
Abstract
Background
The GLA c.337T > C (p.Phe113Leu) is a known pathogenic variant associated to late-onset Fabry disease phenotype with predominant cardiac manifestations. A founder effect was demonstrated in a large cohort in the Portuguese region of Guimarães. Herein we report an in-depth phenotype description of a cluster of five Southern Italy families.
Methods
Family pedigrees of five index males with the p.Phe113Leu variant were obtained and all at-risk relatives underwent biochemical and genetical screening test. Carriers of GLA p.Phe113Leu variant underwent subsequent multidisciplinary clinical and instrumental evaluation.
Results
Thirty-one (16 M, 15 F) individuals with p.Phe113Leu pathogenic variant were identified. Sixteen out of 31 patients (51.6%) had cardiac manifestations. Notably, myocardial fibrosis was found in 7/8 patients, of whom 2 were under 40 years. Stroke occurred in 4 patients. White matter lesions were detected in 12/19 patients and occurred in 2/10 of subjects under 40 years. Seven females complained of acroparesthesias. Renal involvement occurred in 10 patients. Angiokeratomas were evident in 9 subjects. Eyes, ear, gastrointestinal and pulmonary involvement occurred in the minority of subjects.
Conclusion
This study demonstrates that a cluster of subjects with p.Phe113Leu pathogenic variant is also present in Southern Italy. Disease manifestations are frequent in both sexes and may occur early in life. Cardiac involvement represents the core manifestation, but neurological and renal involvement is also frequent, suggesting that extra-cardiac complications deserve clinical attention.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fabry disease (FD; OMIM #301,500) is a rare X-linked inherited lysosomal storage disorder resulting from absent or markedly reduced activity of the α-galactosidase A (α-Gal-A) enzyme due to mutations in the GLA gene (OMIM #300,644) (Germain 2010). The α-Gal A enzymatic deficiency leads to globotriaosylceramides and its deacylated derivative globotriaosylsphingosine (Lyso-Gb3) lysosomal accumulation within tissue of multiple organs including heart, kidney, central and peripheral nervous systems, skin, eyes and gastrointestinal system (Germain 2010; Ortiz et al. 2018). Fabry disease usually affects hemizygous males; however, heterozygous females may exhibit variable phenotype due to the GLA variant or to X-chromosome inactivation profiles in various organs (Echevarria et al. 2016; Germain et al. 2022). Absent or highly reduced (< 1%) α-Gal-A activity determines the “classic” phenotype, with onset in childhood or adolescence and severe multi-systemic involvement (Germain 2010; Germain et al. 2022). Higher residual enzymatic activity leads to milder single organ (mainly heart or kidney) forms of the disease with late onset (“atypical” or “late-onset” FD, LOFD; Germain 2010; Ortiz et al. 2018; Wilcox et al. 2008). Currently, more than 1000 GLA different mutations have been identified (http://www.dbfgp.org/dbFgp/fabry; http://www.fabrygenphen.com).
The GLA missense c.337T > C (p.Phe113Leu ) is a known pathogenic variant, associated with late-onset disease Azevedo et al. 2020a, b; Oliveira et al. 2020). It was firstly identified in an adult male with mild cardiac-variant FD (Eng et al. 1997) and subsequently described in other case reports and population screening studies (Burlina et al. 2008; Favalli et al. 2016; Hagège et al. 2011; Lee et al. 2010; Nowak et al. 2018; Park et al. 2009; Spada et al. 2006; Veroux et al. 2020; Vigneau et al. 2021). Recently, this pathogenic variant was found in a great number of families in the Portuguese region of Guimaraes and a founder effect has been demonstrated (Azevedo et al. 2020b). In all reported cases, disease phenotype was predominantly cardiac Azevedo et al. 2020a, b; Hagège et al. 2011; Oliveira et al. 2020); however, renal, central and peripheral nervous system involvement as well as sensorineural deafness have been reported Azevedo et al. 2020a, b; Burlina et al. 2008; Favalli et al. 2016; Oliveira et al. 2020; Veroux et al. 2020; Vigneau et al. 2021).
