
Abstract
Hepatic glycogen storage diseases constitute a group of disorders due to defects in the enzymes and transporters involved in glycogen breakdown and synthesis in the liver. Although hypoglycemia and hepatomegaly are the primary manifestations of (most of) hepatic GSDs, involvement of the endocrine system has been reported at multiple levels in individuals with hepatic GSDs. While some endocrine abnormalities (e.g., hypothalamic‑pituitary axis dysfunction in GSD I) can be direct consequence of the genetic defect itself, others (e.g., osteopenia in GSD Ib, insulin-resistance in GSD I and GSD III) may be triggered by the (dietary/medical) treatment. Being aware of the endocrine abnormalities occurring in hepatic GSDs is essential (1) to provide optimized medical care to this group of individuals and (2) to drive research aiming at understanding the disease pathophysiology. In this review, a thorough description of the endocrine manifestations in individuals with hepatic GSDs is presented, including pathophysiological and clinical implications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Glycogen storage diseases (GSDs) are rare inherited metabolic disorders due to a specific defect in enzymes or transporters involved in glycogen breakdown and synthesis. More than 12 GSD types are recognized causing various symptoms depending on the location of the defect in the glycogen metabolic pathway. Hepatic GSDs (collective estimated incidence of ~ 3:100,000 newborns) are caused by a specific defect in the liver and include GSD type 0a, Ia, Ib, III, IV, VI, IX, and XI. Based on the ability to perform mitochondrial fatty acid oxidation for ketone body production, hepatic GSDs are further classified as ketotic (GSD 0a, GSD III, GSD VI, GSD IX, GSD XI) or non-ketotic (GSD Ia and GSD Ib). The major symptoms and signs in individuals with (most of the) hepatic GSDs are fasting intolerance, hepatomegaly, growth retardation, elevated transaminases and hyperlipidemia [1,2,3,4]. Additional findings characterize specific hepatic GSD types (Table 1). Clinical, biochemical and imaging features are traditionally employed for monitoring individuals with hepatic GSDs.
Dietary management including frequent feedings, regular uncooked corn starch (UCCS) intake, gastric-drip feeding is the cornerstone of the treatment for hepatic GSDs [5, 6]. Pharmacological therapy (e.g., lipid-lowering drugs, granulocyte colony-stimulating factor, ACE-inhibitors) can correct secondary metabolic disturbances and/or prevent/delay disease complications. Additional treatment options (e.g. radiofrequency ablation, liver transplantation) can be considered when previous options are not sufficiently effective [1].
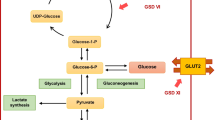
Despite the treatment, individuals with hepatic GSDs can experience metabolic decompensation [7] and develop a number of (long-term) complications, including liver adenomas and renal failure [8, 9]. Among those, disruption of the endocrine system has been extensively reported at multiple levels in hepatic GSDs [10,11,12,13,14]. An overview on the involvement of the different endocrine axes in individuals with hepatic GSDs is provided, including pathophysiological and clinical implications. A summary of major endocrine manifestations observed in hepatic GSDs is presented in Fig. 1; Table 2.

Endocrine manifestations of hepatic glycogen storage diseases (GSDs). For each endocrine component, clinical and biochemical features are presented. Specific GSD subtypes are indicated in brackets. BMD: bone mineral density; GH: growth hormone; IR: insulin-resistance; MS: metabolic syndrome; OGTT: oral glucose tolerance test; PCOs: Polycystic ovaries; T2D: type 2 diabetes *only 1 individual reported
1.1 Hypothalamic-pituitary axis
Failure to thrive and short stature are frequent findings in children with hepatic GSDs [8, 15]. Their prevalence spans from 10% in GSD 0a [16] and GSD III [17] to 17% in GSD IXb [18], 27% in GSD Ia [19], 30% in GSD IXa [18], 38% in GSD Ib [19], 52% in GSD VI [20], 57% in GSD IV patients [21] and up to 90% of GSD XI individuals [22,23,24,25]. Initial growth retardation together with a late growth spurt are common features. A subsequent catch-up growth is usually observed when proper (dietary) treatment is initiated [26]. However, some adult individuals can still experience short stature despite adequate treatment compliance [15, 27].
The underlying mechanism is only partly understood but it is assumed to result from the combination of chronic hypoglycemia, lactic/keto-acidosis, and abnormal hormonal response, including growth hormone (GH)-insulin-like growth factor (IGF-1) axis alteration [14, 28,29,30]. The extent of glucose metabolism derangement may explain why spontaneous catch-up growth can be observed even in untreated children with ketotic GSD types, while it only occurs in children with GSD I upon dietary treatment initiation [10, 31]. In GSD I disruption of gluconeogenesis results in the accumulation of lactate with no (or little) increase in circulating ketones. In ketotic GSDs gluconeogenesis is intact (thus preventing hyperlactatemia) and circulating ketone levels are increased; in these GSD subtypes ketones serve as an alternative energy substrate thus sparing glucose [17]. Nonetheless, chronic ketosis as well as amino acid depletion from gluconeogenesis could worsen growth pattern even in ketotic GSD types [32, 33]. Indeed, chronic hypoglycemia and metabolic (keto)acidosis can decrease the amplitude and frequency of GH pulses in experimental metabolic acidosis [34]. In humans, chronic metabolic acidosis is associated with decreased serum IGF-1 concentration and is related to a resistance to the hepatocellular action of GH [35]. Blunted GH response can also result from elevated circulating free fatty acids (FFA) [36], which is commonly observed in individuals with GSD I who display suboptimal glucose control [10, 37].
Besides the above-mentioned “functional” GH deficiency, growth retardation may also arise from impaired GH secretion. This “structural” GH deficiency likely results from (combination of) deranged glucose metabolism in the pituitary gland or disease-induced pituitary autoimmunity [38]. Indeed, GH deficiency has been variously reported in individuals with inherited metabolic disorders in which phosphorylated, simple carbohydrates accumulate, either due to the primary metabolic defect, or associated with dietary (over)treatment. Examples include GSD Ia [29, 39, 40], GSD Ib [38, 41, 42], GSD III [43], GSD IXa [44], GSD XI [45], phosphoglucomutase 1 deficiency (also called PGM1-CDG) [46] and Fructose-1,6-Bisphosphatase (FBPase) deficiency [47]. Interestingly, similar to the latter two disorders, the authors have observed that individuals with hepatic GSDs may display abnormal carbohydrate-deficient transferrin testing. The question is to what extent an overarching pathophysiology mechanism may play a (partial) role in the development of GH deficiency. Particularly, in GSD Ib higher prevalence of anti-pituitary antibodies has been detected, possibly resulting from immune cell dysfunction observed in this disorder [38].
Additional endocrine imbalance may also contribute to growth failure as an adaptation to glucose metabolism derangement. Indeed, individuals with GSD I and poor growth have been shown to exhibit low serum insulin concentration and higher mean 24-hour plasma cortisol levels as compared to better grown individuals [14]. These findings suggest that chronic hypoglycemia may affect multiple endocrine axes in hepatic GSDs.
Both renal tubular dysfunction and impaired liver glucose homeostasis may play a role in the development of growth failure in GSD XI [11, 22,23,24, 48]. Whether failure to thrive is the result of intestinal malabsorption by impaired glucose transport across enterocytes and/or impairment of monosaccharide transport in renal tubular cells together with hyperaminoaciduria is not fully understood [24]. Furthermore, glucose is an important energy source for the metabolism and growth of chondrocytes. Perturbations of glucose metabolism affect chondrocyte maturation and cartilage matrix production, suggesting a key role for glucose metabolism during endochondral ossification [49]. Interestingly, immunoreactivity of GLUT2 has been detected in the hypertrophic zone of the epiphyseal growth plate in growing rats [50]. However, the impact of pathogenetic GLUT2 variants on cartilage development is yet to be elucidated.
In the medical care, besides traditional biomedical biomarkers routinely employed for monitoring individuals with hepatic GSDs, the following anthropometric parameters should be regularly assessed: (i) height; (ii) weight; (iii) weight/height ratio or body mass index depending on age; v) head circumference in children [5, 51, 52]. Changes in growth trends may reflect either poor metabolic control/overtreatment or disease progression prompting (dietary) treatment adjustments [5, 33, 48, 51, 52]. Close clinical and biochemical monitoring is particularly relevant during periods of rapid growth [33]. Regular evaluation of glucose homeostasis is recommended in patients with all hepatic GSD types. Assessment of baseline IGF-1 levels as well as provocative GH testing should be considered in individuals with unexplained failure to thrive and short stature. Additionally, pituitary autoimmunity should be investigated in individuals with GSD Ib displaying a growth defect [38]. Urine electrolytes and plasma carnitine should be monitored in individuals with GSD XI [48].
Overall, adequate metabolic control together with optimization of dietary treatment are paramount to possibly ensure regular growth in individuals with hepatic GSDs [4, 5, 8, 14, 17, 19, 23, 31, 33, 51,52,53,54,55]. Catch-up growth as well as recovery of height potential has been reported in various hepatic GSD types when proper treatment is initiated [11, 31, 48, 16, 55,56,57,58,59]. The goal of dietary treatment is to maintain normal blood glucose and ketone concentrations by providing appropriate amounts of complex carbohydrates. This can be achieved by (a combination of) frequent feedings, UCCS and (nocturnal) gastric-drip feeding [5, 33, 48]. Children with ketotic GSDs should also be started on a high protein diet to sustain gluconeogenesis [51, 60]. A high-lipid diet may be of benefit in individuals with GSD III [61]. Electrolytes, calcitriol, bicarbonate, and L-carnitine should be supplemented in individuals with GSD XI [48].
Growth hormone (rhGH) therapy is not routinely indicated in hepatic GSDs unless GH deficiency has been proven and only after nutritional therapy has been optimized. Although rhGH can ensure proper growth in GSD I [40, 41], GSD III [43] and GSD XI [45], this treatment is concerning due to the potential increased risk of developing liver adenomas [4, 5, 33, 51, 52]. The possible mechanism remains unresolved but is likely related to promotion of tumor cell migration and/or energy rewiring in metabolically injured hepatocytes [27]. Furthermore, rhGH therapy may exacerbate (extreme) lipid [5, 43, 44] and ketone [4, 33] elevation. Hence, treatment with rhGH should be coupled with strict patient monitoring and use of lipid-lowering agents if needed [41]. Although liver transplantation is a potential treatment option in children with GSD I [6, 62, 64] and GSD IV [52] displaying growth failure, the results after liver transplantation reported in the medical literature point in variable directions [63, 64].
1.2 Thyroid gland
Thyroid involvement has only been reported in GSD I. Thyroid autoimmunity with overt or subclinical hypothyroidism has been described in individuals with GSD Ib [13, 65]. Besides primary thyroid damage, enhanced thyrotropin response to thyrotropin releasing hormone has been observed in both GSD Ia and GSD Ib [13], suggesting that concomitant damage at the level of the hypothalamus or pituitary gland may exist in GSD I. More recently, subclinical hypothyroidism has been reported in one individual with GSD Ia [66].
Although the mechanism underlying the development of hypothyroidism in GSD Ib is not fully understood, it appears to be related to the increased risk of autoimmunity with abnormal T-cell function and neutropenia observed in this disorder [67]. As for GSD Ia, whether the occurrence of hypothyroidism is the result of enzymatic defect per se, chronic liver disease or incidental association remains unclear. Interestingly, decreased hepatic triglycerides content was found in G6pc−/−-deficient mice treated with VK2809 (a liver-specific thyroid hormone receptor β-agonist [68]), suggesting that thyroid dysfunction may concur to the progression of liver disease in GSD I. Future studies elucidating the pathophysiology of hypothyroidism in GSD I are warranted.
Based on the above-mentioned findings, early diagnosis and treatment of thyroid disorders are paramount to improve the prognosis of individuals with GSD I [8]. This is particularly relevant as the risk of autoimmunity increases as patients progress into adulthood [69]. Annual monitoring of TSH and fT4 levels and thyroid hormone supplementation in case of hypothyroidism are recommended in individuals with GSD I [1]. When a pregnancy is possible, pre-conceptional fT4 and TSH should be assessed, taking into account the known influence of even subclinical hypothyroidism on early fetal brain development and long-term cognitive function [69, 70]. Overt hypothyroidism is associated with increased rates of spontaneous abortion, premature delivery and/or low birth weight, fetal distress in labor, and likely gestational hypertension, emphasizing the importance of thyroid balance before and during pregnancy [71].
