
Abstract
Background and aims
The Kiwifruit Vine Decline Syndrome (KVDS) is a disease that is currently a challenge for kiwifruit production in Italy, and it is spreading in new production areas. However, the causal agent of this syndrome has not been clearly identified, and we still know little about the overall effects of KVDS on the interactions between the host plant and its microbiome.
Methods
In this study, we combined metabarcoding and targeted isolation (leaf baiting) to characterize the changes in the rhizosphere and root microbiomes associated with symptoms of KVDS.
Results
Our results suggest that KVDS has little impact on the bacterial, fungal, and oomycete communities associated with soil and roots, and we detected weak signatures of potential dysbiosis. On the other hand, we found a consistent association of the oomycete Phytopythium vexans with samples from plants symptomatic to KVDS, which matches the nucleotide sequences of the isolates obtained through baiting and, partially, the isolates from previous studies.
Conclusion
While our results support the idea that P. vexans might be the major candidate agent of KVDS, there are still several unanswered questions that need to be addressed before being able to provide effective solutions to this emerging challenge in kiwifruit production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Kiwifruit Vine Decline Syndrome (KVDS) is a new challenge for kiwifruit production that has caused great damage in Italy since 2012 and has been reported in several other countries (Donati et al. 2020; Savian et al. 2022). Plants affected by this syndrome quickly die back during summer (Fig. 1A) as consequence of the extensive damage caused to the root system (Fig. 1B and C), which cannot support the water demand from the canopy and leads to the collapse of the entire plant (Savian et al. 2020, 2022). This generates costs due to both the complete loss of production and the replacement of diseased plants. One of the most recent estimates suggests a loss of 300 million EUR only for the year 2020 in Italy, with likely higher values for the following years (Tacconi et al. 2020). Although KVDS is challenging for kiwifruit production, little is known about the mechanisms underlying its induction and spread.

Typical symptoms of Kiwifruit Vine Decline Syndrome (KVDS). (A) A defoliated plant. (B) and (C) Extensive damage to the root system
Initially, it has been proposed that KVDS is induced by abiotic stresses, including waterlogging and high soil temperature, as a consequence of climate change (Tacconi et al. 2014; Sorrenti et al. 2016; Bardi 2020; Bardi et al. 2020, 2022). However, Savian et al. (2020) grew kiwifruit plants on soil from a field where KVDS symptoms were frequent, and this soil was either sterilized by autoclaving or unsterilized, and the results clearly showed that KVDS is solely induced when the soil is not sterilized, demonstrating that KVDS has a biotic origin. Although several studies have attempted to identify the potential microorganisms responsible for KVDS, the causative agents of this syndrome have still not been clearly identified.
Several microorganisms have been identified as potential agents of KVDS. Early studies suggested the potential involvement of bacteria (Clostridium bifermentans and Clostridium subterminale) (Spigaglia et al. 2020) or fungi (e.g., Desarmillaria tabescens) (Donati et al. 2020). However, after more than 10 years of investigations, there is wide consensus on the association of diverse oomycetes with KVDS, as species of Phytopythium, Pythium, and Phytophthora are much more frequently isolated from symptomatic plants in different geographic areas (Donati et al. 2020; Prencipe et al. 2020; Savian et al. 2020) and can induce KVDS under controlled conditions (Prencipe et al. 2023). Metabarcoding of fungal and oomycete communities also suggested associations of species of Phytopythium and Phytophthora with symptomatic kiwifruit plants (Savian et al. 2022; Mian et al. 2023a; Guaschino et al. 2024).
While previous studies have shown that oomycetes might be responsible for KVDS, we still lack a comprehensive analysis of the microbial communities of healthy and symptomatic plants in field conditions, which can help in identifying potential agents of KVDS. To address this knowledge gap, in this study, we surveyed a wide area where KVDS is currently spreading, characterized the bacterial, fungal, and oomycete communities of roots and soil from symptomatic and asymptomatic KVDS plants, and performed targeted isolation of oomycetes potentially responsible for this syndrome. We hypothesize to observe changes in the structure of the microbial communities in symptomatic KVDS plants and shifts in the ecological processes underlying plant microbiome assembly. We also hypothesized that there would be a strong association between symptoms and the abundance of potentially pathogenic species of oomycetes, including for example, species of Phytopythium, Pythium, and Phytophthora. To further test this last hypothesis, we combined metagenomics data with the targeted isolation of oomycetes from the soil and roots of symptomatic plants using the leaf baiting method.
Materials and methods
Field sampling
Samples were collected in 2022 in three fields located in Gioia Tauro (Calabria, Italy – field 1: 38°28′51.2"N 16°01′05.3"E; field 2: 38°30′32.3"N 16°02′04.7"E; field 3: 38°31′35.0"N 16°05′07.4"E; distance between fields 3.4–7.7 km), which is a major kiwifruit production area in Italy. All three fields were planted with kiwifruit vines (Actinidia chinensis var. deliciosa, variety ‘Hayward’) of similar age and included plants showing the classical symptoms of KVDS. In June and October, from each site, we collected samples from the roots and rhizosphere soil of 10 plants showing clear symptoms of KVDS in the root system and 10 asymptomatic plants. Symptomatic plants were selected among those that were not collapsed but showed damage to the root system. Also, within the root system of symptomatic plants, we collected samples from the undamaged area. The samples were immediately transferred to the laboratory, and each sample was (i) immediately used for the isolation of potential pathogenic oomycetes and (ii) prepared for metabarcoding analyses to characterize the bacterial, fungal, and oomycete communities.
