
Abstract
The genetic transcription profile and underlying molecular mechanisms of ischemic stroke (IS) remain elusive. To address this issue, four mRNA and one miRNA expression profile of rats with middle cerebral artery occlusion (MCAO) were acquired from the Gene Expression Omnibus (GEO) database. A total of 780 differentially expressed genes (DEGs) and 56 miRNAs (DEMs) were screened. Gene set and functional enrichment analysis revealed that a substantial number of immune-inflammation–related pathways were abnormally activated in IS. Through weighted gene co-expression network analysis, the turquoise module was identified as meaningful. By taking the intersection of the turquoise module genes, DEM-target genes, and all DEGs, 354 genes were subsequently obtained as key IS-related genes. Among them, six characteristic genes were identified using the least absolute shrinkage and selection operator. After validation with three external datasets, transforming growth factor beta 1 (Tgfb1) was selected as the hub gene. This finding was further confirmed by gene expression pattern analysis in both the MCAO model rats and clinical IS patients. Moreover, the expression of the hub genes exhibited a negative correlation with the modified Rankin scale score (P < 0.05). Collectively, these results expand our knowledge of the genetic profile and molecular mechanisms involved in IS and suggest that the Tgfb1 gene is a potential biomarker of this disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke (IS) is a frequently diagnosed disease and one of the main causes of death and permanent disability worldwide, exerting a heavy burden on both patients and public health systems [1]. Despite the volume of research on this devastating disease that has been performed around the world, no breakthroughs have been translated into therapeutic use [2]. The difficulties in neural regeneration and functional recovery after IS may be attributed to its complex pathological alterations. Therefore, elucidating the underlying pathological mechanisms of IS may optimize treatment strategies, thus improving patient prognosis [3].
Accumulating evidence has suggested that unbalanced immunity and inflammation play critical roles in the progression of IS [4]. Because the brain heavily relies on the continuous supply of oxygen and the delivery of glucose through the bloodstream, a sudden interruption in the cerebral blood supply and subsequent reperfusion can lead to irreversible brain damage and quickly trigger sterile inflammation [5, 6]. During the development of IS, various adaptive and innate immune cells can more easily access ischemic brain regions from the peripheral circulation because of disruption of the blood–brain barrier, further initiating a pro-inflammatory cascade that results in additional and extensive neurological injury. In recent years, some studies have revealed that neurological damage can be limited and that functional recovery can be promoted by controlling the immune-inflammatory response in the central nervous system (CNS) microenvironment [7]. However, reliable biomarkers for IS remain uncertain. Therefore, it is necessary to further investigate the genetic alteration patterns of ischemic regions to identify more promising targets.
In recent years, high-throughput technologies have become a powerful way to explore molecular mechanisms and identify the key genes involved in IS [7, 8]. However, it is challenging to obtain ischemic brain tissue from patients for RNA sequencing or microarray analysis because of certain ethical issues. As an alternative, brain tissue can be collected from model organisms under optimal conditions. The middle cerebral artery occlusion (MCAO) models in animals have been developed to mimic human stroke injury. Rats are commonly exploited in the field of stroke research due to their cerebral vasculature and physiology similarities to human beings [9,10,11,12]. Thus far, large quantities of microarray datasets from rats with MCAO are available in public online data repositories [9, 10, 13]. However, existing single datasets normally include a limited number of samples. Integrating multiple datasets from different experiments not only increases the sample size, but also improves the robustness of the results, which is of great significance to explore the molecular changes after IS in depth [7, 14, 15].
For researchers, screening out the molecular targets that play vital roles in IS progression from the vast amount of high-throughput data is undoubtedly a challenge [16]. As a valuable tool in systems biology, weighted gene co-expression network analysis (WGCNA) has gradually been applied to generate co-expressed gene modules linked to specific disease mechanisms from massive amounts of gene expression data [17]. However, least absolute shrinkage and selection operator (LASSO) regression is a broadly applied machine learning approach that is suitable for advancing the predictive value and accuracy of key genes discovered based on high-throughput data [18]. Therefore, the integration of WGCNA and LASSO regression can effectively screen for important genes associated with IS.
MicroRNAs (miRNAs) are a class of small noncoding RNAs that negatively regulate gene messenger RNA (mRNA) expression at the posttranscriptional level through base pairing with the 3′UTR of target mRNAs [19]. As widely reported, the dysregulation of the expression of numerous miRNAs is related to pathophysiological processes after IS [20]. Therefore, elucidating miRNA expression profiles and constructing miRNA‒mRNA networks are important for discovering molecular markers and improving the understanding of the mechanisms underlying IS.
Therefore, the objective of this study was to identify potential biomarkers associated with IS that exhibit coordinated expression in both the brain and peripheral blood. To achieve this, four mRNA datasets and one miRNA dataset from MCAO rats were collected from the Gene Expression Omnibus (GEO) database. Integrated bioinformatics approaches were utilized to identify differentially expressed genes (DEGs) and miRNAs (DEMs), and a regulatory miRNA‒mRNA network was constructed. Furthermore, WGCNA and the LASSO algorithm were combined to identify characteristic IS-related genes. Finally, the diagnostic efficacy and expression level of these candidate genes were verified by the original training set, three external datasets, rat models, and clinical samples.
Results
Identification of DEGs and DEMs
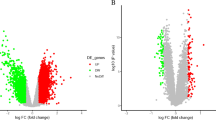
The expression matrix of each dataset was obtained after preprocessing, including background correction and normalization. The training sets included the expression profiles of four mRNAs (GSE36010, GSE78731, GSE61616, and GSE106931) and one miRNA (GSE46266). Then, the four mRNA datasets were combined after batch effect removal. The combined expression matrix of 7684 genes was obtained from 46 samples (21 MCAO samples versus 25 control samples). Through differential expression analysis, a total of 780 DEGs were obtained. Moreover, 56 DEMs were identified from GSE46266. The overall distribution of DEGs and DEMs has been visualized using volcano plots (Fig. 1a and 1b).

Identification of differentially expressed mRNAs (DEGs) and miRNAs (DEMs) in the brains of middle cerebral artery occlusion (MCAO) model rats. a Volcano plot showing DEGs between the MCAO group and the control group. b Volcano plot of DEMs in the GSE46266 dataset. In a and b, red plots indicate upregulation, blue plots represent downregulation, and gray plots indicate nondifferential expression between the two groups
Gene Set Enrichment Analysis (GSEA)
GSEA was conducted to obtain a preliminary understanding of the main roles of all 7684 genes in the training set. The results showed that the top five biological process (BP) terms enriched in these genes included leukocyte proliferation, B-cell activation, symbiotic interaction, synaptic plasticity, positive regulation of locomotion, and regulation of synaptic plasticity (Fig. 2a). In addition, the top five Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were involved in the cell cycle, hematopoietic cell lineage, systemic lupus erythematosus, cytokine‒cytokine receptor interaction, and primary immunodeficiency (Fig. 2b). The top five items are shown in the ridgeline plots (Fig. 2c and 2d). These items were associated with immune-inflammation responses and neural plasticity. The GSEA results with a cutoff value of adjusted P < 0.05 and a value of |normalized enrichment score (NES)|> 1 are presented in Supplementary Table S1.

