
Abstract
Vertical profiles of radiocesium (134Cs and 137Cs) were measured in the western subarctic area of the North Pacific in 2017. The highest concentration of 134Cs, which was derived from the accident of the Fukushima Dai-ichi Nuclear Power Plant in 2011, was 0.14 Bq m−3 (or 1.19 Bq m−3 after the decay correction to the accident date). Although the vertical inventory of 134Cs decreased between 2014 and 2017, the inventory in 2017 was larger than that expected. That was probably arose from the return of some portion of the high-concentration water mass along with the anticlockwise subarctic gyre current.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A massive earthquake in the North Pacific Ocean off Japan on 11 March 2011 resulted in serious damage to the Fukushima Dai-ichi Nuclear Power Plant (FNPP1) operated by Tokyo Electric Power Company (TEPCO). And the consequent giant tsunami enlarged the damage because the FNPP1 is located on the North Pacific shoreline of the Japanese island of Honshu. Radiocesium (134Cs and 137Cs) released from the damaged FNPP1 to the atmosphere in March and April 2011 was deposited on land in the eastern area of Honshu island and sea surface in the North Pacific [1, 2]. The total depositions of FNPP1-derived 134Cs and 137Cs on land were equivalent [3] and have been estimated to be 2.4 PBq (1015 Bq), respectively [4]. On the other hand, the estimate of the total deposition of 134Cs or 137Cs in the ocean ranges between 10 and 20 PBq [5,6,7,8,9]. That suggests that 10–20% of the radiocesium released to the atmosphere was deposited on land in the eastern area of Honshu island and the rest (80–90%) was deposited in the North Pacific Ocean.
The radiocesium measurements in the coastal area by TEPCO [10] and the Japanese Government [11] revealed that another major source of radiocesium in the North Pacific was the direct discharge of radioactive water from the FNPP1 between late March and early April. The 134Cs/137Cs ratio in seawater collected during the coastal observations was almost 1, which suggests that the activity concentrations of 134Cs and 137Cs in the discharged water were also equivalent as same as those atmospheric-deposited. The total release of the directly-discharged radiocesium (134Cs or 137Cs) was estimated to be 2–6 PBq in many previous studies [5, 6, 12,13,14]. As a result, the total (atmospheric deposition and direct discharge) release of 134Cs or 137Cs into the North Pacific Ocean was calculated to be 12–26 PBq approximately.
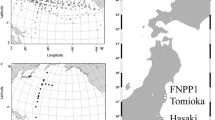
The FNPP1 (37.4°N/141°E) is situated to the north of the Kuroshio Extension Current around 35°N, which corresponds to the Kuroshio Front (Fig. 1). The FNPP1-derived radiocesium directly-discharged and atmospheric-deposited north of the Kuroshio Front was transported eastward along with surface currents. In summer 2012, about one and half years after the accident, High activity concentrations of radiocesium were observed at the sea surface around 165°E–170°W between 40°N and 50°N [15]. This high-concentration water mass then reached stations in the Gulf of Alaska or the eastern subarctic area in the North Pacific in 2015 [16]. A research paper recently published [17] suggested that the water mass had turned to the west and arrived in the northern Bering Sea by summer 2017 along with the anticlockwise subarctic gyre current.

The sampling station (station CL1, 41°N/150°E) in June 2017 (the circle, this work). Those in previous works in June 2011 (the diamond) [18], February 2012 (the triangle) [19], September 2012 (the squares with a dot) [15], October 2012 (the square) [20], July 2014 (the inverted triangle) [21], and October 2014 (the inverted triangle with a dot) [22] are also shown in this figure. At the stations in September 2012 and October 2014, only surface seawater was collected. The star indicates the Fukushima Dai-ichi Nuclear Power Plant (FNPP1). This figure was drawn using Ocean Data View software [23]
As the high-concentration water mass was transported eastward from Japan to the North American Continent, the activity concentration and total inventory of the FNPP1-derived radiocesium increased in the eastern subarctic area of the North Pacific [16]. On the other hand, these in the western subarctic area decreased between 2011 and 2014 [21]. If the high-concentration water mass also returned to the western subarctic area along with the anticlockwise subarctic gyre current in 2017, the FNPP1-derived radiocesium concentration increased between 2014 and 2017. However, its temporal change after 2014 in the western subarctic area is unknown. Here we present vertical profiles of 134Cs and 137Cs in the western subarctic area of the North Pacific Ocean obtained in summer 2017. We discuss the temporal change in the FNPP1-derived radiocesium after 2014 and the time scale of the anticlockwise subarctic gyre circulation in the North Pacific. These discussions are conducive not only to the physical oceanography but also the ongoing monitoring of radionuclide concentrations in seawater at stations in the coastal area of the FNPP1 [10, 11].