Oomycete-targeted isolation and molecular identification
We isolated potential pathogenic oomycetes using leaf baiting as described in Pérez-Sierra et al. (2022) with some modifications. Briefly, soil and root samples from symptomatic and asymptomatic plants were submerged in sterile distilled water inside a sterilized plastic box. Inside each box, we placed one-year old leaves of carob (Ceratonia siliqua) to float on the water surface. Boxes were then left at room temperature (~ 20 °C) for approximately 1 week and checked every 1–2 days for signs of necrosis on the floating leaves. Leaves showing dark spots were removed from the baiting box, dried with a paper towel, and the necrotic area was dissected and placed on PARPNH-PDA selective media (Jung et al. 2021) and incubated at 24 °C in the dark. Oomycete-like colonies growing on the selective media were then transferred to PARPNH-PDA and subsequently grown at 20 °C on V8-juice agar. After 7–10 days of growth, the cultures were examined at 10 × magnification under a light microscope and grouped into morphotypes according to their morphological features: colony growth patterns, hyphal characteristics, and, when possible, sporangia size and shape. For each morphotype, we placed a single isolate into our collection for further analyses.
Total DNA was extracted from each isolate in our collection using the commercial kit Norgen Fungi/Yeast Genomic DNA Isolation Kit (Norgen Biotek Corp., Canada) following the manufacturer’s instructions. DNA was checked using a Nanodrop spectrophotometer (Thermo, USA), and high-quality samples were used to prepare libraries for molecular barcoding. The ITS region was PCR-amplified using the primers ITS6-ITS4 (White et al. 1990; Cooke et al. 2000) and KAPA HiFi HotStart ReadyMix (Roche, USA) as recommended by the manufacturer. Amplifications were performed in an Eppendorf Mastercycler Ep Gradient S thermocycler (Eppendorf, Germany) using the following program: 95 °C for 3 min; 35 cycles of 98 °C for 20 s, 55 °C for 30 s and 72 °C for 45 s; and a 72 °C final extension for 5 min. PCR products were checked for correct size through electrophoresis on a 2% agarose gel stained with GelRed nucleic acid gel stain (Biotium, CA, USA). PCR products were purified using AMPure XP beads (Beckman Coulter Genomics, USA), and amplicons were sequenced in both directions by Macrogen Europe (Amsterdam, The Netherlands). The sequences were then manually edited using CHROMASPRO v. 1.7.6 (http://www.technelysium.com.au/), and low-quality samples were resequenced. The first identification was performed by querying the consensus of forward and reverse reads of each sample to NCBI GenBank using BLAST.
Metabarcoding
From each sample, roots were cleared from the loose surrounding soil, and ~ 100 mg of rhizosphere soil was collected by vigorously shaking the roots. Roots were then carefully washed with sterilized water. All samples were lyophilized and powdered. DNA extraction and library preparation from rhizosphere soil and roots were performed according to our previous study (Malacrinò et al. 2021). Briefly, ~ 25 mg of each sample were lysed in extraction buffer, and total DNA was extracted using a phenol‒chloroform protocol. After a quality check, we prepared libraries targeting the bacterial community (primers 515f/806rB, Caporaso et al. 2012), the fungal community (primers ITS3/ITS4, Toju et al. 2012), and the community of oomycetes (primers ITS3oom/ITS4, Riit et al. 2016). Amplifications were also carried out on DNA extracted from nontemplate controls, where the sample was replaced with nuclease-free water to account for possible contamination of instruments, reagents, and consumables used for DNA extraction. After this first PCR, the samples were purified using an Agencourt AMPure XP kit (Beckman Coulter, USA) and subjected to a second short-term PCR to ligate Illumina adaptors. Libraries were then purified again, quantified using a Qubit spectrophotometer (Thermo Fisher Scientific Inc., USA), normalized using nuclease-free water, pooled together, checked for the correct size on an TapeStation instrument (Agilent, USA), and sequenced on an Illumina MiSeq instrument (Illumina, USA) on a 300PE flow cell according to the manufacturer’s instructions.
The raw data were quality-checked, and adapters were trimmed with TrimGalore (Krueger 2023). Reads were then processed in R v4.3.2 (R Core Team 2020) with the DADA2 pipeline v1.22 (Callahan et al. 2016) to perform quality filtering, ASV identification, chimera removal and taxonomy assignment using the SILVA database v138 (Quast et al. 2013) for bacteria, the UNITE database (Nilsson et al. 2019) for fungi, and a custom database for oomycetes built using the R package refdb (Keck and Altermatt 2023) on the BOLD database (Ratnasingham and Hebert 2007) (accessed on December 6th, 2023). Representative sequences of each ASV were aligned using MAFFT v7.505 (Katoh et al. 2002), and a phylogenetic tree was built using FastTree v2.1.10 (Price et al. 2009). The ASV table, the taxonomic information for each ASV, the sample metadata, and the ASV phylogenetic tree were grouped using phyloseq v1.46 (McMurdie and Holmes 2013). Singletons and sequences identified as “chloroplast” or “mitochondria” were discarded before downstream analyses. Potential contaminants were removed using the data from negative control samples and the package decontam (Davis et al. 2018). Samples with fewer than 1,000 reads were then discarded. Microbiome data were also normalized using Wrench (Kumar et al. 2018) before calculating relative abundances.
