
Abstract
Monogenic forms of Alzheimer’s disease (AD) have been identified through mutations in genes such as APP, PSEN1, and PSEN2, whilst other genetic markers such as the APOE ε carrier allele status have been shown to increase the likelihood of having the disease. Mutations in these genes are not limited to AD, as APP mutations can also cause an amyloid form of cerebral small vessel disease (CSVD) known as cerebral amyloid angiopathy, whilst PSEN1 and PSEN2 are involved in NOTCH3 signalling, a process known to be dysregulated in the monogenic CSVD, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). The overlap between AD genes and causes of CSVD led to the hypothesis that mutations in other genes within the PANTHER AD–presenilin pathway may be novel causes of CSVD in a cohort of clinically suspicious CADASIL patients without a pathogenic NOTCH3 mutation. To investigate this, whole exome sequencing was performed on 50 suspected CADASIL patients with no NOTCH3 mutations, and a targeted gene analysis was completed on the PANTHER. ERN1 was identified as a novel candidate CSVD gene following predicted pathogenic gene mutation analysis. Rare variant burden testing failed to identify an association with any gene; however, it did show a nominally significant link with ERN1 and TRPC3. This study provides evidence to support a genetic overlap between CSVD and Alzheimer’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
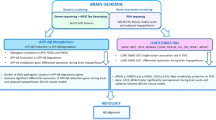
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is the most common monogenic form of cerebral small vessel disease (CSVD) [1]. It has been originally estimated to have a prevalence of 2–4 per 100,000; however, later studies of population data from the Genome Aggregation Database (gnomAD) estimate that it may be up to 1 in 300 [2, 3]. CADASIL predominantly affects the central nervous system (CNS) and symptoms include recurrent ischaemic events, migraine, mood disturbances, progressive cognitive decline/vascular dementia, and sometimes seizures [4, 5].
Mutations in NOTCH3 which alter cysteines within exons 2–24 encompassing the epidermal growth factor-like repeats (EGFRs) were originally identified as causal in CADASIL patients [6]. The theorised pathogenic role is that these mutations result in an odd number of cysteine residues in one of the EGFRs, resulting in disrupted disulphide bond formation which impacts NOTCH3 signalling. Studies have shown that NOTCH3 signalling is important in brain and retinal vasculature, where Notch3 deficient adult mice showed loss of vessel integrity resulting from vascular smooth muscle cell (VSMC) degradation and apoptosis [7]. Furthermore, these Notch3 deficient mice showed an increased likelihood of haemorrhages and a loss of blood–brain barrier function [7, 8]. Notch signalling is highly conserved across species and involves interactions with ligands (JAG1, DLL1, DLL2) binding to the extracellular domain of the NOTCH3 protein. The protein then undergoes a cleavage event by ADAM10 at the S2 cleavage site, and then by the ɣ-secretase complex made up of Presenilin-1 (PSEN1), Presenilin-2 (PSEN2), Nicastrin (NCSTN), anterior pharynx defective 1 (APH-1), and presenilin enhancer 2 (PEN2) [9]. This second cleavage event results in the release of the NOTCH3 intracellular domain (N3ICD) into the cytoplasm where it is trafficked into the nucleus and can act as a transcriptional regulator. NOTCH3 signalling is primarily limited to the VSMCs and as such mutations in NOTCH3 cause a gradual degeneration of these cells leading to recurrent infarcts and gradual development of vascular dementia [10].
Interestingly, the ɣ-secretase complex that is involved in the NOTCH cleavage steps and is also known to play a role in the cleavage of apolipoprotein precursor protein (APP). Along with mutations in PSEN1 and PSEN2, APP is one of the most studied genes known to cause Alzheimer’s disease. Mutations in APP have also been associated with cerebral amyloid angiopathy (CAA), a condition that results in amyloid deposits accumulating around VSMCs in the abluminal aspect of the intimal layers of the blood vessels [11, 12]. In a similar mechanism to CADASIL, this condition can cause cerebral haemorrhages, brain bleeds, and dementia [13,14,15]. Due to the phenotypic overlap of these conditions and that the pathways involved are linked via the ɣ-secretase complex, we hypothesised that there are potential causative mutations in genes within the AD-presenilin pathway that may be causing a CADASIL-like phenotype, or a novel form of CADASIL-related CSVD. Therefore, we investigated whether rare and functional variants in Alzheimer-related genes from the protein analysis through evolutionary relationships (PANTHER) AD-Presenilin pathway were present in patients with suspected CADASIL, but without NOTCH3 mutations.