Experimental
The seawater samples for radiocesium measurement were collected during a cruise of R/V Hakuho-maru, KH-17-3 in June 2017. Our sampling station locates in the western subarctic area of the North Pacific Ocean, about 1000 km away from the FNPP1 (Fig. 1). The seawater samples were collected from eight layers between the surface (23 m) to 782 m depth using a large-volume sampler (N12-1000, Nichiyu-Giken-Kogyo) that consists of four 250 L PVC samplers, a transponder, a motor-driven trigger, pressure/temperature sensors (RBRduo-TD-Ti sensor, RBR), and a battery unit. Thus, we conducted two casts with the large-volume sampler for shallow (23, 63, 102, and 152 m) and deep samplings (201, 396, 591, and 782 m). The salinity of the seawater sample was measured using a salinometer (Autosal Salinometer 8400B, Ocean Scientific International Ltd). The 250 L of seawater was pumped out using an impeller water pump and filtered through a 0.5 µm pore size wind-cartridge filter (TCW-05 N-PPS, Advantec). Then, about 40 L of seawater for the radiocesium measurement was collected in two plastic 20 L-containers.
To save the shipping costs of the seawater samples and the time for sample preparation in our on-shore laboratory, we conducted cesium separation from the 40 L seawater sample on board using a preparation device (Fig. 2). 1 mL of CsCl (07185-08, Kanto Chemical) solution (2.5 mg g−1) was added into each container and the seawater in it was mixed well. The concentration of stable 133Cs, as a carrier for radiocesium, was about 0.1 Cs-mg L−1. The dissolved (stable and radioactive) cesium in the seawater sample was separated using potassium nickel ferrocyanate-polyacrylonitrile resin (KNiFC-PAN Resin-B NC-B200-M, Lot FNCM170321, Triskem International) without adjustment of sample pH. The PAN resin has been used for the separation of radiocesium from seawater samples in previous studies [24,25,26,27]. The volume of the resin bed in a cartridge was about 5 mL (or about 1 g dry weight) and the flow rate of the seawater sample was 50 mL min−1. The seawater sample was injected from the bottom of the cartridge to stir up the resin bed. Before and after the separation, a small volume aliquot (about 50 mL) of the seawater sample was withdrawn, respectively. After measurements of the volume and weight of the seawater sample in a drain tank (50 L), the seawater samples were spilled on board. And the resin in the cartridges and the small aliquots were carried back to our on-shore laboratory.

The preparation device on board. a The device contains four separation lines, each of which consists of a seawater sample (40 L) with the CsCl carrier, a resin bed (5 mL), a drain tank (50 L), and a peristaltic pump. b The two 20 L containers for the seawater sample were connected using a Y-shaped tube. c The KNiFC-PAN resin bed (5 mL or 1 g dry weight) in a 20 mL plastic cartridge (AC-20-R12, Triskem International)
The carrier (stable cesium) concentration in the aliquot was measured using an ICP mass spectrometer (ELEMENT XR, Thermo Scientific). The difference in the concentration in the aliquot between before and after the separation determined the separation efficiency (or yield rate) of each resin bed. The yield rate depends on the resin bed volume, flow rate, and carrier concentration. Because we used well-type gamma-ray detectors, the bed volume should be less than 5 mL. We measured the yield rates in ranges of the flow rate (50–200 mL min−1) and the carrier concentration (0.1–4 mg L−1) for the 5 mL bed volume and got the highest yield rate (about 95%) at 50 mL min−1 of the flow rate and 0.1 mg L−1 of the carrier concentration (Fig. 3).