1.3 Bone
Low bone mineral density (BMD) and higher risk of developing osteopenia (i.e. Z-score < 1.0) [72], osteoporosis (i.e. Z-score < 2.0) [73] and fractures have been reported in both children and adult individuals with GSD type 0a [16], I [1], III [2], VI [4, 33], IX [33, 74] and XI [23]. Fractures have been observed in up to 17% of individuals with hepatic GSDs [75, 76]. Hypoposphatemic rickets are commonly found in untreated patients with GSD XI between 3 and 10 months of age [77]. Despite showing normal circulating calcium levels, studies performed in individuals with GSD I and GSD III suggest the presence of both reduced bone deposition and increased bone remodeling [12, 78].
The pathophysiology of bone involvement in hepatic GSDs appears to be multifactorial, stemming from the combination of abnormal metabolic environment (e.g. elevated lactate, elevated ketones), nutritional deficiency, and possibly hormonal imbalance as well as altered muscle physiology [76, 78]. Both ketosis and hyperlactatemia exert a detrimental effect on bone [79]. Hyperlipidemia is also known to blunt bone anabolism [80]. Indeed, a correlation between BMD and circulating lactate and/or triglycerides has been reported in patients with GSD I and GSD III [12, 75, 78]. Notably, optimized metabolic control is associated with improved BMD [12]. Dietary treatment may also contribute to low BMD. Reduced dietary calcium intake [81, 82] as well as decreased circulating 25-OH vitamin D levels have been observed [18, 75, 83]. Historical studies reported that higher protein intake worsened bone health [84, 85]. Yet, growing literature suggests that high-protein diet may not have adverse effects on BMD [86,87,88]. Endocrine imbalance may also contribute to decreased BMD. Decreased circulating parathyroid hormone (PTH), calcitonin and osteocalcin have been reported in individuals with GSD I and GSD III [12, 78]. Similarly, GH deficiency [27, 29, 38, 40, 43], hypogonadism [89] as well as chronically low insulin [14, 78] and/or elevated cortisol levels [90] may play a role in the development of osteopenia/osteoporosis. Furthermore, failure of glucose supply to the exercising muscle together with impairment of the (endocrine regulation of the) muscle-bone unit appear to be major contributors to low BMD in GSD III [78]. In these disorder IGF-1/Insulin-like growth factor-binding protein 3 (IGFBP3) ratio appears to be a reliable biomarker of reduced BMD [78]. A correlation between BMD and age at start and duration of granulocyte colony-stimulating factor (G-CSF) treatment was found in individuals with GSD Ib [12].
The pathophysiology of bone demineralization is different in GSD XI, where patients are more prone to develop hypocalcemia and hypercalciuria, hyperphosphaturia with or without hypophosphatemic rickets in early childhood, osteoporosis and osteomalacia [23, 24, 91,92,93,94]. Ketosis, chronic metabolic acidosis with or without diarrhea, proximal renal tubular dysfunction, aberrant interplay among PTH, vitamin D and FGF23 are possible contributing factors [77, 91].
In the medical care, measurement of BMD together with circulating 25-OH vitamin D and dietary calcium and vitamin D intake is recommended at the diagnosis in all individuals with hepatic GSDs [5]. Subsequent evaluations are usually performed every 3–5 years or as clinically indicated [1, 2]. Assessment of circulating 25-OH vitamin D levels is indicated annually, or more frequently as needed [1, 2, 77]. Regular assessment of alkaline phosphatase, total calcium, PTH, calciuria, and phosphaturia may be useful for treatment monitoring [95]. Additional endocrine work-up should be performed if clinically indicated. Dual-emission X-ray absorptiometry (DXA) is the gold standard technique for BMD assessment being usually performed at the hip. L1-L4 vetrebrae should be considered in growing children as the hip is not a reliable site. Being a safe, inexpensive and nonradiation method for bone density assessment, Quantitative Ultrasound (QUS) has been proposed as an alternative method for low BMD diagnosis and follow-up in children [12, 78]. Signs of hypophosphatemic rickets should be regularly checked and promptly identified in all children, particularly those with GSD XI including: (i) swelling of joints; (ii) bowing of the legs; (iii) pathological fractures; (iv) teeth problems with a susceptibility to develop severe caries [24].
Good metabolic control, including adequate dietary compliance has been shown to improve BMD in individuals with hepatic GSDs [2, 5, 96]. Given the restricted dietary regimen, supplementation with calcium and/or multivitamins is strongly recommended to prevent osteopenia/osteoporosis in GSD I [5, 97]. Recommendations for vitamin and mineral supplementation in other GSD types should be based on individual patient diet and nutrient needs [33, 51]. Calcium supplementation should be tailored based on renal function, given the risk of kidney stone formation [98]. If necessary, supplementation of vitamin D can be prescribed to ameliorate bone mineralization [2]. 1,25-dihydroxy vitamin D is indicated in individuals with GSD XI [91, 92]. Particularly in individuals with GSD III, physical activity should be encouraged in order to protect the bone [51, 81].
In individuals with GSD Ib under G-CSF treatment, the risk of osteopenia/osteoporosis should be carefully monitored. The demonstration of an association between osteopenia and G-CSF treatment suggests using the minimally effective G-CSF dose. This association also adds to the growing evidence pointing in favour of the use of empagliflozin as a first line treatment for neutropenia/neutrophil dysfunction in individuals with GSD Ib [99, 100]. In individuals with GSD XI sodium bicarbonate and phosphate supplementation are additionally indicated to prevent bone loss and hypophosphatemic rickets [24, 91] and to enhance growth velocity [22, 24, 77]. Alkali supplementation (e.g. in form of Shohl’s solution or bicarbonate solution) can be considered to minimize the hypercalciuria [24, 77]. Phosphate should be supplemented as oral Joulie’s solution [92].
In principle, low BMD can lead to (recurrent) fractures in both children and adults [101]. In the general population bisphosphonates (BP) are indicated in children with osteoporosis and pathological fractures or vertebral fractures regardless of Z-score [102]. However, an “acute phase reaction” (e.g. fever, malaise, back pain, body pains, nausea, and vomiting) following initial dose of BP is commonly observed. Moreover, hypocalcemia can occur as a short-term side effect related to BP therapy [103]. Hence, the role of BP in asymptomatic individuals with hepatic GSDs and decreased BMD is still controversial and currently there is no recommendation for their use [78, 81]. However, there is evidence of improvement of BMD in single individuals with hepatic GSDs treated with BP [75, 76]. When considering whether to start a patient with hepatic GSD on BP therapy, the following factors should be considered: (i) individual’s age (currently there is no evidence-based data to support their use in children); (ii) evidence of increased bone destruction [75, 76].
1.4 Pancreas
Increased prevalence of insulin resistance (IR) and metabolic syndrome (MS) has been reported in individuals with GSD Ia [11, 104,105,106,107]. Type 2 diabetes (T2D) mellitus has been reported in individuals with GSD Ia [108,109,110,111,112] and GSD Ib [113, 114] and found in up to 9% of individuals with GSD III [17, 115,116,117,118]. However, diabetic ketoacidosis secondary to T2D has been observed in only one young girl with GSD Ia [112]. Historical studies have also reported increased glucagon levels in GSD I [60, 119,120,121]. Postprandial hyperglycemia and hyperlactatemia are common findings in individuals with GSD 0a [16]. A combination of chronic glycosuria and postprandial hyperglycemia together with post-oral glucose tolerance test (OGTT) hyperglycemia can be detected in GSD XI especially in the younger patients [22, 77, 122]. Individuals with GSD XI may also develop transient or permanent neonatal diabetes [123,124,125,126,127].
IR may result from the combination of several factors. Dietary overtreatment (i.e. high carbohydrate/UCCS intake) may lead to hyperglycemia, hyperinsulinaemia, obesity and rebound hypoglycaemia [6, 82, 128]. In GSD Ia IR may also develop as a consequence of the G6Pase-α deficiency per se. G6P excess in endoplasmatic reticulum, may upregulate the activity of 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) which results in increased conversion of inactive cortisone in active cortisol [90, 106]. Increased circulating cortisol levels may lead to metabolic syndrome [129]. Furthermore, mitochondrial dysfunction as well as accumulation of lipid metabolism by-products may contribute to IR in GSD Ia [107, 130, 131]. Downregulation of the glucose receptor on the ß-cell membrane (GLUT2) as an adaptation to hypoglycemic events may also accur leading to blunted insulin secretion in response to transient elevations of blood glucose [114]. Although pathophysiology of T2D in GSD I and GSD III is strictly correlated to IR [108,109,110, 112, 114, 116,117,118, 132], additional determinants may also contribute including (i) injured fatty liver [109, 110]; (ii) pancreatic islet β-cell insufficiency as a results of recurrent pancreatitis [109, 114] and (iii) liver cirrhosis in GSD III [133]. Notably, T2D has been observed in two siblings with GSD Ib even following liver transplantation, supporting this hypothesis [113]. Increased glucagon levels have been found in individuals with GSD I [119,120,121, 134] but not in GSD III [134]. This finding has been associated to hyperlactatemia [119, 120] and may reflect preserved gluconeogenic amino acids availability in GSD III [134].
Postprandial hyperglycemia in GSD 0a results from the inability to store glucose as glycogen in the liver (due to glycogen synthase defect) rather than impaired insulin secretion [16, 135]. Pathogenesis of postprandial/post-OGTT hyperglycaemia in GSD XI has been recently reviewed [136]. While fasting hypoglycemia is due to impaired glucose transport out of the hepatocytes, postprandial hyperglycemia likely results from hypoinsulinemia secondary to altered sensitivity of pancreatic beta cells to glucose [122, 123]. As such, insulin response is decreased but not absent in these individuals [122]. Postprandial/post-OGTT hyperglycaemia has been especially observed in younger patients. Likely glucose transport improves in older patients through GLUT2-independent mechanisms (e.g. GLUT1, GLUT3) [122, 123, 137]. This may also explain transient or permanent neonatal diabetes which has been rarely reported in GSD XI [123,124,125,126,127]. However it may be possible that some cases with transient neonatal diabetes remain undiagnosed [137]. More recently overexpression of circulating miRNAs correlated with type 1 diabetes mellitus has been found in one individual with GSD XI [136].
In the medical care, besides traditional biomedical monitoring biomarkers the following parameters should be regularly evaluated: (i) height; (ii) weight; (iii) weight/height ratio or body mass index depending on age; ; (iv) circulating insulin levels. Evaluation of circulating cortisol and ACTH may be performed in individuals with GSD Ia who display IR despite dietary optimization [90]. For early detection and management of glucose intolerance an OGTT may be considered [118]. Yet, OGTT remains contraindicated in women with hepatic GSDs due to the increased risk of hypoglycaemia [138].
Reaching appropriate diagnosis in a patient with hyperglycemia and glycosuria is essential. GSD 0a rather than diabetes mellitus should be considered in the differential diagnosis of postprandial hyperglycaemia when polyuria and polydipsia are absent [139]. Given the association of postprandial hyperglycaemia alternating with ketotic hypoglycaemia, GSD 0a and GSD XI could be reciprocally misdiagnosed. However, postprandial hyperlactatemia is observed in GSD 0a but not GSD XI [139].
Prevention of IR is paramount in hepatic GSDs. Due to the risk of iatrogenic hyperinsulinism, regular diet assessment is recommended and excess feeding/UCCS intake should be avoided [5]. In this respect, a metabolic dietitian should work closely with the patients to refine the dietary plan. Continuous glucose monitoring (CGM) appears particularly helpful in optimizing dietary treatment. Indeed, CGM allows unveiling both hypoglycemia and hyperglycemia which may be missed by traditional capillary glucose monitoring [108,109,110, 140,141,142,143,144,145,146]. Overall, adequate metabolic control together with regular reassessment of dietary plan aim to ensure optimal outcome in individuals with hepatic GSDs [6, 8, 17, 128]. Providing appropriate amounts of UCCS and complex carbohydrates is particularly relevant as glucose requirements decrease with age in individuals with hepatic GSDs [128]. IR may worsen (cardio)myopathy in GSD III by depleting energy substrates (i.e. fatty acids and ketone bodies) and promoting glycogen storage [147]. Hence, dietary treatment paradigm is being revised for this disorder with accumulating evidence indicating a benefit of a high-fat low-carbohydrate diet [61, 148]. Whether simple sugars (e.g. fructose, sucrose and galactose) should be life-long restricted in GSD I to avoid rapid insulin secretion [5, 6, 149] is still controversial.