Data analysis
All data analyses were performed with R v4.3.2 (R Core Team 2020). An overview of the taxonomical composition was obtained by grouping ASVs by genus and discarding genera with relative abundances lower than 1%. Phylogenetic diversity within each sample was calculated using the package picante (Kembel et al. 2010), and differences between asymptomatic and symptomatic samples were assessed by building a linear mixed effect model for each community using the function lmer within the package lme4 (Bates et al. 2015) with compartment (root, rhizosphere) and category (symptomatic, asymptomatic) as fixed factors and field ID and timepoint as random effects. From each model, differences between asymptomatic and symptomatic samples were determined by extracting false discovery rate (FDR)-corrected post hoc contrasts using emmeans (Lenth 2022). The influence of KVDS on the assembly of each community type (bacteria, fungi, and oomycetes) in the roots and rhizosphere samples was also assessed using PERMANOVA with 999 permutations stratified at the field ID and timepoint levels. We also tested the influence of KVDS symptoms on the processes driving the assembly of microbial communities at each compartment and community type by calculating the beta nearest taxon index (b-NTI), which quantifies the deviation of the mean nearest taxon distances from null expectations. This index was calculated using the package picante and used to test whether microbial communities assembled following deterministic or stochastic assembly processes (Arnault et al. 2022; Larsen et al. 2023). ASVs with differential abundance between asymptomatic and symptomatic samples for each compartment (root, rhizosphere) and community type (bacteria, fungi, oomycetes) were identified using the DESeq2 package (Love et al. 2014).
We then compared our results with previously published data by performing a meta-phylogenetic analysis. Our dataset included all the sequences obtained from isolating oomycetes from field samples (baiting) and the oomycete ASV sequences that are enriched in symptomatic samples (metabarcoding). We then collected data from previously published studies that isolated and identified oomycetes from the kiwifruit plant (Akilli et al. 2011; Kurbetli and Ozan 2013; Rahman et al. 2014; Wang et al. 2015; Çiftçi et al. 2016; Polat et al. 2017, 2023; Prencipe et al. 2020, 2023; Savian et al. 2020; Türkkan et al. 2022) (Table S1). We also collected sequences from other studies focusing on reference isolates, so we could use these sequences as a reference for the identification of other sequences within the same cluster (Matsumoto et al. 1999; Schurko et al. 2003; Lévesque and de Cock 2004; Belbahri et al. 2006; Senda et al. 2009; Uzuhashi et al. 2010, 2019; Robideau et al. 2011; Baten et al. 2014; de Cock et al. 2015; Veterano et al. 2018; Rezaei et al. 2021; Nguyen et al. 2022; Eggertson et al. 2023) (Table S1). Sequences were aligned with DECIPHER (Wright 2016), and the bootstrapped tree was built on a UPGMA distance matrix using phangorn (Schliep 2011) (JC69 substitution model and stochastic rearrangement) and visualized using ggtree (Yu et al. 2017).
Results
Sequencing overview
After cleanup, the sequencing dataset included 1,199,677 reads for 16S (8,756.8 ± 544.2 reads/sample), 2,538,681 reads for ITS (17,878.1 ± 1,330.5 reads/sample), and 1,257,147 reads for ITS-OOM (11,859.9 ± 1,063.1 reads/sample). While using strict filters for sequencing quality reduced the number of samples, each combination of sampling timepoint (June, October), compartment (root, soil), and sample type (symptomatic, asymptomatic) was represented by at least 5 independent samples for any of the three marker genes (Table S2, Supplementary Material).
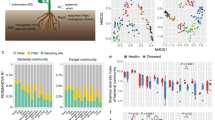
Analysis of the microbial communities of roots and soils from symptomatic and asymptomatic plants revealed that the root bacterial community was dominated by rhizobia, Bacillus, Novosphingobium, Rhodanobacter, and Streptomyces (Fig. 2A). However, the soil bacterial community was dominated by the genera Chujaibacter and Rhodanobacter and unidentified taxa (Fig. 2A). The root fungal community was mainly represented by the genera Acrocalymma, Dactylonectria, and Fusarium (Fig. 2B). Similarly, the soil fungal community was dominated by Fusarium and Pseudaleuria (Fig. 2B). The community of oomycetes in the root and soil samples was dominated by the genera Apodachlya, Phytopythium, and Pythium, with Phytopythium being highly abundant only in the symptomatic root and soil samples (Fig. 2C).

Relative abundances of bacterial (A), fungal (B), and oomycete (C) genera in root and rhizosphere soil samples from symptomatic and asymptomatic plants. Within the overall dataset, > 1% of the plotted genera had a relative abundance > 1%. The high proportion of unidentified oomycete ASVs (Amplicon Sequence Variants) might be the result of using a reference dataset that includes only oomycetes, while the primers are able to amplify also other microorganisms (e.g., fungi)
KVDS has little impact on the soil and plant microbiome
The diversity of the bacterial and fungal communities associated with KVDS-symptomatic plants was not different from that associated with asymptomatic plants (Fig. 3A and B, Table S3 Supplementary Material). However, the community of oomycetes in the soil from symptomatic plants was significantly more diverse than that in the soil from asymptomatic plants (Fig. 3C, Table S3 Supplementary Material). By comparing the structure of the microbial communities of symptomatic and asymptomatic plants, we identified differences in the bacterial community of the roots and the fungal and oomycete communities of the soil (Table 1). Instead, when comparing the microbial structure to a null model, we found that only the bacterial communities of symptomatic plants had a partial distribution with b-NTI values lower than 2 (Fig. 3D, E, and F), and both the bacterial and oomycete communities of the symptomatic root samples had values different from those of the asymptomatic plants (Fig. 3D, Table S4 Supplementary Material).

