
Abstract
ABP 980 was developed as a biosimilar to trastuzumab, a monoclonal antibody targeting human epidermal growth factor receptor 2 (HER2), that is indicated for the treatment of HER2-positive metastatic breast cancer, early breast cancer (EBC), and metastatic gastric cancer. ABP 980 is approved in the United States, European Union, and Japan for all the indications of trastuzumab, based on the totality of evidence (TOE) gathered by the systematic step-wise accumulation of comparative analytical, preclinical, and clinical (pharmacokinetics [PK], efficacy, safety and immunogenicity) data for ABP 980 and trastuzumab reference product (RP). As a key first step of the ABP 980 biosimilar program, comprehensive analytical characterization of critical quality attributes established that ABP 980 is structurally and functionally similar to trastuzumab RP. Complementing these data, results of non-clinical pharmacology, toxicology, and toxicokinetic studies supported similarity between ABP 980 and trastuzumab RP. A randomized study in healthy subjects demonstrated clinical PK equivalence of ABP 980 relative to trastuzumab RP in these subjects. In the final clinical evaluation step, a randomized comparative study (LILAC) confirmed the lack of clinically meaningful differences between ABP 980 and trastuzumab RP in efficacy, safety, and immunogenicity in women with HER2-positive EBC in the neoadjuvant–adjuvant setting. Neoadjuvant EBC represented a sensitive homogenous population for biosimilar demonstrations, and the primary endpoint of pathologic complete response served as a sensitive surrogate endpoint. An important aspect of the LILAC study design is that it is the only study that evaluated the effect of switching from the trastuzumab RP to a trastuzumab biosimilar during the adjuvant phase. No new or unexpected safety signals emerged in the clinical evaluations, with the safety profile of ABP 980 consistent with that previously described for trastuzumab. Overall, the TOE data generated for ABP 980 support the conclusion that it is highly similar to trastuzumab RP, thus providing the scientific justification for extrapolation to all the approved indications of trastuzumab.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Biosimilars are a new therapeutic category that has potential to increase access to important biologic therapies while mitigating cost barriers. |
Trastuzumab reference product (RP) is an integral treatment component in the management of HER2-positive breast cancer and gastric cancer. |
ABP 980, a biosimilar for trastuzumab, represents an alternative treatment option for all the current indications of trastuzumab, based on the totality of evidence supporting clinical similarity in efficacy, safety, and immunogenicity, which is maintained after a single switch from trastuzumab RP to ABP 980. |
1 Introduction
ABP 980 is a biosimilar of trastuzumab (Herceptin®), a recombinant immunoglobulin G1 monoclonal antibody that targets human epidermal growth factor receptor 2 (HER2). Trastuzumab is approved for the treatment of HER2-overexpressing metastatic breast cancer (MBC), early breast cancer (EBC), and metastatic gastric cancer [1]. The HER family of transmembrane receptor tyrosine kinases is implicated in the regulation of several pathways involved in cell growth, proliferation, and survival; overexpression of HER2 results in activation of such cellular pathways, which ultimately confers cancer cells with a growth and survival advantage [2, 3]. Upon binding to the extracellular domain of HER2 overexpressed on cancer cells, trastuzumab mediates its effects, primarily via inhibition of receptor activation and suppression of subsequent downstream cellular effects. Secondary mechanisms of action (MOAs) include antibody-dependent cellular cytotoxicity (ADCC) and suppression of constitutive HER2 cleavage/shedding, although their relative contributions to the clinical efficacy and safety of trastuzumab are unclear [2, 3].
A biosimilar product is highly similar to a licensed biologic, i.e., its reference product (RP), and shows no clinically meaningful differences when compared to the RP in terms of safety, purity, and potency [4,5,6,7,8,9,10]. There is increasing interest in biosimilars as alternatives to their originator RP given their potential to improve patient access to important biologic treatments that are safe and efficacious. Several biologics, including monoclonal antibodies and growth factors, are approved or in development for the treatment and supportive care of cancer [10]. The impending expiration of patents for several biologics in oncology is expected to lead to an increased availability of biosimilars. Given the recent advent of and unfamiliarity with biosimilars, their adoption in clinical practice may be hindered by lack of understanding of terminology, evolving regulatory guidance, and uncertainty about how biosimilars may be prescribed and dispensed, creating the need for education among stakeholders regarding the process of biosimilar development, including their abbreviated clinical testing and their appropriate use.