The results of the gene set enrichment analysis (GSEA). a Three representative biological processes (BPs). b Three representative Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. c The ridgeline plot shows the enrichment results of the top five gene sets of BPs. d The ridgeline plot shows the enrichment results of the top five gene sets of KEGG pathways
Identifying WGCNA Module Genes Associated with IS
WGCNA was used to identify gene clusters strongly related to IS in the combined dataset. We found that when the soft threshold was set at 22 after calculation, the scale independence of the topology network reached 0.85, and the adjacency matrix obtained a comparatively high value for the average connectivity (Fig. 3a). Furthermore, five co-expressed gene modules were identified using a hierarchical tree algorithm (Fig. 3b). After relating the modules to disease state traits by correlation analysis, we found that the turquoise module had the highest absolute correlation with the IS (Fig. 3c). Figure 3d shows the correlation analysis between module membership and gene significance in the turquoise module and the correlation coefficient between the 2157 genes in the turquoise module; this figure also shows that the importance of IS genes was 0.69 (P < 1e − 200).

Weighted gene co-expression network analysis. a Analysis of network topology for various soft-threshold powers. b Hierarchical tree algorithm of genes. The gene hierarchical clustering tree displays the network and the five identified modules. c Heatmap of module-feature associations. d Scatter plot between module members and IS gene salience in the turquoise module
Exploration for the Target Gene of DEMs
In total, 10,849 target mRNAs were predicted by the miRWalk database to have binding sites for 14 of the 56 DEMs mentioned above. Among the 14 DEMs, eight were upregulated, and six were downregulated. We intersected these 10,849 genes with the 780 DEGs and 2157 turquoise module genes, retaining 354 overlapping genes as IS-related key genes for further analysis (Fig. 4a). Subsequently, these 354 intersecting genes were used to construct miRNA‒mRNA subnetworks (Fig. 4b and 4c; Supplementary Table S2).

Exploration of the key ischemic stroke-related genes. a Venn diagram of the intersection of differentially expressed mRNAs (DEGs), genes of the turquoise module, and target mRNAs of differentially expressed miRNAs. b miRNA‒mRNA subnetworks for upregulated DEMs. c miRNA‒mRNA subnetworks for downregulated DEMs
Functional Enrichment Analysis
A functional enrichment analysis of the 354 intersecting genes was carried out. The BP terms included leukocyte-mediated immunity, positive regulation of leukocyte activation, adaptive immune response, leukocyte migration, and leukocyte activation involved in an unbalanced immune response. In terms of cellular components (CCs), there was predominant enrichment in the external side of the plasma membrane, membrane rafts, membrane microdomains, extracellular matrix, external encapsulating structure, and so on. In relation to molecular function (MF) terms, the genes were markedly enriched in cell adhesion molecule binding, cytokine receptor binding, kinase regulator activity, integrin binding, immune receptor activity, and so on (Supplementary Table S3). The top 10 items are shown in the star bar plots (Fig. 5a–5c). The enriched KEGG terms included viral protein interaction with cytokine and cytokine receptor, NF-κB signaling pathway, lipids and atherosclerosis, and cytokine‒cytokine receptor interaction (Supplementary Table S4). The top 10 KEGG pathways are shown in bubble plots (Fig. 5d). The majority of the results of the functional enrichment analysis were associated with immune and inflammatory responses.

Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses. a–c Top 10 terms of GO functional enrichment analysis results, including biological processes, cellular components, and molecular functions. d Top 10 results for KEGG pathway analysis
Screening and Diagnostic Performance of IS Biomarkers
To identify IS-related characteristic genes, LASSO regression analysis identified six genes, namely, Tgfb1, Fc gamma receptor 2 b (Fcgr2b), Vcan versican (Vcan), leukocyte immunoglobulin-like receptor B4 (Lilrb4), cholinergic receptor, muscarinic 1 (Chrm1), and chymotrypsin-like elastase 1 (Cela1), from the 354 intersection genes for subsequent research (Fig. 6a and b). The receiver operating characteristic (ROC) curve results showed that the area under the ROC curve (AUC) values of the six genes were greater than 0.9 in the training set (Fig. 7). After that, ROC curve analysis was applied to assess the diagnostic value of these six genes in the GSE97537, GSE97533, and GSE66724 validation sets (Fig. 8a–c). Finally, only one mRNA, Tgfb1, had an AUC greater than 0.7 in all validation sets. These results indicate that Tgfb1 has good diagnostic performance in distinguishing between the IS group and control group, both in rats and participants.

Identification of biomarkers for ischemic stroke by least absolute shrinkage and selection operator (LASSO) regression. a LASSO regression analysis identified six characteristic genes associated with ischemic stroke with minimum lambda values and nonzero parameters. b The LASSO coefficient profile plot was produced against the log(lambda) sequence in the metadata cohort. Each curve corresponds to the trajectory of each gene

Receiver operating characteristic (ROC) curve analysis of characteristic ischemic stroke-related genes in the training sets GSE36010 combined with GSE78731, GSE61616, and GSE106931. ROC curve analysis of a Tgfb1, b Fcgr2b, c Vcan, d Lilrb4, e Chrm1, and f Cela1

Validation of characteristic ischemic stroke-related genes in the validation sets. Receiver operating characteristic curve analysis of these six genes in a GSE97537, b GSE97533, and c GSE66724
Validation in MCAO Rat Model
The modified neurological severity scores (mNSSs) were similar between the two groups before the operation (P > 0.05). At 1 day and 3 days after the operation, the mNSSs were greater in the MCAO group than in the control group (P < 0.05) (Fig. 9). To verify the expression level of Tgfb1 mRNA in the MCAO rat model, we collected brain and blood tissue 3 days after reperfusion. Hematoxylin and eosin (H&E) staining and Nissl staining confirmed the successful construction of the model (Fig. 10a and b). The RNA extraction and quantitative real-time polymerase chain reaction (qRT‒PCR) results revealed a significant increase in Tgfb1 mRNA in both the brain and blood tissue in the MCAO group compared with that in the control group (P < 0.05 and P < 0.01, respectively) (Fig. 11a–11b).

