
Abstract
Background
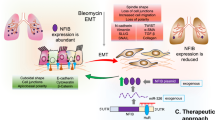
Pulmonary fibrosis is a debilitating condition with limited therapeutic avenues. The pathogenicity of pulmonary fibrosis constitutes involvement of cellular proliferation, activation, and transformational changes of fibroblast to myofibroblasts. It is a progressive lung disease and is primarily characterized by aberrant accumulation of extracellular matrix proteins in the lungs with poor prognosis. The inflammatory response in the pathogenesis of lung fibrosis is suggested because of release of several cytokines; however, the underlying mechanism remains undefined. A genetic model is the appropriate way to delineate the underlying mechanism of pulmonary fibrosis.
Methods and results
In this report, we have used cc-10 promoter based IκBα mutant mice (IKBM, an inhibitor of NF-κB) which were challenged with bleomycin (BLM). Compared to wild-type (WT) mice, the IKBM mice showed significant reduction in several fibrotic, vascular, and inflammatory genes. Moreover, we have identified a new set of dysregulated microRNAs (miRNAs) by miRNA array analysis in BLM-induced WT mice. Among these miRNAs, let-7a-5p and miR-503-5p were further analyzed. Our data showed that these two miRNAs were upregulated in WT-BLM and were reduced in IKBM-BLM mice. Bioinformatic analyses showed that let-7a-5p and miR-503-5p target for endothelin1 and bone morphogenic receptor 1A (BMPR1A), respectively, and were downregulated in WT-BLM mice indicating a link in pulmonary fibrosis.
Conclusion
We concluded that inhibition of NF-κB and modulation of let-7a-5p and miR-503-5p contribute a pivotal role in pulmonary fibrosis and may be considered as possible therapeutic target for the clinical management of lung fibrosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pulmonary fibrosis, a severe condition resulting from an injury to the lung parenchyma, causes increased proliferation and migration of fibroblasts and excessive accumulation of matrix proteins in the lung [1,2,3]. It is a progressive lung disease and primarily characterized by deposition of uncontrolled extracellular matrix (ECM) components including collagens, matrix metalloproteinases (MMPs) and tissue inhibitor of matrix proteinases (TIMPS) [4,5,6]. Fibroblasts play a vital role in fibrotic processes and are regulated by transforming growth factor-β (TGFβ) [7]. During the fibrotic process, the fibroblast becomes activated and transformed into myofibroblast by expressing α-smooth muscle actin (α-SMA or Acta) which is proliferative in nature. The key events of myofibroblast promotes apoptosis and fibrosis [8,9,10]. The myofibroblast is pivotal in pulmonary fibrosis and is linked to structural alteration caused by remodeling of ECM, eventually leading to tissue stiffness and, impaired lung function [11]. Treatment regimens for pulmonary fibrosis are limited. Therefore, identifying key molecule is required for the development of new targeted therapeutics for pulmonary fibrosis.
Accumulative evidence suggested that pro‐inflammatory and pro‐fibrotic cytokines are significantly involved in the pathogenesis of pulmonary fibrosis [12, 13]. Furthermore, it was suggested that excessive inflammation imparted the growth of fibroblasts in the formation of pulmonary fibrosis [14]. Nuclear Factor kappa B (NF-κB) is a pleiotropic transcription factor which regulates a set of genes responsible for both innate and adaptive immune responses [15, 16]. Apart from its’ vital role in immune modulation, NF-κB contributes a critical role in pathogenesis of many lung diseases including pulmonary fibrosis [17, 18]. We demonstrated previously that inhibition of NF-κB in the lungs using IκBα mutant (IKBM) gene, prevented monocrotaline-induced pulmonary hypertension in mice [19]. The IKBM mouse was made using Club (Clara) cell-10 (cc-10) promoter that drives IκBα mutant gene, an inhibitor of NF-κB. Although cc-10 promoter inhibits NF-κB primarily in airway epithelial cells, the existence of Clara cells was observed in the rodent lungs [20]. Therefore, we used this model to understand the role of NF-κB in lung (pulmonary) fibrosis which is currently unknown.
Recently, a small conserved non-coding RNA designated as microRNAs (miRNAs) were implicated in several vascular pathologies including pulmonary fibrosis [21,22,23,24,25]. Therapeutic strategies based on modulation of miRNA hold a great promise in disease pathology managements. MiRNAs are ~22 nucleotides in length which bind to the 3' untranslated region of specific target genes and suppress/inhibit their translation [26, 27]. Currently, NF-κB-mediated miRNA regulation in lung fibrosis remains unknown. In this conceptual setting, determination of miRNAs will provide new strategies to advance our current understanding of the pathogenesis of lung fibrosis. Although, a recent study has shown a signature of miRNA in bleomycin (BLM)-induced lung fibrosis [28]; a major gap remains in understanding the therapeutic benefits of miRNAs and NF-κB-linked miRNAs during pulmonary fibrosis. Despite significant progress in the understanding of pathological mechanisms of persistent fibrosis, effective therapeutic interventions are yet to be identified.