Comparison of phylogenetic diversity (A, B, C) and b-NTI (D, E, F) indices between symptomatic (red) and asymptomatic (green) root and rhizosphere soil samples, focusing on communities of bacteria (A, D), fungi (B, E), and oomycetes (C, F). Red dotted line in panels D, E, and F (beta-NTI = 2) represent the threshold identifying dissimilarity (beta-NTI > 2 or beta-NTI < -2) or similarity (-2 < beta-NTI > 2) compared to the null-model
Phytopythium is commonly associated with symptomatic plants
We also identified the ASVs that were abundant among symptomatic plants. When focusing on bacteria, we did not find any ASV significantly enriched in symptomatic plants (Fig. 4A and B). In fungi, we found two ASVs identified as Zopfiella enriched in the roots of symptomatic plants (Fig. 4C) and 21 ASVs enriched in the soil of symptomatic plants, including ASVs identified as Apiotrichum, Ceratobasidium, Cladophialophora, Epicoccum, Leptodontidium (3 ASVs), Mortierella (4 ASVs), Natantispora, Poaceascoma, Preussia (2 ASVs), Pyrenochaetopsis, Rhizoctonia (2 ASVs), Talaromyces (2 ASVs), and Thanatephorus (Fig. 4D). When looking at oomycetes, we found that the ASVs identified as Phytopythium vexans were enriched in both the roots (n = 3) and soil samples (n = 4) from symptomatic plants. Instead, in asymptomatic plants, we found a greater abundance of Pythium (n = 15) and other oomycete ASVs (n = 27).

ASVs differentially abundant between samples from symptomatic (log2FC > 0) and asymptomatic (log2FC < 0) plants, for communities of bacteria (A, B), fungi (C, D), and oomycetes (E, F) in root (A, C, E) and rhizosphere soil samples (B, D, F)
During our experiment, we isolated several oomycetes, including Phytopythium vexans (n = 50), Phytopythium litorale (n = 35), Phytopythium chamaehyphon (n = 7), Pythium spp. (n = 16), Globisporangium sp. (n = 8), and Achlya sp. (n = 1), using a targeted approach (Table S1, Supplementary Material). We compared the sequence from the ITS region of those isolates with the ASVs identified to be enriched in roots and soil from symptomatic plants with the sequences reported in previous studies on KVDS and the reference sequences of the genera Phytopytium, Pythium, and Globisporangium. The results showed that the ASVs we identified to be differentially abundant in symptomatic samples closely matched the P. vexans isolates we were able to isolate from the field (Fig. 5). Although they belonged to the same cluster, the sequences of P. vexans and P. litorale did not cluster closely together with the sequences obtained from previous studies (Fig. 5).

The phylogenetic tree including ITS2 sequences from targeted isolation (red, black circle on tip), metabarcoding (orange, back square on tip), and reference (green) sequences obtained from previous studies on kiwifruit vines (yellow). Branches highlighted in green were identified as Phytopythium vexans, while branches colored in blue were identified as Phytopythium litorale. See Table S1 (Supplementary Material) for details.
Discussion
KVDS is causing damages in Italy since 2012, and syndromes with similar symptoms have been previously reported in other countries since 1991 (Reid et al. 1991; Savian et al. 2022). Research on KVDS has been quite active during the past 5 years, and several microorganisms, mostly oomycetes, have been identified as potential agents of KVDS. Our study provides a wider analysis over a large area where KVDS is spreading, and by integrating targeted in vitro isolation with metabarcoding, our results support the hypothesis that oomycetes might be involved in the induction of KVDS, with Phytopythium vexans as a major potential agent.
Our analyses revealed a constant association of P. vexans with samples from symptomatic plants. When looking at the composition of the bacterial and fungal communities in symptomatic and asymptomatic samples, there was no visible difference in the presence/absence of potential pathogens. However, when looking at the community of oomycetes, we observed a consistent presence of Phytopythium sp. in samples from symptomatic plants. Additionally, the community of oomycetes associated with soil from symptomatic plants was more diverse than that associated with soil from control plants. When we identified the differentially abundant ASVs between symptomatic and asymptomatic plants, we also observed a clear association between P. vexans and symptomatic plants. This observation was further supported by the clustering of differentially abundant ASVs with the ITS sequences of baited isolates and reference isolates of P. vexans. This species was previously isolated from kiwifruit plants showing symptoms of KVDS, and once reinoculated on healthy plants, it was able to induce KVDS (Polat et al. 2017; Prencipe et al. 2020, 2023; Savian et al. 2021; Türkkan et al. 2022; Mian et al. 2023b). Similarly, other species of Phytopythium (Donati et al. 2020; Savian et al. 2020, 2021, 2022; Prencipe et al. 2023; Polat et al. 2023; Mian et al. 2023b), including P. litorale, which was often isolated from our samples, were reported to induce KVDS. Phytopythium vexans and other species of Phytopythium were reported to cause root rot on other plant species (Yang et al. 2013; Yin et al. 2016; Fichtner et al. 2016; Chen et al. 2016; Radmer et al. 2017; Jabiri et al. 2020; Baysal-Gurel et al. 2021; Panth et al. 2021). Together, this evidence supports the role of Phytopythium spp. and particularly P. vexans as agent of KVDS.