Methods
Patient Cohort
The study cohort comprised patients who were initially referred by neurologists to the Genomics Research Centre (GRC) diagnostic testing facility for CADASIL testing (Targeted NOTCH3 next-generation sequencing). From these, 50 samples were selected based on the previous testing using the GRC custom 5-gene panel (CACNA1A, ATP1A2, SCN1A, NOTCH3, KCNK18) where no causative mutation was identified in NOTCH3 or in any of the other genes on the panel [16]. Ethical approval through the Queensland University of Technology (QUT) human research ethic council (HREC), and appropriate consents for the patient cohort, are already in place (approval number 1800000611).
DNA Extraction and Whole Exome Sequencing
Genomic DNA was extracted from peripheral blood lymphocytes using the QIAGEN QIAcube™ (Venlo, Netherlands). Aliquots of DNA were quantified and checked for quality using a Thermo Fisher Scientific Nanodrop Spectrophotometer 8000 (Waltham, MA, USA) and diluted to a concentration of ~ 20 ng/µL for whole exome sequencing using the Ion AmpliSeq™ Exome RDY-kits (Carlsbad, CA., USA) for library preparation, according to manufactures’ instructions (MAN0010084). Completed libraries were quantified using QIAGEN Qubit™ v.3 (Venlo, Netherlands) and combined at an equimolar concentration of 100 pM. Template preparation, enrichment, and chip loading were performed using the Ion P1™ Hi-Q™ Chef Kit (Cat. Number A27198) and 540 Chips (Cat. Number A30011) on the Applied Biosystems Ion Chef (Carlsbad, CA, USA) targeted at 200 bp lengths. Sequencing was performed using the Ion Proton™ and Ion S5 + platforms with sequencing alignment (Hg19) and variant calling was completed via the Ion Torrent™ software (Carlsbad, CA, USA).
Targeted Candidate Mutation Analysis and Curation
Analysis was completed through merging 50 variant call format (vcf) files using the bcf-tools vcf-merge function and completing variant annotation using ensemble-vep [17]. The merged vcf file was then filtered based on a list of 131 genes that are part of the PANTHER Alzheimer disease – presenilin pathway (Table 1) from (http://www.pantherdb.org/list/list.do?numPerPage=200&save=yes&searchModType=numperpage&listType=1). These genes were identified through selecting the components part of the Alzheimer’s disease – presenilin pathway (Accession: P00004) and converting the list to genes and filtering for homo sapiens. Candidate variants were filtered based on pathogenic in silico prediction tools such as MutationTaster, SIFT, PolyPhen, and PredictSNP2 (which includes CADD, DANN, FATHMM, FunSeq2, and GWAVA scores) as well as an overall gnomAD MAF < 0.001 [18,19,20,21,22]. Variants were excluded if there were two or more in silico tools that identified the variant as benign/tolerated. Variants were further checked in disease and variant databases such as ClinVar, dbSNP, and for previous pathogenicity and disease-causing curations. Finally, variants were checked for the candidacy of causing disease based on gene ontology, genetic interactions, and gene expression.
An additional analysis of APOE ε carrier allele status was also completed. This involved extracting the rs429358 and rs7412 variants identified in APOE from WES data for each individual [23]. The rate of each genotype identified was then used to determine if there is an increased risk for Alzheimer’s disease in this cohort and was compared to gnomAD population data.
Burden Testing the Alzheimer’s Disease Gene Pathway
Following candidate mutation identification, a rare-variant association test using the TRAPD software was also completed to determine if there was any increase in the number of rare variants in the Alzheimer’s disease—presenilin pathway genes [24]. This test involved using a modified analysis pipeline (Fig. 1) of the merged and annotated vcf file with just the AD-presenilin genes and counting the number of rare variants (gnomAD global MAF < 0.001).