The optimal pharmacological treatment for IR in individuals with hepatic GSDs is as yet undefined. Hypoglycemic agents (e.g. insulin and insulin secretagogues) are not routinely indicated as they can precipitate hypoglycemia [16, 77, 150]. Nonetheless, single patients successfully treated with voglibose [117], acarbose [110], insulin [112, 115, 116, 118], canagliflozin [110] and luseogliflozin [132] have been reported. The use of the sodium-glucose co-transporter 2 (SGLT2) inhibitors (e.g. empagliflozin, dapagliflozin) is widely spreading for treatment of neutropenia-related symptoms in GSD Ib [99, 100, 151,152,153,154,155]. Additionally, one adult patient with GSD XI treated with dapagliflozin displayed reduced glycogen content in shed urinary cells and improved serum potassium and phosphate concentrations [156]. Notably, side effects of SGLT2 inhibitors include elevated lactate and ketoacidosis, especially under stress conditions (e.g. intercurrent infections and major surgery) [157] prompting careful monitoring.
1.5 Adrenal cortex
Limited data on adrenal cortex hormones are available in hepatic GSDs. Two historical studies revealed an inverse correlation between plasma cortisol levels and growth parameters [10, 14] in GSD I. More recently, systematic adrenal cortex assessment has been performed in individuals with GSD I [90]. During normoglycemia, increased baseline and ACTH-stimulated serum cortisol levels were found in individuals with GSD Ia, while those with GSD Ib exhibited decreased baseline serum cortisol levels [90]. Furthermore increased plasma corticosterone and epinephrine levels have been found in fasted GSD Ia mouse model [158]. High midnight serum cortisol concentrations have been detected in one untreated boy with GSD IXa who presented with Cushing-like appearance [159].
The mechanism leading to imbalanced cortisol levels in GSD I is yet to be elucidated. Disrupted cortisol metabolism may result from the G6P modulation of the ER-bound enzyme 11β-HSD1, which activates cortisone to cortisol [160]. 11β-HSD1 is typically expressed in glucocorticoid receptor-rich tissues, such as the liver (where G6Pase-α is also expressed), adipose tissue, lung and brain [161]. The otherwise preserved adrenal cortex function suggests that disrupted cortisol metabolism might be secondary to local deregulation rather than hypothalamic-pituitary-adrenal axis dysfunction. Increased cortisol regeneration may represent a potential mechanism to divert lipid excess in GSDIa [90]. Indeed, administration of glucocorticoid receptor- antagonist mifepristone, has been shown to prevent Very-low-density lipoprotein (VLDL) accumulation in g6pc−/− mouse [158]. In addition, recurrent hypoglycemia may likely result in a “stress-induced Cushing syndrome” [159]. These observations warrant mechanistic studies, especially in light of the ongoing, experimental AAV8-mediated gene therapy treatment, which is currently in phase 3, in which temporary treatment with corticosteroids is indicated (NCT05139316). Currently, there are no recommendations on monitoring of adrenal cortex function in patients with hepatic GSDs. Hence, the need for such assessments remains on an individual basis. Evaluation of adrenal cortex function in individuals with hepatic GSDs displaying poor metabolic control is worthy. Reaching good metabolic control may contribute to reverse hypercortisolism [159]. Future studies investigating the effects of agents modulating glucocorticoid metabolism are warranted.
1.6 Gonads
Gonadal involvement has been documented in GSD type I [89, 105, 162, 163], III [105, 133, 164], VI [11], IX [11] and XI [23, 24], including delayed puberty, hypogonadotropic hypogonadism and Polycystic ovaries (PCOs).
Functional delayed puberty is a recognized feature of untreated chronic diseases [165]. Consistently, delayed puberty has been reported in GSD type I, III, VI and IX and XI, likely due to suboptimal metabolic control secondary to poor dietary compliance [4, 5, 17, 22, 23, 31, 33, 51]. A relationship between dietary treatment and pubertal development has been described in several individuals [31, 55, 166, 167]. Failure to thrive together with delayed puberty has been reported in a boy with GSD Ia following voluntary discontinuation of UCCS [166]. Catch-up growth and pubertal development together with normalization of blood testosterone levels were noticed in a 16-year-old boy diagnosed with GSD Ia following institution of dietary treatment [167]. Although dietary treatment plays a role in growth and sexual development, the mechanism underlying delayed puberty in hepatic GSDs is still not fully understood. At least in theory, delayed puberty may also result from hormonal imbalance observed in hepatic GSDs, involving circulating insulin and cortisol levels [10]. A correlation between serum insulin and cortisol levels and growth has been demonstrated in individuals with GSD I [14]. Whether such hormonal imbalance results from the enzyme defect per se or is secondary to (poor) dietary treatment remains to be elucidated.
Hypogonadotropic hypogonadism has been described in males with GSD I, showing low luteinizing hormone (LH) and follicular stimulating hormone (FSH), and correspondingly low total testosterone levels [89]. All individuals displayed recurrent hypoglycaemia and elevated lactate levels, suggesting a possible relation with suboptimal metabolic control [89]. Indeed, chronic recurrent elevations of cortisol in response to hypoglycemia may lead to suppression of gonadotropin-releasing hormone (GnRH), LH and FSH release [168].
PCOs are more commonly observed in women with GSD I, in whom they have been documented as early as 5 years of age [5, 11, 105, 163]. Less frequently PCOs are reported in other GSD types [11, 17, 33, 51, 105, 133]. Although PCOs are main features of Polycystic ovary syndrome together with hyperandogenism and irregular mensens, hyperandrogenism is an infrequent finding in hepatic GSDs [11, 105] being hirsutism reported in some women with GSDIII [17, 51, 133]. Conversely, irregular menses and menorrhagia are commonly associated with PCOs in GSD type I [162, 163] and less frequently in other GSD subtypes [17, 51, 133, 162].The mechanism underlying the development of PCOs in hepatic GSDs remains incompletely understood. Lower serum sex hormone-binding globulin (SHBG) levels have been reported in individuals with GSD Ia displaying an inverse association with intrahepatic lipid content, thus supporting a connection between metabolic (im)balance and circulating sex hormone levels [169]. Hyperinsulinism is commonly observed in suboptimally treated individuals [105] indicating a potential role for the diet in the development of PCOs. Whether good dietary compliance is sufficient to ensure adequate ovarian function in hepatic GSDs is, however, unclear [163]. Interestingly, PCOs are also observed in patients with Cushing’s syndrome [170]. Therefore, imbalanced cortisol levels may also concur to PCOs development in hepatic GSDs [90]. Future studies elucidating the underling mechanisms of PCOs are warranted.
In the medical care, besides traditional biomedical monitoring biomarkers, the following assessments should be regularly performed: (i) pubertal development in children and adolescents; (ii) frequency and regularity of menses, uncovering possible menorrhagia or irregular menstrual bleeding; (iii) signs of hyperinsulinism and/or hypercortisolism (e.g. increased weight and/or waist circumference and altered systolic and/or diastolic blood pressure); (iv) signs of hyperandrogenism, (e.g. acne, alopecia, and hirsutism) [171]. Incorporating clinical and/or biochemical screening of the hypothalamic-pituitary–gonadal axis is be important in the management of hypogonadism in males with hepatic GSDs [89]. Women with hepatic GSDs should be made aware of the increasing risk of severe hypoglycaemia in the premenstrual and luteal phase [172]. Pelvic ultrasonography should be performed regularly in women with hepatic GSDs to document PCOs [133].
Overall, adequate metabolic control is paramount to possibly ensure regular gonadal function in hepatic GSDs [4, 5, 17, 22, 23, 31, 33, 51, 163]. Indeed, puberty can be near normal with appropriate metabolic control [8, 31]. Sex hormone replacement is the most commonly employed treatment for delayed puberty in the general population [173]. Yet, estrogen therapy is not routinely indicated in women with GSD I as estrogens contribute to development of liver neoplasms [174]. Although testosterone replacement therapy allows development or maintenance of secondary sexual characteristics in males with hypogonadotropic hypogonadism [89], patients with hepatic GSDs should be carefully monitored due to the stimulation of hepatocyte proliferation by androgens [89, 174]. When indicated, transdermal estrogens are preferred over oral preparations due to hepatic first-pass metabolism [173]. Estrogen therapy in postmenopausal women may increase the risk of venous thromboembolism and stroke whereas reduces the risk of breast cancer and bone fractures [175]. Conversely, testosterone replacement therapy has not been associated with a significant elevation in the rates of venous thromboembolism and cardiovascular events [176, 177]. As oral testosterone may increase cardiovascular risk [178], intramuscular or transdermal administration should be preferred [178].
Classical combined estrogen-progestogen contraception as well as oral estrogens should be avoided in young women with hepatic GSDs, given the high risk of adenomas onset [5, 51, 179]. Progestin-only contraceptives may be considered [5, 51]. However, clinicians should be aware of the risk for reduced BMD, which needs to be monitored [5, 51]. The use of an intrauterine device should be avoided in GSD Ib, given the high risk of infection [5].
Successful pregnancies have been reported in women with GSD 0a [180], GSD I [151, 181,182,183,184,185], GSD III [186,187,188,189], GSD VI [190] and GSD XI [191] either spontaneously or after fertility treatment [190]. Male individuals with GSD I, GSD III [56] and GSD XI [192] who became fathers have been reported.
Pregnancy should be planned ahead of time and a careful management by a multidisciplinary health care team is required [184]. Good metabolic control together with close blood glucose monitoring and regular adjustments in diet and UCCS dosing are required before conception and throughout pregnancy to ensure successful outcomes [138]. Indeed, maternal hypoglycemia may be associated with intrauterine growth restriction and low birth weight [189]. Increasing protein intake may be necessary to provide an alternate source for glucose via gluconeogenesis in ketotic GSD types [33, 51]. Given the association between high estrogen state during pregnancy and adenoma onset [179], women with GSD I should be made aware of the increased risk of enlargement and rupture of adenomas [163].
2 Conclusions
Hepatic GSDs are complex disorders, requiring a highly specialized multidisciplinary team to achieve treatment goals [2, 5, 6, 20, 33, 51, 52]. Their multisystem involvement raises significant organizational, logistic, and financial obstacles for affected families and healthcare providers. The potentially life-threatening nature of hepatic GSDs symptoms and high variability in patients’ phenotypes, treatment interventions and outcomes emphasize the need and urgency for improved monitoring options.
The progress in dietary treatment as well as the availability of appropriate tools to manage acute metabolic decompensation [7] has shifted the clinical focus from “mortality” to “morbidity”. As a result, a number of long-term complications have emerged, including those affecting the endocrine system. In this review we provided a comprehensive summary of endocrine involvement in hepatic GSDs. Being aware of the endocrine manifestations of hepatic GSDs would have two main benefits: (i) optimized disease management, improving patient outcome and possibly allowing standardization of clinical care; (ii) earlier identification of hepatic GSDs in individuals displaying milder phenotypes; this appears particularly relevant as such individuals may first come to the (pediatric) endocrinologist attention without having been referred by a metabolic specialist.
Disruption of the endocrine system may occur at multiple levels in hepatic GSDs resulting in various (serious) clinical conditions. These include short stature, hypothyroidism, osteopenia/osteoporosis, IR and PCOs, among others (Fig. 1; Table 2). Currently available evidence argues in favour of regular screening for endocrine function in individuals diagnosed with hepatic GSDs in order to start prompt treatment. Appropriate treatment stems from the exact knowledge of the mechanisms underlying each endocrine condition. Many endocrine manifestations (e.g. failure to thrive, osteopenia/osteoporosis, IR, delayed puberty) share a multifactorial pathogenesis, thus complicating the use of targeted approaches. In these cases, current management strategy relies on optimization of (dietary) treatment for hepatic GSDs. In specific cases (e.g. short stature, hypogonadism) a distinct hormone deficiency can be identified, supporting hormone replacement therapy. At least in theory additional mechanisms may concur to endocrine dysfunction in hepatic GSDs, including relationship between energy production and hormone synthesis, effect of toxic metabolite accumulation or hormone/receptor glycosylation. Indeed, depletion of gluconeogenic amino acid precursors (which are employed for endogenous glucose production) may contribute to growth failure in ketotic GSDs [134, 149, 193]. Nonetheless, (glycogen-derived) UDP-glucose is required for glycosylation of glycoprotein hormones such as TSH, FSH and LH [194]. Elucidating such mechanisms may improve current knowledge of disease pathophysiology and potentially develop novel monitoring and treatment tools.