ABP 980 has the same amino acid sequence as trastuzumab and, like trastuzumab, it binds to the extracellular domain of HER2, resulting in suppression of receptor activation and subsequent proliferation of HER2-overexpressing cells. ABP 980 has been developed for the same indications, dosages, and route of administration as trastuzumab. It is approved in the European Union (EU) and United States (US) (Kanjinti™ [trastuzumab]) for the treatment of adult patients with HER2-positive breast cancer [11, 12]. It is also indicated (in combination with capecitabine or 5-fluorouracil and cisplatin) for the treatment of adult patients with HER2-positive metastatic adenocarcinoma of the stomach or gastroesophageal junction who have not received prior anti-cancer treatment for their metastatic disease. ABP 980 is also approved in Japan {trastuzumab (genetical recombination) [trastuzumab biosimilar 2]} for treatment of HER2-overexpressing breast cancer and unresectable advanced or recurrent gastric cancer [13]. Other trastuzumab biosimilars approved in the US, EU, and/or Japan include Ogivri® (MYL-1401O; Mylan and Biocon), Trazimera™ (PF-05280014; Pfizer), Herzuma® (CT-P6; Celltrion), and Ontruzant® (SB3; Samsung Bioepis).
This review outlines the step-wise biosimilar development process used to generate the totality of evidence (TOE) for ABP 980, which led to its approval.
1.1 Step-Wise Development of Biosimilars and Approval Pathway
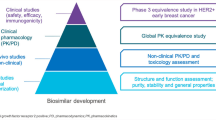
Unlike small-molecule generics that are chemically defined and identical to the originator RP, biosimilars are not identical to their RPs; this is likely due to the unavoidable minor structural and functional differences that arise as a result of the development of a unique cell line and complex manufacturing processes. These differences are deemed acceptable if they do not impact the efficacy, safety, purity, and potency of the biosimilar; therefore, more extensive scientific rigor is applied for a biosimilar than for small-molecule generics. Biosimilars development is focused towards establishing no clinically meaningful differences between the proposed biosimilar and the originator RP. The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) guidelines describing non-clinical and clinical considerations for the development of biosimilars recommend a step-wise TOE approach that begins with comprehensive analytical characterization, which forms the foundation of biosimilar development. This is followed by preclinical assessments and clinical pharmacology (pharmacokinetics [PK] and pharmacodynamics [PD]) evaluations and, finally, comparative clinical evaluation of efficacy, safety, and immunogenicity between the proposed biosimilar and the RP (Fig. 1) [4,5,6,7,8,9,10].

ABP 980 development program: stepwise “totality-of-evidence” approach for demonstration of biosimilarity to trastuzumab RP [modified from Markus R, McBride HJ, Ramchandani M, et al. A review of the totality of evidence supporting the development of the first adalimumab biosimilar ABP 501. Adv Ther. 2019;36(8):1833–1850]. HER2+ human epidermal growth factor receptor 2 positive, PD pharmacodynamics, PK pharmacokinetics, RP reference product
It is important to note that regulatory agencies require that a proposed biosimilar must be compared with RP that is locally sourced. Thus, for approval in the US, the proposed biosimilar must be shown to be similar to the RP approved in the US, and for approval in the EU, it must be shown to be similar to the RP approved in the European Economic Area [4, 9]. From a regulatory perspective, acceptability of clinical data generated utilizing a foreign-sourced comparator is contingent upon successful establishment of a “scientific bridge” [4, 9]. The scientific bridge should demonstrate that the foreign-sourced RP is structurally and functionally similar to the locally sourced RP using comprehensive analytical similarity assessments and also demonstrate PK equivalence. However, in acknowledgement of the challenges and costs associated with conducting clinical trials using RP sourced from multiple regions, regulatory agencies in the US and EU have provisions for using foreign-sourced comparators in clinical studies if a scientific rationale or “scientific bridge” is established [4, 9]. According to the Japanese regulatory guidelines, the use of foreign-approved RPs is acceptable in clinical evaluations if Japanese patients are included in either a comparative PK or comparative clinical efficacy study [14,15,16].
2 Analytical Characterization
For the ABP 980 studies reviewed here, both US-approved and EU-authorized trastuzumab RPs were used in the comparative assessments. Following identification of the critical quality attributes that influence PK, efficacy, safety, and immunogenicity, a comprehensive analytical characterization (structural and functional) was conducted to evaluate these attributes using a range of complementary state-of-the-art analytical techniques, in order to identify any potential structural and functional differences between ABP 980 and trastuzumab RP [17, 18].
For structural characterization, key product quality attributes assessed included primary (e.g., intact mass, peptide sequence, and post-translational glycosylation) and higher-order protein structure (e.g., secondary and tertiary), product-related substances and impurities (e.g., size and charge variants), process-related impurities, particles and aggregates, physicochemical product stability before and after reconstitution, and general properties (e.g., protein content and concentration) [17, 18]. Using this systematic approach, the similarity assessments demonstrated that ABP 980 was highly analytically similar to trastuzumab RP, with minor differences that were not expected to alter biological activity or clinical performance.