Here, we investigated whether NF-κB inhibition in the lungs using IKBM mice had any effect in BLM-induced pulmonary fibrosis. The generation of IKBM mouse was described previously [19, 29]. BLM is a cytotoxic agent used to treat cancer and induce inflammatory and fibrotic reactions in lungs mimicking idiopathic pulmonary fibrosis [30]. In this study, we hypothesized that the blocking of NF-κB in the lungs could mitigate BLM-induced lung fibrosis.
We determined the inflammatory and fibrotic gene expressions pattern in BLM-induced wild type (WT) and IKBM mice lungs. We report for the first time that BLM-induced pulmonary fibrosis is associated with NF-κB activation (RelA), upregulation of fibrotic genes, enhanced inflammatory response and altered expression of bone morphogenic protein receptor A (BMPR1A) in WT mice; and all these alterations were prevented in IKBM mice treated with BLM. Furthermore, several dysregulated miRNAs were identified in BLM-induced lung fibrosis and were restored in IKBM mice. Our data suggests that NF-κB-miR-503-5p-Col1A1-BMPR1A axis is an important regulator of pulmonary fibrosis and may serve as a target for promising therapeutic intervention.
Materials and methods
Generation of transgenic mice overexpressing the IκBα mutant gene (IKBM)
Generation of the cc-10 promoter driven IKBM mice has been previously described [29]. Age- and sex-matched wild-type (WT) mice of C57BL/6 background served as controls. Eight to ten-week-old mice (~25 g) were used for the experiments. The studies were conducted with the approval of Institutional Animal Care and Use Committee (IACUC) at Baylor University.
Mouse model of bleomycin-induced pulmonary fibrosis
Eight to ten weeks old littermate WT mice (C57BL/6) and IKBM mice were used in this study. To induce pulmonary fibrosis, bleomycin (Sigma-Aldrich, St Louis, MO) was dissolved in sterile saline at 1 U/ml. After inducing anesthesia with intraperitoneal ketamine (80 mg/kg) and xylazine (15 mg/kg), bleomycin was injected intratracheally at a dose of 1.5 U/kg body weight. Control groups received the same volume of sterile saline only. Groups of 5–8 mice were sacrificed at day 21 after BLM treatment. The lungs were removed, washed with cold PBS and were snap frozen for mRNA and miRNA isolation. The tissue was used for miRNA microarray and real-time PCR analyses.
Microarray processing and analysis
The miRNA isolation and preparation for MiRNA array were performed as described previously [31]. In brief, miRNA was isolated from the lungs of WT and IKBM mice using miRNeasy Kit (Qiagen, California, USA) as per manufacturer’s instructions. MiRNA array was performed and analyzed by LC Bioscience (Austin, TX, USA). The data was represented as a log2 value. Paired t-test or ANOVA were performed to find statistically significant differences between the groups and P < 0.05 was considered significant. The miRNA array data is MIAME compliant and was submitted to the gene expression omnibus (GEO) server at NCBI vide GEO no GSE155823.
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
RNA was extracted from the lungs of WT, IKBM, WT + BLM and IKBM + BLM mice using RNEasy kit (Qiagen, Valencia, CA, USA), following the manufacturer’s instructions. The qRT-PCR was performed as described previously [32, 33]. All primers used in the study were purchased from OriGene Technologies Inc. (Rockville, MD, USA) and follow manufacturer’s instruction for primer dilution and PCR amplification cycle.
Statistical analysis
All experiments were performed using at least three to five separate mice lung tissue samples for each group. The RT-PCR analyses were performed in triplicates for each sample. All experiments were performed at least three times for each determination. Data was expressed as the means ± SE and was analyzed using one-way ANOVA using Prism 5.0 GraphPad software (GraphPad, San Diego, CA). P < 0.05 was considered as significant.
Results
BLM-induced lung fibrotic genes are prevented in IKBM mice
The mRNA expression of several fibrotic genes, Col1A1, CTGF, MMP2, Acta2 and TGFβ1 was increased by 3.15 ± 0.83-, 1.74 ± 0.24-, 2.3 ± 0.19-, 1.91 ± 0.36 and 3.44 ± 0.39 (P < 0.05)-fold, respectively, in lungs of BLM-treated WT mice, compared with the untreated WT mice (Fig. 1A–E). The BLM-treated IKBM mice showed a significant down regulation of these genes, compared with BLM-treated WT mice (Col1A1, 1.31 ± 0.04, CTGF, 0.67 ± 0.15, MMP2, 1.14 ± 0.08-, Acta 2, 0.64 ± 0.07 and TGFβ1, 1.15 ± 0.2; P < 0.05) (Fig. 1A–E).