The yield rate (%) of the 133Cs carrier against the flow rate (mL min−1) of 3%-NaCl solution (40 L). The concentrations of 133Cs in the NaCl solution were 0.1 (circles), 0.4 (triangles), and 4.0 (squares) mg-Cs L−1. The resin bed volume was about 5 mL (or 1 g dry weight)
The resin in the cartridge was rinsed with pure water at 50 mL min−1 using a peristaltic pump overnight and then dried at 50 °C in an oven. The dried resin was mixed well using a vibrator to avoid the inhomogeneous distribution of radiocesium in the resin. The radiocesium in the resin bed was measured using low-background gamma-ray spectrometers in the underground laboratory of Low Level Radioactivity Laboratory, Kanazawa University [28]. The well-type spectrometers were calibrated with gamma-ray volume sources (Eckert & Ziegler Isotope Products) certificated by Deutscher Kalibrierdienst. The activities of 134Cs and 137Cs were evaluated from gamma-ray peaks at 605 and 661 keV, respectively. The efficiency and background counting in the energy range was about 20% and less than 5 × 10−5 cps, respectively. The gamma counting time was about 10 days. For the peak from 134Cs, the cascade summing effect was corrected. The factor for the effect was about 2, which was calculated as the difference between the 134Cs/137Cs ratios at a distance of 15 cm from the detector and in the well hole of the detector. The averages of the analytical uncertainties (standard deviations) for the 134Cs and 137Cs measurements were calculated to be about 26% and 17%, respectively. This total uncertainty was the sum of those from the gamma counting, the calibration, and the correction for the summing effect. The large uncertainties more than 15% arose from the small number of the total gamma counting. The background concentrations of 134Cs and 137Cs in the KNiFC-PAN resin were below the detection limit (1.1 mBq g−1) and 1.6 mBq g−1, respectively. The detection limits of 134Cs and 137Cs measurements were about 0.05 and 0.01 Bq m−3, respectively, which corresponds to about 0.4 and 0.01 Bq m−3 for the concentrations decay-corrected to the date of the FNPP1 accident (11 March 2011), respectively. The decay-corrected detection limit only depends on the half-life and corrects the underestimation of the detection limit for the decay-corrected activity concentration. The results of the radiocesium measurements are listed in Table S1.
Results
Before the FNPP1 accident, radiocesium was also released into the North Pacific Ocean by the atmospheric testing of nuclear weapons mainly in the 1950s and 1960s. The bomb-derived 137Cs remained in the North Pacific at the time of the FNPP1 accident because its half-life is 30.0 y. The activity concentration at the surface in the western subarctic area just before the FNPP1 accident was about 1.5 Bq m−3 [29]. In contrast, virtually all the 134Cs released into the North Pacific before the FNPP1 accident had disappeared because the half-life of 134Cs is only 2.07 y. Consequently, 134Cs is a more suitable tracer than 137Cs for the FNPP1-derived radiocesium. Therefore, we discuss mainly the results of 134Cs in the following sections.
The FNPP1-derived 134Cs was still observed in the western subarctic area in June 2017, about 6 years after the FNPP1 accident. The highest (0.14 Bq m−3) and lowest (0.05 Bq m−3) activity concentrations were observed at the surface (23 m depth) and 152 m depth (Table S1), respectively. Below 201 m depth, the 134Cs concentration was less than the detection limit (about 0.05 Bq m−3). These low concentrations resulted from the radioactive decay and dilution due to the water mixing. The objective of this study is to elucidate the temporal change in the FNPP1-derived 134Cs due to the water mixing and transport. Therefore, we discuss the activity concentration of 134Cs (and 137Cs) that decay-corrected to the date of the FNPP1 accident in the following sections.