Several innovative treatment strategies are currently being investigated for hepatic GSDs, including gene replacement/base editing (NCT05139316, NCT05095727) [195], anaplerotic therapy (NCT03665636) and drug repurposing (NCT04138251; NCT05960617; NCT04986735). The aim of such approaches is to either restore energy balance or prevent the accumulation of a toxic metabolite. Growing evidence supports the efficacy of these treatments on “classical” disease manifestations (e.g. fasting intolerance, neutrophil dysfunction) [152, 196]. Whether these approaches are also effective on (long-term) endocrine manifestations in hepatic GSDs is yet to be determined.
Delivering standardized high-quality healthcare to patients worldwide is among the top priorities for hepatic GSDs [142, 197]. To this aim, current evidence on endocrine involvement in hepatic GSDs as well as management suggestions were presented in this review. This work also underlines the compelling need to strengthen multistakeholder collaborative networks including both metabolic and endocrine experts to optimize patient care.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- GSDs:
-
Glycogen storage diseases
- BMD:
-
Low bone mineral density
- BP:
-
Bisphosphonates
- CGM:
-
Continuous glucose monitoring
- DXA:
-
Dual-emission X-ray absorptiometry
- FBPase:
-
Fructose-1,6-Bisphosphatase
- FFA:
-
Free fatty acids
- FSH:
-
Follicular stimulating hormone
- G-CSF:
-
Granulocyte colony-stimulating factor
- GH:
-
Growth hormone
- GLUT2:
-
Glucose receptor on the ß-cell membrane
- GnRH:
-
Gonadotropin-releasing hormone
- IGF-1:
-
Insulin-like growth factor
- IR:
-
Insulin resistance
- LH:
-
Luteinizing hormone
- OGTT:
-
Post-oral glucose tolerance test
- PCOs:
-
Polycystic ovaries
- PGM1-CDG:
-
Phosphoglucomutase 1 deficiency
- PTH:
-
Parathyroid hormone
- QUS:
-
Quantitative Ultrasound
- rhGH:
-
Recombinant human growth hormone
- SGLT2:
-
Sodium-glucose co-transporter 2
- SHBG:
-
Serum sex hormone-binding globulin
- T2D:
-
Type 2 diabetes
- UCCS:
-
Uncooked corn starch
- VLDL:
-
Very-low-density lipoprotein
- 11βHSD1:
-
11β-hydroxysteroid dehydrogenase type 1
References
Bali DS, El-Gharbawy A, Austin S, Pendyal S, Kishnani PS. Glycogen Storage Disease Type I. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle, Seattle (WA): University of Washington; 1993.
Schreuder AB, Rossi A, Grünert SC, Derks TG. Glycogen Storage Disease Type III. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle, Seattle (WA): University of Washington; 1993.
Herbert M, Goldstein JL, Rehder C, Austin S, Kishnani PS, Bali DS. Phosphorylase kinase Deficiency. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle, Seattle (WA): University of Washington; 1993.
Labrador E, Weinstein DA. Glycogen Storage Disease Type VI. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, Amemiya A, editors. GeneReviews®. Seattle, Seattle (WA): University of Washington; 1993.
Kishnani PS, Austin SL, Abdenur JE, Arn P, Bali DS, Boney A, Chung WK, Dagli AI, Dale D, Koeberl D, Somers MJ, Wechsler SB, Weinstein DA, Wolfsdorf JI, Watson MS. American College of Medical Genetics and Genomics: diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 2014;16. https://doi.org/10.1038/gim.2014.128. e1.
Derks TGJ, Rodriguez-Buritica DF, Ahmad A, de Boer F, Couce ML, Grünert SC, Labrune P, López Maldonado N, Fischinger Moura de Souza C, Riba-Wolman R, Rossi A, Saavedra H, Gupta RN, Valayannopoulos V, Mitchell J. Glycogen Storage Disease Type Ia: current management options, Burden and Unmet needs. Nutrients. 2021;13(3828). https://doi.org/10.3390/nu13113828.
Rossi A, Hoogeveen IJ, Lubout CMA, de Boer F, Fokkert-Wilts MJ, Rodenburg IL, van Dam E, Grünert SC, Martinelli D, Scarpa M, CONNECT MetabERN Collaboration Group, Dekker H, Te Boekhorst ST, van Spronsen FJ, Derks TGJ. A generic emergency protocol for patients with inborn errors of metabolism causing fasting intolerance: a retrospective, single-center study and the generation of www.emergencyprotocol.net. J Inherit Metab Dis. 2021;44(5):1124–35. https://doi.org/10.1002/jimd.12386.
Dambska M, Labrador EB, Kuo CL, Weinstein DA. Prevention of complications in glycogen storage disease type Ia with optimization of metabolic control. Pediatr Diabetes. 2017;18:327–31. https://doi.org/10.1111/pedi.12540.
Ross KM, Ferrecchia IA, Dahlberg KR, Dambska M, Ryan PT, Weinstein DA. Dietary Management of the glycogen Storage diseases: evolution of treatment and ongoing controversies. Adv Nutr. 2020;11:439–46. https://doi.org/10.1093/advances/nmz092.
Dunger DB, Holder AT, Leonard JV, Okae J, Preece MA. Growth and endocrine changes in the hepatic glycogenoses. Eur J Pediatr. 1982;138:226–30. https://doi.org/10.1007/BF00441207.
Lee PJ, Leonard JV. The hepatic glycogen storage diseases–problems beyond childhood. J Inherit Metab Dis. 1995;18:462–72. https://doi.org/10.1007/BF00710057.
Melis D, Pivonello R, Cozzolino M, Della Casa R, Balivo F, Del Puente A, Dionisi-Vici C, Cotugno G, Zuppaldi C, Rigoldi M, Parini R, Colao A, Andria G, Parenti G. Impaired bone metabolism in glycogen storage disease type 1 is associated with poor metabolic control in type 1a and with granulocyte colony-stimulating factor therapy in type 1b. Horm Res Paediatr. 2014;81:55–62. https://doi.org/10.1159/000351022.
Melis D, Pivonello R, Parenti G, Della Casa R, Salerno M, Lombardi G, Sebastio G, Colao A, Andria G. Increased prevalence of thyroid autoimmunity and hypothyroidism in patients with glycogen storage disease type I. J Pediatr. 2007;150:300–5. https://doi.org/10.1016/j.jpeds.2006.11.056.
Mundy HR, Hindmarsh PC, Matthews DR, Leonard JV, Lee PJ. The regulation of growth in glycogen storage disease type 1. Clin Endocrinol (Oxf). 2003;58:332–9. https://doi.org/10.1046/j.1365-2265.2003.01717.x.
Szymańska E, Lipiński P, Rokicki D, Książyk J, Tylki-Szymańska A. Over 20-Year follow-up of patients with hepatic glycogen Storage diseases: single-center experience. Diagnostics (Basel). 2020;10:297. https://doi.org/10.3390/diagnostics10050297.
Weinstein DA, Correia CE, Saunders AC, Wolfsdorf JI. Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycemia. Mol Genet Metab. 2006;87:284–8. https://doi.org/10.1016/j.ymgme.2005.10.006.
Sentner CP, Hoogeveen IJ, Weinstein DA, Santer R, Murphy E, McKiernan PJ, Steuerwald U, Beauchamp NJ, Taybert J, Laforêt P, Petit FM, Hubert A, Labrune P, Smit GPA, Derks TGJ. Glycogen storage disease type III: diagnosis, genotype, management, clinical course and outcome. J Inherit Metab Dis. 2016;39:697–704. https://doi.org/10.1007/s10545-016-9932-2.
Fernandes SA, Cooper GE, Gibson RA, Kishnani PS. Benign or not benign? Deep phenotyping of liver glycogen storage disease IX. Mol Genet Metab. 2020;131:299–305. https://doi.org/10.1016/j.ymgme.2020.10.004.
Rake J, Visser G, Labrune P, Leonard J, Ullrich K, Smit P. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European study on glycogen Storage Disease Type I (ESGSD I). Eur J Pediatrics. 2002;161:S20–34. https://doi.org/10.1007/s00431-002-0999-4.
Luo X, Duan Y, Fang D, Sun Y, Xiao B, Zhang H, Han L, Liang L, Gong Z, Gu X, Yu Y, Qiu W. Diagnosis and follow-up of glycogen storage disease (GSD) type VI from the largest GSD center in China. Hum Mutat. 2022;43:557–67. https://doi.org/10.1002/humu.24345.
Derks TGJ, Peeks F, de Boer F, van der Fokkert-Wilts M, Szymańska E, Rokicki D, Ryan PT, Weinstein DA. The potential of dietary treatment in patients with glycogen storage disease type IV. J Inherit Metab Dis. 2021;44(3):693–704. https://doi.org/10.1002/jimd.12339.
Manz F, Bickel H, Brodehl J, Feist D, Gellissen K, Geschöll-Bauer B, Gilli G, Harms E, Helwig H, Nützenadel W, Waldherr R. Fanconi-Bickel syndrome. Pediatr Nephrol. 1987;1:509–18. https://doi.org/10.1007/BF00849262.
Santer R, Schneppenheim R, Suter D, Schaub J, Steinmann B. Fanconi-Bickel syndrome - the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature. Eur J Pediatrics. 1998;157:783–97. https://doi.org/10.1007/s004310050937.
Santer R, Steinmann B, Schaub J. Fanconi-Bickel syndrome - A Congenital Defect of Facilitative Glucose Transport. CMM. 2002;2:213–27. https://doi.org/10.2174/1566524024605743.
Govindarajan S, Khandelwal P, Sharma S, Agarwala A, Sinha A, Hari P, Bagga A. Clinical features and genetic sequencing of children with Fanconi–Bickel Syndrome. Indian J Pediatr. 2023;90:178–80. https://doi.org/10.1007/s12098-022-04372-0.
Schippers HM, Smit GPA, Rake JP, Visser G. Characteristic growth pattern in male X-linked phosphorylase‐b kinase deficiency (GSD IX). J Inher Metab Disea. 2003;26:43–7. https://doi.org/10.1023/A:1024071328772.
Jackson DG, Koch RL, Pendyal S, Benjamin R, Kishnani PS. Development of hepatocellular adenomas in a patient with glycogen storage disease Ia treated with growth hormone therapy. JIMD Rep. 2023;64:303–11. https://doi.org/10.1002/jmd2.12381.
Nuoffer JM, Mullis PE, Wiesmann UN. Treatment with low-dose diazoxide in two growth‐retarded prepubertal girls with glycogen storage disease type Ia resulted in catch‐up growth. J Inher Metab Disea. 1997;20:790–8. https://doi.org/10.1023/A:1005319818015.
Dagdelen S, Atmaca A, Alikasifoglu A, Erbas T. Pituitary hypoplasia and growth hormone deficiency in a woman with glycogen storage disease type Ia: a case report. J Med Case Rep. 2008;2:210. https://doi.org/10.1186/1752-1947-2-210.
Karnsakul W, Gillespie S, Skitarelic K, Hummel M. Obesity and reversed growth retardation in a child with type Ia glycogen Storage Disease. J Pediatr Endocrinol Metab. 2010;23. https://doi.org/10.1515/jpem.2010.083.
Dunger DB, Leonard JV, Preece MA. Patterns of growth in the hepatic glycogenoses. Arch Dis Child. 1984;59:657–60. https://doi.org/10.1136/adc.59.7.657.
Brown LM, Van Der Corrado MM, Derks TGJ, Chen MA, Siegel S, Hoyt K, Correia CE, Lumpkin C, Flanagan TB, Carreras CT, Weinstein DA. Evaluation of glycogen storage disease as a cause of ketotic hypoglycemia in children. J Inher Metab Disea. 2015;38:489–93. https://doi.org/10.1007/s10545-014-9744-1.
Kishnani PS, Goldstein J, Austin SL, Arn P, Bachrach B, Bali DS, Chung WK, El-Gharbawy A, Brown LM, Kahler S, Pendyal S, Ross KM, Tsilianidis L, Weinstein DA, Watson MS. Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Sci. 2019;21:772–89. https://doi.org/10.1038/s41436-018-0364-2.
Kuemmerle N, Krieg RJ, Latta K, Challa A, Hanna JD, Chan JC. Growth hormone and insulin-like growth factor in non-uremic acidosis and uremic acidosis. Kidney Int Suppl. 1997;58:S102–105.
Brüngger M, Hulter HN, Krapf R. Effect of chronic metabolic acidosis on the growth hormone/IGF-1 endocrine axis: new cause of growth hormone insensitivity in humans. Kidney Int. 1997;51:216–21. https://doi.org/10.1038/ki.1997.26.