BLM-induced fibrotic genes are prevented in IKBM mice. The mRNA expression of fibrotic genes: Col A1a (A), CTGF (B), Tgfβ1(C), Acta2 (D) and MMP2 (E) in BLM-treated WT and IKBM mice was determined by quantitative RT-PCR. Data are expressed as means ± SE from 3 independent mice. *P < 0.05 compared with the WT mice. **P < 0.05 compared with the WT-BLM mice
BLM-induced inflammatory cytokines and RelA were prevented in IKBM mice
The mRNA expression of proinflammatory cytokine genes; IL6, IL1β and RelA were increased by 2.52 ± 0.31-, 2.32 ± 0.06 and 3.01 ± 0.5 (P < 0.05)-fold, respectively, in the lungs of BLM-treated WT mice, compared with the untreated WT mice (Fig. 2A–C). The BLM-treated IKBM mice showed significant downregulation of the above genes, compared with the BLM-treated WT mice (1.30 ± 0.6-, 1.46 ± 0.25- and 0.91 ± 0.17-fold, P < 0.05, respectively (Fig. 2A–C).

BLM-induced proinflammatory genes are prevented in IKBM mice. The mRNA expression of proinflammatory cytokine IL1β (A), IL6 (B), and RelA (C) in BLM-treated WT and IKBM mice was determined by quantitative RT-PCR. Data are expressed as means ± SE from 3 independent mice. *P < 0.05 compared with the WT mice. **P < 0.05 compared with the WT-BLM mice
BLM treatment alters the expression of BMPR1A, Cav1, Edn1 and PECAM1 genes in the lungs
The mRNA expression of BMPR1A, Cav1 and Edn1 genes were reduced by 0.27 ± 0.04, 0.18 ± 0.804 and 0.95 ± 0.1-fold (P < 0.05), respectively, in BLM-treated WT mice, compared to untreated WT mice. The reduction of BMPR1A, Cav1 and Edn1 were restored in BLM-treated IKBM mice, compared with the BLM-treated WT mice (1.36 ± 0.12, 0.35 ± 0.01 and 0.29 ± 0.04, P < 0.05) (Fig. 3A–C). The restoration of these genes in IKBM group imply their regulation by NF-κB. Additionally, we have determined the expression of PECAM 1 gene. Our data showed an increase of 2.96 ± 0.40-fold in BLM-treated WT mice compared with the untreated WT mice (Fig. 3D). The IKBM mice showed a significant reduction of PECAM1 by 1.5 ± 0.27-fold (P < 0.05) compared to WT-BLM mice.

BLM-induced reduction of BMPR2, Cav1 and Edn1 are restored in IKBM mice. The mRNA expression of BMPR2 (A), Cav1 (B) and Edn1 (C) and PECAM1 (D) in BLM-treated WT and IKBM mice was determined by quantitative RT-PCR. Data are expressed as means ± SE from 3 independent mice. *P < 0.05 compared with the WT mice. **P < 0.05 compared with the WT-BLM mice
Effect of expression of apoptotic gene in BLM-induced lung fibrosis
Expression of the pro-apoptotic Bcl2 family gene, Bak is increased by 2.31 ± 0.30-fold in BLM-treated WT mice, compared with the untreated WT mice. The expression of Bak was reduced to 1.17 ± 0.11 (P < 0.05) in the BLM-treated IKBM mice (Fig. 4).

BLM treatment alters the expression of apoptotic family gene, Bak1 in the lung. The mRNA expression of pro-apoptotic gene Bak1 in BLM-exposed WT and IKBM mice were determined by quantitative RT-PCR. Data are expressed as means ± SE from 3 independent mice. *P < 0.05 compared with the WT mice. **P < 0.05 compared with the WT-BLM mice
MiRNA profiling in the lungs of BLM-treated WT and IKBM mice
MiRNA profiling analyses were performed using lungs of WT and IKBM mice treated with and without BLM by LC Sciences, Houston, TX. The arrays are based on Sanger miR Base release 14.0 databases. Color and spot diameter depicted information about mean expression level and percent detected were shown in the heat maps. For two-color experiments, the ratio of the two sets of detected signals (log2 transformed, balanced) and, the p values of the t-test were calculated (Fig. 5A). Our analysis revealed several novel dysregulated miRNAs in the lungs of WT-BLM group, compared to the untreated WT mice (Table 1).