Analysis pipeline for using the TRAPD burden test protocol with added pre-processing steps. Values in brackets are where there are minor differences completed for each file
Variants were also filtered in if there was a score of ≤ 0.05 for SIFT and a score of ≥ 0.8 for PolyPhen had a coverage depth of ≥ 20X. To further remove the likelihood of artefactual results, there was also a threshold set for an overall allele count (AC) < 5 for each variant in the merged vcf file. To ensure the same regions of the genes were covered, both the gnomAD and CADASIL-related CSVD vcf files were filtered to only include non-intronic variants. The TRAPD burden test scripts initially involved creating a variant (SNP) file for both the gnomAD v2.1.1 filtered vcf and the CADASIL merged filtered vcf file. The number of alleles for each SNP identified was then counted and collated into its associated gene count, where a second filter for the gnomAD controls was added to make sure that only variants seen in less than 1 in 1000 gnomAD cases were counted. This was done in accordance with the TRAPD burden test guidelines to further remove bias from the system. Finally, a burden test was run using Fisher’s exact test to identify a greater burden in variants identified across genes in the CADASIL case-cohort versus the control cohort. There were two p values calculated for each gene based on either a predicted Mendelian autosomal dominant or autosomal recessive inheritance pattern.
Results
WES was conducted for 50 patients referred for CADASIL testing, but negative for mutations in NOTCH3. Overall, the initial analysis of the WES data identified n = 20 mutations across n = 15 PANTHER Alzheimer’s disease – presenilin pathway genes (Table 2). This included a rare mutation in APP in DGR349, where mutations in this gene are known to cause Alzheimer’s disease and/or cerebral amyloid angiopathy. There were n = 2 mutations in SMPD1 across two separate samples (DGR330, DGR332) which encodes for Sphingomyelin phosphodiesterase 1. There were also three samples with mutations in ERN1 (DGR024, DGR037, and DGR323) which encodes for Endoplasmic Reticulum to Nucleus Signalling and TRPC4 (DGR321, DGR343, DGR366) which encodes for Transient Receptor Potential Cation Channel Subfamily C Member 4. Mutations in NOTCH1 and NOTCH2 were also identified in this analysis showing potential variants of interest in other NOTCH family genes.
The ApoE homozygous rs429358 CC variants were seen at 10 times the rate of the gnomAD population control (Table 3) and heterozygous rs7412 CT variants occurred 1.66 times more frequently in the case-cohort compared to the general gnomAD population observing this genotype [24]. There was a total of 10% (n = 5/50) CADASIL referred individuals that have the APOE-ε4/ε4 genotype (Table 4). The remaining genotypes identified that the ε3/ε4 and ε3/ε3 were observed in 42% (n = 21/50) and 38% (n = 19/50) respectively of the CADASIL-CSVD population, while the ε2/ε3 and ε2/ε4 genotypes were seen in 8% (n = 4/50) and 2% (n = 1/50) respectively (Table 4). Interestingly, this showed that 50% of the CADASIL-related CSVD cohort had at least one APOE4 AD risk allele.
Rare variant association testing using the TRAPD software failed to identify an increased pathogenic rare variant burden in the AD-Presenilin genes within the CADASIL-like CSVD cohort compared to the GnomAD population that passed multiple testing. However, two genes (ERN1 p = 0.0015 FDR = 0.38; TRPC3 p = 0.015 FDR = 1) were found to be nominally significant using the autosomal dominant model (Table 5). There was no nominal association identified in the recessive model.
Discussion
Candidate mutation analysis based on MAF thresholds and in silico prediction tools of the AD-presenilin genes identified 20 mutations across 15 genes which were further investigated as novel candidates for CSVD. ERN1 may be the strongest novel candidate for CSVD pathology as there were five variants identified across the CADASIL-CSVD cohort that were considered significant, that included the three candidate mutations (Table 2). The ERN1 association identified from the TRAPD burden test also mimics an exome-wide association study of APOE ε4 non-carrier Alzheimer’s disease which found a rare synonymous variant in ERN1 (rs56201815) as being associated with late-onset Alzheimer’s disease (LOAD) [25]. ERN1 encodes for a resident transmembrane endoplasmic reticulum (ER) protein which has both kinase and endonuclease domains (Fig. 2) that works as a key sensor for the ER unfolded protein response [26, 27]. This response activates the genes involved in the degradation of misfolded proteins, regulating protein synthesis and activating molecular chaperones [27].