Herein we provide an in-depth phenotype description of five Italian families living in Southern Italy, with LOFD caused by p.Phe113Leu pathogenic variant.
Materials and methods
Subjects
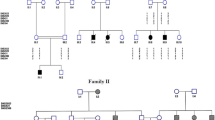
Four men with symptomatic left ventricular hypertrophy (LVH) and one man with ischemic stroke occurred at age 58 years, underwent screening test for FD (α-Gal-A enzymatic activity and plasmatic Lyso-Gb3 levels) with subsequent identification of p.Phe113Leu variant in GLA gene in all of them. They referred to the Rare Disease Center of our hospital and received complete clinical evaluation. Family pedigree was obtained for all the five probands (Fig. 1). All at-risk relatives were invited to undergo screening test (Gal et al. 2011; Michaud et al. 2020; Stiles et al. 2020) for FD (α-Gal-A enzymatic activity for males, plasmatic Lyso-Gb3 level and genetic study for both males and females). Subjects identified as carriers of GLA pathogenic variant underwent subsequent detailed clinical evaluation.

Family pedigrees from the five patients with GLA p.Phe113Leu variant
Index patients are marked with a black arrow. Numbers within symbols denote number of male or female relatives; question mark denotes subjects unavailable for biochemical and genetic evaluation due to refusal or death
Biochemical and genetic evaluation
Enzymatic activity of α-Gal-A was measured trough dried blood spot technique (Chamoles et al. 2001), using high performance liquid chromatography (HPLC) and tandem mass. Normal value of α-Gal-A enzymatic activity and plasmatic Lyso-Gb3 were > 15.3 µ/l/h and < 1.8 ng/ml, respectively. Genetic study was achieved by DNA isolation from whole blood and application of DNA next-generation sequencing (NGS) of the target region (NM_000169.2).
Clinical and instrumental evaluation
The predefined protocol comprised multidisciplinary evaluation. Cardiac evaluation comprised standard ECG or Holter-ECG monitoring, echocardiography, cardiac magnetic resonance imaging (C-MRI). Diagnosis of LVH was defined as the presence of LV wall thickness ≥ 15 mm (Azevedo et al. 2021). Neurological investigation included brain MRI (or brain CT, when MRI was unavailable), color-doppler ultrasound (or angio-TC) of carotid arteries, electroneurography when clinically indicated. Nephrological evaluation included blood and urine analysis (serum creatinine, proteinuria, albuminuria on 24 h-urine and albumin/creatinine ratio on random urine, estimation of glomerular filtration rate (eGFR) by chronic kidney disease-epidemiology collaboration (CKD-EPI) formula) and kidney ultrasound (if indicated). The protocol also included ophthalmologic, dermatologic, gastroenterological, pneumological, psychological/psychiatric and otorhinolaryngology examination, with instrumental evaluation when needed. Symptomatic patients underwent 6 months follow-up evaluation, while asymptomatic individuals without relevant alterations at clinical and instrumental evaluations were followed yearly.
Results
Screening of at risk relatives of the probands resulted in identification of 31 individuals (16 M, 15 F) with GLA p.Phe113Leu variant (Fig. 1). Main demographic, clinical and biochemical features of patients are summarized in Table 1. Mean age at diagnosis was 53.8 years both in male and female. Of the 31 subjects, 7 were members of family I, 4 of family II, 5 of family III, 11 of family IV and 4 of family V. Seventeen of the 31 patients fulfilled diagnostic criteria for late-onset FD diagnosis, while the other 14 were asymptomatic carriers. The α-Gal-A activity levels were < 2.8 μm/l/h in all tested subjects. Plasmatic Lyso-Gb3 levels were available for 29 patients and pathological values were found in 19 (65.5%) subjects. Sixteen (51.6%) patients manifested cardiac involvement (Table 1). Cardiac imaging was performed in 20 patients. Left ventricular hypertrophy was found in 12 (60%) subjects, with slight predominance in males (6/16 M and 6/15 F); all males but one were over 40-year-old. Diastolic dysfunction occurred in 6 (30%) subjects. Eight patients performed C-MRI and late gadolinium enhancement (LGE) was revealed in 7 (87.5%); three of them had concomitant LVH. Moreover, 2 out of 7 subjects with LGE were under 40 years. History of myocardial infarction was reported in 2 males. Heart conduction disturbances were found in 7 (22.5%) subjects, leading to the implantation of a pacemaker in 3 of them.