Heat map showing the expression of dysregulated miRNAs in WT and BLM treated mice lungs. A MiRNA microarray data was prepared using data from the miRNA microarray performed using samples from WT and BLM treated mice lungs and compared with untreated WT mice lungs. The signal difference (log2) is plotted versus the level of statistical significance (2log10 p-value). Color and spot diameter depicted information about mean expression level and percent detected, respectively, in static plots. B Validation of miRNAs in the lungs of WT and BLM treated mice. The mature miR-503-5p (B) and let-7a-5p (C) expressions were determined by qRT-PCR. U6 was used as an internal control. Data are expressed as means ± SE from 3 independent mice. *P < 0.05 compared with the WT mice. **P < 0.05 compared with the WT-BLM mice. D BMR1A and Edn1genes are the target for miR-503-5p and let-7a-5p. An alignment between BMPR1A and miR-503-5p as predicted by TargetScan 7.2 and miRBase analyses as shown in the upper panel by vertical bars. The sequence alignment of putative miR-503-5p and its targeting site on 3’-UTR of BMPR1A shows a high level of complementarity. An alignment between Edn1 and let-7a-5p as predicted by TargetScan 7.2 and miRBase analyses as shown in the bottom panel by vertical bars. The sequence alignment of putative let-7a-5p and its targeting site on 3’-UTR of Edn1 shows a high level of complementarity
Validation of NF-κB-dependent miRNAs in BLM-induced lung fibrosis mouse model
The miR-503-5p and let-7a-5p expression were determined in the WT and IKBM mice lungs treated with BLM. The qRT-PCR analysis demonstrated a 2.65 ± 0.16-fold and 2.05 ± 0.25-fold increase in miR-503-5p and let-7a-5p, respectively (Fig. 5B, C); in the lungs of WT + BLM mice compared to untreated WT mice (P < 0.001). The expressions of both miRNAs were significantly reduced in BLM-treated IKBM mice (1.11 ± 0.05 and 1.04 ± 0.07) compared to WT + BLM treated mice (P < 0.05), indicating the regulation of miR-503-5p by NF-κB (Fig. 5 B, C).
To identify putative binding sites of miR‐503-5p in the 3′‐UTR, we used the miRNA target predicted search engine, TargetScan 7.2. As shown in miRNA: messenger RNA alignment analysis, 3′‐UTR of BMPR1A gene contained miR‐503-5p binding sites which were highly conserved among different species (Fig. 5D). The 7-mer sits on 128–134 nucleotide region of BMPR1A 3′UTR region. Our analysis of upregulation of miR-503-5p as shown in Fig. 5A and downregulation of BMPR1A as shown in Fig. 3A suggested a potential link between these molecules. Our data also demonstrated an upregulation of let-7a-5p shown in Fig. 5C and down regulation of Edn1 shown in Fig. 3C suggested a potential connection between them.
Discussion
Our results demonstrate for the first time that inhibition of NF-κB in the lungs attenuated BLM-induced lung fibrosis. The mechanism is likely to be associated with restoration of BMPR1A-Acta2-Col I-axis molecules and, inhibition of the inflammatory response. Additionally, miRNA array analysis revealed the involvement of the novel miRNAs, miR-503-5p and let-7a-5p in BLM-induced lung fibrosis. There are few reports indicating that inhibition of NF-κB is effective in pulmonary fibrosis. However, there is no report showing a direct link between genetic blockade of NF-κB in the lungs and pulmonary fibrosis [34,35,36]. This report provides for the first time that the genetic inhibition of NF-κB in the lungs prevents the BLM-induced lung fibrosis.
Our study evaluated the hallmark genes for fibrosis like Col1A1, Acta2, CTGF, MMP2 and TGFβ1, which are critical in extracellular matrix (ECM) deposition, a key process in fibrosis. All the above genes are significantly reduced in IKBM mice challenged with BLM indicating that they are regulated by NF-κB. It is reported that CTGF has NF-κB binding element in the promoter region, and this may suggest a direct regulation of CTGF by NF-κB [37]. The regulation of Col1A1 by NF-κB may involve the interaction with other transcription factors binding sites that were present in Col1A1 promoter since Col1A1 promoter did not show any NF-κB binding element [38]. Also, our study showed a significant increase in TGFβ1 mRNA expression. It is known that TGFβ has many physiological functions which are associated with airway remodeling and fibrosis and is a triggering factor for ECM deposition [39]. The TGFβ pathway is pivotal in the pathogenesis of lung fibrosis [40]. It has been reported that deletion of the TGFβ receptor type II is protective against BLM-induced pulmonary fibrosis in mice [41]. Moreover, TGFβ activation triggers fibroblast activation leading to the formation of myofibroblast by irreversible acquisition of alpha-smooth muscle actin, Acta2 protein, another important player in fibrosis [42]. Differentiation of lung fibroblasts to myofibroblasts is a key event in the pathogenesis of several fibrotic diseases including pulmonary fibrosis [43]. Our study shows that inhibition of NF-κB in the lungs significantly reduced Acta2 expression in BLM treated IKBM mice indicating an effective therapeutic approach for the treatment of pulmonary fibrosis.
NF-κB is a key regulator of inflammatory response. The transcription of many inflammatory cytokines, such as TNFα, IL6, IL1β and TGFβ, are vital in development of lung fibrosis [44, 45]. Our data showed that both IL1β and RelA was significantly reduced in BLM-induced IKBM mice indicating a NF-κB-mediated regulation. Activation of inflammatory molecules particularly TNFα and TGFβ promotes inflammation; damaging ECM protein components and triggering myofibroblast differentiation that leads to the development of lung fibrosis [46, 47].