While previous work has mostly focused on oomycetes, other microorganisms have also been reported to induce symptoms resembling those of KVDS under controlled conditions, including Desarmillaria tabescens (Donati et al. 2020), Clostridium bifermentans and Clostridium subterminale (Spigaglia et al. 2020), Cylindrocarpon pauciseptatum, Cylindrocladiella parva, and different species of Ilyonectria (Erper et al. 2013). In our study, none of these taxa were associated with KVDS or were found in high abundance within our samples enabling to exclude their involvement in KVDS in the investigated area. This discrepancy might be explained in two different ways. First, those previous studies performed reinoculation experiments under heavily controlled conditions with high loads of microbial inoculum and high soil water content, which might have created the conditions for weak pathogens to induce KVDS. Second, we might hypothesize that KVDS is not the result of a single pathogen or pathogen group but rather the result of detrimental interactions between the host plant and its microbiome. Indeed, previous studies speculate that KVDS might be the consequence of dysbiosis (Savian et al. 2022) as a temporary loss of the capacity of the host plant to control the assembly of its microbiota (Arnault et al. 2022). If KVDS were to be induced by dysbiosis, we would have expected to observe a strong reduction in the selective forces driving the assembly of the plant microbiome in favor of stochastic processes. Instead, our results only show a weak shift from deterministic processes driving the assembly of the bacterial root microbiome in favor of stochastic processes. We did not observe the same pattern in the fungal and oomycete communities. Overall, we found little impact of KVDS on the diversity and structure of the plant rhizosphere and root microbiomes. This suggests that KVDS might not be the result of dysbiosis. This contrasts with the results from Guaschino et al. (2024), where they found that stochastic processes dominate the assembly of rhizosphere bacterial, fungal, and oomycete communities, regardless the health status of the plant. This discrepancy in the results might be driven by different factors, including sampling timepoint and environmental conditions, thus more tests are needed to understand the effect of KVDS on the processes driving the assembly of the kiwifruit plant microbiomes.
Although our results and those of previous studies provide strong evidence that oomycetes are involved in the induction of KVDS, particularly the genera Phytopythium and Phytophthora, this evidence needs to be compared with a series of alternative hypotheses. A first alternative hypothesis is that KVDS is not caused by a specific pathogen, but rather by abiotic stresses, mainly waterlogging and high soil temperature which reduce natural plant resistance, create conducive disease conditions and enable the infection of weak opportunistic plant pathogens commonly present in soils. This idea supports the fact that a live microbial community is required for the induction of KVDS (Savian et al. 2020), and that several microorganisms have been found to induce this syndrome. Although this hypothesis is worth of further investigation, if this is the case, we should observe a generalized decline in kiwifruit plants all over the areas where they are cultivated, as a generalized effect of climate changes. However, KVDS is still spreading, and it is not present in all kiwifruit production areas and fields, even when plant (e.g., plant genotype and age) and environmental conditions seem to be very similar. Similarly, our data show that it is unlikely that KVDS is generated by dysbiosis. Thus, the results from this and previous studies fully support the idea that KVDS is induced by pathogens under field conditions, and oomycetes seem to be the best candidates. This conclusion can lead us to a second alternative hypothesis, where P. vexans might not be the only or main causal agent of KVDS. Indeed, previous studies isolated several oomycetes, and most of them were able to induce KVDS under controlled conditions. Similarly, previous studies using metabarcoding suggested the association of KVDS with Phytopythium and other genera of oomycetes (Savian et al. 2022; Guaschino et al. 2024). Our data also show that isolates from our study and previous studies do not cluster together, particularly within the branches of P. vexans and P. litorale. Also previous research has not investigated the possibility that KVDS might be caused by a hybrid of oomycetes and hybrids are often reported in oomycetes (Safaiefarahani et al. 2016) and might contribute to the evolution of novel pathogenic strains (Schardl and Craven 2003; Corredor-Moreno and Saunders 2020). The next challenge is to dig deeper into the genomes of potential agents of KVDS and test for signatures of hybridization across isolates. It is also possible that KVDS is the results of consecutive infection processes by different pathogens, where P. vexans may not be the most important causal agent. This might explain why, although different studies on KVDS suggest the role of a similar group of oomycetes, we still find discrepancies in the identity of the potential agent(s) of KVDS. A third alternative hypothesis that still needs to be addressed relies on one of the limitations of the studies that have been carried out thus far, in that KVDS might be induced by an agent that has not been investigated at all. Nematodes, for example, were recently identified as agents of a new disease of beech plants (Ewing et al. 2021; Carta et al. 2023), and several viral pathogens are currently emerging and spreading to new locations (Jeger et al. 2023). None of the studies conducted thus far on KVDS have tested the potential contributions of nematodes or viruses, and these contributions need to be directly tested to narrow the list of potential agents of this syndrome.
While research on KVDS is progressing rapidly, there is still little consensus on the agent(s) and mechanisms responsible for its induction. Our results in agreement with previous reports support a major role of oomycetes, particularly P. vexans. However, the mechanisms underlying KVDS are not yet clear. On the one hand, P. vexans might be the KVDS agent currently spreading throughout kiwifruit cultivation areas. On the other hand, P. vexans might not be the main agent, and other plant pathogenic organisms might be involved, which might not have been identified because difficult to isolate or because current observations focused on a limited timeframe or targeted other members of the microbiome. Another possible alternative, is that KVDS might be induced by the assembly of a pathobiome (Bass et al. 2019; Lv et al. 2023), a microbiome structure that is associated with a detrimental plant health status. KVDS will likely be the next major challenge of kiwifruit production worldwide after bacterial canker, and thus, it is timely to prioritize the identification of the agents and mechanisms of disease, together with outlining potential low-impact solutions that can be implemented to prevent its emergence and spread in other major kiwifruit cultivation areas.