ERN1 protein structure and potential impact of identified variants. Panel of ERN1 protein structure and role in the unfolded protein response (UPR) cycle. Panel A. shows the ERN1 protein with its topological domains (Lumenal, Transmembrane, and Cytoplasmic), functional domains (Protein Kinase and Ribonuc_5_2A), and the CDS exonic locations that encode for each domain. The two variants identified in this study show the Ribonuc_5_2A functional domain may be affected. Panel B. shows the role of ERN1 in the UPR cycle and is adapted from Li et. al. (2019) [28]. The Ribonuc_5_2A domain at the C-terminal of the protein is involved in splicing XBP1 mRNA allowing for the protein to be translated to act as a transcriptional regulator of UPR genes. Panel C. predicts the potential downstream functional effects of the ERN1 gene variants in splicing XBP1 mRNA and altering the transcriptional regulation of UPR genes
Misfolded proteins that aggregate are a common feature of CSVDs such as CADASIL (NOTCH3 mutations), Collagen, type IV, alpha-related small vessel disease (COL4A1 and COL4A2 mutations), and CAA (APP – mutations) [29,30,31,32]. As such, ERN1 dysfunction may result in increased formation and accumulation of misfolded proteins, and aggregation in the intra- or extracellular spaces in the vessel walls contributing to the pathology of CADASIL and related CSVD pathologies. Interestingly, a previous study found that inhibited expression of ERN1 reduced the levels of APP in cortical and hippocampal areas in AD mice, restoring their learning and memory capacity [33]. Whilst the mutations identified have not been confirmed to affect function, these studies indicate that dysfunctional ERN1 may have a contributory or causative role in CSVD pathology.
In humans, pharmacological modulation of the ERN1 protein can counteract metaflammation (ubiquitous low levels of inflammation throughout the body) a process that is associated with blood vessel–affecting diseases such as atherosclerosis, chronic heart disease, and diabetes mellitus [34]. ERN1 signalling has also been identified to increase cell cytotoxicity and apoptosis in neuronal ischaemic injury [35]. However, some studies have also identified that ERN1 signalling also exerts a neuroprotective response which supports that the ERN1-pathway may be pro-survival in ischaemic stroke through the activation of chaperone proteins [36]. While this gene did not pass multiple testing using TRAPD burden software, this analysis was based on a low sample size and ERN1 remains an interesting candidate to investigate further in CSVD and neurodegenerative phenotypes [37, 38].
The amyloid cascade hypothesis for AD is based on the accumulation of amyloid-β peptide as the key driver of early-onset AD [39]. This hypothesis is based on disease-causing mutations identified in PSEN1, PSEN2, and APP, as well as the ApoE variants found to increase the risk of developing early onset AD. The role of PSEN1 and PSEN2 in the ɣ-secretase complex, as well as APP being a known cause of the CAA made these genes interesting targets to investigate further in this study. Furthermore, the link between CSVD and AD is constantly expanding, with some studies showing that CSVD can occur with AD, indicating that the two diseases are interconnected [40, 41]. Within this suspected CADASIL cohort, it was observed that five participants (10%) had the APOE-ε4/ε4 genotype and 25 participants (50%) had at least one APOE-ε4 allele. Interestingly, the percentage of APOE-ε4 carriers in the CADASIL-related CSVD cohort is in line with what is expected to see in global AD patient cohorts which can range from 38.9–64.4% [42, 43]. This may represent a cause of some of the CADASIL-related symptoms, as APOE-ε4 carriers show a significant decline in cognitive function by age 69, with the strongest association being seen in APOE-ε4/ε4 individuals [44]. This may mean that some suspected CADASIL patients are presenting with Alzheimer’s disease and may have comorbidities that mimic a CADASIL phenotype. It would be interesting to investigate this further in other cohorts.