Bone morphogenic proteins are the member of TGFβ family members and contributed a critical role in pulmonary hypertension, a proliferative vascular disease [19, 48, 49]. But the role of BMPR1A in BLM-induced pulmonary fibrosis is currently unknown. Previously, we have shown that monocrotaline-induced pulmonary hypertension was attenuated in IKBM mice by restoring BMPR2 (in part) indicated NF-κB- mediated regulation [19]. The current study also showed a significant reduction in BMPR2 in BLM-induced pulmonary fibrosis and was restored in IKBM mice indicating a NF-κB-mediated regulation. There is a report for inappropriate NF-κB signaling in BMPRII-deficient pulmonary arterial smooth muscle cells and increased IL6 secretion [50]. Our data also revealed a significant increase in IL6 expression that was reduced in IKBM mice which may correlate an association between BMPR1A-IL6 and NF-κB in pulmonary fibrosis.
Furthermore, the study revealed an upregulation of Acta2 in BLM treated WT mice which was reduced in IKBM mice. The phenomenon of endothelial-to-mesenchymal transition (EndoMT) is critical in lung fibrosis [51,52,53] which is characterized by acquiring mesenchymal phenotype, such as α-SMA (Acta2). This is the first report showing NF-κB regulated this critical phenotypic switch in the pathogenesis of pulmonary fibrosis. The study also showed Cav1, a highly expressed membrane protein in the lungs, is downregulated in BLM-induced pulmonary fibrosis. The Cav1 contributes an essential role in tissue repair and fibrosis by modulating ECM turnover [54]. Furthermore, Cav1 is shown to be a critical regulator of idiopathic lung fibrosis [55]. Our data is consistent with a previous observation that reduced Cav1 promotes fibrosis [55]. This new finding shows that restoration of Cav1 in IKBM mice suggest a potential connection between NF-κB and Cav1 in lung fibrosis which is unknown but needs further investigation.
Finally, we have identified a panel of dysregulated miRNAs in BLM-treated WT mice and the current study validated let-7a-5p and miR-503-5p as potential miRNAs in pulmonary fibrosis. Interestingly, these two miRNAs were upregulated in BLM treated WT group and were reduced in BLM treated IKBM group suggested a possible regulation by NF-κB. Modulation of miRNA is suggested to be a possible therapeutic target in several fibrotic diseases including pulmonary fibrosis [21,22,23,24,25, 56]. Previously, we have demonstrated NF-κB-mediated miR-130a regulation in TGFβ1 stimulated lung microvascular remodeling [33]. However, the association between let-7a-5p, miR-503-5p and NF-κB in pulmonary fibrosis is unknown. Our study provides evidence for the first time that let-7a-5p and miR-503-5p may be regulated by NF-κB. Furthermore, bioinformatic tool for miRNA target analysis showed that miR-503-5p has potential target for BMPR1A. Downregulation of BMPR1A and upregulation of miR-503-5p in BLM treated WT mice may suggest a possible underlying mechanism of pulmonary fibrosis. The identification of miR-503-5p and let-7a-5p along with their target genes are preliminary observations documented in the current form of the study. Further research is underway to delineate detail mechanism in pulmonary fibrosis.
In conclusion, our study identifies the importance of NF-κB-miR-503-5p-Col1A1-BMPR1A- axis and provide new mechanistic information in BLM-induced lung fibrosis. The cytokine released followed by EndMT provide new insights for the progression of lung fibrosis. Our data further suggests that let-7a-5p and miR-503-5p play a critical role in lung fibrosis and may be considered as possible therapeutic targets.
Data availability
Data will be made available on reasonable request.
