known to be crucial to cancer initiation and progression [26, 27]. Román-Pérez et al. proposed a method for extratumoral microenvironment subtyping based on gene expression patterns, classifying tumor adjacent normal tissue as active or inactive [28]. The active subtype is characterized by features such as inflammatory response, fibrosis and cellular movement; features similar to the claudin-low breast cancer subtype, proposed by Herschkowitz et al. [29]. The inactive subtype is characterized by maturation, differentiation of epithelial cells, and high cell adhesion. This subtype was later shown to correlate with high MD [21]. These microenvironment subtypes have not been explored in individuals without cancer, but if present in healthy breast tissue, they could potentially influence breast cancer initiation differently.
The aim of this study was to investigate the changes in gene expression that take place in normal breast tissue over a time period of several years, especially in relation to changes in MD and to validate correlations between gene expression and MD identified in our previous study. We validated a negative correlation between RBL1 expression and mammographic density in postmenopausal women and found an association between change in MD and change in expression of histone-related genes. We also demonstrated that the previously defined active and inactive microenvironment subtypes are present in normal breast tissue.
Methods
Subjects
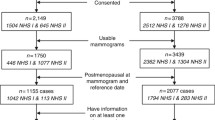
Two separate breast biopsies from healthy volunteering women (i.e. without cancer disease) were obtained with 5–8 years between sample times. The present study is based on our previous study, Mammographic Density and Genetics 1 (MDG1) [22], of women attending the National Breast Cancer Screening Program. The included women were referred to one of several breast diagnostic centers for biopsies due to suspicious findings on mammograms or abnormal clinical findings, and biopsies from breasts without any malignant disease were obtained. Only women without signs of malignant disease were included in this study and biopsies were taken from the contralateral breast of the suspected lesions. MD was determined from mammograms. A total of 120 healthy women were included. Of these, gene expression profiles were available for 79 and MD for 113, with overlapping data for 65 women. Five to eight years later, women who revisited the breast diagnostic center were invited to participate in a follow-up study (MDG2) where new biopsies were obtained, new mammograms taken and new MD assessments performed. A total of 25 women revisited the center at the second time point. All women agreed to participate and completed a questionnaire providing information like height, weight and menopausal status. With regard to menopause status in MDG1, the women were estimated to be pre-, post- or peri-menopausal based on serum levels of FSH, LH and estradiol as previously described [30]. All women provided a signed informed consent. The study was approved by the local ethical committee and local authorities (IRB approval no S-02036).
Biopsies
In both studies, biopsies were obtained as previously described [22]. Briefly, ultrasound guided core biopsies using a 14 gauge needle was performed in an area of some MD to avoid biopsies consisting purely of adipose tissue. Most biopsies were sampled in the upper, lateral quadrant at both time points. The biopsies were snap frozen and stored in -80 °C until RNA isolation. Since healthy breast tissue express less mRNA than tumor tissue, the entire biopsy was required for mRNA extraction. Therefore, no tissue was left for histological or immunohistochemical evaluation.
RNA Isolation and Expression Arrays
Gene expression data for the samples from the previous study (MDG1) are deposited in NCBI’s Gene Expression Omnibus [31] and are accessible through GEO Series accession number GSE18672 [32]. Two additional gene expression datasets were retrieved from GEO: GSE72644 comprises data from breast cancer patients, where multiple biopsies from unaffected normal ducts in the same breast were retrieved for several patients [33]. GSE4823 [34] contains data from normal breast tissue microdissected into epithelium and stroma cellular compartments. The platform used for all three datasets was Agilent Human Gene Expression 4x44K microarrays (G4110A, two colors) (Agilent, Technologies, Santa Clare, USA).
From the new set of biopsies (MDG2), total RNA was isolated using Qiagen miRNeasy Mini kit (Qiagen, Hilden, Germany). The tissue was homogenized by manually mincing on ice with a scalpel followed by Mixer Mill for 40 seconds until complete homogenization. RNA extraction including DNAse treatment was performed according to the protocol provided by the supplier. RNA concentrations were measured by NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) and RNA quality was analyzed using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA).
To obtain whole genome expression data, Agilent Sureprint G3 Human Gene Expression 8x60K microarrays (G4851A) (Agilent, Technologies, Santa Clare, USA) with Low Input Quick Amp Labeling protocol were used. RNA input was 40 ng and Cy3 was used as fluorophore. Quality Control was performed in Agilent’s Feature Extraction software. The microarray expression data for MDG2 have been deposited in the ArrayExpress database at EMBL-EBI under accession number E-MTAB-5885 [35, 36].
In the current study, a total of 25 biopsies were obtained. Of these, three samples were excluded due to too low RNA concentration for expression analysis, and three samples failed Cy3-labeling. Nineteen samples were successfully run on arrays and passed all quality control criteria. For controls, one sample of commercially available normal breast RNA (Ambion Human Breast Total RNA, Thermo Fisher Scientific, Wilmington, DE, USA) and one tumor sample were included throughout the whole pipeline. Two of the samples had no associated MD data. In total, data from 17 samples were complete with both gene expression and MD data. From the previous study (MDG1), 65 samples were complete with gene expression and MD data. For six women, gene expression data were obtained at time point two only. In total, paired data were available from 11 women.
Mammographic Density
Digital craniocaudal mammograms were obtained at routine mammographic centers using a standard protocol. Mammographic density was estimated using the University of Southern California Madena assessment method as described by Ursin et al. [37]: Using the Madena computer software, the reader (GU) outlined the total area of the breast, and the number of pixels was counted by the software. This represents the total breast area. MD was assessed as follows: First, a region of interest that includes all dense areas except those representing the pectoralis muscle or scanning artifacts was identified. Then, a yellow tint was applied to all pixels within the region of interest shaded at or above a threshold intensity of gray. The software then counted the tinted pixels, which represent the area of absolute density. Percent density was determined by dividing the absolute dense area by the total breast area, and multiplying by 100 [5]. Test-retest reliability was 0.99 for absolute density. For cases with mammograms for both breasts available (14 out of 17), the correlation of MD was very high between the right and left breast (Pearson correlation r = 0.97, p < 0,001, n = 14), thus, for these women the average MD was used. For the remaining three women, MD was calculated for the breast with available scans. As a measure of MD change, both absolute change (MD2 − MD1) and relative change (\( \frac{\ MD2- MD1\ }{MD1} \)) was calculated. Since women with low MD in the first study may potentially have a lower absolute decrease than women with high MD, relative change was used for comparison to gene expression and clinical parameters.