The vertical profile of decay-corrected 134Cs concentration in June 2017 is shown in Fig. 4a (the circles). The observed highest and lowest concentrations correspond to 1.19 and 0.43 Bq m−3 of the decay-corrected concentrations, respectively. Below 201 m depth, the decay-corrected 134Cs concentration was less than the detection limit (about 0.4 Bq m−3). The FNPP1-derived 134Cs had penetrated to 200 m depth at least. The deeper penetration, however, was uncertain because the detection limit was high. This vertical profile observed in 2017 in the western subarctic area, north of the Kuroshio Front, was different from those in the western subtropical area, south of the Kuroshio Front. In the south, the FNPP1-derived 134Cs had been penetrated to 600 m depth by 2017 [30]. The deeper penetration of the FNPP1-derived 134Cs in the south was derived from the sinking (subduction) of the subtropical mode water into the subsurface layer (about 300–400 m) [31], which did not occur in the north of the front.

Vertical profiles of decay-corrected 134Cs (a) and 137Cs (b) activity concentration (Bq m−3) between June 2011 and June 2017 in the western subarctic area of the North Pacific Ocean. The symbols are the same as those in Fig. 1 (the new data in 2017 in this work are indicated by the circles). The activity concentrations are decay-corrected to the date of the FNPP1 accident. The error bar is the standard deviation of each measurement. BDL is the below detection level
The decay-corrected 137Cs activity concentration decreased with depth from 2.61 Bq m−3 at the sea surface to 0.27 Bq m−3 at 800 m depth in 2017 (the circles in Fig. 4b), which indicates that the bomb-derived 137Cs had been penetrated to 800 m depth. In the layers shallower than 200 m depth, where the FNPP1-derived 134Cs was detected, the measured 137Cs included both the bomb-derived and FNPP1-derived 137Cs. The difference between the decay-corrected 134Cs (Fig. 4a) and 137Cs (Fig. 4b) activity concentrations indicates the decay-corrected concentration of the bomb-derived 137Cs (Table S1) because the decay-corrected concentrations of the FNPP1-derived 134Cs and 137Cs were equivalent. The averaged decay-corrected concentration of the bomb-derived 137Cs in the surface layer shallower than 100 m depth is about 1.3 Bq m−3. The averaged value agrees with the observed concentrations just before the FNPP1 accident [29].
Discussion
Changes in the vertical profile
In Fig. 4a, the vertical profiles and surface concentrations of decay-corrected 134Cs at stations about 500 km away from the FNPP1 (Fig. 1) in previous works [15, 18,19,20,21,22] are also shown. In June 2011, about 3 months after the accident, the FNPP1-derived 134Cs concentration in the surface layer shallower than 100 m depth ranged between about 27 and 78 Bq m−3. In April–May 2011, the similar high concentrations of decay-corrected 134Cs between 20 and 80 Bq m−3 approximately were also observed at the sea surface between 143ºE and 148ºE in the western subarctic area [19]. These high concentrations observed within 2 months after the accident at the stations 500–1000 km away from the FNPP1 were probably derived from the atmospheric deposition. In June–August 2011, the decay-corrected concentration at the sea surface increased to 50–110 Bq m−3 approximately due to the eastward transport of the high-concentration water mass of the directly-discharged 134Cs from the coastal area of the FNPP1 to the sampling area [19]. Then those decreased to 10–40 Bq m−3 approximately in September–December 2011 due to the further eastward transport of the high-concentration water mass [19]. Therefore, the 134Cs observed in June 2011 probably has the two sources, the atmospheric-deposited and directly-discharged 134Cs.
In February and September–October 2012, the decay-corrected 134C concentrations in the mixed surface layer above 200 m depth were about 25 and 2–14 Bq m−3, respectively (Fig. 4a). The decay-corrected concentration of 134Cs in the shallower layers than 100 m depth in October 2012 was less than the detection limit. Therefore, we lined between the concentration at 100 m depth (5.27 Bq m−3) in October 2012 and the surface concentration (2.25 Bq m−3) in September 2012 at the station (40.4ºN/146.8ºE) close to the station in October 2012 (Fig. 1). In July 2014, the decay-corrected 134C concentration in the surface mixed layer above 200 m depth ranged between 0.90 and 1.47 Bq m−3 (Fig. 4a). These low concentrations resulted from that the main body of the high-concentration water mass had been transported to the central North Pacific Ocean by the summer of 2012 [15]. The stations between June 2011 and July 2014 were close to each other (Fig. 1). Thus, the observed changes in the vertical profile during this period indicate the temporal decrease of the FNPP1-derived 134Cs concentration at the sampling area (38–41ºN/146–147ºE) in the western subarctic area.