References
Okamoto M, Izuhara K, Ohta S, Ono J, Hoshino T (2019) Ability of periostin as a new biomarker of idiopathic pulmonary fibrosis. Adv Exp Med Biol 1132:79–87. https://doi.org/10.1007/978-981-13-6657-4_9
Datta A, Scotton CJ, Chambers RC (2011) Novel therapeutic approaches for pulmonary fibrosis. Br J Pharmacol 163(1):141–172. https://doi.org/10.1111/j.1476-5381.2011.01247.x
Yin W, Han J, Zhang Z, Han Z, Wang S (2018) Aloperine protects mice against bleomycin-induced pulmonary fibrosis by attenuating fibroblast proliferation and differentiation. Sci Rep 8(1):6265. https://doi.org/10.1038/s41598-018-24565-y
Hardie WD, Glasser SW, Hagood JS (2009) Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol 175(1):3–16. https://doi.org/10.2353/ajpath.2009.081170
Upagupta C, Shimbori C, Alsilmi R, Kolb M (2018) Matrix abnormalities in pulmonary fibrosis. Eur Respir Rev 27(148):180033. https://doi.org/10.1183/16000617.0033-2018
Shimbori C, Gauldie J, Kolb M (2013) Extracellular matrix microenvironment contributes actively to pulmonary fibrosis. Curr Opin Pulm Med 19(5):446–452. https://doi.org/10.1097/MCP.0b013e328363f4de
Noguchi S, Saito A, Mikami Y, Urushiyama H, Horie M, Matsuzaki H, Takeshima H, Makita K, Miyashita N, Mitani A, Jo T, Yamauchi Y, Terasaki Y, Nagase T (2017) TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci Rep 7:42595. https://doi.org/10.1038/srep42595
Kendall RT, Feghali-Bostwick CA (2014) Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 5:123. https://doi.org/10.3389/fphar.2014.00123
Liu P, Miao K, Zhang L, Mou Y, Xu Y, Xiong W, Yu J, Wang Y (2020) Curdione ameliorates bleomycin-induced pulmonary fibrosis by repressing TGF-β-induced fibroblast to myofibroblast differentiation. Respir Res 21(1):58. https://doi.org/10.1186/s12931-020-1300-y
Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, Locy ML, Ravi S, Deshane J, Mannon RB, Abraham E, Darley-Usmar V, Thannickal VJ, Zmijewski JW (2018) Metformin reverses established lung fibrosis in a bleomycin model. Nat Med 24(8):1121–1127. https://doi.org/10.1038/s41591-018-0087-6
Skibba M, Drelich A, Poellmann M, Hong S, Brasier AR (2020) Nano approaches to modifying epigenetics of epithelial mesenchymal transition for treatment of pulmonary fibrosis. Front Pharmacol 11:607689. https://doi.org/10.3389/fphar.2020.607689
Bringardner BD, Baran CP, Eubank TD, Marsh CB (2018) The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal 10:287–301. https://doi.org/10.1089/ars.2007
Desai O, Winkler J, Minasyan M, Herzog EL (2018) The role of immune and inflammatory cells in idiopathic pulmonary fibrosis. Front Med 5:43. https://doi.org/10.3389/fmed.2018.00043
Wynn TA, Vannella KM (2016) Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44:450–462. https://doi.org/10.1016/j.immuni.2016.02.015
Mankan AK, Lawless MW, Gray SG, Kelleher D, McManus R (2009) NF-kappaB regulation: the nuclear response. J Cell Mol Med 13(4):631–643. https://doi.org/10.1111/j.1582-4934.2009.00632.x
Liang Y, Zhou Y, Shen P (2014) NF-kappaB and its regulation on the immune system. Cell Mol Immunol 1(5):343–350 (PMID: 16285893)
Wright JG, Christman JW (2013) The role of nuclear factor kappa B in the pathogenesis of pulmonary diseases: implications for therapy. Am J Respir Med 2(3):211–219. https://doi.org/10.1007/BF03256650
Christman JW, Sadikot RT, Blackwell TS (2000) The role of nuclear factor-kappa B in pulmonary diseases. Chest 117(5):1482–1487. https://doi.org/10.1378/chest.117.5.1482
Li L, Chuanyu W, Il-Kwon K, Janssen-Heininger Y, Gupta S (2014) Inhibition of NF-kB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension 63(6):1260–1269. https://doi.org/10.1161/HYPERTENSIONAHA.114.03220
Temann UA, Geba GP, Rankin JA, Flavell RA (1998) Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med 188:1307–1320. https://doi.org/10.1084/jem.188.7.1307
Li C, Wang Z, Zhang J, Zhao X, Xu P, Liu X, Li M, Lv C, Song X (2019) Crosstalk of mRNA, miRNA, lncRNA, and circRNA and their regulatory pattern in pulmonary fibrosis. Mol Ther Nucleic Acids 18:204–218. https://doi.org/10.1016/j.omtn.2019.08.018
Miao C, Xiong Y, Zhang G, Chang J (2018) MicroRNAs in idiopathic pulmonary fibrosis, new research progress and their pathophysiological implication. Exp Lung Res 44(3):178–190. https://doi.org/10.1080/01902148.2018.1455927
Ge L, Habiel DM, Hansbro PM, Kim RY, Gharib SA, Edelman JD, Königshoff M, Parimon T, Brauer R, Huang Y, Allen J, Jiang D, Kurkciyan AA, Mizuno T, Stripp BR, Noble PW, Hogaboam CM, Chen P (2016) miR-323a-3p regulates lung fibrosis by targeting multiple profibrotic pathways. JCI Insight 1(20):e90301. https://doi.org/10.1172/jci.insight.90301