Statistical Analysis
Analysis of the relationship between MD at time point one (MD1) and two (MD2) was performed using Pearson correlation. Out of the 24 genes whose expression were identified as significantly associated with MD1 in our first study (MDG1), 16 genes, represented by 23 probes, were present on the array used in the second study (MDG2). To investigate the association between gene expression of these genes and MD in the second study, Pearson correlation was used. Different versions of whole genome expression arrays were used for MDG1 and MDG2; notably Agilent Human Gene Expression 4x44K and 8x60K. To avoid introducing bias, the two expression datasets were analyzed separately and then compared using a rank based approach: For the 11 samples with expression data at both time points, after collapsing to gene level using the median expression of the probes, the genes overlapping in both datasets (n = 15,107) were extracted. For each time point separately, the genes in each sample were ranked based on their expression value. We then calculated rank change as a proxy for change in expression. Spearman correlation analysis was performed to investigate the association between relative change in MD (\( \frac{MD2- MD1}{MD1} \)) and relative change in rank of gene expression (\( \frac{Rank2- Rank1}{Rank1} \)). The top and bottom 200 genes from this analysis were used for gene ontology analyses. As a sensitivity analysis, we checked gene ontology terms associated with the top 500 genes as well.
The breast microenvironment subtypes (active/inactive) were calculated using the Chreighton correlation method as described in Sun et al. [21, 28, 38], separately for the two datasets: The signature consisting of 3194 genes was retrieved from Sun et al. with +1 assigned to up-regulated and − 1 to down-regulated genes. Expression values for genes overlapping with the signature were extracted (for MDG1 2786 genes, for MDG2 2444 genes) and the Pearson correlation coefficients to the signature were calculated. The samples were classified as active if the correlation coefficient was positive, and inactive if it was negative. A Welch two sample t-test was used to find differentially expressed genes between active and inactive subtype in MDG1 followed by gene ontology analyses. The association between microenvironment subtypes and MD was tested using the non-parametric Wilcoxon-Mann-Whitney test. This test was also used when exploring associations between microenvironment subtypes and relevant genes in both MDG1 and MDG2. All statistical tests were two-sided with significance level α = 0.05. Spearman correlation was used where associations between ranks were explored, otherwise Pearson correlation was used accompanied by visualization of the data. All statistical analysis were performed in Rstudio version 1.0.136 [39]. PAM50-subtypes were estimated using the R Package “genefu” [40] and for power analyses the R Package “pwr” was used [41]. To be able to discover similarly strong correlations between MD and age/BMI as previously reported (−0.56/−0.21) [42] with a power above 0.8 and significance level α = 0.05, at least 174/21 samples would be needed. Thus, the size of our cohort is too small to draw any firm conclusions of an association between MD, BMI and age (Online resource 1: Fig.S1 A and B). These parameters were therefore not adjusted for in the analyses to prevent introducing unnecessary noise. A power of 0.66 was obtained in the analyses of the association between microenvironment subtype and MD (n1 = 28, n2 = 37, d = 0.61 (effect size as reported in Sun et al. [21]), α = 0.05). Gene Ontology (GO) analyses were performed in the web-based functional annotation tool DAVID 6.8 [43, 44].which performs enrichment analyses on gene sets enabling exploration of biological systems and pathways.
Scores for epithelial-to-mesenchymal transition (EMT) [28], proliferation [45] and fibrosis (gene signature associated with desmoid type fibromatosis) [46] were calculated using a standard (Z) score approach: For every gene in each signature, a standardized expression value was calculated by subtracting the mean across all samples, then dividing by the standard deviation. The sample’s score was calculated by taking the mean of the standardized expression values of all genes in the signature (Online resource 2).
Gene Set Enrichment Analyses
Gene set enrichment analyses were carried out using the Hallmark gene sets from the Molecular Signatures Database (MSigDB [47, 48]) on the MDG1 dataset: For each sample, genes were ranked by their expression values. Wilcoxon-Mann-Whitney test was used to test difference in rank between the genes in each gene set compared to those not in the gene set. The resulting p value was transformed using this formula: −10 x log10(p value) and the sign was changed according to the direction of enrichment of genes (i.e. whether the genes were highly or lowly expressed) resulting in an enrichment score for each sample and each gene set (Online resource 3). This enrichment score was used for subsequent statistical testing.
Results
Cohort Description
A total of 24 women included in the first MDG study accepted participation in the second study. For 17 of these, both MD and gene expression data was available and used for further analyses. None of the women experienced breast cancer after they were included in the first study. Relevant clinical information is presented in Table 1. Age at second biopsy ranged from 55 to 66 and all the women were at this time point postmenopausal. Mammograms were obtained and MD was estimated as described in Methods. As expected, MD1 and MD2 were highly dependent (Pearson correlation r = 0.80, p < 0.001, n = 17) (Online resource 1: Fig.S1 C). MD decreased from the first to the second measurement for all but one woman. There was no difference in relative MD change between women who had passed menopause between sampling times (n = 5, mean relative MD change = −41.7%) compared to those who already were postmenopausal at the first time point (n = 8, mean relative MD change = −42.6%).