Data availability
The raw sequencing data are available at NCBI SRA under Bioprojects PRJNA1137840 (16S), PRJNA1137841 (ITS), and PRJNA1137842 (ITS-OOM). ITS2 sequences from individual isolates are available at NCBI GenBank under accession numbers PQ050083-PQ050192 (see also Table S1, supplementary materials).
References
Akilli S, Serçe ÇU, Zekaİ Katircioğlu Y et al (2011) Involvement of Phytophthora citrophthora in Kiwifruit Decline in Turkey. J Phytopathol (1986) 159:579–581
Arnault G, Mony C, Vandenkoornhuyse P (2022) Plant microbiota dysbiosis and the Anna Karenina Principle. Trends Plant Sci. https://doi.org/10.1016/j.tplants.2022.08.012
Bardi L (2020) Early kiwifruit decline: a soil-borne disease syndrome or a climate change effect on plant–soil relations? Front Agron 2. https://doi.org/10.3389/fagro.2020.00003
Bardi L, Nari L, Morone C et al (2020) Possible role of high temperature and soil biological fertility on kiwifruit early decline syndrome. Front Agron 2. https://doi.org/10.3389/fagro.2020.580659
Bardi L, Nari L, Morone C et al (2022) Kiwifruit adaptation to rising vapor pressure deficit increases the risk of kiwifruit decline syndrome occurrence. Horticulturae 8:906
Bass D, Stentiford GD, Wang H-C et al (2019) The pathobiome in animal and plant diseases. Trends Ecol Evol 34:996–1008
Baten MA, Asano T, Motohashi K et al (2014) Phylogenetic relationships among Phytopythium species, and re-evaluation of Phytopythium fagopyri comb. nov., recovered from damped-off buckwheat seedlings in Japan. Mycol Prog 13. https://doi.org/10.1007/s11557-014-1003-1
Bates D, Mächler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48
Baysal-Gurel F, Liyanapathiranage P, Panth M et al (2021) First report of Phytopythium vexans causing root and crown rot on flowering cherry in Tennessee. Plant Dis 105:232
Belbahri L, Calmin G, Sanchez-Hernandez E et al (2006) Pythium sterilum sp. nov. isolated from Poland, Spain and France: its morphology and molecular phylogenetic position. FEMS Microbiol Lett 255:209–214
Callahan BJ, McMurdie PJ, Rosen MJ et al (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583
Caporaso JG, Lauber CL, Walters WA et al (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624
Carta LK, Li S, Mowery J (2023) Beech leaf disease (BLD), Litylenchus crenatae and its potential microbial virulence factors. In: Asiegbu FO and Kovalchuk S (eds) Forest microbiology: tree diseases and pests. Elsevier, pp 183–192
Chen XR, Liu BB, Xing YP et al (2016) Identification and characterization of Phytopythium helicoides causing stem rot of Shatangju mandarin seedlings in China. Eur J Plant Pathol 146:715–727
Çiftçi O, Serçe ÇU, Türkölmez Ş, Derviş S (2016) First report of Phytophthora palmivora causing crown and root rot of kiwifruit (Actinidia deliciosa) in Turkey. Plant Dis 100:210
Cooke DE, Drenth A, Duncan JM et al (2000) A molecular phylogeny of Phytophthora and related oomycetes. Fungal Genet Biol 30:17–32
Corredor-Moreno P, Saunders DGO (2020) Expecting the unexpected: factors influencing the emergence of fungal and oomycete plant pathogens. New Phytol 225:118–125
Davis NM, Proctor DM, Holmes SP et al (2018) Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6:226
de Cock AWAM, Lodhi AM, Rintoul TL et al (2015) Phytopythium: molecular phylogeny and systematics. Persoonia 34:25–39
Donati I, Cellini A, Sangiorgio D et al (2020) Pathogens associated to kiwifruit vine decline in Italy. Agriculture 10:119
Eggertson QA, Rintoul TL, Lévesque CA (2023) Resolving the Globisporangium ultimum (Pythium ultimum) species complex. Mycologia 115:768–786
Erper I, Agustí-Brisach C, Tunali B, Armengol J (2013) Characterization of root rot disease of kiwifruit in the Black Sea region of Turkey. Eur J Plant Pathol 136:291–300
Ewing CJ, Slot J, Benítez M-S et al (2021) The foliar microbiome suggests that fungal and bacterial agents may be involved in the beech leaf disease pathosystem. Phytobiomes J 5:335–349
Fichtner EJ, Browne GT, Mortaz M et al (2016) First report of root rot caused by Phytopythium helicoides on pistachio rootstock in California. Plant Dis 100:2337–2337
Guaschino M, Garello M, Nari L et al (2024) Soil, rhizosphere, and root microbiome in kiwifruit vine decline, emerging multifactorial disease. Front Microbiol 15. https://doi.org/10.3389/fmicb.2024.1330865
Jabiri S, Lahlali R, Bahra C et al (2020) First report of Phytopythium vexans associated with dieback disease of apple trees in Morocco. J Plant Pathol 102:1319–1319
Jeger MJ, Fielder H, Beale T et al (2023) What can be learned by a synoptic review of plant disease epidemics and outbreaks published in 2021? Phytopathology 113:1141–1158
Jung T, Horta Jung M, Webber JF et al (2021) The destructive tree pathogen Phytophthora ramorum originates from the laurosilva forests of east Asia. J Fungi (Basel) 7:226