Casanueva FF, Villanueva L, Dieguez C, Diaz Y, Cabranes JA, Szoke B, Scanlon MF, Schally AV, Fernandez-Cruz A. Free fatty acids Block Growth hormone (GH) releasing hormone-stimulated GH Secretion in Man directly at the Pituitary*. J Clin Endocrinol Metabolism. 1987;65:634–42. https://doi.org/10.1210/jcem-65-4-634.
Derks TGJ, Van Rijn M. Lipids in hepatic glycogen storage diseases: pathophysiology, monitoring of dietary management and future directions. J Inher Metab Disea. 2015;38:537–43. https://doi.org/10.1007/s10545-015-9811-2.
Melis D, Pivonello R, Parenti G, Della Casa R, Salerno M, Balivo F, Piccolo P, Di Somma C, Colao A, Andria G. The growth hormone-insulin-like growth factor Axis in Glycogen Storage Disease Type 1: evidence of different growth patterns and insulin-like growth factor levels in patients with glycogen storage disease type 1a and 1b. J Pediatr. 2010;156:663–e6701. https://doi.org/10.1016/j.jpeds.2009.10.032.
Bierich JR. Growth hormone therapy in short children without classical growth hormone deficiency. J Endocrinol Invest. 1989;12:25–33.
Wu S, Guo S, Fu L, Du C, Luo X. Case Report: Glycogen Storage Disease Type Ia in a Chinese child treated with growth hormone. Front Pediatr. 2022;10:921323. https://doi.org/10.3389/fped.2022.921323.
Noto RA, Vijayaraghavan V, Timoshin A, Sansobrino D. Improved growth with growth hormone therapy in a child with glycogen storage disease ib. Acta Paediatr. 2003;92:977–9.
Zhang Y, Sun H, Wan N. Mutation analysis of SLC37A4 in a patient with glycogen storage disease-type ib. J Int Med Res. 2019;47:5996–6003. https://doi.org/10.1177/0300060519867819.
Larizza D, Maggiore G, Marzani D, Maghnie M, Ciceri R. Difficult hGH treatment in a patient with type III glycogen storage disease. Eur J Pediatr. 1986;145:84–5. https://doi.org/10.1007/BF00441862.
Hodax JK, Uysal S, Quintos JB, Phornphutkul C. Glycogen storage disease type IX and growth hormone deficiency presenting as severe ketotic hypoglycemia. J Pediatr Endocrinol Metab. 2017;30. https://doi.org/10.1515/jpem-2016-0342.
Scully KJ, Wolfsdorf J, Dedekian M. Acquired growth hormone deficiency in Fanconi-Bickel syndrome. BMJ Case Rep. 2021;14:e246212. https://doi.org/10.1136/bcr-2021-246212.
Altassan R, Radenkovic S, Edmondson AC, Barone R, Brasil S, Cechova A, Coman D, Donoghue S, Falkenstein K, Ferreira V, Ferreira C, Fiumara A, Francisco R, Freeze H, Grunewald S, Honzik T, Jaeken J, Krasnewich D, Lam C, Lee J, Lefeber D, Marques-da-Silva D, Pascoal C, Quelhas D, Raymond KM, Rymen D, Seroczynska M, Serrano M, Sykut-Cegielska J, Thiel C, Tort F, Vals M-A, Videira P, Voermans N, Witters P, Morava E. International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): diagnosis, follow-up, and management. J Inherit Metab Dis. 2021;44:148–63. https://doi.org/10.1002/jimd.12286.
Takagi D, Ben-Ari J, Nemet D, Zeharia A, Eliakim A. Recurrent infantile hypoglycemia due to combined fructose-1,6-diphosphatase deficiency and growth hormone deficiency. J Pediatr Endocrinol Metab. 2013;26:761–3. https://doi.org/10.1515/jpem-2012-0119.
Pennisi A, Maranda B, Benoist J, Baudouin V, Rigal O, Pichard S, Santer R, Romana Lepri F, Novelli A, De Baulny O, Dionisi-Vici H, Schiff C. Nocturnal enteral nutrition is therapeutic for growth failure in Fanconi‐Bickel syndrome. J Inher Metab Disea. 2020;43:540–8. https://doi.org/10.1002/jimd.12203.
Hollander JM, Zeng L. The emerging role of glucose metabolism in Cartilage Development. Curr Osteoporos Rep. 2019;17:59–69. https://doi.org/10.1007/s11914-019-00506-0.
Ohara H, Tamayama T, Maemura K, Kanbara K, Hayasaki H, Abe M, Watanabe M. Immunocytochemical demonstration of glucose transporters in epiphyseal growth plate chondrocytes of young rats in correlation with autoradiographic distribution of 2-deoxyglucose in chondrocytes of mice. Acta Histochem. 2001;103:365–78. https://doi.org/10.1078/0065-1281-00604.
Kishnani PS, Austin SL, Arn P, Bali DS, Boney A, Case LE, Chung WK, Desai DM, El-Gharbawy A, Haller R, Smit GPA, Smith AD, Hobson-Webb LD, Wechsler SB, Weinstein DA, Watson MS. Glycogen Storage Disease Type III diagnosis and management guidelines. Genet Sci. 2010;12:446–63. https://doi.org/10.1097/GIM.0b013e3181e655b6.
Koch RL, Soler-Alfonso C, Kiely BT, Asai A, Smith AL, Bali DS, Kang PB, Landstrom AP, Akman HO, Burrow TA, Orthmann-Murphy JL, Goldman DS, Pendyal S, El-Gharbawy AH, Austin SL, Case LE, Schiffmann R, Hirano M, Kishnani PS. Diagnosis and management of glycogen storage disease type IV, including adult polyglucosan body disease: a clinical practice resource. Mol Genet Metab. 2023;138:107525. https://doi.org/10.1016/j.ymgme.2023.107525.
Rake JP, Visser G, Huismans D, Van Der Huitema S, Piers DA, Smit GPA. Bone mineral density in children, adolescents and adults with glycogen storage disease type Ia: a cross-sectional and longitudinal study. J Inher Metab Disea. 2003;26:371–84. https://doi.org/10.1023/A:1025111220095.
Wolfsdorf J, Rudlin C, Crigler J. Physical growth and development of children with type 1 glycogen-storage disease: comparison of the effects of long-term use of dextrose and uncooked cornstarch. Am J Clin Nutr. 1990;52:1051–7. https://doi.org/10.1093/ajcn/52.6.1051.
Talente GM. Glycogen storage disease in adults. Ann Intern Med. 1994;120:218. https://doi.org/10.7326/0003-4819-120-3-199402010-00008.
Greene HL, Ghishan FK, Brown B, McClenathan DT, Freese D. Hypoglycemia in type IV glycogenosis: hepatic improvement in two patients with nutritional management. J Pediatr. 1988;112:55–8. https://doi.org/10.1016/S0022-3476(88)80121-5.
Korula S, Danda S, Paul PG, Mathai S, Simon A. Hepatic glycogenoses among children—clinical and biochemical characterization: single-center study. J Clin Experimental Hepatol. 2020;10:222–7. https://doi.org/10.1016/j.jceh.2019.07.007.
Goldberg T, Slonim AE. Nutrition therapy for hepatic glycogen storage diseases. J Am Diet Assoc. 1993;93:1423–30. https://doi.org/10.1016/0002-8223(93)92246-T.
Nakai A, Shigematsu Y, Takano T, Kikawa Y, Sudo M. Uncooked cornstarch treatment for hepatic phosphorylase kinase deficiency. Eur J Pediatr. 1994;153:581–3. https://doi.org/10.1007/BF02190663.
Slonim AE, Coleman RA, Moses WS. Myopathy and growth failure in debrancher enzyme deficiency: improvement with high-protein nocturnal enteral therapy. J Pediatr. 1984;105:906–11. https://doi.org/10.1016/S0022-3476(84)80075-X.
Rossi A, Hoogeveen IJ, Bastek VB, De Boer F, Montanari C, Meyer U, Maiorana A, Bordugo A, Dianin A, Campana C, Rigoldi M, Kishnani PS, Pendyal S, Strisciuglio P, Gasperini S, Parenti G, Parini R, Paci S, Melis D, Derks TGJ. Dietary lipids in glycogen storage disease type III: a systematic literature study, case studies, and future recommendations. J Inher Metab Disea. 2020;43:770–7. https://doi.org/10.1002/jimd.12224.
Labrune P. Glycogen storage disease type I: indications for liver and/or kidney transplantation. Eur J Pediatrics. 2002;161:S53–5. https://doi.org/10.1007/s00431-002-1004-y.
Davis MK, Weinstein DA. Liver transplantation in children with glycogen storage disease: controversies and evaluation of the risk/benefit of this procedure. Pediatr Transplant. 2008;12:137–45. https://doi.org/10.1111/j.1399-3046.2007.00803.x.
Matern D, Starzl TE, Arnaout W, Barnard J, Bynon JS, Dhawan A, Emond J, Haagsma EB, Hug G, Lachaux A, Smit GPA, Chen Y-T. Liver transplantation for glycogen storage disease types I, III, and IV. Eur J Pediatr. 1999;158:S043–8. https://doi.org/10.1007/PL00014320.
Melis D, Della Casa R, Balivo F, Minopoli G, Rossi A, Salerno M, Andria G, Parenti G. Involvement of endocrine system in a patient affected by glycogen storage disease 1b: speculation on the role of autoimmunity. Ital J Pediatr. 2014;40(1):30. https://doi.org/10.1186/1824-7288-40-30.
Ön ŞŞ, Acar S, Demir K, Abacı A, Öztürk Y, Kahveci Çelik S, Böber E. Evaluation of thyroid function tests in children with Chronic Liver diseases. Jcrpe. 2020;12:143–9. https://doi.org/10.4274/jcrpe.galenos.2019.2019.0029.
Melis D, Carbone F, Minopoli G, La Rocca C, Perna F, De Rosa V, Galgani M, Andria G, Parenti G, Matarese G. Cutting Edge: increased autoimmunity risk in glycogen storage disease type 1b is Associated with a reduced Engagement of Glycolysis in T cells and an impaired Regulatory T cell function. J Immunol. 2017;198:3803–8. https://doi.org/10.4049/jimmunol.1601946.
Zhou J, Waskowicz LR, Lim A, Liao X-H, Lian B, Masamune H, Refetoff S, Tran B, Koeberl DD, Yen PM. A liver-specific Thyromimetic, VK2809, decreases Hepatosteatosis in Glycogen Storage Disease Type Ia. Thyroid. 2019;29:1158–67. https://doi.org/10.1089/thy.2019.0007.
Vantyghem M-C, Dobbelaere D, Mention K, Wemeau J-L, Saudubray J-M, Douillard C. Endocrine manifestations related to inherited metabolic diseases in adults. Orphanet J Rare Dis. 2012;7:11. https://doi.org/10.1186/1750-1172-7-11.
Krassas GE, Poppe K, Glinoer D. Thyroid function and human Reproductive Health. Endocr Rev. 2010;31:702–55. https://doi.org/10.1210/er.2009-0041.
Glinoer D, Delange F. The potential repercussions of maternal, fetal, and neonatal hypothyroxinemia on the progeny. Thyroid. 2000;10:871–87. https://doi.org/10.1089/thy.2000.10.871.
Varacallo M, Seaman TJ, Jandu JS, Pizzutillo P. Osteopenia. Treasure Island (FL): In: StatPearls. StatPearls Publishing; 2024.
Gordon CM, Leonard MB, Zemel BS. 2013 Pediatric Position Development Conference: Executive Summary and Reflections. Journal of Clinical Densitometry. 17, 219–224 (2014). https://doi.org/10.1016/j.jocd.2014.01.007.
İnci A, Kılıç Yıldırım G, Ergin C, Sarı FB, Eğritaş Gürkan S, Okur Ö, Biberoğlu İ, Bükülmez G, Ezgü A, Dalgıç FS, Tümer B. Expected or unexpected clinical findings in liver glycogen storage disease type IX: distinct clinical and molecular variability. J Pediatr Endocrinol Metab. 2022;35:451–62. https://doi.org/10.1515/jpem-2021-0278.
Minarich LA, Kirpich A, Fiske LM, Weinstein DA. Bone mineral density in glycogen storage disease type Ia and ib. Genet Sci. 2012;14:737–41. https://doi.org/10.1038/gim.2012.36.
Cabrera-Abreu J, Crabtree NJ, Elias E, Fraser W, Cramb R, Alger S. Bone mineral density and markers of bone turnover in patients with glycogen storage disease types I, III and IX. J Inher Metab Disea. 2004;27:1–9. https://doi.org/10.1023/B:BOLI.0000016632.13234.56.