Associations between RBL1 Expression and Mammographic Density Were Validated in the Second Biopsies
Probes for two of the genes identified in our previous study, were significantly associated to MD also in our second study: Retinoblastoma-like protein 1 (RBL1) and Leucine-rich repeat-containing 2 (LRRC2) (Table 2). RBL1 was represented by two probes and both confirmed the previously identified negative association to MD, however, only one of these reached statistical significance (Fig. 1). For the UGT genes that were found to be negatively correlated to MD in MDG1, a negative association was found at the second time point as well, although statistical significance was not reached. (Online resource 1: Fig.S2).

Expression of RBL1 as a function of mammographic density in MDG1 (stratified by menopause status) and MDG2 (all post-menopausal). Pearson correlation: MDG1: Post-menopausal (n = 28), r = −0.51, p = 0.0061; pre/peri-menopausal (n = 22), r = 0.0039, p = 0.99. MDG2 (n = 17): r = −0.49, p = 0.045
When stratifying MDG1 based on menopause status, the correlation between RBL1 expression and MD was evident in post-menopausal women only (Fig. 1), indicating that processes relevant for breast tissue composition may change with menopause. To assess the effect of menopausal status on overall gene expression, we identified differentially expressed genes between biopsies from post- and pre/peri-menopausal women in the largest cohort (MDG1), and found only five differentially expressed genes. Next, we correlated overall gene expression with MD separately in the two menopausal groups, and found substantially more genes associated to MD in the postmenopausal group than the pre−/peri-menopausal (1169 vs. 436 genes) with only 14 genes overlapping between the two groups.
Seeing that RBL1-expression showed a consistent negative correlation to MD over time, prompted us to examine the association between RBL1 expression and enrichment of Hallmark gene sets from the Molecular Signature Database (Online resource 3). We correlated expression values for RBL1 in the MDG1 dataset with Gene Set Enrichment analysis (GSEA) enrichment scores and found that the enrichment scores of WNT/β-catenin signaling and MYC-targets were significantly negatively correlated to RBL1 expression (Spearman correlation, rho = −0.397, p = 0.0011/rho = −0.259, p = 0.037). Further, we wanted to investigate whether MD could be associated with processes relevant for cancer development. To this end, we correlated enrichment scores from GSEA to MD for the samples in the MDG1 dataset and found that gene sets related to Apoptosis and Estrogen response were significantly negatively correlated to MD (Spearman correlation, p = 0.0268/0.0343, rho = −0.277/−0.265), while TGFβ-signaling was marginally not significant (Spearman correlation, p = 0.0638, rho = −0.233).
Intra-individual variation of gene expression may be a complicating factor in all studies where only one biopsy is analyzed. To assess the intra-individual variability of RBL1 expression, we made use of a separate dataset (GSE72644) with gene expression data from two biopsies of normal ductal tissue obtained from different parts of the breast from several patients. We found low correlation between RBL1 expression in different ducts of the same patient (Spearman correlation, p = 0.67, rho = 0.167), indicating some degree of intra-individual variability of RBL1 expression; however the inter-individual variability was small, as demonstrated by a low standard deviation of RBL1 (SD for RBL1: 0.21 vs. mean SD for all genes: 0.68).
Gene Expression in Normal Breasts Changes over Time
From 11 of the women, tissue biopsies were obtained at both time points. A rank-based approach (see Methods section) was taken to overcome the challenge of analyzing gene expression data from two different platforms. To identify biological processes changing in breast tissue over time in parallel with changes in MD, normalized gene expression values were ranked from lowest to highest within each sample, separately for time point one and two. This was followed by Spearman correlation to identify genes with positive or negative correlation between relative change of MD and relative change in gene expression ranks (Online resource 4). Gene ontology analysis of the top 200 genes with a negative correlation between change in gene expression and relative change in MD, revealed involvement of several genes in the histone family H4. Sensitivity analysis using the top 500 genes confirmed these results. In other words, breast tissue samples with a large decrease in MD from the first to the second time point sustained a high expression of these genes (Online resource 1: Fig.S3).
Identifying Microenvironment Subtypes in Normal Breast Tissue
To investigate whether the microenvironment subtypes proposed by Román-Pérez [28] could be identified in normal tissue from healthy breasts, we assigned all tissue samples to a microenvironment subtype (active/inactive) (Online resource 5). In the MDG1 study, 27 samples (41.5%) were of the active subtype, while 38 samples (58.5%) were assigned to the inactive subtype, whereas for the MDG2 study, 8 samples (47.1%) were active, and 9 (52.9%) were inactive. Of the 11 samples with data at both time points, five kept their subtype (45.5%) (one active and four inactive), while six samples (44.5%) changed subtype (three from active to inactive, and three from inactive to active). There was no difference in distribution of menopause status between the two subtypes (Fisher exact test, p = 0.314).
As previously noted by Sun et al., the two microenvironment subtypes may differ in characteristics such as adhesion, stem cell features and TGFβ-signalling. We confirmed these results in our data from normal breasts. In our largest study, the MDG1 study, 3104 genes were significantly differentially expressed between the two subtypes (1390 up and 1714 down in active vs. inactive, Welch two sample t-test, FDR <1%). Gene ontology (GO) analysis showed enrichment of GO-terms related to cell-cell adhesion and tight junctions among the genes that were lower expressed in the active subtype compared to the inactive, while for genes higher expressed in the active subtype, we found GO-terms related to stem cell-like features such as Aldehyde dehydrogenase and Wnt-signaling (Online resource 6). There was a clear distinction between the subtypes in both cohorts with regard to the expression of genes relevant for the claudin-low tumor subtype [28, 49]; the adhesion genes (e.g. CLDN3, CLDN4, CLDN7, CDH1 and OCLN) were lower expressed in the active subtype compared to the inactive, while the EMT-related genes (e.g. TWIST, ZEB1 and ZEB2) were higher expressed (Fig. 2). To consolidate these findings, we tested whether gene signatures from the GSEA analyses were differently enriched between active and inactive microenvironment subtypes in the MDG1 dataset. As many as 28 (out of 50) Hallmark gene sets were differently enriched, confirming the extensive differences between the subtypes (Online resource 3). Most notably were genes involved in Adipogenesis, TGFβ-signaling and Epithelial to Mesenchymal Transition higher expressed in the active subtype compared to the inactive (Mann Whitney U tests, p < 0.001).