Stroke occurred in 4 patients (age range 58–70 years). Brain imaging was performed in 19 patients. Singular, multiple or confluent T2-weighted hyperintense MRI lesions (white matter lesions, WML) were detected in 12 (63.1%, Table 1) subjects and represented an early finding, as they were found in 2/5 (40%) of the subjects under 40 years. Interestingly, 4/12 patients with WML had no cerebrovascular risk factors. Peripheral nervous system (PNS) involvement was found in 5 subjects (Table 1). In addition, autonomic dysfunction (hypohidrosis, dysfunctional gastrointestinal motility) was found in 6 patients. Seven females complained with acroparesthesias. Neuropsychiatric manifestations (i.e. depression, anxiety disorder) occurred in 3 subjects.
Renal involvement (albuminuria and/or proteinuria) was found in 10 patients (Table 1); 3/10 were younger than 40 years. Renal insufficiency was rare: stage 1 chronic kidney failure was evident only in 3 females (all older than 60 years). Angiokeratomas were found in 9 (29%) patients. Ocular and hearing manifestations were uncommon (Table 1).
Discussion
This study demonstrated the presence of a FD cluster due to GLA c.337T > C (p.Phe113Leu) pathogenic variant in Southern Italy. The GLA c.337T > C mutation is known to be responsible for late-onset cardiac phenotype since it was found in patients with predominantly cardiac manifestations Azevedo et al. 2020a, b, 2021; Eng et al. 1997; Hagège et al. 2011; Oliveira et al. 2020). Clinical, biochemical and instrumental data confirmed that GLA c.337T > C (p.Phe113Leu) pathogenic variant results in a LOFD with a predominant cardiac phenotype. Clinical or instrumental signs of cardiac involvement were found in about two thirds of subjects. The main cardiac abnormality was LVH, which was found in 60% of our patients and in 40.4% of a large cohort of Portuguese FD subjects (Azevedo et al. 2020b). These results seem to confirm a higher prevalence of LVH in GLA c.337T > C variant as compared to other variants with predominant cardiac involvement such as IVS4 and p.N215S (Hsu et al. 2016; Lavalle et al. 2018). C-MRI has been demonstrated to be a valuable tool for an early assessment of cardiac fibrosis through demonstration of LGE (Perry et al. 2019). Studies providing C-MRI data on cardiac predominant FD phenotypes are scant. We found LGE in 7 out of 8 of our subjects, thus confirming the usefulness of C-MRI. Although an age-dependent incidence of LGE is known (Aquaro et al. 2022), we found LGE in two patients aged under 40 years, suggesting that myocardial fibrosis can occur early in p.Phe113Leu FD variant. Moreover, LVH was found only in 3 patients with LGE, suggesting that LGE may precede LVH (Niemann et al. 2011). In agreement with Azevedo et al. (2020b), hearth failure developed in about a quarter of our patients, whereas myocardial ischemia was less frequent and occurred exclusively in two males. Occurrence of heart arrhythmias is common in classic as well as late-onset variant FD (Azevedo et al. 2021; Hagège et al. 2019; Linhart et al. 2007; Pieroni et al. 2021). In the p.Phe113Leu variant, rhythm and conduction disorders, including atrial fibrillation and flutter, atrioventricular block, fascicular block and non-sustained ventricular tachycardia, have been described Azevedo et al. 2020a, b; Oliveira et al. 2020). Accordingly, cardiac rhythm disturbances were frequent in our cohort too, occurring in 22.5% of subjects, with necessity of pacemaker implantation in 9.6% of subjects.