Functionally, similarity assessments for ABP 980 and trastuzumab RP compared target binding, in vitro potency via disruption of HER2 ligand-independent cell proliferation, and effector functions (ADCC and antibody-dependent cellular phagocytosis [ADCP]) via their Fc binding properties. In these studies, ABP 980 and trastuzumab RP exhibited similar kinetic binding properties to HER2 and showed similar potency and ADCC activities in experimental cell-based systems [17, 19,20,21].
Trastuzumab is used clinically in combination with multiple chemotherapeutic agents; therefore, it was of interest to compare the relative synergies of ABP 980 and trastuzumab RP with docetaxel [22, 23]. In in vitro combination studies using gastric cancer cells, ABP 980 and trastuzumab RP exhibited similar synergistic activity in the presence and absence of docetaxel [19]. Specifically, the combination of docetaxel and ABP 980 in gastric cancer cells exhibited a robust and clinically relevant, synergistic effect [19]. Overall, functional assessments supported the conclusion of biosimilarity with respect to ABP 980 and trastuzumab RP.
3 In Vivo Pharmacology and Toxicokinetics
In non-clinical in vivo studies, the pharmacology and toxicokinetic (TK) similarity of ABP 980 was compared to trastuzumab RP [19, 21]. Pharmacology studies showed that ABP 980 and trastuzumab RP have similar dose-dependent antitumor activity in tumor xenograft models overexpressing HER2, using either breast or gastric cancer cells. Furthermore, TK assessments using a multiple-dose cynomolgus monkey toxicology study demonstrated similar in vivo exposure and dose accumulation between the ABP 980 and trastuzumab RP treatment groups [19]. Taken together, the in vivo pharmacology, toxicology, and TK studies conducted support the overall conclusion of similarity between ABP 980 and trastuzumab RP.
4 Clinical Pharmacology
The PK equivalence of ABP 980 relative to trastuzumab RP was demonstrated in healthy male subjects [24, 25]. In a randomized, single-blind, single-dose, three-arm, parallel-group PK study, a total of 157 subjects were randomized 1:1:1 to receive a single 6-mg/kg intravenous (IV) infusion of ABP 980 (n = 50) or trastuzumab RP (FDA-licensed, n = 52; EU-authorized, n = 55). Trastuzumab RP was acquired from both the EU and the US to complete the scientific bridge, as previously discussed. PK equivalence was considered to be established if the 90% confidence intervals (CIs) for the ratio of least square geometric means (GMs) of primary PK parameters of ABP 980 versus trastuzumab RP fell within the standard bioequivalence criteria of 0.80 and 1.25.
Mean serum concentration–time profiles were similar between ABP 980 and trastuzumab RP over the entire course of sampling [24]. The primary PK parameters area under the curve (AUC) from time 0 extrapolated to infinity (AUCinf) and the maximum observed concentration (Cmax) as well as the secondary PK parameter of AUC from time 0 to the last quantifiable concentration (AUClast) were similar across groups. The 90% CIs of the ratios of the GMs were fully contained within the prespecified bioequivalence criteria of 0.80–1.25, confirming the PK similarity of ABP 980 and trastuzumab RP (both EU and US).
The frequency of treatment-emergent adverse events (TEAEs) of all grades, regardless of causality, was comparable across the three groups [24]. The safety profiles of ABP 980 and trastuzumab RP were similar, including headache, upper respiratory tract infection, chills, pyrexia, myalgia, nausea, epistaxis, and arthralgia; these were consistent with adverse events (AEs) previously described for the trastuzumab RP. No TEAEs or serious adverse events (SAEs) related to the study drug led to study discontinuation; there were no SAEs that resulted in death. Immunogenicity assessments, conducted at baseline prior to dosing (day 1) and at the end of study (day 64) or upon early study discontinuation, found no pre-existing antidrug antibodies (ADAs) at baseline; no subjects developed ADAs during the study.
5 Clinical Evaluation
Considering the demonstrated high similarity of ABP 980 to trastuzumab RP with respect to structure, function, and PK, no clinically meaningful differences between these agents in efficacy, safety, or immunogenicity were expected. In order to confirm the clinical similarity of ABP 980 to trastuzumab RP, a clinical study, the LILAC study, was conducted to compare the treatment effect of ABP 980 with trastuzumab RP regarding pathologic complete response (pCR) in women with HER2+ EBC [26].
In biosimilar clinical studies, it is critical to choose sensitive patient populations and robust study endpoints in order to confirm there are no clinically meaningful differences versus the RP [7, 27]. Accordingly, in the LILAC study, EBC was selected as a sensitive patient population to demonstrate clinical equivalence of ABP 980 versus trastuzumab RP. Clinical equivalence testing in EBC allows for a careful and thorough biosimilarity assessment. In contrast, MBC can be considered a less sensitive and less homogenous population since prior treatment, line of therapy, disease burden and comorbidities, and risk of secondary cancers may potentially confound the results [27, 28]. Moreover, the metastatic setting does not allow comprehensive immunogenicity characterization, owing to the lack of a treatment-free follow-up phase and the immunosuppressive effects of common treatment modalities, such as chemotherapy and radiation [28, 29].