Modified neurological severity scores (mNSSs) at various times following the operation, where a higher score indicates more severe neurological dysfunction. nsP > 0.05, compared with the control group; **P < 0.01, compared with the control group

Representative H&E- and Nissl-stained micrographs of the brain (scale bars = 1000 or 100 μm). a Representative H&E-stained transverse sections. b Nissl bodies in neurons in the brain tissue of the two groups were observed by Nissl staining. Blue staining represents a Nissl body, where the darker the color of the Nissl body or the tabby shape, the better the neuronal state. Hematoxylin and eosin (H&E). **P < 0.05, compared with the control group

Animal experimental validation of ischemic stroke-related biomarkers. a Expression level of Tgfb1 mRNA in the brains of rats. b Relative mRNA expression of Tgfb1 in the blood of rats in the middle cerebral artery occlusion (MCAO) group versus the control group. *P < 0.05. **P < 0.01, compared with the control group
Verification in Clinical Cohort
Next, we performed qRT‒PCR to validate the expression of TGF-β1 mRNA in peripheral blood samples obtained from both IS patients and healthy controls (HCs). The demographic features of the participants are summarized in Table 1. These IS patients and HCs did not significantly differ in age or sex (P > 0.05). Consistent with the trend of bioinformatics analysis, qRT‒PCR revealed that TGF-β1 mRNA expression in the blood of the IS patients was greater than that in the blood of the HCs (P < 0.001) (Fig. 12a). The AUC of TGF-β1 in the clinical samples was 0.739 (95% confidence interval, 0.590–0.888) (Fig. 12b).

Clinical experimental validation of ischemic stroke (IS)–related biomarkers. a Relative mRNA expression of TGF-β1 in IS patients versus healthy controls. **P < 0.01. b Receiver operating characteristic curve analysis of TGF-β1 expression
Correction Between TGF-β1 Level and Functional Outcomes
The functional outcomes of the IS patients were quantified using the modified Rankin scale (mRS) score. The mRS score separated the IS patients into groups with good (mRS ≤ 2) and poor functional outcome (mRS > 2). The characteristics of patients are summarized in Table 2. The level of TGF-β1 in patients with good functional outcomes was significantly greater than that in patients with poor functional outcomes (P < 0.05) (Fig. 13a). There was a negative correlation between the TGF-β1 level and the mRS score (r = − 0.442, P < 0.05) (Fig. 13b).

The relationship between the TGF-β1 level and modified Rankin scale (mRS) score. a Blood TGF-β1 levels in patients with good functional outcomes and poor functional outcomes. *P < 0.05. b The correlation between the TGF-β1 level and the mRS score was analyzed using Spearman’s linear correlation
Discussion
Despite substantial advances in the treatment and rehabilitation of IS in recent decades, a considerable number of survivors remain severely disabled [21, 22]. Dysregulation of the immune-inflammatory response is one of the barriers to IS recovery; this phenomenon has been supported by many studies. Therefore, identifying immune-inflammation–related pathways and providing suggestions for further studies will greatly increase the chances for IS patients to receive effective rehabilitation. In the present study, six genes (Tgfb1, Fcgr2b, Vcan, Lilrb4, Chrm1, and Cela1) were revealed to be significantly correlated with IS by WGCNA and LASSO logistic regression. Finally, only one candidate gene, the Tgfb1 gene, was identified as a potential target by ROC curve analysis in the external validation sets, and this finding was statistically supported in samples from both the MCAO models and clinical cohorts.
IS is defined as the sudden interruption of arterial blood flow into the brain parenchyma and is accompanied by a loss of integrity of the blood–brain barrier [5]. In response to ischemia‒reperfusion injury, innate and adaptive immune cells are quickly recruited to ischemic brain tissue, where they produce pro-inflammatory cytokines, chemokines, and reactive oxygen species, leading to amplification of the local inflammatory response. This early activation of the immune system leads to secondary injury after IS [23]. In our study, the GSEA results implied that the genes in the combined set were primarily enriched in BPs, including the regulation of synaptic plasticity, leukocyte proliferation, and B-cell activation. Leukocytes, also known as white blood cells, are markers of inflammation and have been confirmed to be closely related to the occurrence of cerebrovascular diseases [24]. In the pathophysiological state, leukocytes can be recruited and adhere to the arterial vessel wall, thus initiating and mediating the progression of atherosclerosis. Elevated levels of leukocytes may contribute to IS either through an effect on chronic atherosclerosis or by inducing acute thrombosis (possibly by increasing the likelihood of plaque rupture) [25]. Under physiological conditions, the number of detectable B cells is very low in the brain parenchyma, but B cells are frequently trafficked to CNS tissues in response to ischemic injury following a stroke. The function of B cells in the context of stroke has not been thoroughly elucidated, although it has been proposed that B-cell–mediated responses can interfere with neuronal function and could contribute to the development of poststroke cognitive impairment [26, 27]. In addition, neuroinflammation affects the ability of neurons to exhibit synaptic plasticity. These excessive inflammatory responses can be detrimental and may lead to hindered neural repair and expanded infarct volume [28,29,30]. In the present study, we performed KEGG enrichment analyses of the 354 IS-related key genes related to NF-κB signaling pathways, lipids and atherosclerosis, cytokine–cytokine receptor interactions, and so on. Some of these pathways have been well studied in the field of IS and have been shown to be associated with immune-inflammatory responses. For example, the transcription factor NF-κB is a core part of the regulation of inflammatory responses post-IS [31]. The NF-κB signaling pathway can be triggered following IS, causing the expression of target genes associated with the inflammatory response and upregulating the level of cytokines necessary for inflammation [32]. Li et al. showed that inhibiting the entry of the NF-kappa B signaling pathway into the ischemic brain can alleviate neuroinflammation-induced damage and attenuate neurologic function deficits in MCAO rats [33]; numerous studies have shown that the inhibition of NF-κB activation can be considered a critical target and an effective way to curb the immune-inflammatory response after IS [34]. The understanding of atherosclerosis has been redefined by the concept of an ongoing inflammatory response. Inflammation not only drives the formation and development of atherosclerotic plaques, but also, importantly, plays a central role in the destabilization of artery plaques, including erosion and fissuring, thus transforming chronic atherosclerosis into an acute thromboembolic disease [35]. Therefore, approaches that attenuate the inflammatory response may be effective therapeutic strategies to combat the development of atherosclerosis [36]. Typically, following IS, multiple cytokines are released from damaged cerebral tissue. Cytokines, such as tumor necrosis factor, interleukin-1, and interleukin-6, have been proven to modulate infarct evolution in experimental stroke (focal cerebral ischemia) rodent models. Hence, therapeutic strategies related to cytokines, such as the administration of cytokine antibodies or cytokine receptors, may be promising adjuvants for IS management [37, 38].