Rajasekaran S, Rajaguru P, Sudhakar Gandhi PS (2015) MicroRNAs as potential targets for progressive pulmonary fibrosis. Front Pharmacol 5(6):254
Pandit KV, Milosevic J, Kaminski N (2011) MicroRNAs in idiopathic pulmonary fibrosis. Transl Res 157(4):191–199. https://doi.org/10.1016/j.trsl.2011.01.012
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(281–297):2004. https://doi.org/10.1016/s0092-8674(04)00045-5
Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP (2008) The impact of microRNAs on protein output. Nature 455:64–71. https://doi.org/10.1038/nature07242
Xie T, Liang J, Guo R, Liu N, Noble PW, Jiang D (2011) Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol Genomics 43:479–487. https://doi.org/10.1152/physiolgenomics.00222.2010
Poynter ME, Cloots R, van Woerkom T, Butnor KJ, Vacek P, Taatjes DJ, Irvin CG, Janssen-Heininger YM (2004) NF-κB activation in airways modulates allergic inflammation but not hyperresponsiveness. J Immunol 173:7003–7009. https://doi.org/10.4049/jimmunol.173.11.7003
Mouratis MA, Aidinis V (2011) Modeling pulmonary fibrosis with bleomycin. Curr Opin Pulm Med 17:355–361. https://doi.org/10.1097/MCP.0b013e328349ac2b
Kumar S, Wei C, Thomas CM, Kim IK, Seqqat R, Kumar R, Baker KM, Jones WK, Gupta S (2012) Cardiac-specific genetic inhibition of nuclear factor-κB prevents right ventricular hypertrophy induced by monocrotaline. Am J Physiol Heart Circ Physiol 302:H1655-1666. https://doi.org/10.1152/ajpheart.00756.2011
Wei C, Kim IK, Kumar S, Jayasinghe S, Hong N, Castoldi G, Catalucci D, Jones WK, Gupta S (2013) NF-κB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol 228:1433–1442. https://doi.org/10.1002/jcp.24296
Li L, Kim IK, Chiasson V, Chatterjee P, Gupta S (2017) NF-κB mediated miR-130a modulation in lung microvascular cell remodeling: Implication in pulmonary hypertension. Exp Cell Res 359:235–242. https://doi.org/10.1016/j.yexcr.2017.07.024
Yoshida K, Abe K, Ishikawa M, Saku K, Shinoda-Sakamoto M, Ishikawa T, Watanabe T, Oka M, Sunagawa K, Tsutsui H (2019) Inhibition of TLR9-NF-κB-mediated sterile inflammation improves pressure overload-induced right ventricular dysfunction in rats. Cardiovasc Res 115(3):658–668. https://doi.org/10.1093/cvr/cvy209
Sun X, Chen E, Dong R, Chen W, Hu Y (2015) Nuclear factor (NF)-κB p65 regulates differentiation of human and mouse lung fibroblasts mediated by TGF-β. Life Sci 122:8–14. https://doi.org/10.1016/j.lfs.2014.11.033
Fujimoto H, D’Alessandro-Gabazza CN, Palanki MS, Erdman PE, Takagi T, Gabazza EC, Bruno NE, Yano Y, Hayashi T, Tamaki S, Sumida Y, Adachi Y, Suzuki K, Taguchi O (2007) Inhibition of nuclear factor-kappaB in T cells suppresses lung fibrosis. Am J Respir Crit Care Med 176(12):1251–1260. https://doi.org/10.1164/rccm.200609-1288OC
Chaqour B, Yang R, Sha Q (2006) Mechanical stretch modulates the promoter activity of the profibrotic factor CCN2 through increased actin polymerization and NF-kappaB activation. J Biol Chem 281(29):20608–20622. https://doi.org/10.1074/jbc.M600214200
Beauchef G, Bigot N, Kypriotou M, Renard E, Poree B, Widom R, Dompmartin-Blanchere A, Oddos T, Maquart FX, Demoor M, Boumediene K, Galera P (2012) The p65 subunit of NF-kappaB inhibits COL1A1 gene transcription in human dermal and scleroderma fibroblasts through its recruitment on promoter by protein interaction with transcriptional activators (c-Krox, Sp1, and Sp3). J Biol Chem 287(5):3462–3478. https://doi.org/10.1074/jbc.M111.286443
Ambalavanan N, Nicola T, Hagood J, Bulger A, Serra R, Murphy-Ullrich J, Oparil S, Chen YF (2008) Transforming growth factor-beta signaling mediates hypoxia-induced pulmonary arterial remodeling and inhibition of alveolar development in newborn mouse lung. Am J Physiol Lung Cell Mol Physiol 295(1):L86-95. https://doi.org/10.1152/ajplung.00534.2007
Willis BC, Borok Z (2007) TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol 293(3):L525-534. https://doi.org/10.1152/ajplung.00163.2007
Li M, Krishnaveni MS, Li C, Zhou B, Xing Y, Banfalvi A, Li A, Lombardi V, Akbari O, Borok Z, Minoo P (2011) Epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest 121(1):277–287. https://doi.org/10.1172/JCI42090
Koo HY, El-Baz LM, House S, Cilvik SN, Dorry SJ, Shoukry NM, Salem ML, Hafez HS, Dulin NO, Ornitz DM, Guzy RD (2018) Fibroblast growth factor 2 decreases bleomycin-induced pulmonary fibrosis and inhibits fibroblast collagen production and myofibroblast differentiation. J Pathol 246(1):54–66. https://doi.org/10.1002/path.5106