Expression of selected claudin-low and EMT relevant genes in MDG1 and MDG2. Menopausal status, mammographic density and microenvironment subtype are indicated in the colored boxes above the heatmaps
In contrast to the findings in Sun et al. [21], we did not find a significant association between microenvironment subtype and MD in any of the cohorts (Online resource 1: Fig. S4). Neither was RBL1 differently expressed between the subtypes (Fig. 3). However, since we found a negative correlation between RBL1 and MD, we wanted to investigate whether genes that may be influenced by RBL1 expression (through its role as a co-repressor together with the transcriptional repressor E2F4) were differentially expressed between the two subtypes. In this context, MYC is particularly interesting, as it is highly relevant in cancer and involved in proliferation [50]. We found that MYC was significantly differently expressed between the subtypes in both cohorts (Fig. 3). There was, however, no significant correlation between MYC and RBL1 (Spearman correlation, MDG1: p = 0.114, rho = −0.198, MDG2: 0,503, rho = 0.174). We therefore wanted to identify E2F4 target genes that were both differentially expressed between the microenvironment subtypes and negatively correlated to RBL1 expression. Platelet derived growth factor subunit A (PDGFA) fulfilled both these criteria in MDG1 (Fig. 3, Spearman correlation PDGFA vs. RBL1, p value = 0.007, rho = −0.33). PDGFA was also differentially expressed between the subtypes in MDG2. In both cohorts, PDGFA was higher expressed in the inactive compared to the active subtype.

RBL1, MYC and PDGFA expression in the microenvironment subtypes active and inactive in MDG1 and MDG2. P-values from Wilcoxon-Mann-Whitney tests
To further explore the differences in properties between the microenvironment subtypes that could be relevant for mammographic density, we calculated standardized z-scores for EMT, fibrosis and proliferation (Online resource 1, Fig. S5 and Online resource 2). Both EMT- and fibrosis signatures were significantly higher in the active compared to the inactive subtype. In MDG2, there were significantly higher proliferation scores in the inactive subtype and the same tendency was also seen in MDG1, however not significant. In addition, scores for EMT and fibrosis in MDG1 were significantly positively correlated in active, but not for the inactive samples (Fig. 4).

Fibrosis score vs. EMT score in MDG1 and MDG2. P-values and rho from Spearman correlation tests
Since there were extensive differences between the microenvironment subtypes in the Gene Set Enrichment Analyses, we wanted to investigate whether the GSEA enrichment scores were differentially associated with MD between the two subtypes in MDG1 (Online resource 3). We found that, in the active subtype, the enrichment scores for the pathway MYC-targets were significantly positively correlated with MD (Spearman correlation, rho = 0.385, p = 0.0476). For the inactive subtype, several gene sets involved in hormonal processes (i.e. Estrogen response and Androgen response) were negatively correlated with MD. In addition, TGFβ-signaling was negatively correlated to MD in the inactive subtype, although significance was not reached (p = 0.063). These results suggest that target genes of the TGFβ pathway may be involved in processes relevant for MD in both microenvironment subtypes.
Spatial Distribution of RBL1 in Normal Breast Tissue
The microenvironment subtypes most likely reflect interplay between stromal and epithelial cells. In this context, it was of interest to investigate whether there was a spatial difference in gene expression of relevant genes between epithelial and stromal cellular compartments. As additional tissue from our cohort was not available for analyses, the spatial distribution of RBL1-expression was studied in a separate dataset (GSE4823) comprising data from normal breast tissue microdissected into epithelial and stromal cellular compartments. These data showed higher expression of RBL1 and a trend toward high expression of PDGFA in the epithelial cells compared to the stromal cells (Online resource 1, Fig. S6). For MYC, there was substantial variation of expression in the epithelial cell compartment, with higher expression in stromal cells in general. This difference was, however, not significant. The spatial distribution of the corresponding proteins were validated using the Human Protein Atlas where protein expression in epithelial cells were confirmed for all three proteins [51,52,53].
Discussion
Our study confirmed that low expression of RBL1 in normal breast tissue in repeated measurements years apart was associated with high MD in postmenopausal women. RBL1 closely resembles RB1 and functions as a tumor suppressor gene involved in cell cycle regulation [54, 55]. The inverse relationship between RBL1 expression and MD harmonizes with its presumptive role as a tumor suppressor through regulation of epithelial cell proliferation and modification of the ECM. RBL1 acts as a co-repressor of transcription as part of the SMAD complex downstream of TGFβ in the TGFβ-signaling pathway [56, 57]. TGFβ has a pleiotropic role in cancer development, contributing to regulating cell proliferation, epithelial-to-mesenchymal transition (EMT) and ECM formation in a highly context dependent manner [58]. Increased TGFβ-signaling in the normal breast is known to inhibit proliferation of epithelial cells [59] and TGFβ-signaling has previously been shown to be reduced in dense mammary tissue [60]. Paradoxically, TGFβ enhances the synthesis of collagen crosslinking enzymes, which increases the rigidity of the collagen network in the ECM [61] and contribute to MD [62, 63]. Adding to the complexity, is the fact that high activity of the TGFβ pathway may have a tumor suppressive role in the initiation and early progression of cancer, and later switch to have a pro-tumorigenic and pro-metastatic role [64]. Reduced TGFβ-signaling may lead to decreased repression of several target genes involved in cell proliferation (possibly affecting MD) and neoplastic transformation [50, 54]. We did not find a significant correlation between RBL1 and MYC expression. However, Gene Set Enrichment Analyses (GSEA) indicated a relationship between RBL1 and MYC-related pathways as both WNT/β-catenin and MYC–target gene sets were negatively correlated to RBL1 expression. These pathways are involved in epithelial proliferation [50, 65].