The pCR of both the breast tissue and axillary lymph nodes was chosen as a sensitive endpoint in the LILAC trial since it has been validated to be clinically meaningful in the neoadjuvant EBC setting. FDA guidelines indicate that pCR in the neoadjuvant setting and in patients with operable breast cancer is the preferred primary endpoint to support accelerated drug approval [27]. This recommendation was supported by results of a meta-analysis of 12 randomized, controlled trials in neoadjuvant EBC, which showed that patients who achieved a pCR had a more favorable long-term survival outcome in terms of event-free survival, although the analysis could not establish a trial-level correlation between pCR and long-term outcome [30]. While long-term outcome, e.g., overall survival, as the primary endpoint is regarded as the gold standard in oncology clinical trials for innovator molecules, it is not considered appropriate for biosimilar trials since it might be confounded by several factors, such as tumor burden, disease status, and previous and subsequent lines of therapy. The use of objective response rate (ORR) is not preferred since it does not correlate reliably with long-term efficacy outcomes [28, 29].
Taken together, demonstration of clinical similarity between the trastuzumab RP and its biosimilar in the neoadjuvant–adjuvant setting using pCR as the primary endpoint is preferred. This also represents a more appropriate approach for extrapolation to other indications, such as metastatic disease [29]. Therefore, the LILAC study was conducted in the more homogeneous neoadjuvant setting using a sensitive population and endpoint, making this an ideal study design for evaluating a biosimilar for use in breast cancer.
The LILAC study was a randomized, multicenter, double-blind, active-controlled, two-phase, equivalence trial conducted in both the neoadjuvant and adjuvant settings, with a single-switch transition phase design in the adjuvant phase [26]. Eligible patients were randomized to one of three treatment arms: ABP 980, trastuzumab RP, or neoadjuvant trastuzumab RP followed by adjuvant ABP 980. Prior to randomization, patients received 12-week run-in chemotherapy consisting of epirubicin plus cyclophosphamide every 3 weeks for four cycles. Patients then received ABP 980 or trastuzumab RP, at a loading dose of 8 mg/kg plus paclitaxel 175 mg/m2 in a 90-min IV infusion, followed by three cycles of 6 mg/kg IV ABP 980 or trastuzumab RP plus paclitaxel 175 mg/m2 every 3 weeks in 30-min IV infusions (or 80 mg/m2 paclitaxel once per week for 12 cycles if that was the local standard of care) [26]. Surgery was performed 3–7 weeks after the last dose of neoadjuvant therapy. During the adjuvant treatment phase, patients either continued ABP 980 or trastuzumab RP (6 mg/kg), or switched from trastuzumab RP to ABP 980 every 3 weeks for up to 1 year after the first dose in the study.
The co-primary efficacy endpoints of the LILAC trial were risk difference (ABP 980 minus trastuzumab RP) and risk ratio (RR) (ABP 980 vs trastuzumab RP) of pCR, which was defined as the absence of invasive tumor cells in the breast tissue and in axillary lymph nodes regardless of ductal carcinoma in situ. This was the first multicenter neoadjuvant breast cancer study that incorporated an independent central pathology review of pCR (sensitivity analysis) in addition to the local pathology review (primary analysis); both were conducted in all patients who were randomized and received neoadjuvant treatment followed by surgery.
A total of 827 patients were enrolled in the trial; 725 patients were randomized to receive ABP 980 (n = 364) or trastuzumab RP (n = 361) [26]. By protocol-defined criteria, similarity between ABP 980 and trastuzumab RP was established if the two-sided 90% CI for the risk difference between the ABP 980 and trastuzumab RP groups was within the statistical equivalence margins of − 13% and 13%. If the risk difference was successful, similarity was then tested by the RR of pCR by comparing the two-sided 90% CI between the ABP 980 and trastuzumab groups, with statistical margins of 0.759 and 1.318.
The primary analysis (local pathology review) found that a higher proportion of patients treated with ABP 980 achieved pCR compared to those treated with trastuzumab RP (48% [95% CI 43–53] vs 41% [95% CI 35–46]), with a risk difference of 7.3% (90% CI 1.2–13.4) and an RR of 1.188 (90% CI 1.033–1.366). It was concluded that the non-inferiority for the primary endpoint was met; however, non-superiority could not be excluded since the upper bounds of the 90% CIs for RR and risk difference exceeded the predefined equivalence margins (Fig. 2) [26]. However, sensitivity analyses from central laboratory review confirmed clinical similarity of ABP 980 and trastuzumab RP in terms of pCR (48% vs 42%; risk difference 5.8% [90% CI − 0.5 to 12.0]; RR 1.142 [90% CI 0.993–1.312]) (Fig. 2) [26].