Gupta N, Nambam B, Weinstein DA, Shoemaker LR. Late diagnosis of Fanconi-Bickel syndrome: challenges with the diagnosis and literature review. J Inborn Errors Metabolism Screen. 2016;4:232640981667943. https://doi.org/10.1177/2326409816679430.
Melis D, Rossi A, Pivonello R, Del Puente A, Pivonello C, Cangemi G, Negri M, Colao A, Andria G, Parenti G. Reduced bone mineral density in glycogen storage disease type III: evidence for a possible connection between metabolic imbalance and bone homeostasis. Bone. 2016;86:79–85. https://doi.org/10.1016/j.bone.2016.02.012.
Kraut JA. The role of metabolic acidosis in the Pathogenesis of Renal Osteodystrophy. Adv Renal Replace Ther. 1995;2:40–51. https://doi.org/10.1016/S1073-4449(12)80070-7.
Pirih F, Lu J, Ye F, Bezouglaia O, Atti E, Ascenzi M, Tetradis S, Demer L, Aghaloo T, Tintut Y. Adverse effects of hyperlipidemia on bone regeneration and strength. J Bone Mineral Res. 2012;27:309–18. https://doi.org/10.1002/jbmr.541.
Mundy HR, Williams JE, Lee PJ, Fewtrell MS. Reduction in bone mineral density in glycogenosis type III may be due to a mixed muscle and bone deficit. J Inher Metab Disea. 2008;31:418–23. https://doi.org/10.1007/s10545-008-0830-0.
Bhattacharya K. Dietary dilemmas in the management of glycogen storage disease type I. J Inher Metab Disea. 2011;34:621–9. https://doi.org/10.1007/s10545-011-9322-8.
Jacoby JT, Bento Dos Santos B, Nalin T, Colonetti K, Farret Refosco L, De Souza FM, Spritzer C, Poloni PM, Hack-Mendes S, Schwartz R. Bone Mineral density in patients with hepatic glycogen Storage diseases. Nutrients. 2021;13:2987. https://doi.org/10.3390/nu13092987.
Barzel US, Massey LK. Excess dietary protein can adversely affect bone. J Nutr. 1998;128:1051–3. https://doi.org/10.1093/jn/128.6.1051.
Ince BA, Anderson EJ, Neer RM. Lowering Dietary protein to U.S. recommended Dietary Allowance levels reduces urinary calcium excretion and bone resorption in Young Women. J Clin Endocrinol Metabolism. 2004;89:3801–7. https://doi.org/10.1210/jc.2003-032016.
Bertoli S, Trentani C, Ferraris C, De Giorgis V, Veggiotti P, Tagliabue A. Long-term effects of a ketogenic diet on body composition and bone mineralization in GLUT-1 deficiency syndrome: a case series. Nutrition. 2014;30:726–8. https://doi.org/10.1016/j.nut.2014.01.005.
Colica C, Merra G, Gasbarrini A, De Lorenzo A, Cioccoloni G, Gualtieri P, Perrone MA, Bernardini S, Bernardo V, Di Renzo L, Marchetti M. Efficacy and safety of very-low-calorie ketogenic diet: a double blind randomized crossover study. Eur Rev Med Pharmacol Sci. 2017;21:2274–89.
Yu D, Chen W, Zhang J, Wei L, Qin J, Lei M, Tang H, Wang Y, Xue S, Dong J, Chen Y, Xie L, Di H. Effects of weight loss on bone turnover, inflammatory cytokines, and adipokines in Chinese overweight and obese adults. J Endocrinol Invest. 2022;45:1757–67. https://doi.org/10.1007/s40618-022-01815-5.
Wong EM, Lehman A, Acott P, Gillis J, Metzger DL, Sirrs S. Hypogonadotropic hypogonadism in males with Glycogen Storage Disease Type 1. In: Morava E, Baumgartner M, Patterson M, Rahman S, Zschocke J, Peters V, editors. JIMD reports. Volume 36. Berlin, Heidelberg: Springer Berlin Heidelberg; 2017. pp. 79–84.
Rossi A, Simeoli C, Salerno M, Ferrigno R, Della Casa R, Colao A, Strisciuglio P, Parenti G, Pivonello R, Melis D. Imbalanced cortisol concentrations in glycogen storage disease type I: evidence for a possible link between endocrine regulation and metabolic derangement. Orphanet J Rare Dis. 2020;15:99. https://doi.org/10.1186/s13023-020-01377-w.
Roy M, Bose K, Paul DK, Anand P. Hypophosphatemic Rickets: Presenting Features of Fanconi—Bickel Syndrome. Case Reports in Pathology. 2011, 1–3 (2011). https://doi.org/10.1155/2011/314696.
Afroze B, Chen M. Fanconi-Bickel syndrome: two Pakistani patients presenting with hypophosphatemic rickets. J Pediatr Genet. 2016;5:161–6. https://doi.org/10.1055/s-0036-1584360.
Mohandas Nair K, Sakamoto O, Jagadeesh S, Nampoothiri S. Fanconi–Bickel Syndrome. Indian J Pediatr. 2012;79:112–4. https://doi.org/10.1007/s12098-011-0373-5.
Şahin F, Sipahi T, Doğan H, Oksal A, Ertan Ü. Pathological case of the Month. Arch Pediatr Adolesc Med. 2000;154:1165. https://doi.org/10.1001/archpedi.154.11.1165.
Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, Kamenicky P, Nevoux J, Prié D, Rothenbuhler A, Wicart P, Harvengt P. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connections. 2014;3:R13–30. https://doi.org/10.1530/EC-13-0103.
Wolfsdorf JI. Bones benefit from better biochemical control in type 1 glycogen storage disease. J Pediatr. 2002;141:308–10. https://doi.org/10.1067/mpd.2002.127504.
Chen MA, Weinstein DA. Glycogen storage diseases: diagnosis, treatment and outcome. Translational Sci Rare Dis. 2016;1:45–72. https://doi.org/10.3233/TRD-160006.
Weinstein DA, Somers MJG, Wolfsdorf JI. Decreased urinary citrate excretion in type 1a glycogen storage disease. J Pediatr. 2001;138:378–82. https://doi.org/10.1067/mpd.2001.111322.
Rossi A, Miele E, Fecarotta S, Veiga-da-Cunha M, Martinelli M, Mollica C, D’Armiento M, Mozzillo E, Strisciuglio P, Derks TGJ, Staiano A, Parenti G. Crohn disease-like enterocolitis remission after empagliflozin treatment in a child with glycogen storage disease type ib: a case report. Ital J Pediatr. 2021;47:149. https://doi.org/10.1186/s13052-021-01100-w.
Grünert SC, Derks TGJ, Adrian K, Al-Thihli K, Ballhausen D, Bidiuk J, Bordugo A, Boyer M, Bratkovic D, Brunner-Krainz M, Burlina A, Chakrapani A, Corpeleijn W, Cozens A, Dawson C, Dhamko H, Milosevic MD, Eiroa H, Finezilber Y, De Souza M, Garcia-Jiménez CF, Gasperini MC, Haas S, Häberle D, Halligan J, Fung R, Hörbe-Blindt LH, Horka A, Huemer LM, Uçar M, Kecman SK, Kilavuz B, Kriván S, Lindner G, Lüsebrink M, Makrilakis N, Mei-Kwun Kwok K, Maier A, Maiorana EM, McCandless A, Mitchell SE, Mizumoto JJ, Mundy H, Ochoa H, Pierce C, Fraile K, Regier PQ, Rossi D, Santer A, Schuman R, Sobieraj HC, Spenger P, Spiegel J, Stepien R, Tal KM, Tanšek G, Torkar MZ, Tchan AD, Thyagu M, Vergano SS, Vucko SA, Weinhold E, Zsidegh N, Wortmann P. Efficacy and safety of empagliflozin in glycogen storage disease type ib: data from an international questionnaire. Genet Sci. 2022;24:1781–8. https://doi.org/10.1016/j.gim.2022.04.001.
Sopher AB, Fennoy I, Oberfield SE. An update on childhood bone health: mineral accrual, assessment and treatment. Curr Opin Endocrinol Diabetes Obes. 2015;22:35–40. https://doi.org/10.1097/MED.0000000000000124.
Galindo-Zavala R, Bou-Torrent R, Magallares-López B, Mir-Perelló C, Palmou-Fontana N, Sevilla-Pérez B, Medrano-San Ildefonso M, González-Fernández MI, Román-Pascual A, Alcañiz-Rodríguez P, Nieto-Gonzalez JC, López-Corbeto M, Graña-Gil J. Expert panel consensus recommendations for diagnosis and treatment of secondary osteoporosis in children. Pediatr Rheumatol. 2020;18:20. https://doi.org/10.1186/s12969-020-0411-9.
Ward LM, Konji VN, Ma J. The management of osteoporosis in children. Osteoporos Int. 2016;27:2147–79. https://doi.org/10.1007/s00198-016-3515-9.
Lockwood DH, Merimee TJ, Edgar PJ, Greene ML, Fujimoto WY, Seegmiller JE, Howell RR. Insulin secretion in type I glycogen Storage Disease. Diabetes. 1969;18:755–8. https://doi.org/10.2337/diab.18.11.755.
Lee PJ, Patel A, Hindmarsh PC, Mowat AP, Leonard JV. The prevalence of polycystic ovaries in the hepatic glycogen storage diseases: its association with hyperinsulinism. Clin Endocrinol. 1995;42:601–6. https://doi.org/10.1111/j.1365-2265.1995.tb02686.x.
Melis D, Rossi A, Pivonello R, Salerno M, Balivo F, Spadarella S, Muscogiuri G, Casa RD, Formisano P, Andria G, Colao A, Parenti G. Glycogen storage disease type Ia (GSDIa) but not glycogen storage disease type ib (GSDIb) is associated to an increased risk of metabolic syndrome: possible role of microsomal glucose 6-phosphate accumulation. Orphanet J Rare Dis. 2015;10:91. https://doi.org/10.1186/s13023-015-0301-2.
Rossi A, Ruoppolo M, Formisano P, Villani G, Albano L, Gallo G, Crisci D, Moccia A, Parenti G, Strisciuglio P, Melis D. Insulin-resistance in glycogen storage disease type Ia: linking carbohydrates and mitochondria? J Inher Metab Disea. 2018;41:985–95. https://doi.org/10.1007/s10545-018-0149-4.
Marcalo J, Oliveira A, Nunes PA, Do Vale S. Type la glycogen storage disease complicated with diabetes mellitus: the role of flash continuous glucose monitoring. BMJ Case Rep. 2021;14:e240489. https://doi.org/10.1136/bcr-2020-240489.
Sun Y, Qiang W, Wu R, Yin T, Yuan J, Yuan J, Gu Y. A glycogen storage disease type 1a patient with type 2 diabetes. BMC Med Genomics. 2022;15:205. https://doi.org/10.1186/s12920-022-01344-3.
Cohn A, Ohri A. Diabetes mellitus in a patient with glycogen storage disease type Ia: a case report. J Med Case Rep. 2017;11:319. https://doi.org/10.1186/s13256-017-1462-5.
Sechi A, Ellerton C, Peters C, Gissen P, Mundy H, Lachmann R, Murphy E. Insulin resistance and diabetes in glycogen storage disease: presentation of 5 clinical cases. J Inherit Metab Dis. 2012;35:S77–77.
Aggarwal A, Patel D, Kulshreshtha B. Secondary diabetes as a rare complication of glycogen storage disease 1a: case report and review of literature. pedm. 2021;27:283–6. https://doi.org/10.5114/pedm.2021.109121.
Kumar K, Sachdev P, Randell T, Denvir L. Diabetes Mellitus a late complication in glycogen Storage Disease Type 1b |. ESPE Abstracts (2014) 82 P-D-3-2-717.
Spiegel R, Rakover-Tenenbaum Y, Mandel H, Lumelski D, Admoni O, Horovitz Y. Secondary diabetes Mellitus: late complication of glycogen Storage Disease Type 1b. J Pediatr Endocrinol Metab. 2005;18. https://doi.org/10.1515/JPEM.2005.18.6.617.
Moe PJ, Waaler PE, Garatun-Tjeldstø O, GLYCOGEN STORAGE DISEASE TYPE III AND DIABETES MELLITUS. Acta Paediatrica. 61, 483–486 (1972). https://doi.org/10.1111/j.1651-2227.1972.tb15869.x.
Spengos K, Michelakakis H, Vontzalidis A, Zouvelou V, Manta P. Diabetes mellitus associated with glycogen storage disease type III. Muscle Nerve. 2009;39:876–7. https://doi.org/10.1002/mus.21201.