Katoh K, Misawa K, Kuma K-I, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Keck F, Altermatt F (2023) Management of DNA reference libraries for barcoding and metabarcoding studies with the R package refdb. Mol Ecol Resour 23:511–518
Kembel SW, Cowan PD, Helmus MR et al (2010) Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26:1463–1464
Krueger F (2023) TrimGalore. https://doi.org/10.5281/zenodo.7598955
Kumar MS, Slud EV, Okrah K et al (2018) Analysis and correction of compositional bias in sparse sequencing count data. BMC Genomics 19:799
Kurbetli İ, Ozan S (2013) Occurrence of Phytophthora root and stem rot of kiwifruit in Turkey. J Phytopathol 161:887–889
Larsen S, Albanese D, Stegen J et al (2023) Distinct and temporally stable assembly mechanisms shape bacterial and fungal communities in vineyard soils. Microb Ecol 86:337–349
Lenth RV (2022) emmeans: Estimated marginal means, aka least-squares means. https://rvlenth.github.io/emmeans/
Lévesque CA, de Cock AWAM (2004) Molecular phylogeny and taxonomy of the genus Pythium. Mycol Res 108:1363–1383
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Lv T, Zhan C, Pan Q et al (2023) Plant pathogenesis: toward multidimensional understanding of the microbiome. iMeta 2:e129
Malacrinò A, Karley AJ, Schena L, Bennett AE (2021) Soil microbial diversity impacts plant microbiota more than herbivory. Phytobiomes J 5:408–417
Matsumoto C, Kageyama K, Suga H, Hyakumachi M (1999) Phylogenetic relationships of Pythium species based on ITS and 5.8S sequences of the ribosomal DNA. Mycoscience 40:321–331
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217
Mian G, Cipriani G, Firrao G et al (2023a) Genetic diversity of Actinidia spp. shapes the oomycete pattern associated with Kiwifruit Vine Decline Syndrome (KVDS). Sci Rep 13. https://doi.org/10.1038/s41598-023-43754-y
Mian G, Zuiderduin K, Barnes LS et al (2023b) In vitro application of Eruca vesicaria subsp. sativa leaf extracts and associated metabolites reduces the growth of Oomycota species involved in Kiwifruit Vine Decline Syndrome. Front Plant Sci 14:1292290
Nguyen HDT, Dodge A, Dadej K et al (2022) Whole genome sequencing and phylogenomic analysis show support for the splitting of genus Pythium. Mycologia 114:501–515
Nilsson RH, Larsson K-H, Taylor AFS et al (2019) The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res 47:D259–D264
Panth M, Baysal-Gurel F, Avin FA, Simmons T (2021) Identification and chemical and biological management of Phytopythium vexans, the causal agent of Phytopythium root and crown rot of woody ornamentals. Plant Dis 105:1091–1100
Pérez-Sierra A, Jung MH, Jung T (2022) Survey and monitoring of Phytophthora species in natural ecosystems: methods for sampling, isolation, purification, storage, and pathogenicity tests. Methods Mol Biol 2536:13–49
Polat Z, Awan QN, Hussain M, Akgül DS (2017) First report of Phytopythium vexans causing root and collar rot of kiwifruit in Turkey. Plant Dis 101:1058
Polat Z, Kaymak S, Gültekin MA et al (2023) First report of Phytopythium litorale associated with dieback disease of kiwifruit in Turkey. J Plant Pathol 105:1761–1762
Prencipe S, Savian F, Nari L et al (2020) First report of Phytopythium vexans causing decline syndrome of Actinidia deliciosa ‘Hayward’ in Italy. Plant Dis 104:2032
Prencipe S, Schiavon G, Rosati M et al (2023) Characterization of Phytopythium species involved in the establishment and development of kiwifruit vine decline syndrome. Microorganisms 11:216
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26:1641–1650
Quast C, Pruesse E, Yilmaz P et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Radmer L, Anderson G, Malvick DM et al (2017) Pythium, Phytophthora, and Phytopythium spp. Associated with soybean in Minnesota, their relative aggressiveness on soybean and corn, and their sensitivity to seed treatment fungicides. Plant Dis 101:62–72
Rahman MZ, Uematsu S, Coffey MD et al (2014) Re-evaluation of Japanese Phytophthora isolates based on molecular phylogenetic analyses. Mycoscience 55:314–327
Ratnasingham S, Hebert PDN (2007) bold: the Barcode of Life Data System (http://www.barcodinglife.org). Mol Ecol Notes 7:355–364
Reid JB, Tate KG, Brown NS, Cheah LH (1991) Effects of flooding and alluvium deposition on kiwifruit (Actinidia deliciosa): 1. Early vine decline. N Z J Crop Hortic Sci 19:247–257
Rezaei S, Abrinbana M, Ghosta Y (2021) Taxonomic and pathogenic characterization of Phytopythium species from West Azarbaijan, Iran, and description of two new species. Mycologia 113:612–628
Riit T, Tedersoo L, Drenkhan R et al (2016) Oomycete-specific ITS primers for identification and metabarcoding. MycoKeys 14:17–30
Robideau GP, De Cock AWAM, Coffey MD et al (2011) DNA barcoding of oomycetes with cytochrome c oxidase subunit I and internal transcribed spacer. Mol Ecol Resour 11:1002–1011
Safaiefarahani B, Mostowfizadeh-Ghalamfarsa R, Hardy GESJ, Burgess TI (2016) Species from within the Phytophthora cryptogea complex and related species, P. erythroseptica and P. sansomeana, readily hybridize. Fungal Biol 120:975–987