The heterozygous APP p.Gly326Ser mutation may be causative of the amyloid-type CSVD, CAA—an autosomal dominant condition where amyloid progressively deposits in the cerebral blood vessel walls causing degenerative vascular changes and spontaneous cerebral haemorrhages, ischaemic lesions, and dementia [12]. Furthermore, mutations in APP are also well documented to cause AD [45, 46]. The mutation was predicted as disease-causing across all forms of in silico pathogenicity prediction tools (SIFT, PolyPhen2, MutationTaster, PredictSNP2, CADD, DANN, FATHMM, and FunSeq2) and has a MAF of 2.4e−5. The computational evidence together with the MAF may be indicative of CAA/AD. The presence of the white matter hyperintensities that were “characteristic of CADASIL” make CAA the most-plausible cause of CSVD in DGR349. Interestingly, this individual also had the heterozygous APOE-ε3/ε4 genotype. Together, this may have increased the likelihood of AD or CAA in this individual.
Interestingly, three TRPC gene paralogs (TRPC3, TRPC4, and TRPC6) were identified within this study. TRPC3 was identified as nominally significant through the TRAPD burden test although didn’t pass multiple testing. Interestingly, this was due to three individuals which had the same variant identified (4:122872901A > G) in an untranslated region of the gene, so the functional consequence of these is not clear. In contrast, TRPC4 and TRPC6 were identified with candidate predicted pathogenic variants (TRPC4 rs377554360 -NP_057263.1:p.Lys758Gln and rs146807006—NP_057263.1:p.Leu753Ser; TRPC6 rs117273916—NP_004612.2:p.Arg58Trp). Further investigation of these mutations via MutationTaster classified the TRPC6 rs117273916 as polymorphism and therefore as likely harmless, despite it recognising that splice sites may be affected, whilst the TRPC4 mutations were classified as disease-causing by MutationTaster software. The two TRPC4 mutations (NP_057263.1:p.Lys758Gln and NP_057263.1:p.Leu753Ser) were seen in three separate individuals where NP_057263.1:p.Leu753Ser was identified as pathogenic across all in silico prediction tools and NP_057263.1:p.Lys758Gln was identified as pathogenic across all tools, apart from DANN. Both mutations in TRPC4 are within the topological domain of the protein in the region that binds to ITPR1, ITPR2, and ITPR3. A role for TRPC4 has not been implicated CSVD; however, it has been found to play a role in regulating blood–brain barrier function and is hypothesised to be crucial for oedema formation in ischaemic stroke [47,48,49].
Similarly, there is currently no implicated role of TRPC6 in CSVD; however, some studies have shown that PSEN2 is known to interfere with the activity of TRPC6 without affecting agonist-induced Ca2+ release of other Ca2+ channels in HEK 293 T cells [47, 50]. As there were three paralogous TRPC genes identified either through TRAPD association or the candidate mutations approach, it shows a potential role of these genes in CSVD and neurodegenerative phenotypes. This may be related to the role they play in Ca2+ homeostasis in cells, something that is found to be dysregulated in AD-related pathologies. Furthermore, members of the TRPC family are found in rodent and human cerebrovascular tissue, cerebral arteries, and VSMCs, although clinical evidence linking these proteins to CSVD is limited [47, 51, 52].
Only ERN1 was identified using the WES candidate mutation identification approach. The use of rare variant association testing using the TRAPD software was unsuccessful in identifying novel associations. In part, this is due to one of the limitations of this type of testing in this study as there were only 50 probands available for research. Future genetic investigations should involve segregation studies within families, functional studies using cell lines or animal studies to further confirm overlapping molecular processes in these diseases. Furthermore, increasing the population of the CADASIL-CSVD cohort should also be completed and other statistical burden tests should be repeated to utilise a more specific control dataset such as the UK Biobank or ASPREE dataset [53, 54]. This would allow for a better control population and may even aid in identifying other novel causes of disease [55, 56].
Conclusion
In conclusion, analysis of the PANTHER Alzheimer’s disease – presenilin pathway genes in the CADASIL-CSVD cohort was successful in identifying novel candidate mutations that may be contributing to patient phenotypes. In particular, the mutations identified in ERN1 across three separate unrelated individuals show a promising novel CSVD gene. There was also further evidence for potential links between AD and CSVD through the proportion of individuals shown as carriers for APOE-ε4. This work builds on theories of an overlapping genetic mechanism between Alzheimer’s disease and CSVD, however further studies need to be completed to truly start to elucidate an overlapping role between these two diseases.