Central or peripheral nervous system involvement was demonstrated in half of our patients. Stroke is a common and important manifestation in both “classic” and “late-onset” variant FD (Arends et al. 2017; Buechner et al. 2008; Cianci et al. 2022; Brouns et al. 2010; Sims et al. 2009), including prevalent cardiac FD phenotype Alharbi et al. 2018; Azevedo et al. 2020b; Germain DP et al.2018; Lee et al. 2016, 2017). Stroke represented the first FD manifestation in one of our index cases and occurred in 12.9% of whole cohort, mainly in male. A lower incidence of stroke (3.0%) was found in Azevedo et al. series (Azevedo et al. 2020a); this may be explained by differences in the two population’s characteristics, such as higher mean age at stroke and smaller sample of our study.WML occurred in 63.1% of our subjects and were evident also in young patients. Our findings are in line with previous reports Azevedo et al. 2020a, b) and confirm that a small vessel brain disease represents a common finding of FD and may arise early in the course of disease (Kolodny et al. 2015). PNS involvement frequently occur in FD (Cianci et al. 2022; Ortiz et al. 2018; Ranieri et al. 2016). We found acroparesthesias in nearly half of our patients and carpal tunnel syndrome exclusively in one woman. Our findings confirm the female predominance of acroparesthesias and carpal tunnel syndrome previously observed Azevedo et al. 2020a, b; Oliveira et al. 2020).
We found kidney involvement in about a third of our patients. However, the main signs of renal disease were increased albuminuria or proteinuria, whereas low grade renal insufficiency was a rare event. No subject in our series underwent renal dialysis or transplant. Kidney involvement has been reported in p.Phe113Leu variant Azevedo et al. 2020b, b; Burlina et al. 2008; Favalli et al. 2016; Oliveira et al. 2020; Veroux et al. 2020; Vigneau et al. 2021) as well as other prevalently cardiac LOFD (Di Stefano et al. 2021; Germain et al. 2018; Lavalle et al. 2018), rarely requiring renal dialysis or transplant (Arends et al. 2017; Germain et al. 2018; Veroux et al. 2020; Vigneau et al. 2021). Our findings confirms that kidney involvement is often mild and slowly progressive in the p.Phe113Leu LOFD variant Azevedo et al. 2020a, b). Interestingly, it still remains to be elucidated whether kidney involvement in FD due to p.Phe113Leu variant is due to the disease itself or to the presence of other risk factors such as age, hypertension or diabetes mellitus (Azevedo et al. 2020b; Oliveira et al. 2020; Smirnova et al. 2020). In our cohort, albuminuria/proteinuria was also found in subjects under 40 years and none of these patients had major additional risk factors. Our findings corroborate the hypothesis that FD itself may play a major role in kidney disease pathogenesis.
Conclusions
In conclusion, we provide a description of FD phenotype due to the GLA p.Phe113Leu pathogenic variant in a large cohort of subjects belonging to five Italian families. Our findings confirm that the GLA missense c.337T > C (p.Phe113Leu) variant causes a LOFD with a predominant cardiac phenotype. Central and peripheral nervous system as well as kidney involvement may often occur even if the cause-effect relationship with the GLA p.Phe113Leu variant remains to be fully elucidated.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Alharbi FJ, Baig S, Auray-Blais C, Boutin M, Ward DG, Wheeldon N, Steed R, Dawson C, Hughes D, Geberhiwot T (2018) Globotriaosylsphingosine (Lyso-Gb3) as a biomarker for cardiac variant (N215S) fabry disease. J Inherit Metab Dis 41:239–247. https://doi.org/10.1007/s10545-017-0127-2