Phan SH (2003) Fibroblast phenotypes in pulmonary fibrosis. Am J Respir Cell Mol Biol 29(3 Suppl):S87-92 (PMID: 14503563)
Zank DC, Bueno M, Al M, Rojas M (2018) Idiopathic pulmonary fibrosis: aging, mitochondrial dysfunction and cellular bioenergetics. Front Med 5(10):1–9. https://doi.org/10.3389/fmed.2018.00010
Chen T, Nie H, Gao X, Yang J, Pu J, Chen Z, Cui X, Wang Y, Wang H, Jia G (2014) Epithelial–mesenchymal transition involved in pulmonary fibrosis induced by multi-walled carbon nanotubes via TGF-beta/Smad signaling pathway. Toxicol Lett 226(2):150–162. https://doi.org/10.1016/j.toxlet.2014.02.004
Sivakumar P, Ntolios P, Jenkins G, Laurent G (2012) 2016) Into the matrix: targeting fibroblasts in pulmonary fibrosis. Curr Opin Pulm Med 18(5):462–469. https://doi.org/10.1097/MCP.0b013e328356800f
Darby IA, Zakuan N, Billet F, Desmoulière A (2015) The myofibroblast, a key cell in normal and pathological tissue repair. Cell Mol Life Sci 73(6):1145–1157. https://doi.org/10.1007/s00018-015-2110-0
Orriols M, Gomez-Puerto MC, Ten Dijke P (2017) BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell Mol Life Sci 74(16):2979–2995. https://doi.org/10.1007/s00018-017-2510-4
Morrell NW (2010) Role of bone morphogenetic protein receptors in the development of pulmonary arterial hypertension. Adv Exp Med Biol 661:251–264. https://doi.org/10.1007/978-1-60761-500-2_16
Davies RJ, Holmes AM, Deighton J, Long L, Yang X, Barker L, Walker C, Budd DC, Upton PD, Morrell NW (2012) BMP type II receptor deficiency confers resistance to growth inhibition by TGF-β in pulmonary artery smooth muscle cells: role of proinflammatory cytokines. Am J Physiol Lung Cell Mol Physiol 302(6):L604-615. https://doi.org/10.1152/ajplung.00309.2011
Willis BC, Borok Z (2007) TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol 293:L525–L534. https://doi.org/10.1152/ajplung.00163.2007
Goumans MJ, van Zonneveld AJ, ten Dijke P (2008) Transforming growth factor beta-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis? Trends Cardiovasc Med 18:293–298. https://doi.org/10.1016/j.tcm.2009.01.001
Sato Y, Nakanuma Y (2013) Role of endothelial-mesenchymal transition in idiopathic portal hypertension. Histol Histopathol 28:145–154. https://doi.org/10.14670/HH-28.145
Gvaramia D, Blaauboer MB, Hanemaaijer R, Everts V (2013) Role of caveolin-1 in fibrotic diseases. Matrix Biol 32:307–315. https://doi.org/10.1016/j.matbio.2013.03.005
Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, Ifedigbo E, Xu X, Oury TD, Kaminski N, Choi AM (2006) Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 203(13):2895–2906. https://doi.org/10.1084/jem.20061536
Boateng E, Krauss-Etschmann S (2020) MiRNAs in lung development and diseases. Int J Mol Sci 21(8):2765. https://doi.org/10.3390/ijms21082765
Acknowledgements
The authors acknowledge Dr. Dwayne Simmons for his support and providing his laboratory facility to perform experiments and his team members to assist us whenever needed. The authors acknowledge Dr. Yvonne Janssen-Heininger, Department of Pathology and Laboratory Medicine, University of Vermont, Burlington, VT, USA; for providing IKBM mice.
Funding
This study was partly supported by American Heart Association-Grant-in-Aid (17GRNT33670897) to S. Gupta (SG). The authors declared that they do not have any financial or proprietary interest in any material discussed in this article.
Author information
Authors and Affiliations
Contributions
SG conceived the project, designed experiments and funded the project. DT, OT and MA performed qRT-PCR experiments. DT maintained IKBM colonies. RS performed lung instillation experiment. RSG provided intellectual input and critical comments on the manuscript. SG analyzed the data and drafted manuscript. All authors edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they do not have any potential conflict of interest that are relevant to the article.
Ethical approval
Animals were handled according to NIH guidelines for animal research. The lung instillation was performed under anesthesia.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thakur, D., Taliaferro, O., Atkinson, M. et al. Inhibition of nuclear factor κB in the lungs protect bleomycin-induced lung fibrosis in mice. Mol Biol Rep 49, 3481–3490 (2022). https://doi.org/10.1007/s11033-022-07185-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07185-8