References
Joutel A et al (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383(6602):707–710
Narayan SK et al (2012) The minimum prevalence of CADASIL in northeast England. Neurology 78(13):1025–1027
Rutten JW et al (2016) Archetypal NOTCH3 mutations frequent in public exome: implications for CADASIL. Ann Clin Transl Neurol 3(11):844–853
Di Donato I et al (2017) Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med 15(1):41
Mancuso M et al (2020) Monogenic cerebral small-vessel diseases: diagnosis and therapy. Consensus recommendations of the European Academy of Neurology. Eur J Neurol 27(6):909–927
Joutel A et al (1997) Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 350(9090):1511–1515
Hosseini-Alghaderi S, Baron M (2020) Notch3 in development, health and disease. Biomolecules 10(3):485
Belin de Chantemèle EJ et al (2008) Notch3 is a major regulator of vascular tone in cerebral and tail resistance arteries. Arterioscler Thromb Vasc Biol 28(12):2216–2224
Zhang X et al (2014) The γ-secretase complex: from structure to function. Front Cell Neurosci 8:427–427
Wu J, Bresnick EH (2007) Bare rudiments of notch signaling: how receptor levels are regulated. Trends Biochem Sci 32(10):477–485
Revesz T et al (2009) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 118(1):115–130
Revesz T et al (2003) Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 62(9):885–898
Sellal F et al (2017) APP Mutations in cerebral amyloid angiopathy with or without cortical calcifications: report of three families and a literature review. J Alzheimers Dis 56(1):37–46
Biffi A, Greenberg SM (2011) Cerebral amyloid angiopathy: a systematic review. J Clin Neurol (Seoul Korea) 7(1):1–9
Biffi A et al (2010) Screening for familial APP mutations in sporadic cerebral amyloid angiopathy. PLoS ONE 5(11):e13949–e13949
Maksemous N et al (2016) Targeted next generation sequencing identifies novel NOTCH3 gene mutations in CADASIL diagnostics patients. Hum Genomics 10(1):38
McLaren W et al (2016) The Ensembl Variant Effect Predictor. Genome Biol 17(1):122
Schwarz JM et al (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7:575
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 76:7.20.1–7.20.41
Sim N-L et al (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40(Web Server issue):W452–W457
Bendl J et al (2016) PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput Biol 12(5):e1004962
Karczewski KJ et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809):434–443
Tsai MS et al (1994) Apolipoprotein E: risk factor for Alzheimer disease. Am J Hum Genet 54(4):643–649
Guo MH et al (2018) Burden testing of rare variants identified through exome sequencing via publicly available control data. Am J Hum Genet 103(4):522–534
He L et al (2021) Exome-wide age-of-onset analysis reveals exonic variants in ERN1 and SPPL2C associated with Alzheimer’s disease. Transl Psychiatry 11(1):146
Liu CY, Xu Z, Kaufman RJ (2003) Structure and intermolecular interactions of the luminal dimerization domain of human IRE1alpha. J Biol Chem 278(20):17680–17687
Moore K, Hollien J (2015) Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol Biol Cell 26(16):2873–2884
Li A et al (2019) The emerging roles of endoplasmic reticulum stress in balancing immunity and tolerance in health and diseases: mechanisms and opportunities. Front Immunol 10:3154
Papakonstantinou E et al (2019) NOTCH3 and CADASIL syndrome: a genetic and structural overview. EMBnet J 24:e921
Rutten JW et al (2014) Interpretation of NOTCH3 mutations in the diagnosis of CADASIL. Expert Rev Mol Diagn 14(5):593–603
Kuo DS, Labelle-Dumais C, Gould DB (2012) COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet 21(R1):R97-110
Ghiso J, Fossati S, Rostagno A (2014) Amyloidosis associated with cerebral amyloid angiopathy: cell signaling pathways elicited in cerebral endothelial cells. J Alzheimers Dis: JAD 42 Suppl 3(0 3):S167–S176
Duran-Aniotz C et al (2017) IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol 134(3):489–506