As a class of important pervasive genes, a substantial body of evidence has indicated that miRNAs are key players in the regulation of the IS-induced immune-inflammatory response [17]. MiRNAs are noncoding 18–25-nucleotide-long RNA molecules that can negatively regulate the expression of specific target genes at the posttranscriptional level [39]. In the current study, a total of 14 miRNAs were identified from the constructed miRNA‒mRNA regulatory networks. Of these miRNAs, eight were upregulated, and six were downregulated. Several of these miRNAs have been confirmed to participate in the pathophysiological processes of IS in previous studies. For example, Zhang et al. found that the expression of miR-324-3p was downregulated both in patients with IS and in in vitro and in vivo models; they also found that silencing miR-324-3p can decrease the inflammatory response to protect against cerebral ischemic damage in MCAO rat models [40]. Mehta recently reported that the level of miR-7b was significantly reduced in the brain following transient focal ischemia in rodent models and that miR-7 supplementation promoted poststroke recovery by targeting α-Syn [41]. MiR-342-5p was reported to prevent neuronal apoptosis and attenuate cerebral ischemia–induced injuries in MCAO-treated mice via the inhibition of the Akt/NF-κB signaling pathway [42]. Additionally, several miRNA‒mRNA pairs in regulatory networks, such as miR-324-5p/TGFBR1 and miR-330-5p/CD44, have been confirmed through dual-luciferase reporter assays in previous studies [43,44,45]. Therefore, more experimental studies are warranted in the future to verify the biological functions of these miRNAs in IS and to clarify miRNA‒mRNA interactions. Therefore, more experimental studies are warranted in the future to verify the biological functions of these miRNAs in IS and to clarify miRNA‒mRNA interactions.
Microglia, also called brain macrophages, are the primary immune cells in the CNS and are essential for preserving brain homeostasis. These immune cells can be activated within minutes of a damaging event following IS, reach peak amounts of activity 2–3 days after the injury, and linger for weeks [46, 47]. The relationship between microglia and the inflammatory response to stroke is complicated. Intralesional microglia and newly recruited microglia assume an anti-inflammatory phenotype in the initial stages of ischemia but gradually transform into a pro-inflammatory phenotype in areas next to the lesions [48]. It is important to note that, although the M1/M2 paradigm has been criticized as an oversimplification of neuroinflammation, it continues to be the model that is used most to explain the function of microglia [28]. M1-type microglia secrete pro-inflammatory cytokines, whereas M2-type microglia alter the cellular microenvironment of the ischemic brain by clearing cell debris and releasing an anti-inflammatory cytokine panel, including Tgfb1 [28, 49]. Tgfb1 belongs to the Tgfb superfamily, which consists of several highly conserved multifunctional cell–cell signaling proteins that are important regulators of cell survival, immunomodulation, and post-injury repair and that play important roles in controlling tissue homeostasis [48, 50, 51]. In the current study, Tgfb1 was identified as a promising biomarker for IS. A significant increase in the expression and release of Tgfb1 has been observed in response to CNS lesions. Such elevated levels of Tgfb1 under early stroke conditions may, in turn, induce anti-inflammatory signaling cascades and drive microglia toward an anti-inflammatory M2 polarization shift from the pro-inflammatory M1 phenotype within the ischemic brain [51]. Xin et al. reported that inhibition of the TGF-β1 signaling pathway resulted in an augmented inflammatory response, while the administration of exogenous TGF-β1 protein increased the expression level of anti-inflammatory cytokines and reduced the release of pro-inflammatory cytokines in in vitro experimental models of IS [48]. At the signaling level, TGF-β1 protein induced the persistent anti-inflammatory effects on microglia, and this event may also be dependent on the activation of the canonical small mother against decapentaplegic-dependent signaling pathway, the prevention of the phosphorylation of IκB kinase, and the suppression of the NF-κB-dependent pathway [52]. Li et al. found that the crosstalk exists between TGFβ1 and NF-κB signaling pathway in multiple contexts [53]. A study by Gao et al. also showed evidence that the suppression on pro-inflammatory cytokines/chemokines and critical activators of inflammation exerted by TGF-β1 is partially mediated by the blockade on NF-κB pathways [54]. In this study, the NF-κB signaling pathway was enriched by KEGG analysis, implicating this pathway as a potential downstream mechanism for the anti-inflammatory effects of TGF-β1. TGF-β1 was also suggested to be anti-atherogenic by decreasing inflammation [55]. Inhibition of TGF-β1 signaling has been shown to accelerate plaque formation and its progression toward an unstable phenotype. Our study found that the lipid and atherosclerotic pathways were also significantly enriched by KEGG analysis. Therefore, lipids and atherosclerosis pathways may be a critical molecular pathway for TGF-β1 to participate in the pathological processes after IS. In another study, Sugimoto et al. reported that Tgfb1 expression can be highly induced in the ischemic brain of a rat stroke model [56]. Krupinski et al. showed that infarcted penumbra tissue has greater levels of TGF-β1 protein and TGF-β1 mRNA in human specimens than normal contralateral hemisphere tissue [57]. Zhang et al. also demonstrated that elevated TGF-β1 levels correlate with the extent of tissue injury in MCAO models [51]. In a prospective cohort study, Taylor observed that the concentrations of TGF-β1 in the peripheral blood of patients with strokes were associated with long-term outcomes [46]. As expected, a similar trend in TGF-β1 levels was observed in the MCAO model and the clinical patients in the current study. We also noted that patients with higher levels of TGF-β1 in peripheral blood may have better outcomes in the recovery phase after IS.
In addition to its anti-inflammatory effects, the Tgfb1 protein has been demonstrated to exert neurotrophic and neuroprotective effects [58]. Wen et al. presented data suggesting that peripheral Tgfb1 protein administration can reduce damage to the blood‒brain barrier and decrease the infiltration of neutrophils and macrophages, contributing to a reduction in neuronal damage and increasing neurological recovery [59]. In addition, several in vitro studies have linked Tgfb1 treatment to the promotion of axonal growth in several subtypes of neurons and have causally implicated Tgfb1 in the rehabilitation after strokes [46]. It has also been reported that the TGF-β1 protein may reactivate chronically denervated Schwann cells in vivo and could be used to prolong regenerative responses to promote axonal regeneration [60]. There is also evidence that the TGF-β1 protein is required for the full neuroprotective activity of nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and glial-derived neurotrophic factor (GDNF). Therefore, TGF-β1 might also promote neuronal survival and neurogenesis by acting synergistically with these neurotrophins [50]. In our study, results using the GSEA method supported this interpretation because the BP term of synaptic plasticity was significantly enriched. Therefore, synaptic plasticity may be one of the specific downstream mechanisms by which TGF-β1 exerts neuroprotective effects, and it is worth further investigation through experiments. Furthermore, a component of the neuroprotective action of Tgfb1 is mediated by inhibiting the activation of apoptotic proteins such as caspase-3, increasing the expression of antiapoptotic proteins such as Bcl-2 and Bcl-xl, and maintaining mitochondrial membrane potential [61, 62]. Blocking TGF-β1 signaling has been shown to increase the extent of infarction and deteriorate the homeostatic function of the CNS following brain ischemia in animal stroke models, whereas delivery of TGF-β1 via intranasal administration has been shown to enhance neurogenesis and improve functional outcomes [63], whereas delivery of TGF-β1 via intranasal administration has been shown to enhance neurogenesis and improve functional outcomes [56]. Wang et al. indicated that electroacupuncture at Baihui and Shuigou acupoints can further increase the level of Tgfb1 in MCAO rats to alleviate cerebral edema, attenuate infarct volume, and promote the recovery of neurological function. Hence, TGF-β1 may be a feasible and attractive molecular target for the management of IS [64].