Because our sampling station in June 2017 is about 500 km away from the stations in the previous studies (Fig. 1), we should consider the spatial variation when we compare the vertical profiles in June 2017 to those in the past. In September 2012, the spatial variation in decay-corrected 134Cs concentration at the surface was small (2.25–2.62 Bq m−3) between 146 and 153ºE (the squares with a dot in Fig. 1). Also, the decay-corrected concentrations at the surface in July and October 2014 were almost equal between 146 and 150ºE (the inverted triangle and inverted triangle with a dot in Fig. 1, respectively). These results suggest a small spatial variation in decay-corrected 134Cs concentration at the surface between our sampling station in 2017 and those in the previous studies. Therefore, we concluded that the decay-corrected 134Cs in the surface mixed layer above 200 m depth in the western subarctic area decreased from 0.90–1.47 Bq m−3 in July 2014 to 0.43–1.19 Bq m−3 in June 2017 significantly (Fig. 4a).
The temporal change in the penetration depth of 134Cs is not clear because the detection limits of the 134Cs data in Fig. 4a are different. However, the small temporal variation in 137Cs concentration in layers deeper than 400 m depth between 2012 and 2017 (Fig. 4b) implies that the FNPP1-derived 134Cs have not penetrated to 400 m depth by June 2017. As seen in the vertical profile in February 2012, the surface mixed layer deepens to about 200 m depth in the midwinter in the western subarctic area. As a result, the FNPP1-derived radiocesium penetrates to the bottom of the surface mixed layer. However, the pycnocline between the surface mixed and deeper layers restricts the deeper penetration. In addition, no mode water transports the FNPP1-derived radiocesium to the subsurface layer below about 200 m depth in the western subarctic area. Thereby, the penetration depth and its temporal change in the western subarctic area have been shallower and smaller than those in the western subtropical area, respectively. The small temporal change in the penetration depth implies that the dilution of the FNPP1-derived radiocesium in the surface mixed layer above about 200 m depth in the western subarctic area has been primarily caused by the horizontal water mixing rather than the vertical mixing.
Changes in the vertical inventory
The vertical water-column inventory (or vertical-accumulated activity per square meter) of the decay-corrected 134Cs activity concentration from the surface (0 m) to 800 m depth was calculated from the vertical profiles shown in Fig. 4a using a simple trapezoid method. The vertical inventory (Bq m−2) is identical to an area surrounded by the lines of the 134Cs concentration and zero (below detection level) in the figure. The vertical inventory decreased exponentially from 7459 ± 239 Bq m−2 in 2011 to 283 ± 45 Bq m−2 in 2014 (Fig. 5). The half-valued period for this decrease was calculated to be 227 days. If this exponential decrease had continued to 2017, the vertical inventory would have been about 10 Bq m−2 in 2017. However, a much larger vertical inventory, 186 ± 45 Bq m−2 was observed in June 2017, which suggests additional sources of the FNPP1-derived 134Cs in the sampling area between 2014 and 2017. At stations closed to the FNPP1, 134Cs activity concentration at the sea surface ranged from 10 to 100 Bq m−3 between 2014 and 2017 [11]. These concentrations near the FNPP1 were 10–100 times higher than those in our observational area (Fig. 4a). Although TEPCO denies any leakage of the radioactive water from the FNPP1 during that period, the higher concentrations could be explained by the continuous discharge of the contaminated water. However, these high concentration water at the stations near the FNPP1 probably cannot be the additional source at our sampling station about 1000 km away from the FNPP1 (Fig. 1).