Tufanli O et al (2017) Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc Natl Acad Sci 114(8):E1395
Maier PJ et al (2014) Ischemia-like oxygen and glucose deprivation mediates down-regulation of cell surface γ-aminobutyric acidB receptors via the endoplasmic reticulum (ER) stress-induced transcription factor CCAAT/enhancer-binding protein (C/EBP)-homologous protein (CHOP). J Biol Chem 289(18):12896–12907
Nakka VP, Gusain A, Raghubir R (2010) Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res 17(2):189–202
Ito D et al (2001) Up-regulation of the Ire1-mediated signaling molecule, Bip, in ischemic rat brain. NeuroReport 12(18):4023–4028
Koh SH, Park HH (2017) Neurogenesis in stroke recovery. Transl Stroke Res 8(1):3–13
Karran E, Mercken M, Strooper BD (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discovery 10(9):698–712
Cai Z et al (2015) Cerebral small vessel disease and Alzheimer’s disease. Clin Interv Aging 10:1695–1704
Kalaria RN, Sepulveda-Falla D (2021) Cerebral Small Vessel Disease in Sporadic and Familial Alzheimer Disease. Am J Pathol 191(11):1888–1905
Crean S et al (2011) Apolipoprotein E & #949;4 prevalence in Alzheimer’s disease patients varies across global populations: a systematic literature review and meta-analysis. Dement Geriatr Cogn Disord 31(1):20–30
Heffernan AL et al (2016) The neurobiology and age-related prevalence of the ε4 allele of apolipoprotein E in Alzheimer’s disease cohorts. J Mol Neurosci: MN 60(3):316–324
Rawle MJ et al (2018) Apolipoprotein-E (Apoe) ε4 and cognitive decline over the adult life course. Transl Psychiatry 8(1):18
Lanoiselée H-M et al (2017) APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med 14(3):e1002270
Hardy J (2017) The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis.” FEBS J 284(7):1040–1044
Zeng C, Tian F, Xiao B (2016) TRPC channels: prominent candidates of underlying mechanism in neuropsychiatric diseases. Mol Neurobiol 53(1):631–647
Ahmmed GU, Malik AB (2005) Functional role of TRPC channels in the regulation of endothelial permeability. Pflugers Arch 451(1):131–142
Simard JM et al (2007) Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 6(3):258–268
Lessard CB et al (2005) The overexpression of presenilin2 and Alzheimer’s-disease-linked presenilin2 variants influences TRPC6-enhanced Ca2+ entry into HEK293 cells. Cell Signal 17(4):437–445
Chen J et al (2009) Inhibition of TRPC1/TRPC3 by PKG contributes to NO-mediated vasorelaxation. Am J Physiol Heart Circ Physiol 297(1):H417–H424
Thilo F et al (2011) Decreased expression of transient receptor potential channels in cerebral vascular tissue from patients after hypertensive intracerebral hemorrhage. Clin Exp Hypertens 33(8):533–537
ASPREE Investigator Group (2013) Study design of ASPirin in Reducing Events in the Elderly (ASPREE): a randomized, controlled trial. Contemp Clin Trials 36(2):555–564
O’Connell J et al (2016) Haplotype estimation for biobank-scale data sets. Nat Genet 48(7):817–820
Rutten JW et al (2019) The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1–6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7–34 pathogenic variant. Genet Med 21(3):676–682
Malik R et al (2018) Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 50(4):524–537
Acknowledgements
Acknowledgement goes to the Australian National Health and Medical Research Council (NHMRC) for funding this project through the Dora Lush Biomedical Sciences Postgraduate Scholarship.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This work was supported through the National Health and Medical Research Council of Australia (GNT1168601) Dora Lush Biomedical Postgraduate Research scholarship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dunn, P.J., Lea, R.A., Maksemous, N. et al. Investigating a Genetic Link Between Alzheimer’s Disease and CADASIL-Related Cerebral Small Vessel Disease. Mol Neurobiol 59, 7293–7302 (2022). https://doi.org/10.1007/s12035-022-03039-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-03039-3