Despite these promising findings, the current study has several limitations. First, the data analyzed were obtained from online databases and experimental rat models. Because of disease characteristics, we could not collect brain tissue from IS patients to validate our findings. Second, we integrated four datasets from different sequencing platforms, which could have biased our results. The results were also influenced by the timing, occlusion process (permanent or transient MCAO model), and severity of the animal models. Third, although we screened one candidate hub gene, its specific function and underling mechanism have not yet been elucidated in depth. Fourth, dual-luciferase gene reporter assays and other experiments should be used to further examine miRNA‒mRNA interactions in the regulatory network. Fifth, although we performed external validation, the sample size of the validation group was relatively small. Finally, longitudinal comparisons were needed to assess the dynamics of Tgfb1 expression change over time. Therefore, bioinformatics analyses demand further evaluation in larger prospective multicenter cohort studies, and there is also an urgent need for several in vitro and in vivo functional assays to support our findings.
Conclusions
The hub gene, Tgfb1, identified in this study exhibited stable expression patterns in both MCAO rat models and clinical IS patients. Moreover, accumulating evidence has highlighted the close association between this hub gene and IS. Notably, Tgfb1 demonstrated promising diagnostic efficacy for IS and the relationship with disease servility. These findings underscore the potential utility of Tgfb1 as a promising biomarker for IS diagnosis.
Materials and Methods
Data Acquisition
The mRNA and miRNA expression profile data of the IS in the original training and the external validation sets were downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo) into R software (version 4.2.3) using the GEOquery package (version 2.64.2) [65]. The training sets included the expression profiles of four mRNAs (GSE36010, GSE78731, GSE61616, and GSE106931) and one miRNA (GSE46266). The sequencing information in the training sets came from the brain tissue of MCAO rats. The GSE97537, GSE97533, and GSE66724 datasets were used for validation. GSE97537 and GSE97533 were sourced from the brain tissue and blood samples of MCAO rats, respectively. The GSE66724 dataset contained 16 blood samples from eight IS patients and eight HCs. More detailed information about the eight datasets is presented in Supplementary Table S5.
Data Preprocessing and Study Design
Raw data from each set were converted into an expression matrix and then processed with a standard workflow, including background correction and normalization, here by using the limma package (version 3.52.4) [66]. Afterward, the four training sets, including 21 MCAO model samples and 25 control samples, were merged and adjusted for batch effects using the ComBat function from the sva package (version 3.44.0) in R [67]. The specific flow diagram is shown in Fig. 14.

Flow chart of the analysis
Analysis of DEGs and DEMs
The limma package was used to explore DEGs and DEMs between the MCAO group and the control group, with a selection value of |log fold change (FC)|> 0.58 and a significance threshold of P < 0.05. Thereafter, volcano plots were generated based on the ggplot2 package (version 3.4.2) to visualize the results [60].
GSEA
The clusterProfiler package (version 4.8.3) [68] was used to perform GSEA on all gene expression matrices in the training set to find significantly enriched gene sets and their change trends. The enrichment was considered significant, with a value of |NES|> 1 and adjusted P < 0.05. The enrichment results, here arranged according to the NES, were visualized by ridgeline plots and GSEA enrichment plots using the ggplot2 package.
Construction of WGCNA and Identification of Modules Significantly Associated with IS
For the construction of the gene co-expression matrix, the Pearson correlation coefficients between gene pairs were calculated via the WGCNA package (version 1.72–1) [69]. The top 25% of genes with high expression variances were selected for analysis by the WGCNA method. Then, soft power = 22 and R2 = 0.85 were selected as the thresholding parameters based on the principle of a scale-free network to construct a biological network, and the resulting adjacency matrix was further transformed into a topological overlap matrix before being assigned to gene modules by hierarchical clustering analysis. The gene modules were defined as branches of the hierarchical clustering tree, and each module was assigned a color. Finally, the correlation between each module and the characteristics of the samples was measured by gene significance values and module membership values to identify key modules for subsequent research.
Predictions of miRNA‒mRNA Interactions
The target genes of the DEMs were predicted using miRWalk 2.0 (http://mirwalk.umm.uniheidelberg.de/) [70]. The predicted DEM-target genes overlapped with all DEGs as well as the genes in the WGCNA key module to obtain the intersecting genes. The intersection genes were used as the IS-related key genes in the current study for further analysis. The miRNA‒mRNA interactions based on the DEMs and IS-related key genes were visualized using Cytoscape (version 3.9.0) [71].
Functional Enrichment Analysis
Gene ontology (GO) analysis and KEGG pathway analysis were carried out to determine the biological functions and physiological functions of the above IS-related key genes using the clusterProfiler package. The GO terms included BP, cellular component, and MF. The significantly enriched results were filtered with P < 0.05 and visualized using the ggplot2 package.
Screening Potential Biomarkers Based on LASSO Regression
LASSO is a machine learning algorithm for regression that introduces a penalty coefficient λ to find the optimal model for reducing data dimensions. LASSO regression was performed using the glmnet package (version 4.7–1) in R to pick out the characteristic genes from the IS-related genes mentioned above, with tenfold cross-validation to select the optimal regularization parameter λ [72]. The parameters were set as follows: family = “binomial,” alpha = “1,” and “lambda. min” was chosen as the optimal lambda. Next, the prediction performance of these genes in the training and validation sets was measured by ROC curve analysis and the AUC. If a gene had an AUC greater than 0.7 in all validation sets, it was retained as an IS biomarker.
Establishment of the Rat MCAO Model
Rat is one of the most commonly used laboratory animal species in studies of IS. To verify the authenticity of the IS biomarkers, an MCAO model was induced in specific pathogen-free (SPF)-grade male Sprague–Dawley rats (6–8 weeks old, approximately 250–280 g per rat) purchased from Guangdong Weitong Lihua Experimental Technology Co., Ltd. (license no. SCXK [Yue] 2022–0063) and maintained in a constant environment (22–24 °C, 12/12-h light and dark cycle and 50–70% humidity) with food and water ad libitum. All animal care and experimental procedures were approved by the Guangxi Medical University Animal Research Ethics Committee (approval no. 202210004). All rats were allowed to adapt for 1 week prior to the operation.
A total of 16 rats were divided into the following two groups of eight rats each: the control group and the MCAO group. The rats in the MCAO group were induced by transient MCAO surgery under pentobarbital anesthesia, as previously described [73]. A silicone-coated nylon monofilament (diameter, 0.35–0.37 mm; Jialing Biotech Co. Ltd., Guangzhou, China) was inserted into the right middle cerebral artery from the external carotid artery and left in situ for 90 min. The rats were kept on a heating pad at 37 °C during the entire surgical procedure. The rats in the control group underwent the same operation process without filament insertion. After sufficient anesthesia, the rats were sent to clean cages. Rats with neurological deficit scores of 1 to 3 points according to a Longa 5-point scale at 24 h after reperfusion were considered effective in establishing an IS model and were included in the study [74]. The rats were sacrificed 3 days after reperfusion. Blood samples from circulation and brain samples from the ischemic hemisphere of the rats were collected for further experiments.