The temporal change in vertical water-column inventory of decay-corrected 134Cs (Bq m−2) between June 2011 and June 2017. The symbols are the same as those in Fig. 1 (the new data in 2017 in this work are indicated by the circle). The inventory in October 2012 was calculated using the data in September and October 2012. The inventories are decay-corrected to the date of the FNPP1 accident. The error bar at each station was calculated from the standard deviations of the data at each station (Fig. 4a)
The second candidate for the additional source of 134Cs is the westward transport of the high-134Cs water mass into the western subarctic area. The main body of the high-134Cs water mass circulated along with the anticlockwise subarctic gyre current in the northern North Pacific. As mentioned above, that reached the eastern subarctic area in 2015 [16] and then the Bering Sea in 2017 [17]. If the forefront of the water mass had reached the western subarctic area by June 2017, that could be the additional source at our sampling station. In the eastern subarctic area, the arrival of the water mass in 2015 resulted in significant increases in the decay-corrected 134Cs concentration and vertical inventory [16]. The concentration and vertical inventory in the western subarctic area, however, did not increase between 2014 and 2017 (Figs. 4a, 5). That suggests that the main body of the high-134Cs water mass had not reached the western subarctic area by summer 2017.
The North Pacific subarctic gyre consists of two anticlockwise circulations, the western and eastern gyres. The western gyre current circulates within the western subarctic area between 140ºE and 180º approximately in a time scale of several years. That is shorter than the time scale of the whole (western and eastern) subarctic gyre circulation, about 10 years, which was suggested by the transport of the FNPP1-derived 134Cs as discussed above [15,16,17]. Therefore, some portions of the high-concentration water mass probably circulated in the western subarctic area along with the anticlockwise western gyre current and retuned our sampling area in the western marge of the gyre within several years after the FNPP1 accident. In the southern Okhotsk Sea, the FNPP1-derived 134Cs concentration and inventory increased between 2013 and 2017 [32]. This increase in the Okhotsk Sea adjacent to the western subarctic area (Fig. 1) also suggests the return of the FNPP1-derived 134Cs to the western marge of the western subarctic area and its consequent transport into the Okhotsk Sea by 2017. Therefore, we concluded that the smaller decrease in 134Cs concentration than expected at our observational station in 2017 was probably derived from the return of some portions (not the main body) of the high-134Cs water mass in the western subarctic area about 6 years after the FNPP1 accident.
Conclusions
The radiocesium derived from the FNPP1 accident in 2011 was still observed in the surface mixed layer above about 200 m depth in the western subarctic area in June 2017, about 6 years after the accident. The decay-corrected activity concentration of the FNPP1-derived radiocesium decreased from 0.90–1.47 Bq m−3 in 2014 to 0.43–1.19 Bq m−3 in 2017 although its penetration depth did not change significantly. That implies that the FNPP1-derived radiocesium in the subarctic area was diluted due to the horizontal water mixing rather than the vertical mixing. The vertical inventory of the decay-corrected FNPP1-derived radiocesium also decreased from 283 ± 45 Bq m−2 in 2014 to 186 ± 46 Bq m−2 in 2017. However, the inventory in 2017 was one-order larger than that expected by the exponential decrease of the inventory between 2011 and 2014, which indicates additional sources of the FNPP1-derived radiocesium in the western subarctic area in 2017. We concluded that the additional source was derived from the return of some portion of the high-concentration water mass to the western subarctic area about 6 years after the accident. The main body of the high-concentration water mass, however, had been circulating along with the whole (western and eastern) subarctic gyre current and had not returned yet to the western subarctic area by summer 2017.