Oki Y, Okubo M, Tanaka S, Nakanishi K, Kobayashi T, Murase T. Diabetes mellitus secondary to glycogen storage disease type III. Diabet Med. 2000;17:810–2. https://doi.org/10.1046/j.1464-5491.2000.00378.x.
Ismail H. Glycogen storage disease type III presenting with secondary diabetes and managed with insulin: a case report. Cases J. 2009. https://doi.org/10.4076/1757-1627-2-6891.
Okada S, Seino Y, Kodama H, Yutaka T, Inui K, Ishida M, Yabuuchi H, Seino Y. Insulin and glucagon secretion in hepatic glycogenoses. Acta Paediatr. 1979;68:735–8. https://doi.org/10.1111/j.1651-2227.1979.tb18448.x.
Wolfsdorf JI, Crigler JF. Biochemical evidence for the requirement of continuous glucose therapy in young adults with type 1 glycogen storage disease. J Inher Metab Disea. 1994;17:234–41. https://doi.org/10.1007/BF00711624.
Cohen JL, Vinik A, Faller J, Fox IH. Hyperuricemia in glycogen storage disease type I. contributions by hypoglycemia and hyperglucagonemia to increased urate production. J Clin Invest. 1985;75:251–7. https://doi.org/10.1172/JCI111681.
Taha D, Al-Harbi N, Al-Sabban E. Hyperglycemia and hypoinsulinemia in patients with Fanconi-Bickel syndrome. J Pediatr Endocrinol Metab. 2008;21:581–6.
Khandelwal P, Sinha A, Jain V, Houghton J, Hari P, Bagga A. Fanconi syndrome and neonatal diabetes: phenotypic heterogeneity in patients with GLUT2 defects. CEN Case Rep. 2018;7:1–4. https://doi.org/10.1007/s13730-017-0278-x.
Yoo H-W, Shin Y-L, Seo E-J, Kim G-H. Identification of a novel mutation in the GLUT2 gene in a patient with Fanconi-Bickel syndrome presenting with neonatal diabetes mellitus and galactosaemia. Eur J Pediatr. 2002;161:351–3. https://doi.org/10.1007/s00431-002-0931-y.
Setoodeh A, Rabbani A. Transient neonatal diabetes as a presentation of Fanconi- Bickel Syndrome. Acta Med Iran. 2012;50:836–8.
Sansbury FH, Flanagan SE, Houghton JAL, Shuixian Shen FL, Al-Senani AMS, Habeb AM, Abdullah M, Kariminejad A, Ellard S, Hattersley AT. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia. 2012;55:2381–5. https://doi.org/10.1007/s00125-012-2595-0.
Habeb AM, Al-Magamsi MS, Eid IM, Ali MI, Hattersley AT, Hussain K, Ellard S. Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatr Diabetes. 2012;13:499–505. https://doi.org/10.1111/j.1399-5448.2011.00828.x.
Dahlberg KR, Ferrecchia IA, Dambska-Williams M, Resler TE, Ross KM, Butler GL, Kuo C, Ryan PT, Weinstein DA. Cornstarch requirements of the adult glycogen storage disease Ia population: a retrospective review. J Inher Metab Disea. 2020;43:269–78. https://doi.org/10.1002/jimd.12160.
Lundqvist MH, Pereira MJ, Almby K, Hetty S, Eriksson JW. Regulation of the Cortisol Axis, Glucagon, and growth hormone by glucose is altered in Prediabetes and Type 2 diabetes. J Clin Endocrinol Metabolism. 2024;109:e675–88. https://doi.org/10.1210/clinem/dgad549.
Farah BL, Sinha RA, Wu Y, Singh BK, Lim A, Hirayama M, Landau DJ, Bay BH, Koeberl DD, Yen PM. Hepatic mitochondrial dysfunction is a feature of glycogen Storage Disease Type Ia (GSDIa). Sci Rep. 2017;7:44408. https://doi.org/10.1038/srep44408.
Rossi A, Assunto A, Rosano C, Tucci S, Ruoppolo M, Caterino M, Pirozzi F, Strisciuglio P, Parenti G, Melis D. Mitochondrial reprogramming in peripheral blood mononuclear cells of patients with glycogen storage disease type Ia. Genes Nutr. 2023;18:10. https://doi.org/10.1186/s12263-023-00729-y.
Katayama D, Baba H, Kuwabara T, Kido J, Mitsubuchi H, Matsumoto S, Nakamura K. SGLT2 inhibition alleviated hyperglycemia, glucose intolerance, and dumping syndrome-like symptoms in a patient with glycogen storage disease type Ia: a case report. J Med Case Rep. 2021;15:75. https://doi.org/10.1186/s13256-020-02658-5.
Hijazi G, Paschall A, Young SP, Smith B, Case LE, Boggs T, Amarasekara S, Austin SL, Pendyal S, El-Gharbawy A, Deak KL, Muir AJ, Kishnani PS. A retrospective longitudinal study and comprehensive review of adult patients with glycogen storage disease type III. Mol Genet Metabolism Rep. 2021;29:100821. https://doi.org/10.1016/j.ymgmr.2021.100821.
Slonim AE, Coleman RA, Moses S, Bashan N, Shipp E, Mushlin P. Amino acid disturbances in type III glycogenosis: differences from type I glycogenosis. Metabolism. 1983;32:70–4. https://doi.org/10.1016/0026-0495(83)90159-2.
Aynsley-Green A, Williamson DH, Gitzelmann R. Hepatic glycogen synthetase deficiency. Definition of syndrome from metabolic and enzyme studies on a 9-year-old girl. Arch Dis Child. 1977;52:573–9. https://doi.org/10.1136/adc.52.7.573.
Sharari S, Kabeer B, Mohammed I, Haris B, Pavlovski I, Hawari I, Bhat AA, Toufiq M, Tomei S, Mathew R, Syed N, Nisar S, Maacha S, Grivel J-C, Chaussabel D, Ericsson J, Hussain K. Understanding the role of GLUT2 in Dysglycemia Associated with Fanconi-Bickel Syndrome. Biomedicines. 2022;10:2114. https://doi.org/10.3390/biomedicines10092114.
Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia. 2015;58:221–32. https://doi.org/10.1007/s00125-014-3451-1.
Ferrecchia IA, Guenette G, Potocik EA, Weinstein DA. Pregnancy in Women with Glycogen Storage Disease Ia and ib. J Perinat Neonatal Nurs. 2014;28:26–31. https://doi.org/10.1097/JPN.0000000000000017.
Bachrach BE, Weinstein DA, Orho-Melander M, Burgess A, Wolfsdorf JI. Glycogen synthase deficiency (glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: report of three new mutations. J Pediatr. 2002;140:781–3. https://doi.org/10.1067/mpd.2002.124317.
Herbert M, Pendyal S, Rairikar M, Halaby C, Benjamin RW, Kishnani PS. Role of continuous glucose monitoring in the management of glycogen storage disorders. J Inherit Metab Dis. 2018;41(6):917–27. https://doi.org/10.1007/s10545-018-0200-5.
White FJ, Jones SA. The use of continuous glucose monitoring in the practical management of glycogen storage disorders. J Inher Metab Disea. 2011;34:631–42. https://doi.org/10.1007/s10545-011-9335-3.
Peeks F, Hoogeveen IJ, Feldbrugge RL, Burghard R, De Boer F, Fokkert-Wilts MJ, Van Der Klauw MM, Oosterveer MH, Derks TGJ. A retrospective in‐depth analysis of continuous glucose monitoring datasets for patients with hepatic glycogen storage disease: recommended outcome parameters for glucose management. J Inher Metab Disea. 2021;44:1136–50. https://doi.org/10.1002/jimd.12383.
Maran A, Crepaldi C, Avogaro A, Catuogno S, Burlina A, Poscia A, Tiengo A. Continuous glucose monitoring in conditions other than diabetes. Diabetes Metab Res Rev. 2004;20(Suppl 2):50–5. https://doi.org/10.1002/dmrr.518.
Kasapkara ÇS, Aycan Z, Açoğlu E, Senel S, Oguz MM, Ceylaner S. The variable clinical phenotype of three patients with hepatic glycogen synthase deficiency. J Pediatr Endocrinol Metab. 2017;30. https://doi.org/10.1515/jpem-2016-0317.
Rossi A, Venema A, Haarsma P, Feldbrugge L, Burghard R, Rodriguez-Buritica D, Parenti G, Oosterveer MH, Derks TGJ. A prospective study on continuous glucose monitoring in glycogen Storage Disease Type Ia: toward glycemic targets. J Clin Endocrinol Metabolism. 2022;107:e3612–23. https://doi.org/10.1210/clinem/dgac411.
Kaiser N, Gautschi M, Bosanska L, Meienberg F, Baumgartner MR, Spinas GA, Hochuli M. Glycemic control and complications in glycogen storage disease type I: results from the Swiss registry. Mol Genet Metab. 2019;126:355–61. https://doi.org/10.1016/j.ymgme.2019.02.008.
Mayorandan S, Meyer U, Hartmann H, Das AM. Glycogen storage disease type III: modified Atkins diet improves myopathy. Orphanet J Rare Dis. 2014;9:196. https://doi.org/10.1186/s13023-014-0196-3.
Kumru Akin B, Ozturk Hismi B, Daly A. Improvement in hypertrophic cardiomyopathy after using a high-fat, high-protein and low-carbohydrate diet in a non-adherent child with glycogen storage disease type IIIa. Mol Genet Metabolism Rep. 2022;32:100904. https://doi.org/10.1016/j.ymgmr.2022.100904.
Fernandes J, The effect of disaccharides on the hyperlactacidaemia of glucose-6-phosphatase-deficient children. Acta Paediatr. 1974;63:695–8. https://doi.org/10.1111/j.1651-2227.1974.tb16992.x.
Ben Salem C, Fathallah N, Hmouda H, Bouraoui K. Drug-Induced Hypoglycaemia: an update. Drug Saf. 2011;34:21–45. https://doi.org/10.2165/11538290-000000000-00000.
Grünert SC, Rosenbaum-Fabian S, Schumann A, Selbitz A, Merz W, Gieselmann A, Spiekerkoetter U. Two successful pregnancies and first use of empagliflozin during pregnancy in glycogen storage disease type ib. JIMD Rep. 2022;63:303–8. https://doi.org/10.1002/jmd2.12295.
Grünert SC, Derks TGJ, Mundy H, Dalton RN, Donadieu J, Hofbauer P, Jones N, Uçar SK, LaFreniere J, Contreras EL, Pendyal S, Rossi A, Schneider B, Spiegel R, Stepien KM, Wesol-Kucharska D, Veiga-da-Cunha M, Wortmann SB. Treatment recommendations for glycogen storage disease type IB- associated neutropenia and neutrophil dysfunction with empagliflozin: Consensus from an international workshop. Molecular Genetics and Metabolism. 141, 108144 (2024). https://doi.org/10.1016/j.ymgme.2024.108144.
Mikami M, Arai A, Mizumoto H. Empagliflozin ameliorated neutropenia in a girl with glycogen storage disease ib. Pediatr Int. 2021;63:1394–6. https://doi.org/10.1111/ped.14629.
Halligan RK, Dalton RN, Turner C, Lewis KA, Mundy HR. Understanding the role of SGLT2 inhibitors in glycogen storage disease type ib: the experience of one UK centre. Orphanet J Rare Dis. 2022;17:195. https://doi.org/10.1186/s13023-022-02345-2.
Hexner-Erlichman Z, Veiga-da-Cunha M, Zehavi Y, Vadasz Z, Sabag AD, Tatour S, Spiegel R. Favorable outcome of empagliflozin treatment in two pediatric glycogen storage disease type 1b patients. Front Pediatr. 2022;10:1071464. https://doi.org/10.3389/fped.2022.1071464.
Trepiccione F, Iervolino A, D’Acierno M, Siccardi S, Costanzo V, Sardella D, De La Motte LR, D’Apolito L, Miele A, Perna AF, Capolongo G, Zacchia M, Frische S, Nielsen R, Staiano L, Sambri I, De Cegli R, Unwin R, Eladari D, Capasso G. The SGLT2 inhibitor dapagliflozin improves kidney function in glycogen storage disease XI. Sci Transl Med. 2023;15:eabn4214. https://doi.org/10.1126/scitranslmed.abn4214.
Juneja D, Nasa P, Jain R, Singh O. Sodium-glucose Cotransporter-2 inhibitors induced euglycemic diabetic ketoacidosis: a meta summary of case reports. World J Diabetes. 2023;14:1314–22. https://doi.org/10.4239/wjd.v14.i8.1314.