In postmenopausal women, the estrogen-mediated cell proliferation is lower than in pre-menopausal women [66]. In this study, we saw that low expression of RBL1 was associated with high MD only in postmenopausal women. The explanation for this may lie in the cross-talk between the ERα and TGF-β signaling pathways as ERα represses SMAD3-function in an estradiol-dependent manner [67, 68]. There was a distinct difference in the number of genes whose expression correlated to MD in postmenopausal breast tissue compared to the pre/peri-menopausal breast tissue in the MDG1 cohort. This is in contrast to the low number of differentially expressed genes between breast biopsies from post- and pre/peri-menopausal women at a genome-wide level which has also been reported from other studies [60, 69]. The low number of genes whose expression correlated to MD in pre/peri-menopausal women may be a reflection of more heterogeneity in genes relevant for MD as these may fluctuate substantially due to hormonal changes through the menstrual cycle, potentially masking such associations [70].
Low expression of histones may lead to a more open chromatin structure which is thought to cause higher genomic instability and inappropriate gene expression possibly contributing to carcinogenesis [71]. In accordance with this, we found that breast tissue with a large decrease in MD over time showed sustained or higher expression of histone proteins of the H4 family compared to those with a smaller decrease in MD. Interestingly, high expression of histone genes has been shown to slow down the aging process in cells as high availability of histone proteins contributes to a tighter chromatin structure [71].
The microenvironment subtypes proposed by Sun et al. and Román-Pérez et al. [21, 28] were observed in the normal breast samples in our study. The samples classified as active subtype showed features such as high expression of EMT-related genes, low expression of genes involved in cell-cell adhesion and upregulation of GO-terms related to stem cell-like characteristics similar to what is found in the claudin-low breast tumor subtype [49, 72,73,74]. Although the claudin-low subtype was initially discovered in breast tumors, we have previously found evidence of claudin-low characteristics in normal breast tissue from MDG1 [23]. All of these were in the present study determined as active subtype. There was no difference in RBL1 expression between the subtypes, but both MYC and PDGFA were significantly higher expressed in the inactive samples compared to the active, indicating higher activation of the TGFβ pathway in the active compared to the inactive subtype. The samples of the active subtype also showed enrichment of fibrosis-related genes shown by Beck et al. to be enriched in a subset of breast carcinomas associated with longer survival [46]. The presence of increased EMT features, TGFβ activation and fibrosis in the active subtype may indicate the presence of a “wound healing” phenotype even without any tumor initiation [75, 76].
In Sun et al. the inactive subtype was associated with slightly higher MD. We could not detect the same association between MD and microenvironment subtypes in our data. However, this may be a question of insufficient power. The high degree of fibrosis seen in the active subtype does not harmonize with higher MD in the inactive samples as was observed by Sun et al., since one would suspect that a high degree of fibrosis would lead to higher density. However, normal fibroblasts may inhibit proliferation of epithelial cells [77], and as mammographic density is a product of both different cell types and ECM constituents, a higher content of epithelial cells in the inactive subtype could explain this discrepancy. Additionally, the samples analyzed by Sun et al. were tumor adjacent tissue, while in our study, the biopsies were normal breast tissue from healthy individuals. This is an important distinction, as dynamic interactions between tumor cells, tumor adjacent normal epithelium, and stroma may influence gene expression patterns.
Mammographic density is a comprehensive measurement representing the whole breast and may have limited ability of capturing local differences, which may further explain the lack of association between MD and microenvironment subtype in our study. Also, intra-breast heterogeneity, such as presence of stem cell niches [78], may explain differences between two biopsies from the same breast. We found, however, a low degree of intra-individual variability of expression of relevant genes in normal breasts using an external dataset, which strengthens our finding of a negative association between MD and RBL1-expression.
Conclusions
This is the first study of gene expression in two normal breasts biopsies from the same healthy individuals taken several years apart. We have validated a negative correlation between RBL1 expression and mammographic density in postmenopausal women, and found that breast tissue samples from women with a large decrease in mammographic density over time sustained higher expression of histone family genes. We also identified the previously defined active and inactive microenvironment subtypes and characterized their biological differences in normal breast tissue. Our data indicated an association between MD and target genes in the TGFβ-signaling pathway regardless of microenvironment subtype. This study has identified mechanisms relevant for normal breast tissue biology and MD over time that may be of importance for breast cancer risk and tumor initiation.
Abbreviations
- BMI:
-
body mass index
- ECM:
-
extracellular matrix
- EMT:
-
epithelial-to-mesenchymal transtition
- FDR:
-
false discovery rate
- FSH:
-
follicle-stimulating hormone
- GO:
-
gene ontology
- LH:
-
luteinizing hormone
- MD:
-
mammographic density
- MDG:
-
mammographic density and genetics (name of study)
- RBL1:
-
Retinoblastoma-like protein 1
- SD:
-
standard deviation
- TGFβ:
-
Transforming Growth Factor Beta
References
Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. https://doi.org/10.1038/35094059.
Boyd N, Berman H, Zhu J, Martin LJ, Yaffe MJ, Chavez S, et al. The origins of breast cancer associated with mammographic density: a testable biological hypothesis. Breast Cancer Res. 2018;20(17):17. https://doi.org/10.1186/s13058-018-0941-y.
Yaffe MJ. Mammographic density. Measurement of mammographic density Breast Cancer Res. 2008;10:209. https://doi.org/10.1186/bcr2102.
Martin LJ, Boyd NF. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: hypotheses based on epidemiological evidence. Breast Cancer Res. 2008;10:201. https://doi.org/10.1186/bcr1831.
McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidemiol Biomark Prev. 2006;15:1159–69. https://doi.org/10.1158/1055-9965.EPI-06-0034.
Pettersson A, Graff RE, Ursin G, Santos Silva ID, McCormack V, Baglietto L, et al. Mammographic density phenotypes and risk of breast Cancer: a meta-analysis. JNCI J Natl Cancer Inst. 2014;106:dju078–dju078. https://doi.org/10.1093/jnci/dju078.
Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E, et al. Mammographic density and the risk and detection of breast Cancer. N Engl J Med. 2007;356:227–36. https://doi.org/10.1056/NEJMoa062790.
Li T, Sun L, Miller N, Nicklee T, Woo J, Hulse-Smith L, et al. The association of measured breast tissue characteristics with mammographic density and other risk factors for breast cancer. Cancer Epidemiol Biomark Prev. 2005;14:343–9. https://doi.org/10.1158/1055-9965.EPI-04-0490.
Cuzick J, Warwick J, Pinney E, Duffy SW, Cawthorn S, Howell A, et al. Tamoxifen-induced reduction in mammographic density and breast Cancer risk reduction: a nested case-control study. JNCI J Natl Cancer Inst. 2011;103:744–52. https://doi.org/10.1093/jnci/djr079.
Li J, Humphreys K, Eriksson L, Edgren G, Czene K, Hall P. Mammographic density reduction is a prognostic marker of response to adjuvant Tamoxifen therapy in postmenopausal patients with breast Cancer. J Clin Oncol. 2013;31:2249–56. https://doi.org/10.1200/JCO.2012.44.5015.
Pike MC, Krailo MD, Henderson BE, Casagrande JT, Hoel DG. “Hormonal” risk factors, “breast tissue age” and the age-incidence of breast cancer. Nature. 1983;303:767–70.
Ziv E, Shepherd J, Smith-Bindman R, Kerlikowske K. Mammographic breast density and family history of breast cancer. J Natl Cancer Inst. 2003;95:556–8. https://doi.org/10.1093/JNCI/95.7.556.
Boyd NF, Dite GS, Stone J, Gunasekara A, English DR, McCredie MRE, et al. Heritability of mammographic density, a risk factor for breast Cancer. N Engl J Med. 2002;347:886–94. https://doi.org/10.1056/NEJMoa013390.
Couto E, Qureshi SA, Hofvind S, Hilsen M, Aase H, Skaane P, et al. Hormone therapy use and mammographic density in postmenopausal Norwegian women. Breast Cancer Res Treat. 2012;132:297–305. https://doi.org/10.1007/s10549-011-1810-x.
Knight JA, Martin LJ, Greenberg CV, Lockwood GA, Byng JW, Yaffe MJ, et al. Macronutrient intake and change in mammographic density at menopause: results from a randomized trial. Cancer Epidemiol Biomark Prev. 1999;8:123–8.
Ursin G, Sun C-L, Koh W-P, Khoo K-S, Gao F, Wu AH, et al. Associations between soy, diet, reproductive factors, and mammographic density in Singapore Chinese women. Nutr Cancer. 2006;56:128–35. https://doi.org/10.1207/s15327914nc5602_2.
Boyd NF, Martin LJ, Sun L, Guo H, Chiarelli A, Hislop G, et al. Body size, mammographic density, and breast Cancer risk. Cancer Epidemiol Biomark Prev. 2006;15:2086–92. https://doi.org/10.1158/1055-9965.EPI-06-0345.
Checka CM, Chun JE, Schnabel FR, Lee J, Toth H. The relationship of mammographic density and age: implications for breast Cancer screening. Am J Roentgenol. 2012;198:W292–5. https://doi.org/10.2214/AJR.10.6049.
Sterns EE, Zee B. Mammographic density changes in perimenopausal and postmenopausal women: is effect of hormone replacement therapy predictable? Breast Cancer Res Treat. 2000;59:125–32. https://doi.org/10.1023/A:1006326432340.
Boyd N, Martin L, Stone J, Little L, Minkin S, Yaffe M. A longitudinal study of the effects of menopause on mammographic features. Cancer Epidemiol Prev Biomarkers. 2002;11:1048–53.
Sun X, Gierach GL, Sandhu R, Williams T, Midkiff BR, Lissowska J, et al. Relationship of mammographic density and gene expression: analysis of normal breast tissue surrounding breast cancer. Clin Cancer Res. 2013;19:4972–82. https://doi.org/10.1158/1078-0432.CCR-13-0029.
Haakensen VD, Biong M, Lingjærde OC, Holmen MM, Frantzen JO, Chen Y, et al. Expression levels of uridine 5′-diphospho-glucuronosyltransferase genes in breast tissue from healthy women are associated with mammographic density. Breast Cancer Res. 2010;12:R65. https://doi.org/10.1186/bcr2632.
Haakensen VD, Lingjaerde OC, Lüders T, Riis M, Prat A, Troester MA, et al. Gene expression profiles of breast biopsies from healthy women identify a group with claudin-low features. BMC Med Genet. 2011;4(77). https://doi.org/10.1186/1755-8794-4-77.
Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–66. https://doi.org/10.1038/nrd2907.
Indovina P, Marcelli E, Casini N, Rizzo V, Giordano A. Emerging roles of RB family: new defense mechanisms against tumor progression. J Cell Physiol. 2013;228:525–35. https://doi.org/10.1002/jcp.24170.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Place AE, Jin Huh S, Polyak K. The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 2011;13:227. https://doi.org/10.1186/bcr2912.
Román-Pérez E, Casbas-Hernández P, Pirone JR, Rein J, Carey LA, Lubet RA, et al. Gene expression in extratumoral microenvironment predicts clinical outcome in breast cancer patients. Breast Cancer Res. 2012;14:R51. https://doi.org/10.1186/bcr3152.
Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. https://doi.org/10.1186/gb-2007-8-5-r76.
Haakensen VD, Bjøro T, Lüders T, Riis M, Bukholm IK, Kristensen VN, et al. Serum estradiol levels associated with specific gene expression patterns in normal breast tissue and in breast carcinomas. BMC Cancer. 2011;11:332. https://doi.org/10.1186/1471-2407-11-332.
Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. https://doi.org/10.1093/NAR/30.1.207.