References
Buesseler K, Aoyama M, Fukasawa M (2011) Impacts of the Fukushima Nuclear Power Plants on marine radioactivity. Environ Sci Technol 45:9931–9935
Yoshida N, Kanda J (2012) Tracking the Fukushima radionuclides. Science 336:1115–1116
Saito K, Tanihata I, Fujiwara M, Saito T, Shimoura S, Otsuka T, Onda Y, Hoshi M, Ikeuchi Y, Takahashi F, Kinouchi N, Saegusa J, Seki A, Takemiya H, Shibata T (2015) Detailed deposition density maps constructed by large-scale soil sampling for gamma-ray emitting radioactive nuclides from the Fukushima Dai-ichi Nuclear Power Plant accident. J Environ Radioact 129:308–319
Morino Y, Ohara T, Watanabe M, Hayashi S, Nishizawa M (2013) Episode analysis of deposition of radiocesium from the Fukushima Daiichi Nuclear Power Plant accident. Environ Sci Technol 47:2314–2322
Aoyama M, Kajino M, Tanaka TY, Sekiyama TT, Tsumune D, Tsubono T, Hamajima Y, Inomata Y, Gamo T (2016) 134Cs and 137Cs in the North Pacific Ocean derived from the March 2011 TEPCO Fukushima Dai-ichi Nuclear Power Plant accident, Japan. Part two: estimation of 134Cs and 137Cs inventories in the North Pacific Ocean. J Oceanogr 72:67–76
Buesseler K, Dai M, Aoyama M, Benitez-Nelson C, Charmasson S, Higley K, Maderich V, Masqué P, Morris PJ, Oughton D, Smith JN (2017) Fukushima Daiichi-derived radionuclides in the ocean: transport, fate, and impacts. Annu Rev Mar Sci 9:173–203
Hirose K (2016) Fukushima Daiichi Nuclear Plant accident: atmospheric and oceanic impacts over the five years. J Environ Radioact 157:113–130
Inomata Y, Aoyama M, Tsubono T, Tsumune D, Hirose K (2016) Spatial and temporal distributions of 134Cs and 137Cs derived from the TEPCO Fukushima Daiichi Nuclear Power Plant accident in the North Pacific Ocean by using optimal interpolation analysis. Environ Sci Process Impacts 18:126–136
Tsubono T, Misumi K, Tsumune D, Bryan FO, Hirose K, Aoyama M (2016) Evaluation of radioactive cesium impact from atmospheric deposition and direct release fluxes into the North Pacific from the Fukushima Daiichi Nuclear Power Plant. Deep Sea Res Part I 115:10–21
Tokyo Electric Power Company (2011) Influence to surrounding environment. Archives http://www.tepcocojp/en/nu/fukushima-np/f1/index2-ehtml. Accessed 11 Mar 2020
Japan Nuclear Regulation Authority (2019) Readings of sea area monitoring. http://radioactivity.nsr.go.jp/en/list/205/list-1.html. Accessed 11 Mar 2020
Kaeriyama H (2017) Oceanic dispersion of Fukushima-derived radioactive cesium: a review. Fish Oceanogr 26:99–113
Kumamoto Y, Yamada M, Aoyama M, Hamajima Y, Kaeriyama H, Nagai H, Yamagata T, Murata A, Masumoto Y (2019) Radiocesium in North Pacific coastal and offshore areas of Japan within several months after the Fukushima accident. J Environ Radioact 198:79–88
Tsumune D, Tsubono T, Aoyama M, Uematsu M, Misumi K, Maeda Y, Yoshida Y, Hayami H (2013) One-year, regional-scale simulation of 137Cs radioactivity in the ocean following the Fukushima Dai-ichi Nuclear Power Plant accident. Biogeosciences 10:5601–5617
Kumamoto Y, Aoyama M, Hamajima Y, Nishino S, Murata A, Kikuchi T (2016) Meridional distribution of Fukushima-derived radiocesium in surface seawater along a trans-Pacific line from the Arctic to Antarctic Oceans in summer 2012. J Radioanal Nucl Chem 307:1703–1710
Smith JN, Rossi V, Buesseler KO, Cullen JT, Cornett J, Nelson R, Macdonald AM, Robert M, Kellogg J (2017) Recent transport history of Fukushima radioactivity in the Northeast Pacific Ocean. Environ Sci Technol 51:10494–10502