Comparative clinical study results of ABP 980 vs trastuzumab RP in HER2 + EBC: total pCR [26]. CI confidence interval, EBC early breast cancer, HER2+ human epidermal growth factor receptor 2 positive, pCR pathologic complete response, RD risk difference, RP reference product, RR risk ratio
The safety analysis (including both neoadjuvant and adjuvant phases) found that the overall incidences of AEs, events of interest (EOIs), and SAEs were similar between the two treatment groups during both the neoadjuvant and adjuvant phases [26]. The AE profile was consistent with the historical safety profile of trastuzumab, with no emergence of new safety signals during the neoadjuvant or adjuvant phases, or with switching (Table 1) [26]. During the neoadjuvant phase that included combination of investigational product (IP) plus chemotherapy, infusion reaction was the most common EOI in both groups; while the most common EOIs during the adjuvant phase were infections and infestations, neutropenia, and infusion reactions. Not surprisingly, the incidence of AEs was higher in the neoadjuvant phase that was preceded by run-in chemotherapy and comprised of IP plus chemotherapy (paclitaxel).
Cardiac toxicity was an event of particular interest, given the accumulated evidence indicating increased risk of cardiac toxicity with trastuzumab treatment that may be attributable to previous anthracycline exposure [26]. Median left ventricular ejection fraction (LVEF) values did not change in any treatment group over the course of the study. For any time during the study, LVEF declines of ≥ 10% and < 50% occurred in 2.8% (ABP 980) to 3.5% (trastuzumab RP/ABP 980) of patients (Table 2) [26]. The frequency of cardiac disorders was low throughout the study, and none resulted in discontinuation of IP. Only seven patients experienced cardiac failure (all grade 1–2) in the neoadjuvant phase, and four patients experienced this in the adjuvant phase (one grade 3). Moreover, all 11 patients received the planned doses of IPs, which suggests resolution or no worsening of cardiac failure. Only one of the 11 patients with a cardiac failure AE also had clinically significant LVEF findings, which suggests very low cardiac toxicity in this study. While long-term follow-up (median 3.6 years) in a study with trastuzumab RP has shown that the incidence of congestive heart failure and left ventricle dysfunction remain low with trastuzumab treatment, because there is evidence of a possible delayed manifestation of cardiac events with anthracycline, long-term follow-up is still required [31]. In this regard, the similar incidence of events during therapy with ABP 980 and trastuzumab RP is reassuring.
Notably, the safety outcomes following single switch from trastuzumab RP to ABP 980 were similar in terms of incidences of AEs and SAEs or rate of discontinuation due to an AE; common AEs experienced by ≥ 5% of patients were radiation skin injury, neutropenia, arthralgia, asthenia, and anemia [26]. Also, switching from trastuzumab RP to ABP 980 was not associated with an increase in incidence of cardiotoxicity. It should also be noted that cardiac toxicity frequencies were comparable with those previously reported in the literature for trastuzumab [32].
Immunogenicity was similar in patients who received ABP 980 and those who received trastuzumab RP. A total of eight patients developed ADAs at any time during the entire course of the study: two each in the ABP 980 and trastuzumab RP groups, and four in the switch group [23]. No patients tested positive for neutralizing antibodies.
6 Extrapolation
Finally, it is important to understand the concept of extrapolation with respect to the development and approval of biosimilars. This concept is unique to biosimilars and reduces or eliminates the need for clinical studies in multiple indications. Extrapolation is based on the TOE and requires adequate scientific justification, which includes comprehensive studies designed to evaluate MOA as well as structural, functional, non-clinical, PK, and clinical similarity between the biosimilar and the RP, as have been conducted for ABP 980. Based on this, approval for use for ABP 980 was sought and received in all indications for which the trastuzumab RP is approved, without having to perform clinical trials in different disease settings and patient populations, thus enabling an accelerated clinical trial program. A shared MOA is key for the approval of biosimilars across different indications. For example, in HER2-positive breast cancer, the results of biosimilar clinical testing performed in the adjuvant or neoadjuvant setting may be used for extrapolation to MBC or HER2-positive metastatic gastric cancer. It must be emphasized that the scientific justification for extrapolation is based on the historical knowledge and understanding of the originator RP regarding any potential differences in PK, MOA/efficacy, safety or immunogenicity across its approved indications predicated on the conclusion of biosimilarity of the biosimilar candidate.