La Rose AM, Bazioti V, Hoogerland JA, Svendsen AF, Groenen AG, Van Faassen M, Rutten MGS, Kloosterhuis NJ, Dethmers-Ausema B, Nijland JH, Mithieux G, Rajas F, Kuipers F, Lukens MV, Soehnlein O, Oosterveer MH, Westerterp M. Hepatocyte-specific glucose-6-phosphatase deficiency disturbs platelet aggregation and decreases blood monocytes upon fasting-induced hypoglycemia. Mol Metabolism. 2021;53:101265. https://doi.org/10.1016/j.molmet.2021.101265.
Stefater MA, Wolfsdorf JI, Ma NS, Majzoub JA. Glycogen storage disease presenting as Cushing syndrome. JIMD Rep. 2019;47:17–22. https://doi.org/10.1002/jmd2.12031.
Walker EA, Ahmed A, Lavery GG, Tomlinson JW, Kim SY, Cooper MS, Ride JP, Hughes BA, Shackleton CHL, McKiernan P, Elias E, Chou JY, Stewart PM. 11β-Hydroxysteroid dehydrogenase type 1 regulation by intracellular glucose 6-Phosphate provides evidence for a Novel link between glucose metabolism and hypothalamo-pituitary-adrenal Axis function. J Biol Chem. 2007;282:27030–6. https://doi.org/10.1074/jbc.M704144200.
White PC, Rogoff D, McMillan DR. Physiological roles of 11 beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase. Curr Opin Pediatr. 2008;20(4):453–7. https://doi.org/10.1097/MOP.0b013e328305e439.
Austin SL, El-Gharbawy AH, Kasturi VG, James A, Kishnani PS. Menorrhagia in patients with type I glycogen Storage Disease. Obstet Gynecol. 2013;122:1246–54. https://doi.org/10.1097/01.AOG.0000435451.86108.82.
Sechi A, Deroma L, Lapolla A, Paci S, Melis D, Burlina A, Carubbi F, Rigoldi M, Di Rocco M. Fertility and pregnancy in women affected by glycogen storage disease type I, results of a multicenter Italian study. J Inher Metab Disea. 2013;36:83–9. https://doi.org/10.1007/s10545-012-9490-1.
Wolfsdorf JI, Weinstein DA. Glycogen storage diseases. Rev Endocr Metab Disord. 2003;4:95–102. https://doi.org/10.1023/a:1021831621210.
Traggiai C, Stanhope R. Delayed puberty. Best Pract Res Clin Endocrinol Metab. 2002;16:139–51. https://doi.org/10.1053/beem.2001.0186.
Hou J-W, Wang T-R. A 20-year follow-up of a male patient with type Ia glycogen storage disease. Chang Gung Med J. 2003;26:283–7.
Wang W, Yu R, Tan W, Dan Y, Deng G, Xia J. A patient with glycogen storage disease type Ia combined with chronic hepatitis B infection: a case report. BMC Med Genet. 2019;20:85. https://doi.org/10.1186/s12881-019-0816-9.
Gore AC, Attardi B, DeFranco DB. Glucocorticoid repression of the reproductive axis: effects on GnRH and gonadotropin subunit mRNA levels. Mol Cell Endocrinol. 2006;256:40–8. https://doi.org/10.1016/j.mce.2006.06.002.
Simons PIHG, Valkenburg O, van der Telgenkamp I, de Groot DM, Veeraiah P, Bons JaP, Derks TGJ, Schalkwijk CG, Schrauwen-Hinderling VB, Stehouwer CDA, Brouwers MCGJ. Serum sex hormone-binding globulin levels are reduced and inversely associated with intrahepatic lipid content and saturated fatty acid fraction in adult patients with glycogen storage disease type 1a. J Endocrinol Invest. 2022;45:1227–34. https://doi.org/10.1007/s40618-022-01753-2.
Arnaldi G, Martino M. Androgens in Cushing’s syndrome. Front Horm Res. 2019;53:77–91. https://doi.org/10.1159/000494904.
Teede HJ, Misso ML, Costello MF, Dokras A, Laven J, Moran L, Piltonen T, Norman RJ, International PCOS, Network, Andersen M, Azziz R, Balen A, Baye E, Boyle J, Brennan L, Broekmans F, Dabadghao P, Devoto L, Dewailly D, Downes L, Fauser B, Franks S, Garad RM, Gibson-Helm M, Harrison C, Hart R, Hawkes R, Hirschberg A, Hoeger K, Hohmann F, Hutchison S, Joham A, Johnson L, Jordan C, Kulkarni J, Legro RS, Li R, Lujan M, Malhotra J, Mansfield D, Marsh K, McAllister V, Mocanu E, Mol BW, Ng E, Oberfield S, Ottey S, Peña A, Qiao J, Redman L, Rodgers R, Rombauts L, Romualdi D, Shah D, Speight J, Spritzer PM, Stener-Victorin E, Stepto N, Tapanainen JS, Tassone EC, Thangaratinam S, Thondan M, Van Der Tzeng C-R, Vanky E, Vogiatzi M, Wan A, Wijeyaratne C, Witchel S, Woolcock J, Yildiz BO. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome†‡. Hum Reprod. 2018;33:1602–18. https://doi.org/10.1093/humrep/dey256.
Oertelt-Prigione S. Immunology and the menstrual cycle. Autoimmun rev. 2012;11:A486–92. https://doi.org/10.1016/j.autrev.2011.11.023.
Wei C, Crowne EC. Recent advances in the understanding and management of delayed puberty. Arch Dis Child. 2016;101:481–8. https://doi.org/10.1136/archdischild-2014-307963.
Giannitrapani L, Soresi M, La Spada E, Cervello M, D’Alessandro N, Montalto G. Sex hormones and risk of Liver Tumor. Ann N Y Acad Sci. 2006;1089:228–36. https://doi.org/10.1196/annals.1386.044.
Marjoribanks J, Farquhar C, Roberts H, Lethaby A, Lee J. Long-term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database of Systematic Reviews. 2017, (2017). https://doi.org/10.1002/14651858.CD004143.pub5.
Baillargeon J, Urban RJ, Morgentaler A, Glueck CJ, Baillargeon G, Sharma G, Kuo Y-F. Risk of Venous Thromboembolism in Men Receiving Testosterone Therapy. Mayo Clinic Proceedings. 90, 1038–1045 (2015). https://doi.org/10.1016/j.mayocp.2015.05.012.
Corona G, Maseroli E, Rastrelli G, Isidori AM, Sforza A, Mannucci E, Maggi M. Cardiovascular risk associated with testosterone-boosting medications: a systematic review and meta-analysis. Exp Opin Drug Saf. 2014;13:1327–51. https://doi.org/10.1517/14740338.2014.950653.
Borst SE, Shuster JJ, Zou B, Ye F, Jia H, Wokhlu A, Yarrow JF. Cardiovascular risks and elevation of serum DHT vary by route of testosterone administration: a systematic review and meta-analysis. BMC Med. 2014;12:211. https://doi.org/10.1186/s12916-014-0211-5.
Mairovitz V, Labrune P, Fernandez H, Audibert F, Frydman R. Contraception and pregnancy in women affected by glycogen storage diseases. Eur J Pediatr. 2002;161(1):97–101. https://doi.org/10.1007/s00431-002-1013-x.
Grünert SC, Rosenbaum-Fabian S, Hannibal L, Schumann A, Spiekerkötter U. Three successful pregnancies in a patient with glycogen storage disease type 0. JIMD Rep. 2021;57:38–43. https://doi.org/10.1002/jmd2.12178.
Dagli AI, Lee PJ, Correia CE, Rodriguez C, Bhattacharya K, Steinkrauss L, Stanley CA, Weinstein DA. Pregnancy in glycogen storage disease type ib: gestational care and report of first successful deliveries. J Inher Metab Disea. 2010;33:151–7. https://doi.org/10.1007/s10545-010-9054-1.
Farber M, Knuppel RA, Binkiewicz A, Kennison RD. Pregnancy and Von Gierke’s disease. Obstet Gynecol. 1976;47:226–8.
Johnson MP, Compton A, Drugan A, Evans MI. Metabolic control of Von Gierke disease (glycogen storage disease type Ia) in pregnancy: maintenance of euglycemia with cornstarch. Obstet Gynecol. 1990;75:507–10.
Jones AM, Tower C, Green D, Stepien KM. Multidisciplinary management of pregnancy and labour in a patient with glycogen storage disease type 1a. BMJ Case Rep. 2021;14:e241161. https://doi.org/10.1136/bcr-2020-241161.
Ryan IP, Havel RJ, Laros RK. Three consecutive pregnancies in a patient with glycogen storage disease type IA (Von Gierke’s disease). Am J Obstet Gynecol 170, 1687–90; discussion 1690–1691 (1994).
Beneru D, Tchan MC, Billmore K, Nayyar R. Glycogen storage disease type IIIa in pregnant women: a guide to management. JIMD Rep. 2022;63:216–20. https://doi.org/10.1002/jmd2.12282.
Bhatti S, Parry E, Aust. NZ J Obst Gynaeco. 2006;46:168–9. https://doi.org/10.1111/j.1479-828X.2006.00549.x.
Mendoza A, Fisher NC, Duckett J, McKiernan J, Preece MA, Green A, McKiernan PJ, Constantine G, Elias E. Successful pregnancy in a patient with type III glycogen storage disease managed with cornstarch supplements. BJOG. 1998;105:677–80. https://doi.org/10.1111/j.1471-0528.1998.tb10186.x.
Ramachandran R, Wedatilake Y, Coats C, Walker F, Elliott P, Lee PJ, Lachmann RH, Murphy E. Pregnancy and its management in women with GSD type III - a single centre experience. J Inher Metab Disea. 2012;35:245–51. https://doi.org/10.1007/s10545-011-9384-7.
Grünert SC, Rosenbaum-Fabian S, Hannibal L, Schumann A, Spiekerkoetter U. Successful pregnancy in a woman with glycogen storage disease type 6. Mol Genet Metabolism Rep. 2021;27:100770. https://doi.org/10.1016/j.ymgmr.2021.100770.
Pena L, Charrow J. Fanconi–Bickel syndrome: report of life history and successful pregnancy in an affected patient. Am J Med Genet Pt A. 2011;155:415–7. https://doi.org/10.1002/ajmg.a.33822.
Von Schnakenburg C, Santer R. Fanconi–Bickel syndrome and fertility. Am J Med Genet Pt A. 2011;155:2607–2607. https://doi.org/10.1002/ajmg.a.34202.
Fine RN. Growth in glycogen-storage disease type 1: evaluation of endocrine function. Am J Dis Child. 1969;117:169. https://doi.org/10.1001/archpedi.1969.02100030171009.
Willey K. An elusive role for glycosylation in the structure and function of reproductive hormones. Hum Reprod Update. 1999;5:330–55. https://doi.org/10.1093/humupd/5.4.330.
Arnaoutova I, Zhang L, Chen HD, Mansfield BC, Chou JY. Correction of metabolic abnormalities in a mouse model of glycogen storage disease type Ia by CRISPR/Cas9-based gene editing. Mol Ther. 2021;29(4):1602–10. https://doi.org/10.1016/j.ymthe.2020.12.027.
Derks TGJ, et al. Sustained efficacy and safety at week 52 and up to three years in adults with glycogen storage disease type Ia (GSDIa): results from a phase 1/2 clinical trial of DTX401, an AAV8-mediated, liver-directed gene therapy. J Inherit Metab Dis. 2022;45 S1:0141–8955. https://doi.org/10.1002/jimd.12536.
Venema A, Peeks F, Rossi A, Jager EA, Derks TGJ. Towards values-based healthcare for inherited metabolic disorders: an overview of current practices for persons with liver glycogen storage disease and fatty acid oxidation disorders. J Inherit Metab Dis. 2022;45:1018–27. https://doi.org/10.1002/jimd.12555.
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
AR, CS, CR, BB wrote the first version of the manuscript. RP, MS, PS, AC, GP, DM and TGJD critically reviewed the manuscript. AR drafted and wrote the final version of manuscript. All authors substantially contributed to the work and were involved in (a) conception and design of the study and/ or analysis and interpretation of data, and (b) revising the article critically for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rossi, A., Simeoli, C., Pivonello, R. et al. Endocrine involvement in hepatic glycogen storage diseases: pathophysiology and implications for care. Rev Endocr Metab Disord 25, 707–725 (2024). https://doi.org/10.1007/s11154-024-09880-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-024-09880-2