Gene Expression Omnibus Series GSE18672. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18672
Gene Expression Omnibus Series GSE72644. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72644. Accessed 29 Aug 2018.
Gene Expression Omnibus Series GSE4823. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE4823. Accessed 3 Sep 2018.
Kolesnikov N, Hastings E, Keays M, Melnichuk O, Tang YA, Williams E, et al. ArrayExpress update--simplifying data submissions. Nucleic Acids Res. 2015;43:D1113–6. https://doi.org/10.1093/nar/gku1057.
ArrayExpress Database. Accession number E-MTAB-5885. https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5885
Ursin G, Astrahan MA, Salane M, Parisky YR, Pearce JG, Daniels JR, et al. The detection of changes in mammographic densities. Cancer Epidemiol Biomark Prev. 1998;7:43–7.
Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26:4078–85. https://doi.org/10.1200/JCO.2007.13.4429.
Team RS. RStudio: integrated development for R. Boston: RStudio, Inc; 2016.
Gendoo DMA, Ratanasirigulchai N, Schröder MS, Paré L, Parker JS, Prat A, et al. Genefu: an R/bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics. 2016;32:1097–9. https://doi.org/10.1093/bioinformatics/btv693.
Champely S Basic Functions for Power Analysis [R package pwr version 1.2–1].
Skippage P, Wilkinson L, Allen S, Roche N, Dowsett M, a’Hern R. Correlation of age and HRT use with breast density as assessed by Quantra ™. Breast J. 2013;19:79–86. https://doi.org/10.1111/tbj.12046.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. https://doi.org/10.1038/nprot.2008.211.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. https://doi.org/10.1093/nar/gkn923.
Nielsen TO, Parker JS, Leung S, Voduc D, Ebbert M, Vickery T, et al. A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in Tamoxifen-treated estrogen receptor-positive breast Cancer. Clin Cancer Res. 2010;16:5222–32. https://doi.org/10.1158/1078-0432.CCR-10-1282.
Beck AH, Espinosa I, Gilks CB, van de Rijn M, West RB. The fibromatosis signature defines a robust stromal response in breast carcinoma. Lab Investig. 2008;88:591–601. https://doi.org/10.1038/labinvest.2008.31.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–50. https://doi.org/10.1073/pnas.0506580102.
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database Hallmark gene set collection. Cell Syst. 2015;1:417–25. https://doi.org/10.1016/J.CELS.2015.12.004.
Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. https://doi.org/10.1186/bcr2635.
Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. https://doi.org/10.1016/j.cell.2012.03.003.
Tissue expression of RBL1 - Staining in breast - The Human Protein Atlas. https://www.proteinatlas.org/ENSG00000080839-RBL1/tissue/breast. Accessed 12 Nov 2018.
Tissue expression of MYC - Staining in breast - The Human Protein Atlas. https://www.proteinatlas.org/ENSG00000136997-MYC/tissue/breast. Accessed 12 Nov 2018.
Tissue expression of PDGFA - Staining in breast - The Human Protein Atlas. https://www.proteinatlas.org/ENSG00000197461-PDGFA/tissue/breast. Accessed 12 Nov 2018.
O’Connor RJ, Schaley JE, Feeney G, Hearing P. The p107 tumor suppressor induces stable E2F DNA binding to repress target promoters. Oncogene. 2001;20:1882–91. https://doi.org/10.1038/sj.onc.1204278.
Di Fiore R, D’Anneo A, Tesoriere G, Vento R. RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol. 2013;228:1676–87. https://doi.org/10.1002/jcp.24329.
Chen C-R, Kang Y, Siegel PM, Massagué J. E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell. 2002;110:19–32. https://doi.org/10.1016/S0092-8674(02)00801-2.
Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–24. https://doi.org/10.1038/nrc2853.
Papageorgis P, Stylianopoulos T. Role of TGFβ in regulation of the tumor microenvironment and drug delivery (review). Int J Oncol. 2015;46:933–43. https://doi.org/10.3892/ijo.2015.2816.
Moses H, Barcellos-Hoff MH. TGF-beta biology in mammary development and breast cancer. Cold Spring Harb Perspect Biol. 2011;3:a003277. https://doi.org/10.1101/cshperspect.a003277.
Yang WT, Lewis MT, Hess K, Wong H, Tsimelzon A, Karadag N, et al. Decreased TGFβ signaling and increased COX2 expression in high risk women with increased mammographic breast density. Breast Cancer Res Treat. 2010;119:305–14. https://doi.org/10.1007/s10549-009-0350-0.
Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. https://doi.org/10.1016/j.ceb.2010.08.015.
Ironside AJ, Jones JL. Stromal characteristics may hold the key to mammographic density: the evidence to date. Oncotarget. 2016;7:31550–62. https://doi.org/10.18632/oncotarget.6912.
Huo CW, Chew G, Hill P, Huang D, Ingman W, Hodson L, et al. High mammographic density is associated with an increase in stromal collagen and immune cells within the mammary epithelium. Breast Cancer Res. 2015;17(79):79. https://doi.org/10.1186/s13058-015-0592-1.
Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, et al. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–24. https://doi.org/10.1172/JCI18899.
Kaldis P, Pagano M. Wnt Signaling in Mitosis. Dev Cell. 2009;17:749–50. https://doi.org/10.1016/j.devcel.2009.12.001.
Christov K, Chew KL, Ljung BM, Waldman FM, Duarte LA, Goodson WH, et al. Proliferation of normal breast epithelial cells as shown by in vivo labeling with bromodeoxyuridine. Am J Pathol. 1991;138:1371–7.
Cherlet T, Murphy LC. Estrogen receptors inhibit Smad3 transcriptional activity through Ap-1 transcription factors. Mol Cell Biochem. 2007;306:33–42. https://doi.org/10.1007/s11010-007-9551-1.