In the case of trastuzumab, the primary MOA, inhibition of ligand-independent HER2-mediated proliferation, is the same across all its approved indications. Additionally, the secondary MOA, ADCC, is also conserved. There are no differences in PK, safety, or immunogenicity across the approved indications of trastuzumab that would prevent extrapolation to unstudied indications; thus, we conclude that ABP 980 should have the same clinical performance across all approved indications of the RP.
7 Discussion
The TOE generated for ABP 980 from analytical, functional, non-clinical, PK, and clinical studies shows that ABP 980 is highly similar to trastuzumab RP. The results of the LILAC study demonstrated that there are no clinically meaningful differences between ABP 980 and trastuzumab RP with respect to efficacy (based on sensitivity analysis), safety, and immunogenicity. Importantly, no new or unexpected safety signals emerged throughout the study, with the safety profile consistent with that previously described for trastuzumab.
7.1 Clinical Evidence from Other Trastuzumab Biosimilars
Four other biosimilars (CT-P6, MYL-1401O, SB3, and PF-05280014) have been approved by the FDA and EMA and have been shown to be similar to trastuzumab RP in confirmatory clinical studies (Table 3) [26, 33,34,35,36,37,38]. While all four biosimilars were evaluated with the recommended equivalence trial design, the patient population and primary endpoints in these studies differed from those used in the ABP 980 studies. In these five trials, anthracycline/taxane-based chemotherapy was used in the neoadjuvant setting and taxane-based chemotherapy in the metastatic setting; no significant differences between the biosimilar trastuzumab and the trastuzumab RP were reported. The trials evaluating MYL-1401O, PF-05280014, and CT-P6 were conducted in HER2-overexpressing MBC; the primary endpoint in all three trials was ORR (the prespecified equivalence margin was ± 15%) [33, 35, 37]. As discussed earlier, MBC is a less sensitive and less homogenous population to demonstrate clinical equivalence of a biosimilar trastuzumab, and ORR is not the preferred primary endpoint since it is associated with a larger uncertainty in long-term efficacy outcomes. On the other hand, pCR is a direct and objective measure of antitumor activity that may be a potential surrogate for disease-free survival and overall survival in patients with HER2 + EBC treated with neoadjuvant chemotherapy and trastuzumab [39], and therefore, is considered sensitive for use in evaluating potential differences between trastuzumab biosimilar and RP. For these reasons, PF-05280014, and CT-P6 were subsequently evaluated in EBC, using pCR as the primary endpoint [34, 38]. The SB3 equivalence trial was conducted in the sensitive neoadjuvant EBC patient population using pCR as the primary endpoint; however, the SB3 trial evaluated pCR in breast only and did not evaluate lymph node or document ductal carcinoma in situ status [36]. Equivalence was declared in the SB3 trial if the 95% CI of the RR was within 0.785–1.546 or the 95% CI of the difference was within the lower and upper equivalence margin of ± 13% [36]. In the context of these data, it must be noted that the LILAC study was designed to evaluate equivalence in EBC and comprehensively assessed total pCR in both breast tissue and axillary lymph nodes, with an equivalence margin of ± 13%, which provides confidence in the demonstrated similarity between ABP 980 and the trastuzumab RP [26]. Moreover, the unique study design of the LILAC trial in terms of clinical assessments in both the neoadjuvant and adjuvant settings, as well as after switching from trastuzumab RP to ABP 980, allowed demonstration that ABP 980 may be used in each of these settings, which are of particular relevance for clinical practice. Taken together, ABP 980 has shown no significant differences compared to its RP in terms of efficacy, safety, and immunogenicity, thus, supporting its safe and effective use in place of trastuzumab RP, in all the approved indications of trastuzumab, i.e., EBC, MBC, and metastatic gastric cancer [40].
7.2 Other Considerations
It is thought that biosimilars may provide better overall value by offering additional treatment options. A few considerations that would help realize the value of biosimilars include quality, sustainability, and reliability of supply without disruptions or shortages [41]. Given the grave impact of oncology drug shortages, including increased burden of time to deal with the shortage and potentially increased drug costs and, in some cases, elevated risk of medication errors, a reliable supply of high-quality biosimilars is critical for pharmacy operations and delivery of patient care [42].
At this time, it is not known whether biosimilars will provide any cost savings in the US similar to what has been observed in Europe and other global regions [43]. However, it is expected that the expanded treatment choices provided by the availability of biosimilars may favorably impact the overall value-based delivery of care. Moreover, biosimilars might afford patients reduced out-of-pocket costs that might encourage medication adherence [41].
8 Conclusions
The TOE of ABP 980 supported the scientific justification for extrapolation across all approved indications of trastuzumab RP. The current data allow the use of ABP 980 as a biosimilar to trastuzumab RP, thus, providing an additional treatment option for patients with breast and gastric cancers.
References
Herceptin® (trastuzumab) prescribing information. Genentech; 2017.
Vu T, Claret FX. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Front Oncol. 2012;2:62.