Aquaro GD, De Gori C, Faggioni L, Parisella ML, Aringhieri G, Cioni D, Lencioni R, Neri E (2022) Cardiac magnetic resonance in Fabry Disease: morphological, functional, and tissue features. Diagnostics (Basel) 12:2652. https://doi.org/10.3390/diagnostics12112652
Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, Elliott PM, Linthorst GE, Wijburg FA, Biegstraaten M, Hollak CE (2017) Characterization of classical and nonclassical fabry disease: a Multicenter Study. J Am Soc Nephrol 28:1631–1641. https://doi.org/10.1681/ASN.2016090964
Azevedo O, Gago MF, Miltenberger-Miltenyi G, Robles AR, Costa MA, Pereira O, Vide AT, Castelo Branco G, Simões S, Guimarães MJ, Salgado A, Sousa N, Cunha D (2020a) Natural history of the late-onset phenotype of fabry disease due to the p.F113L mutation. Mol Genet Metab Rep 22:100565. 10.1016/j.ymgmr.2020a.100565
Azevedo O, Gal A, Faria R, Gaspar P, Miltenberger-Miltenyi G, Gago MF, Dias F, Martins A, Rodrigues J, Reimão P, Pereira O, Simões S, Lopes E, Guimarães MJ, Sousa N, Cunha D (2020b) Founder effect of fabry disease due to p.F113L mutation: clinical profile of a late-onset phenotype. Mol Genet Metab 129:150–160. 10.1016/j.ymgme.2019.07.012
Azevedo O, Cordeiro F, Gago MF, Miltenberger-Miltenyi G, Ferreira C, Sousa N, Cunha D (2021) Fabry Disease and the heart: a Comprehensive Review. Int J Mol Sci 22:4434. https://doi.org/10.3390/ijms22094434
Brouns R, Thijs V, Eyskens F, Van den Broeck M, Belachew S, Van Broeckhoven C, Redondo P, Hemelsoet D, Fumal A, Jeangette S, Verslegers W, Baker R, Hughes D, De Deyn PP, BeFaS I (2010) Belgian fabry study: prevalence of fabry disease in a cohort of 1000 young patients with cerebrovascular disease. Stroke 41:863–868. https://doi.org/10.1161/STROKEAHA.110.579409
Buechner S, Moretti M, Burlina AP, Cei G, Manara R, Ricci R, Mignani R, Parini R, Di Vito R, Giordano GP, Simonelli P, Siciliano G, Borsini W (2008) Central nervous system involvement in Anderson-Fabry disease: a clinical and MRI retrospective study. J Neurol Neurosurg Psychiatry 79:1249–1254. https://doi.org/10.1136/jnnp.2008.143693
Burlina AP, Manara R, Caillaud C, Laissy JP, Severino M, Klein I, Burlina A, Lidove O (2008) The pulvinar sign: frequency and clinical correlations in fabry disease. J Neurol 255:738–744. https://doi.org/10.1007/s00415-008-0786-x
Chamoles NA, Blanco M, Gaggioli D (2001) Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta 308:195–196. https://doi.org/10.1016/s0009-8981(01)00478-8
Cianci V, Pascarella A, Gasparini S, Donadio V, Liguori R, Incensi A, Rao CM, Franzutti C, Scappatura G, Aguglia U, Ferlazzo E (2022) Late-onset fabry disease due to a new (p.Pro380Leu) pathogenic variant of GLA Gene. Metab Brain Dis 37:3023–3026. https://doi.org/10.1007/s11011-022-01079-1
Di Stefano V, Mancarella M, Camporeale A, Regalia A, Ferraresi M, Pisaniello M, Cassinerio E, Pieruzzi F, Motta I (2021) Migalastat Treatment in a kidney-transplanted patient with Fabry Disease and N215S Mutation: the First Case Report. Pharmaceuticals (Basel) 14:1304. https://doi.org/10.3390/ph14121304
Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, Jabbour F, Beldjord C, De Mazancourt P, Germain DP (2016) X-chromosome inactivation in female patients with fabry disease. Clin Genet 8944–8954. https://doi.org/10.1111/cge.12613