Savian F, Ginaldi F, Musetti R et al (2020) Studies on the aetiology of kiwifruit decline: interaction between soil-borne pathogens and waterlogging. Plant Soil 456:113–128
Savian F, Marroni F, Ermacora P et al (2022) A metabarcoding approach to investigate fungal and Oomycete communities associated with kiwifruit vine decline syndrome in Italy. Phytobiomes J 6:290–304
Savian F, Prencipe S, Filippini N et al (2021) Pathogenicity of Phytopythium chamaehyphon: a new player in Kiwifruit Vine Decline Syndrome of Actinidia chinensis var. deliciosa “Hayward” in Italy. Plant Dis 105:2781–2784
Schardl CL, Craven KD (2003) Interspecific hybridization in plant-associated fungi and oomycetes: a review. Mol Ecol 12:2861–2873
Schliep KP (2011) phangorn: phylogenetic analysis in R. Bioinformatics 27:592–593
Schurko AM, Mendoza L, Lévesque CA et al (2003) A molecular phylogeny of Pythium insidiosum. Mycol Res 107:537–544
Senda M, Kageyama K, Suga H, Lévesque CA (2009) Two new species of Pythium, P. senticosum and P. takayamanum, isolated from cool-temperate forest soil in Japan. Mycologia 101:439–448
Sorrenti G, Toselli M, Reggidori G et al (2016) Implicazioni della gestione idrica nella “moria del kiwi” del veronese [in Italian]. Frutticoltura 3:2–7
Spigaglia P, Barbanti F, Marocchi F et al (2020) Clostridium bifermentans and C. subterminale are associated with kiwifruit vine decline, known as moria, in Italy. Plant Pathol 69:765–774
Tacconi G, Giacopini A, Tosi L (2014) La moria del kiwi nel veronese [in Italian]. Kiwi Informa Aprile/Giugno:5–23
Tacconi G, Tosi L, Giacopini A et al (2020) La moria del kiwi al 2020: lungi dalla soluzione! [in Italian]. Kiwi Informa Aprile/Giugno:6–17
Toju H, Tanabe AS, Yamamoto S, Sato H (2012) High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS One 7:e40863
Türkkan M, Özer G, Karaca G et al (2022) Characterization and pathogenicity of Pythium-like species associated with root and collar rot of kiwifruit in Turkey. Plant Dis 106:854–863
Uzuhashi S, Kakishima M, Tojo M (2010) Phylogeny of the genus Pythium and description of new genera. Mycoscience 51:337–365
Uzuhashi S, Nakagawa S, Abdelzaher HMA, Tojo M (2019) Phylogeny and morphology of new species of Globisporangium. Fungal Syst Evol 3:13–18
Veterano ST, Coffua LS, Mena-Ali JI, Blair JE (2018) Pythium yorkensis sp. nov., a potential soybean pathogen from southeastern Pennsylvania, USA. Plant Pathol 67:619–625
Wang KX, Xie YL, Yuan GQ et al (2015) First report of root and collar rot caused by Phytopythium helicoides on kiwifruit (Actinidia chinensis). Plant Dis 99:725
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis, MA, Gelfand DH, Sninsky JJ and White TJ (eds) PCR protocols: a guide to methods and applications, Academic Press, New York, pp 315–322
Wright E (2016) Using DECIPHER v2.0 to analyze big biological sequence data in R. R J 8:352
Yang X, Richardson PA, Olson HA, Hong CX (2013) Root and stem rot of begonia caused by Phytopythium helicoides in Virginia. Plant Dis 97:1385–1385
Yin X, Li XZ, Yin JJ, Wu X (2016) First report of Phytopythium helicoides causing rhizome rot of Asian lotus in China. Plant Dis 100:532
Yu G, Smith DK, Zhu H et al (2017) ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol 8:28–36
Acknowledgements
We are all thankful for the support provided by the O.P. Orizzonte Soc. Coop. Agr. within the agreement “Indagini sulle cause della moria del kiwi in Calabria e possibili strategie di contenimento”.
Funding
Open access funding provided by Università degli Studi Mediterranea di Reggio Calabria within the CRUI-CARE Agreement. SM was supported by the Italian Ministry of University and Research (MUR) with the project “Studio dell’eziologia della moria del kiwi e sviluppo di strategie di lotta biologica” funded within D.M. 1062/2021—Azione IV.6 “Contratti di ricerca su tematiche green”. LS was supported by MUR through the project PRIN 2022 “Unveiling the plant exposome to dissect a multifactorial disease: the kiwifruit vine decline” (grant no. 2022LLJH7E). AM and DS were supported by Project AGER 3, grant no. 2022–3307.
Author information
Authors and Affiliations
Contributions
Conceptualization: SM, DS, LS, AM.
Methodology: SM, LS, AM.
Investigation: SM, MMA, GP, VV, GV, EF, NZM, MGLDN, GEA.
Data analysis and visualization: AM.
Funding acquisition: SM, LS, AM.
Writing-original draft: SM, AM.
Writing-review and editing: all coauthors.
Corresponding authors
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Responsible Editor: Stéphane Compant.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mosca, S., Aci, M.M., Procopio, G. et al. Integrated analyses of the plant and soil microbiome identify Phytopythium vexans as agent of the Kiwifruit Vine Decline Syndrome. Plant Soil (2024). https://doi.org/10.1007/s11104-024-06891-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11104-024-06891-5