Baselga J, Albanell J, Molina MA. Mechanism of action of trastuzumab and scientific update. Semin Oncol. 2001;28:4–11.
U.S. Department of Health and Human Services. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. 2015. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 7 Apr 2019.
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. Guidance for industry. 2015. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291134.pdf. Accessed 7 Apr 2019.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry: clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Guidance for industry. 2016. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Accessed 7 Apr 2019.
European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. Accessed 7 Apr 2019.
European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/06/WC500167838.pdf. Accessed 7 Apr 2019.
European Medicines Agency. Guideline on similar biological medicinal products (revision 1). 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf. Accessed 7 Apr 2019.
Markus R, Liu J, Ramchandani M, et al. Developing the totality of evidence for biosimilars: regulatory considerations and building confidence for the healthcare community. BioDrugs. 2017;31:175–87.
KANJINTI. Summary of product characteristics. Amgen Europe B.V., the Netherlands: 18 September 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/kanjinti#product-information-section. Accessed 8 July 2019.
KANJINTI (trastuzumab-anns) for injection, for intravenous use. Prescribing information. Amgen Inc., Thousand Oaks, CA: June 2019. https://www.pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/kanjinti/kanjinti_pi.ashx. Accessed 8 July 2019.
Generics and Biosimilars Initiative (GaBi) Online. Biosimilars approved in Japan: 14 December, 2018. http://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Japan. Accessed 8 July 2019.
Arato T. Japanese regulation of biosimilar products: past experience and current challenges. Br J Clin Pharmacol. 2016;82:30–40.
Kuribayashi R, Sawanobori K. Current Japanese regulatory systems for generics and biosimilars. J Pharm Sci. 2018;107:785–7.
Nagai S, Yanagihara R, Kishioka Y. Japanese regulatory authority’s perspective on biosimilars. Lancet Oncol. 2015;16(3):e101.
Hutterer K, Polozova A, Kuhns S, McBride H, Cao X, Liu J. Assessing analytical and functional similarity of proposed Amgen biosimilar ABP 980 to trastuzumab. BioDrugs. 2019;33:321–33.
Crampton S, Polozovab A, Asburyc D. Stability of ABP 980, a trastuzumab biosimilar, after reconstitution of lyophilized drug product. In: Presented at the 21st British Oncology Pharmacy Association (BOPA) annual symposium (2018). Birmingham, UK, October 12–14, 2018.
Jassem S, Wang W, Sweet H, et al. Functional and non-clinical similarity of ABP 980, a biosimilar of trastuzumab. Pharm Res. 2019 (manuscript accepted for publication).
Hanes V, Born T, Chow V, et al. Comparative similarity of ABP 980 and trastuzumab: results of functional similarity and human pharmacokinetic assessment. In: 2015 San Antonio Breast Cancer Conference: Abstract 3147.
Hanes V, Born T, Chow V, et al. Functional and human pharmacokinetic similarity of ABP 980 and trastuzumab. In: European Breast Cancer Conference; 2016: Abstract 3272.
Bullock K, Blackwell K. Clinical efficacy of taxane-trastuzumab combination regimens for HER-2-positive metastatic breast cancer. Oncologist. 2008;13:515–25.
Nahta R. Molecular mechanisms of trastuzumab-based treatment in HER2-overexpressing breast cancer. ISRN Oncol. 2012;2012:428062.
Hanes V, Chow V, Zhang N, Markus R. A randomized single-blind, single-dose study evaluating the pharmacokinetic equivalence of proposed biosimilar ABP 980 and trastuzumab in healthy male subjects. Cancer Chemother Pharmacol. 2017;79:881–8.
Hanes V, Chow V, Zhang N, et al. 2016 Pharmacokinetic similarity of ABP 980 relative to trastuzumab in the Japanese subset: results from a randomized, single-blind, single-dose, parallel group study in healthy subjects. Jpn Soc Clin Oncol. 2016: Abstract P25-1.
von Minckwitz G, Colleoni M, Kolberg H-C, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19:987–98.
US Food and Drug Administration. Guidance for industry—pathologic complete response in neoadjuvant treatment of high-risk early-stage breast cancer: use as an endpoint to support accelerated approval. 2014. https://www.fda.gov/downloads/drugs/guidances/ucm305501.pdf. Accessed 7 Apr 2019.
Cortés J, Curigliano G, Diéras V. Expert perspectives on biosimilar monoclonal antibodies in breast cancer. Breast Cancer Res Treat. 2014;144:233–9.
Jackisch C, Scappaticci FA, Heinzmann D, et al. Neoadjuvant breast cancer treatment as a sensitive setting for trastuzumab biosimilar development and extrapolation. Future Oncol. 2015;11:61–71.
Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384:164–72.