Neurological Deficit Evaluation
The mNSS test was performed to evaluate the neurobehavioral function of the rats, as described in a previous study [75]. All rats were subjected to this assessment before injury at 1 day and 3 days following the operation by two blinded investigators. The mNSS is a composite of four tests (motor, sensory, reflex, and beam balance tests). Function was scored on a scale of 0 to 18, where 0 represents normal and 18 represents maximum neurological dysfunction. Each rat was tested three times, and the average score was recorded.
H&E and Nissl Staining
After being fixed in 4% paraformaldehyde for 24 h at 4 °C, the brain samples were dehydrated and embedded in paraffin blocks. Then, serial 4-μm-thick cross-sections were made on slides and mounted with poly-L-lysine coating. The specimens were sequentially deparaffinized and rehydrated and then subjected to morphological examination via H&E and Nissl staining. The images were captured using an optical microscope (BX53, Olympus, Tokyo, Japan).
Study Population
Blood samples were collected from 10 patients with IS and 10 HCs at the Department of Rehabilitation Medicine of the First Affiliated Hospital of Guangxi Medical University between January 2023 and May 2023. The present study was approved by the ethical committee of the First Affiliated Hospital of Guangxi Medical University (approval no. 2022-K138-01) and identified in the Chinese Clinical Trial Registry (registration no. ChiCTR2200067102) on December 27, 2022. All IS patients underwent detailed and rigorous neurological examinations and brain magnetic resonance imaging (MRI) scans. The inclusion criteria were as follows: first-time IS, anterior circulation infarction, and large artery atherosclerosis confirmed by magnetic resonance angiography (MRA); disease duration ranging from 1 to 3 months; stable vital signs; and consciousness; and aged 18 to 75 years. The exclusion criteria were as follows: prior history of cerebral hemorrhage, severe head trauma or other neurological disease, rheumatic heart disease, heart failure, renal dysfunction, severe liver disease, or malignancy. The National Institutes of Health Stroke Scale (NIHSS) score and modified Rankin scale (mRS) score for each IS patient were collected upon admission. Each HC was age- and sex-matched to the corresponding IS patient. In addition, another 22 patients were later recruited between June 2023 and March 2024 using the same diagnostic and recruitment criteria to explore the correlation between the levels of biomarkers and IS severity evaluated by mRS. All participants provided written informed consent. Samples of approximately 4 mL of peripheral blood were taken from each participant via ethylenediaminetetraacetic acid anticoagulant tubes (Sanli, Liuyang, China) between 7 a.m. and 9 a.m. of the day after admission under 10 h of fasting. All procedures were conducted in accordance with the Declaration of Helsinki. All participants provided written informed consent.
qRT-PCR
Total RNA from brain and blood samples was extracted using NucleoZol RNA reagent (Macherey–Nagel, Duren, Germany) and transcribed into cDNA according to the instructions of the TranScript All-in-One First-Strand cDNA Synthesis SuperMix for qPCR Kit (TransGen, Beijing, China). The qRT‒PCRs were conducted with PerfectStart® Green qPCR SuperMix (TransGen, Beijing, China) on the StepOne Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). Furthermore, the expression of the housekeeping genes GAPDH/Gapdh was used as an internal control. Relative gene expression was quantified using the 2−ΔΔCt method. The experiment was repeated independently five times. The sequences of primers used in the current study were designed and synthesized by Sangon Biotech (Shanghai, China) and are listed in Table 3.
Statistical Analysis
Statistical analysis and image construction were performed using GraphPad Prism software, version 8 (GraphPad Software, La Jolla, CA, USA). Normally distributed continuous data are expressed as the mean ± standard deviation (SD) and were compared using Student’s t test. Nonnormally distributed variables are represented by medians and quartile ranges and were evaluated with the Wilcoxon–Mann–Whitney test. The correlation between the TGF-β1 level and the mRS score was analyzed using Spearman linear regression. P < 0.05 indicated statistical significance.
Data Availability
No datasets were generated or analysed during the current study.
References
Casas AI, Kleikers PW, Geuss E, Langhauser F, Adler T, Busch DH et al (2019) Calcium-dependent blood-brain barrier breakdown by NOX5 limits postreperfusion benefit in stroke. J Clin Invest 129:1772–1778. https://doi.org/10.1172/jci124283
Xing Y, Zhang Y, Li C, Luo L, Hua Y, Hu J et al (2023) Repetitive transcranial magnetic stimulation of the brain after ischemic stroke: mechanisms from animal models. Cell Mol Neurobiol 43:1487–1497. https://doi.org/10.1007/s10571-022-01264-x
Sun Y, Wang J, Han B, Meng K, Han Y, Ding Y (2021) Elucidating the molecular mechanism of ischemic stroke using integrated analysis of miRNA, mRNA, and lncRNA expression profiles. Front Integr Neurosci 15:638114. https://doi.org/10.3389/fnint.2021.638114
Ren P, Wang JY, Chen HL, Lin XW, Zhao YQ, Guo WZ et al (2022) Diagnostic model constructed by nine inflammation-related genes for diagnosing ischemic stroke and reflecting the condition of immune-related cells. Front Immunol 13:1046966. https://doi.org/10.3389/fimmu.2022.1046966
Candelario-Jalil E, Dijkhuizen RM, Magnus T (2022) Neuroinflammation, stroke, blood-brain barrier dysfunction, and imaging modalities. Stroke 53:1473–1486. https://doi.org/10.1161/strokeaha.122.036946
Zheng Y, Hu Y, Han Z, Yan F, Zhang S, Yang Z et al (2022) Lomitapide ameliorates middle cerebral artery occlusion-induced cerebral ischemia/reperfusion injury by promoting neuronal autophagy and inhibiting microglial migration. CNS Neurosci Ther 28:2183–2194. https://doi.org/10.1111/cns.13961
Zheng PF, Chen LZ, Liu P, Pan HW, Fan WJ, Liu ZY (2022) Identification of immune-related key genes in the peripheral blood of ischaemic stroke patients using a weighted gene coexpression network analysis and machine learning. J Transl Med 20:361. https://doi.org/10.1186/s12967-022-03562-w