Eng CM, Ashley GA, Burgert TS, Enriquez AL, D’Souza M, Desnick RJ (1997) Fabry disease: thirty-five mutations in the alpha-galactosidase A gene in patients with classic and variant phenotypes. Mol Med 3:174–182
Favalli V, Disabella E, Molinaro M, Tagliani M, Scarabotto A, Serio A, Grasso M, Narula N, Giorgianni C, Caspani C, Concardi M, Agozzino M, Giordano C, Smirnova A, Kodama T, Giuliani L, Antoniazzi E, Borroni RG, Vassallo C, Mangione F, Scelsi L, Ghio S, Pellegrini C, Zedde M, Fancellu L, Sechi G, Ganau A, Piga S, Colucci A, Concolino D, Di Mascio MT, Toni D, Diomedi M, Rapezzi C, Biagini E, Marini M, Rasura M, Melis M, Nucera A, Guidetti D, Mancuso M, Scoditti U, Cassini P, Narula J, Tavazzi L, Arbustini E (2016) Genetic screening of Anderson-Fabry Disease in Probands Referred from Multispecialty Clinics. J Am Coll Cardiol 68:1037–1050. https://doi.org/10.1016/j.jacc.2016.05.090
Gal A, Hughes DA, Winchester B (2011) Toward a consensus in the laboratory diagnostics of fabry disease - recommendations of a european expert group. J Inherit Metab Dis 34(2):509–514
Germain DP (2010) Fabry disease. Orphanet. J Rare Dis 5:30. https://doi.org/10.1186/1750-1172-5-30
Germain DP, Brand E, Burlina A, Cecchi F, Garman SC, Kempf J, Laney DA, Linhart A, Maródi L, Nicholls K, Ortiz A, Pieruzzi F, Shankar SP, Waldek S, Wanner C, Jovanovic A Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: a multicenter Fabry registry study. Mol. Genet. Genom. Med. 6:492–503. doi:, Oder 389D, Liu D, Hu K, Üçeyler N, Salinger T, Müntze J, Lorenz K, Kandolf R, Gröne HJ, Sommer C, Ertl G, Wanner C (2018) P. Nordbeck, α-Galactosidase A genotype N215S induces a specific cardiac variant of fabry disease, Circ. Cardiovasc. Genet. 10 (2017) e001691. doi: 10.1161/CIRCGENETICS.116.001691
Germain DP, Altarescu G, Barriales-Villa R, Mignani R, Pawlaczyk K, Pieruzzi F, Terryn W, Vujkovac B, Ortiz A (2022) An expert consensus on practical clinical recommendations and guidance for patients with classic fabry disease. Mol Genet Metab 137:49–61. https://doi.org/10.1016/j.ymgme.2022.07.010
Hagège AA, Caudron E, Damy T, Roudaut R, Millaire A, Etchecopar-Chevreuil C, Tran TC, Jabbour F, Boucly C, Prognon P, Charron P, Germain DP, FOCUS study investigators (2011) Screening patients with hypertrophic cardiomyopathy for fabry disease using a filter-paper test: the FOCUS study. Heart 97:131–136. https://doi.org/10.1136/hrt.2010.200188
Hagège A, Réant P, Habib G, Damy T, Barone-Rochette G, Soulat G, Donal E, Germain DP (2019) Fabry disease in cardiology practice: literature review and expert point of view. Arch Cardiovasc Dis 112:278–287. https://doi.org/10.1016/j.acvd.2019.01.002
Hsu TR, Hung SC, Chang FP, Yu WC, Sung SH, Hsu CL, Dzhagalov I, Yang CF, Chu TH, Lee HJ, Lu YH, Chang SK, Liao HC, Lin HY, Liao TC, Lee PC, Li HY, Yang AH, Ho HC, Chiang CC, Lin CY, Desnick RJ, Niu DM (2016) Later onset fabry disease, cardiac damage progress in silence: experience with a highly prevalent mutation. J Am Coll Cardiol 68:2554–2563. https://doi.org/10.1016/j.jacc.2016.09.943
Kolodny E, Fellgiebel A, Hilz MJ, Sims K, Caruso P, Phan TG, Politei J, Manara R, Burlina A (2015) Cerebrovascular involvement in fabry disease: current status of knowledge. Stroke 6:302–313. https://doi.org/10.1161/STROKEAHA.114.006283