Procter M, Suter TM, de Azambuja E, et al. Longer-term assessment of trastuzumab-related cardiac adverse events in the Herceptin Adjuvant (HERA) trial. J Clin Oncol. 2010;28:3422–8.
Burki TK. Trastuzumab cardiotoxicity in early-stage breast cancer. Lancet Oncol. 2016;17(6):e226.
Im YH, Odarchenko P, Grecea D, et al. Double-blind, randomized, parallel group, phase III study to demonstrate equivalent efficacy and comparable safety of CT-P6 and trastuzumab, both in combination with paclitaxel, in patients with metastatic breast cancer (mBC) as first-line treatment. J Clin Oncol. 2013;31:629.
Stebbing J, Baranau Y, Baryash V, et al. CT-P6 compared with reference trastuzumab for HER2-positive breast cancer: a randomised, double-blind, active-controlled, phase 3 equivalence trial. Lancet Oncol. 2017;18:917–28.
Rugo HS, Barve A, Waller CF, et al. Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317:37–47.
Pivot XB, Bondarenko I, Dvorkin M, et al. Phase III, randomized, double-blind study comparing the efficacy, safety, and immunogenicity of SB3 (trastuzumab biosimilar) and reference trastuzumab in patients treated with neoadjuvant therapy for human epidermal growth factor receptor 2-positive early breast cancer. J Clin Oncol. 2018;36:968–74.
Pegram MD, Bondarenko I, Zorzetto MMC, et al. PF-05280014 (a trastuzumab biosimilar) plus paclitaxel compared with reference trastuzumab plus paclitaxel for HER2-positive metastatic breast cancer: a randomised, double-blind study. Br J Cancer. 2019;120:172–82.
Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2 + breast cancer [published online July 13, 2018]. Br J Cancer. 2018;119:266–73.
Gianni L, Eiermann W, Semiglazov V, et al. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet. 2010;375:377–84.
Thill M. Biosimilar trastuzumab in clinical trials: differences or not? Breast Care. 2019;14:17–22.
Smeeding J, Malone DC, Ramchandani M, et al. Considerations for payers. P&T. 2019;44:54–63.
Cournier D, Wong RS, Tiwana S, et al. A qualitative assessment of the impact of an IV cancer drug shortage on US pharmacy operations. ACCP 2019. Poster 4009.
Patel KB, Arantes LH, Tang WY, et al. The role of biosimilars in value-based oncology care. Cancer Manag Res. 2018;10:4591–602.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Sponsorship of ABP 980 studies described in this review and article processing charges were funded by Amgen Inc, Thousand Oaks, CA.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing Assistance
Medical writing assistance was provided by Sabby Muneer, PhD, from Innovation Communications Group, Inc, New York, NY, under the guidance of Monica Ramchandani, PhD, Amgen Inc.
Conflict of interest
Hans-Christian Kolberg has received honoraria from Carl Zeiss Meditec, TEVA, Theraclion, Novartis, Amgen, Astra Zeneca, Pfizer, Janssen-Cilag, GSK, LIV Pharma, Roche and Genomic Health and nonfinancial support from Carl Zeiss Meditec, Novartis, Pfizer, Amgen, Roche, LIV Pharma, Tesaro, Daiichi Sankyo, and Genomic Health. Marco Colleoni is a consultant for AstraZeneca, Celldex, Novartis, OBI Pharma, Pfizer, Pierre Fabre, and Puma Biotechnology, and is also an investigator on the ABP 980 LILAC study. Yasuhiro Fujiwara has received lecture fees from Astra Zeneca KK, Daiichi-Sankyo, Taiho, Chugai, and Novartis Pharma KK. Georgia Savva Demetriou is a consultant for Astra Zeneca and Celgene, and was on speaker bureaus for Merck and Serono. Patricia Santi was an investigator on the ABP 980 LILAC study and has no other conflicts to declare. Miguel Angel Segui-Palmer is a consultant for Pfizer, Novartis, Roche, Amgen, and MSD; has received research funding from Pfizer, Novartis, Lilly, Amgen, and Roche; and is a speaker for Pfizer, Novartis, Roche, and Amgen. Vladimir Hanes is an employee and stockholder of Amgen. Sara A. Hurvitz reports grants and nonfinancial support from Ambryx, Amgen, Bayer, OBI Pharma, BioMarin, Cascadian, Daiichi Sankyo, Dignitana, Genentech, GSK, Lilly, MacroGenics, Medivation, Merrimack, Novartis, OBI Pharma, Pfizer, Pieris, PUMA, Roche, and Seattle Genetics.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kolberg, HC., Colleoni, M., Santi, P. et al. Totality of Scientific Evidence in the Development of ABP 980, a Biosimilar to Trastuzumab. Targ Oncol 14, 647–656 (2019). https://doi.org/10.1007/s11523-019-00675-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-019-00675-z