Takuma A, Abe A, Saito Y, Nito C, Ueda M, Ishimaru Y, et al (2017) Gene expression analysis of the effect of ischemic infarction in whole blood. Int J Mol Sci 18. https://doi.org/10.3390/ijms18112335
Nie QQ, Zheng ZQ, Liao J, Li YC, Chen YT, Wang TY et al (2022) SPP1/AnxA1/TIMP1 as essential genes regulate the inflammatory response in the acute phase of cerebral ischemia-reperfusion in rats. J Inflamm Res 15:4873–4890. https://doi.org/10.2147/jir.S369690
Zhang H, Zhang Q, Liao Z (2019) Microarray data analysis of molecular mechanism associated with stroke progression. J Mol Neurosci 67:424–433. https://doi.org/10.1007/s12031-018-1247-3
Kumar A, Aakriti GV (2016) A review on animal models of stroke: an update. Brain Res Bull 122:35–44. https://doi.org/10.1016/j.brainresbull.2016.02.016
Fluri F, Schuhmann MK, Kleinschnitz C (2015) Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther 9:3445–3454. https://doi.org/10.2147/dddt.S56071
Lv W, Jiang J, Xu Y, Chen Z, Wang Z, Xing A et al (2023) Re-exploring the inflammation-related core genes and modules in cerebral ischemia. Mol Neurobiol 60:3439–3451. https://doi.org/10.1007/s12035-023-03275-1
Lyu M, Yi X, Huang Z, Chen Y, Ai Z, Liang Y, et al (2022) A transcriptomic analysis based on aberrant methylation levels revealed potential novel therapeutic targets for nasopharyngeal carcinoma. Ann Transl Med 10: 47. https://doi.org/10.21037/atm-21-6628
Miao F, Li X, Wang C, Yuan H, Wu Z (2023) Bioinformatics analysis of differentially expressed genes in diabetic foot ulcer and preliminary experimental verification. Ann Transl Med 11:89. https://doi.org/10.21037/atm-22-6437
Wang Z, Liu J, Wang Y, Guo H, Li F, Cao Y, et al (2023) Identification of key biomarkers associated with immunogenic cell death and their regulatory mechanisms in severe acute pancreatitis based on WGCNA and machine learning. Int J Mol Sci 24. https://doi.org/10.3390/ijms24033033
Pu L, Wang M, Li K, Feng T, Zheng P, Li S et al (2020) Identification micro-RNAs functional modules and genes of ischemic stroke based on weighted gene co-expression network analysis (WGCNA). Genomics 112:2748–2754. https://doi.org/10.1016/j.ygeno.2020.03.011
Zheng PF, Zou QC, Chen LZ, Liu P, Liu ZY, Pan HW (2022) Identifying patterns of immune related cells and genes in the peripheral blood of acute myocardial infarction patients using a small cohort. J Transl Med 20:321. https://doi.org/10.1186/s12967-022-03517-1
Lu M, Dong X, Zhang Z, Li W, Khoshnam SE (2020) Non-coding RNAs in ischemic stroke: roles in the neuroinflammation and cell death. Neurotox Res 38:564–578. https://doi.org/10.1007/s12640-020-00236-7
Qu YM, Sun X, Yan XL, Jin H, Guo ZN, Yang Y (2019) Identification of microRNAs and messenger RNAs involved in human umbilical cord mesenchymal stem cell treatment of ischemic cerebral infarction using integrated bioinformatics analysis. Neural Regen Res 14:1610–1616. https://doi.org/10.4103/1673-5374.255998
Li Z, Fang J, Yang J, Qin B, He W, Qi J et al (2022) Site of nerve division affects pain-related behavior and spinal cord glial proliferation after C7 neurotomy in a rat stroke model. Pain Res Manag 2022:7446482. https://doi.org/10.1155/2022/7446482
Tu-Chan AP, Natraj N, Godlove J, Abrams G, Ganguly K (2017) Effects of somatosensory electrical stimulation on motor function and cortical oscillations. J Neuroeng Rehabil 14:113. https://doi.org/10.1186/s12984-017-0323-1
Iadecola C, Buckwalter MS, Anrather J (2020) Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J Clin Invest 130:2777–2788. https://doi.org/10.1172/jci135530
Hughes SF, Cotter MJ, Evans SA, Jones KP, Adams RA (2006) Role of leucocytes in damage to the vascular endothelium during ischaemia-reperfusion injury. Br J Biomed Sci 63:166–170. https://doi.org/10.1080/09674845.2006.11732743
Koren-Morag N, Tanne D, Goldbourt U (2005) White blood cell count and the incidence of ischemic stroke in coronary heart disease patients. Am J Med 118:1004–1009. https://doi.org/10.1016/j.amjmed.2005.03.010
Doyle KP, Buckwalter MS (2017) Does B lymphocyte-mediated autoimmunity contribute to post-stroke dementia? Brain Behav Immun 64:1–8. https://doi.org/10.1016/j.bbi.2016.08.009
Engler-Chiurazzi EB, Monaghan KL, Wan ECK, Ren X (2020) Role of B cells and the aging brain in stroke recovery and treatment. Geroscience 42:1199–1216. https://doi.org/10.1007/s11357-020-00242-9
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P et al (2015) Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol 11:56–64. https://doi.org/10.1038/nrneurol.2014.207
Lenz M, Eichler A, Vlachos A (2021) Monitoring and modulating inflammation-associated alterations in synaptic plasticity: role of brain stimulation and the blood-brain interface. Biomolecules 11. https://doi.org/10.3390/biom11030359
Xu P, Zhang X, Liu Q, Xie Y, Shi X, Chen J et al (2019) Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis 10:555. https://doi.org/10.1038/s41419-019-1777-9
Sivandzade F, Prasad S, Bhalerao A, Cucullo L (2019) NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol 21:101059. https://doi.org/10.1016/j.redox.2018.11.017
Li R, Zhou Y, Zhang S, Li J, Zheng Y, Fan X (2022) The natural (poly)phenols as modulators of microglia polarization via TLR4/NF-κB pathway exert anti-inflammatory activity in ischemic stroke. Eur J Pharmacol 914:174660. https://doi.org/10.1016/j.ejphar.2021.174660
Li F, Xu D, Hou K, Gou X, Lv N, Fang W et al (2021) Pretreatment of indobufen and aspirin and their combinations with clopidogrel or ticagrelor alleviates inflammasome mediated pyroptosis via inhibiting NF-κB/NLRP3 pathway in ischemic stroke. J Neuroimmune Pharmacol 16:835–853. https://doi.org/10.1007/s11481-020-09978-9
Harari OA, Liao JK (2010) NF-κB and innate immunity in ischemic stroke. Ann N Y Acad Sci 1207:32–40. https://doi.org/10.1111/j.1749-6632.2010.05735.x
Stoll G, Bendszus M (2006) Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke 37:1923–1932. https://doi.org/10.1161/01.Str.0000226901.34927.10
Zhang X, Bishawi M, Zhang G, Prasad V, Salmon E, Breithaupt JJ et al (2020) Modeling early stage atherosclerosis in a primary human vascular microphysiological system. Nat Commun 11:5426. https://doi.org/10.1038/s41467-020-19197-8