Matsuda T, Yamamoto T, Muraguchi A, Saatcioglu F. Cross-talk between transforming growth factor-beta and estrogen receptor signaling through Smad3. J Biol Chem. 2001;276:42908–14. https://doi.org/10.1074/jbc.M105316200.
Pirone JR, D’Arcy M, Stewart DA, Hines WC, Johnson M, Gould MN, et al. Age-associated gene expression in normal breast tissue mirrors qualitative age-at-incidence patterns for breast cancer. Cancer Epidemiol Biomark Prev. 2012;21:1735–44. https://doi.org/10.1158/1055-9965.EPI-12-0451.
Atashgaran V, Wrin J, Barry SC, Dasari P, Ingman WV. Dissecting the biology of menstrual cycle-associated breast Cancer risk. Front Oncol. 2016;6:267. https://doi.org/10.3389/fonc.2016.00267.
Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, et al. Elevated histone expression promotes life span extension. Mol Cell. 2010;39:724–35. https://doi.org/10.1016/j.molcel.2010.08.015.
Dias K, Dvorkin-Gheva A, Hallett RM, Wu Y, Hassell J, Pond GR, et al. Claudin-low breast cancer; clinical & pathological characteristics. Clin Pathol Charact PLoS ONE. 2017;12. https://doi.org/10.1371/journal.pone.0168669.
Pohl S-G, Brook N, Agostino M, Arfuso F, Kumar A, Dharmarajan A. Wnt signaling in triple-negative breast cancer. Oncogenesis. 2017;6:e310. https://doi.org/10.1038/oncsis.2017.14.
Ma I, Allan AL. The role of human aldehyde dehydrogenase in Normal and Cancer stem cells. Stem Cell Rev Reports. 2011;7:292–306. https://doi.org/10.1007/s12015-010-9208-4.
Cheng F, Shen Y, Mohanasundaram P, Lindström M, Ivaska J, Ny T, et al. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-β-slug signaling. Proc Natl Acad Sci U S A. 2016;113:E4320–7. https://doi.org/10.1073/pnas.1519197113.
Klass BR, Grobbelaar AO, Rolfe KJ. Transforming growth factor beta1 signalling, wound healing and repair: a multifunctional cytokine with clinical implications for wound repair, a delicate balance. Postgrad Med J. 2009;85:9–14. https://doi.org/10.1136/pgmj.2008.069831.
Sadlonova A, Bowe DB, Novak Z, Mukherjee S, Duncan VE, Page GP, et al. Identification of molecular distinctions between Normal breast-associated fibroblasts and breast Cancer-associated fibroblasts. Cancer Microenviron. 2009;2:9–21. https://doi.org/10.1007/s12307-008-0017-0.
Roberts KJ, Kershner AM, Beachy PA. The stromal niche for epithelial stem cells: a template for regeneration and a brake on malignancy. Cancer Cell. 2017;32:404–10. https://doi.org/10.1016/j.ccell.2017.08.007.
Acknowledgements
We would like to thank the women volunteering for an extra biopsy for their time and cooperation participating in this study, Phuong Vu and Tone Olsen for assistance and support in the lab, and Laxmi Silwal-Pandit for valuable input to the bioinformatic analyses.
Availability of Data and Material
Gene expression data for the samples from the previous study (MDG1) are deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE18672. The microarray expression data for MDG2 have been deposited in the ArrayExpress database at EMBL-EBI (http://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-5885.
Funding
This work was supported by grants from the South-Eastern Norway Regional Health Authority (2012056 to TS and 2010046 to ÅH) and The Norwegian Cancer Society (794926 to ÅH).
Author information
Authors and Affiliations
Contributions
The study was designed by ÅH, GU, MMH and VD. ÅH, VD and TS ensured funding. MMH and VD assisted in data collection. HB performed the laboratory work. GU estimated the amount of mammographic density. HB and TGL performed statistical analyses of the data. HB, TGL, TS and VDH interpreted the results and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
All women provided a signed informed consent. The study was approved by the Norwegian Regional Committee for Medical and Health Research Ethics, Region South-East (IRB approval no S-02036) and the Protocol Committee of Oslo University Hospital, Oncological department (ref: 2002–15) and finally by the Norwegian Centre for Research Data (NSD) (ref: 2004/00008 CBR/−).
Competing Interests
All authors declare that they have no potential conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
Fig.S1 A and B: Age and BMI vs. mammographic density at time point 2 C: Correlation between mammographic density at the two time points. (Pearson correlation r = 0.80, p < 0.001, n = 17). Fig.S2 Expression of three different UGT genes as a function of mammographic density in MDG2 (n = 17). Pearson correlation: UGT2B7, r = −0.28, p = 0.268; UGT2B10, r = −0.18, p = 0.483; UGT2B11, r = −0.14, p = 0.591. Fig.S3 Gene expression rank change of members of the Histone family 4 from time point one to two. Higher rank at time point 2 than time point 1 (red) correlates with a higher reduction of mammographic density. Fig.S4 Mammographic density in the two microenvironment subtypes active and inactive in MDG1 and MDG2. P-values from Wilcoxon-Mann-Whitney tests. Fig.S5 Z-scores for EMT, Fibrosis and Proliferation for MDG1 and MDG2. P-values from Wilcoxon-Mann-Whitney tests. Fig.S6 Gene expression of RBL1, MYC and PDGFA in epithelial and stromal cell compartments microdissected from normal breast tissue (GEO-dataset GSE482).P-values from Wilcoxon-Mann-Whitney tests. (PPTX 652 kb)
ESM 2
(XLS 17 kb)
ESM 3
(XLS 76 kb)
ESM 4
(XLS 1146 kb)
ESM 5
(XLS 136 kb)
ESM 6
(XLS 218 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bergholtz, H., Lien, T.G., Ursin, G. et al. A Longitudinal Study of the Association between Mammographic Density and Gene Expression in Normal Breast Tissue. J Mammary Gland Biol Neoplasia 24, 163–175 (2019). https://doi.org/10.1007/s10911-018-09423-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-018-09423-x