Kumamoto Y, Aoyama M, Hamajima Y, Nishino S, Murata A, Kikuchi T (2019) Radiocesium in the western subarctic area of the North Pacific Ocean, Bering Sea, and Arctic Ocean in 2015 and 2017. Polar Sci 21:228–232
Buesseler K, Jayne SR, Fisher NS, Rypina II, Baumann H, Baumann Z, Breier CF, Douglass EM, George J, Macdonald AM, Miyamoto H, Nishikawa J, Pike SM, Yoshida S (2012) Fukushima-derived radionuclides in the ocean and biota off Japan. Proc Natl Acad Sci USA 109:5984–5988
Kumamoto Y, Aoyama M, Hamajima Y, Aono T, Kouketsu S, Murata A, Kawano T (2014) Southward spreading of the Fukushima-derived radiocesium across the Kuroshio extension in the North Pacific. Sci Rep 4:4276. https://doi.org/10.1038/srep04276
Kaeriyama H, Shimizu Y, Setou T, Kumamoto Y, Okazaki M, Ambe D, Ono T (2016) Intrusion of Fukushima-derived radiocaesium into subsurface water due to formation of mode waters in the North Pacific. Sci Rep 6:22010. https://doi.org/10.1038/srep22010
Kumamoto Y, Aoyama M, Hamajima Y, Nagai H, Yamagata T, Kawai Y, Oka E, Yamaguchi A, Imai K, Murata A (2017) Fukushima-derived radiocesium in the western North Pacific in 2014. J Radioanal Nucl Chem 311:1209–1217
Kumamoto Y, Aoyama M, Hamajima Y, Nishino S, Murata A, Kikuchi T (2017) Radiocesium in the western subarctic area of the North Pacific Ocean, Bering Sea, and Arctic Ocean in 2013 and 2014. Appl Radiat Isot 126:88–92
Schlitzer R (2019) Ocean Data View, https://www.odvawide/. Accessed 11 Mar 2020
Breier CF, Pike SM, Sebesta F, Tradd K, Breier JA, Buesseler KO (2016) New applications of KNiFC-PAN resin for broad scale monitoring of radiocesium following the Fukushima Dai-ichi nuclear disaster. J Radioanal Nucl Chem 307:2193–2200
Kamenik J, Dulaiova H, Sebesta F, Stastna K (2013) Fast concentration of dissolved forms of cesium radioisotopes from large seawater samples. J Radioanal Nucl Chem 296:841–846
Pike SM, Buesseler KO, Breier CF, Dulaiova H, Stastna K, Sebesta F (2013) Extraction of cesium in seawater off Japan using AMP-PAN resin and quantification via gamma spectroscopy and inductively coupled mass spectrometry. J Radioanal Nucl Chem 296:369–374
Šebesta F, Štefula V (1990) Composite ion exchanger with ammonium molybdophosphate and its properties. J Radioanal Nucl Chem 140:15–21
Hamajima Y, Komura K (2004) Background components of Ge detectors in Ogoya underground laboratory. Appl Radiat Isot 61:179–183
Aoyama M, Tsumune D, Hamajima Y (2013) Distribution of 137Cs and 134Cs in the North Pacific Ocean: impacts of the TEPCO Fukushima-Daiichi NPP accident. J Radioanal Nucl Chem 296:535–539
Kumamoto Y, Aoyama M, Hamajima Y, Oka E, Murata A (2018) Time evolution of Fukushima-derived radiocesium in the western subtropical gyre of the North Pacific Ocean by 2017. J Radioanal Nucl Chem 318:2181–2187
Masuzawa J (1969) Subtropical mode water. Deep Sea Res 16:463–472
Inoue M, Morokado T, Fujimoto K, Miki S, Kofuji H, Isoda Y, Nagao S (2018) Vertical profiles of Fukushima Dai-ichi NPP-derived radiocesium concentrations in the waters of the southwestern Okhotsk Sea (2011–2017). J Environ Radioact 192:580–586
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan (KAKENHI), Interdisciplinary Study on Environmental Transfer of Radionuclides from the Fukushima Dai-ichi NPP Accident (#24110004).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumamoto, Y., Aoyama, M., Hamajima, Y. et al. Activity concentration of Fukushima-derived radiocesium in the western subarctic area of the North Pacific Ocean in summer 2017. J Radioanal Nucl Chem 325, 263–270 (2020). https://doi.org/10.1007/s10967-020-07203-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07203-8