Lavalle L, Thomas AS, Beaton B, Ebrahim H, Reed M, Ramaswami U, Elliott P, Mehta AB, Hughes DA (2018) Phenotype and biochemical heterogeneity in late onset fabry disease defined by N215S mutation. PLoS ONE 13:e0193550
Lee BH, Heo SH, Kim GH, Park JY, Kim WS, Kang DH, Choe KH, Kim WH, Yang SH, Yoo HW (2010) Mutations of the GLA gene in korean patients with fabry disease and frequency of the E66Q allele as a functional variant in korean newborns. J Hum Genet 55:512–517. https://doi.org/10.1038/jhg.2010.58
Lee HJ, Hung SC, Hsu TR, Ko SC, Chui-Mei T, Huang CC, Niu DM, Lin CP (2016) Brain MR imaging findings of cardiac-type fabry disease with an IVS4 + 919G > A mutation. AJNR Am J Neuroradiol 37:1044–1049. https://doi.org/10.3174/ajnr.A4677
Lee HJ, Hsu TR, Hung SC, Yu WC, Chu TH, Yang CF, Bizjajeva S, Tiu CM, Niu DM (2017) A comparison of central nervous system involvement in patients with classical fabry disease or the later-onset subtype with the IVS4 + 919G > a mutation. BMC Neurol 17:25. https://doi.org/10.1186/s12883-017-0810-9
Linhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A, Elliott PM (2007) European FOS investigators, cardiac manifestations of Anderson-Fabry disease: results from the international fabry outcome survey. Eur Heart J 28:1228–1235. https://doi.org/10.1093/eurheartj/ehm153
Michaud M, Mauhin W, Belmatoug N, Garnotel R, Bedreddine N, Catros F, Ancellin S, Lidove O, Gaches F (2020) When and how to diagnose Fabry Disease in Clinical Pratice. Am J Med Sci 360:641–649. https://doi.org/10.1016/j.amjms.2020.07.011
Niemann M, Herrmann S, Hu K, Breunig F, Strotmann J, Beer M, Machann W, Voelker W, Ertl G, Wanner C, Weidemann F (2011) Differences in fabry cardiomyopathy between female and male patients: consequences for diagnostic assessment. JACC Cardiovasc Imaging 4:592–601. https://doi.org/10.1016/j.jcmg.2011.01.020
Nowak A, Mechtler TP, Hornemann T, Gawinecka J, Theswet E, Hilz MJ, Kasper DC (2018) Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients with fabry disease. Mol Genet Metab 123:148–153. https://doi.org/10.1016/j.ymgme.2017.07.002
Oliveira JP, Nowak A, Barbey F, Torres M, Nunes JP, Teixeira-E-Costa F, Carvalho F, Sampaio S, Tavares I, Pereira O, Soares AL, Carmona C, Cardoso MT, Jurca-Simina IE, Spada M, Ferreira S, Germain DP (2020) Fabry disease caused by the GLA p.Phe113Leu (p.F113L) variant: natural history in males. Eur J Med Genet 63:103703. https://doi.org/10.1016/j.ejmg.2019.103703
Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, Eng C, Hopkin RJ, Laney D, Linhart A, Waldek S, Wallace E, Weidemann F, Wilcox WR (2018) Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 123:416–427. https://doi.org/10.1016/j.ymgme.2018.02.014
Park JY, Kim GH, Kim SS, Ko JM, Lee JJ, Yoo HW (2009) Effects of a chemical chaperone on genetic mutations in alpha-galactosidase A in korean patients with fabry disease. Exp Mol Med 41:1–7. https://doi.org/10.3858/emm.2009.41.1.001
Perry R, Shah R, Saiedi M, Patil S, Ganesan A, Linhart A, Selvanayagam JB (2019) The role of Cardiac Imaging in the diagnosis and management of Anderson-Fabry Disease. JACC Cardiovasc Imaging 12:1230–1242. https://doi.org/10.1016/j.jcmg.2018.11.039
Pieroni M, Moon JC, Arbustini E, Barriales-Villa R, Camporeale A, Vujkovac AC, Elliott PM, Hagege A, Kuusisto J, Linhart A, Nordbeck P, Olivotto I, Pietilä-Effati P, Namdar M (2021) Cardiac involvement in Fabry Disease: JACC Review topic of the Week. J Am Coll Cardiol 77:922–936. https://doi.org/10.1016/j.jacc.2020.12.024
Ranieri M, Bedini G, Parati EA, Bersano A (2016) Fabry Disease: Recognition, diagnosis, and treatment of neurological features. Curr Treat Options Neurol 18:33. https://doi.org/10.1007/s11940-016-0414-5
Sims K, Politei J, Banikazemi M, Lee P (2009) Stroke in fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke 40:788–794. https://doi.org/10.1161/STROKEAHA.108.526293
Smirnova A, Di Toro A, Giuliani L, Tagliani M, Urtis M, Favalli V, Arbustini E (2020) Renal and brain complications in GLA p.Phe113Leu fabry disease. Comments on “Fabry disease caused by the GLA p.Phe113Leu (p.F113L) variant: natural history in males” by Oliveira. (Eur J Med Genet 2019) Eur J Med Genet 63:103847. https://doi.org/10.1016/j.ejmg.2020.103847
Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ (2006) High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet 79:31–40. https://doi.org/10.1086/504601
Stiles AR, Zhang H, Dai J, McCaw P, Beasley J, Rehder C, Koeberl DD, McDonald M, Bali DS, Young SP (2020) A comprehensive testing algorithm for the diagnosis of fabry disease in males and females. Mol Genet Metab 130:209–214. https://doi.org/10.1016/j.ymgme.2020.04.006
Veroux M, Monte IP, Rodolico MS, Corona D, Bella R, Basile A, Palmucci S, Pistorio ML, Lanza G, De Pasquale C, Veroux P (2020) Screening for fabry disease in kidney transplant recipients: experience of a Multidisciplinary Team. Biomedicines 8:396. https://doi.org/10.3390/biomedicines8100396
Vigneau C, Germain DP, Larmet D, Jabbour F, Hourmant M, SNOUFY Investigators Group (2021) Screening for fabry disease in male patients with end-stage renal disease in western France. Nephrol Ther 17:180–184. https://doi.org/10.1016/j.nephro.2021.03.002
Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F, Wanner C, Warnock DG, Lemay RM, Germain DP, Fabry Registry (2008) Females with fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab 93:112–128. https://doi.org/10.1016/j.ymgme.2007.09.013
http://www.dbfgp.org/dbFgp/fabry. Accessed 15 February 2023
http://www.fabrygenphen.com. Accessed 15 February 2023
Funding
Open access funding provided by Università degli studi “Magna Graecia” di Catanzaro within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
Concept and design: Vittoria Cianci, Angelo Pascarella, Lucia Manzo, Umberto Aguglia. Data collection, analysis and interpretation: Vittoria Cianci, Angelo Pascarella, Sara Gasparini, Oreste Marsico, Anna Mammì, Carmelo Massimiliano Rao, Claudio Franzutti, Umberto Aguglia, Edoardo Ferlazzo. Drafting of the manuscript: Vittoria Cianci, Angelo Pascarella, Lucia Manzo, Umberto Aguglia, Edoardo Ferlazzo. Critical revision of the manuscript for important intellectual content: Umberto Aguglia.
Corresponding author
Ethics declarations
Ethical statement
All procedures performed were in accordance with the ethical standards of the institutional research committee and with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Informed consent and Consent to participate
Written informed consent and consent to participate have been obtained from patients.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cianci, V., Pascarella, A., Manzo, L. et al. Late-onset fabry disease due to the p.Phe113Leu variant: the first italian cluster of five families. Metab Brain Dis 38, 1905–1912 (2023). https://doi.org/10.1007/s11011-023-01216-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-023-01216-4