Lambertsen KL, Biber K, Finsen B (2012) Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 32:1677–1698. https://doi.org/10.1038/jcbfm.2012.88
Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G (2008) Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des 14:3574–3589. https://doi.org/10.2174/138161208786848739
Zhang SS, Zhang JW, Zhang KX, Cui WQ, Zhi HW, Li HT et al (2023) Hsa-miR-877-5p expression in acute ischemic stroke based on bioinformatics analysis and clinical validation. Mol Neurobiol. https://doi.org/10.1007/s12035-023-03675-3
Zhang AQ, Wang L, Wang YX, Hong SS, Zhong YS, Yu RY et al (2022) Silencing miRNA-324-3p protects against cerebral ischemic injury via regulation of the GATA2/A1R axis. Neural Regen Res 17:2504–2511. https://doi.org/10.4103/1673-5374.339009
Mehta SL, Chokkalla AK, Bathula S, Vemuganti R (2023) MicroRNA miR-7 is essential for post-stroke functional recovery. Transl Stroke Res 14:111–115. https://doi.org/10.1007/s12975-021-00981-7
Zhu H, Zhang Y, Zhu Y (2023) MiR-342-5p protects neurons from cerebral ischemia induced-apoptosis through regulation of Akt/NF-κB pathways by targeting CCAR2. J Stroke Cerebrovasc Dis 32:106901. https://doi.org/10.1016/j.jstrokecerebrovasdis.2022.106901
Lang C, Dai Y, Wu Z, Yang Q, He S, Zhang X et al (2020) SMAD3/SP1 complex-mediated constitutive active loop between lncRNA PCAT7 and TGF-β signaling promotes prostate cancer bone metastasis. Mol Oncol 14:808–828. https://doi.org/10.1002/1878-0261.12634
Wang R, Dong HX, Zeng J, Pan J, Jin XY (2018) LncRNA DGCR5 contributes to CSC-like properties via modulating miR-330-5p/CD44 in NSCLC. J Cell Physiol 233:7447–7456. https://doi.org/10.1002/jcp.26590
Huang M, Huang X, Huang N (2022) Exosomal circGSE1 promotes immune escape of hepatocellular carcinoma by inducing the expansion of regulatory T cells. Cancer Sci 113:1968–1983. https://doi.org/10.1111/cas.15365
Taylor RA, Chang CF, Goods BA, Hammond MD, Mac Grory B, Ai Y et al (2017) TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J Clin Invest 127:280–292. https://doi.org/10.1172/jci88647
Dabrowska S, Andrzejewska A, Lukomska B, Janowski M (2019) Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J Neuroinflammation 16:178. https://doi.org/10.1186/s12974-019-1571-8
Xin W, Pan Y, Wei W, Gerner ST, Huber S, Juenemann M, et al (2023) TGF-β1 decreases microglia-mediated neuroinflammation and lipid droplet accumulation in an in vitro stroke model. Int J Mol Sci 24. https://doi.org/10.3390/ijms242417329
Liu C, Liu S, Xiong L, Zhang L, Li X, Cao X et al (2021) Genistein-3′-sodium sulfonate attenuates neuroinflammation in stroke rats by down-regulating microglial M1 polarization through α7nAChR-NF-κB signaling pathway. Int J Biol Sci 17:1088–1100. https://doi.org/10.7150/ijbs.56800
Caraci F, Battaglia G, Bruno V, Bosco P, Carbonaro V, Giuffrida ML et al (2011) TGF-β1 pathway as a new target for neuroprotection in Alzheimer’s disease. CNS Neurosci Ther 17:237–249. https://doi.org/10.1111/j.1755-5949.2009.00115.x
Zhang L, Wei W, Ai X, Kilic E, Hermann DM, Venkataramani V et al (2021) Extracellular vesicles from hypoxia-preconditioned microglia promote angiogenesis and repress apoptosis in stroke mice via the TGF-β/Smad2/3 pathway. Cell Death Dis 12:1068. https://doi.org/10.1038/s41419-021-04363-7
Islam A, Choudhury ME, Kigami Y, Utsunomiya R, Matsumoto S, Watanabe H et al (2018) Sustained anti-inflammatory effects of TGF-β1 on microglia/macrophages. Biochim Biophys Acta Mol Basis Dis 1864:721–734. https://doi.org/10.1016/j.bbadis.2017.12.022
Li F, Kang X, Xin W, Li X (2022) The emerging role of extracellular vesicle derived from neurons/neurogliocytes in central nervous system diseases: novel insights into ischemic stroke. Front Pharmacol 13:890698. https://doi.org/10.3389/fphar.2022.890698
Gao P, Wu W, Ye J, Lu YW, Adam AP, Singer HA et al (2018) Transforming growth factor β1 suppresses proinflammatory gene program independent of its regulation on vascular smooth muscle differentiation and autophagy. Cell Signal 50:160–170. https://doi.org/10.1016/j.cellsig.2018.07.002
Edsfeldt A, Singh P, Matthes F, Tengryd C, Cavalera M, Bengtsson E et al (2023) Transforming growth factor-β2 is associated with atherosclerotic plaque stability and lower risk for cardiovascular events. Cardiovasc Res 119:2061–2073. https://doi.org/10.1093/cvr/cvad079
Sugimoto K, Nishioka R, Ikeda A, Mise A, Takahashi H, Yano H et al (2014) Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-β1. Glia 62:185–198. https://doi.org/10.1002/glia.22598
Krupinski J, Kumar P, Kumar S, Kaluza J (1996) Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke 27:852–857. https://doi.org/10.1161/01.str.27.5.852
Caraci F, Gulisano W, Guida CA, Impellizzeri AA, Drago F, Puzzo D et al (2015) A key role for TGF-β1 in hippocampal synaptic plasticity and memory. Sci Rep 5:11252. https://doi.org/10.1038/srep11252
Wen H, Tan J, Tian M, Wang Y, Gao Y, Gong Y (2023) TGF-β1 ameliorates BBB injury and improves long-term outcomes in mice after ICH. Biochem Biophys Res Commun 654:136–144. https://doi.org/10.1016/j.bbrc.2023.03.007
Wang H, Cao X, Wen X, Li D, Ouyang Y, Bao B, et al (2021) Transforming growth factor‑β1 functions as a competitive endogenous RNA that ameliorates intracranial hemorrhage injury by sponging microRNA‑93‑5p. Mol Med Rep 24. https://doi.org/10.3892/mmr.2021.12138
Zhu Y, Ahlemeyer B, Bauerbach E, Krieglstein J (2001) TGF-beta1 inhibits caspase-3 activation and neuronal apoptosis in rat hippocampal cultures. Neurochem Int 38:227–235. https://doi.org/10.1016/s0197-0186(00)00084-x
Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C et al (2002) Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci 22:3898–3909. https://doi.org/10.1523/jneurosci.22-10-03898.2002
Ruocco A, Nicole O, Docagne F, Ali C, Chazalviel L, Komesli S et al (1999) A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J Cereb Blood Flow Metab 19:1345–1353. https://doi.org/10.1097/00004647-199912000-00008
Wang WB, Yang LF, He QS, Li T, Ma YY, Zhang P et al (2016) Mechanisms of electroacupuncture effects on acute cerebral ischemia/reperfusion injury: possible association with upregulation of transforming growth factor beta 1. Neural Regen Res 11:1099–1101. https://doi.org/10.4103/1673-5374.187042
Davis S, Meltzer PS (2007) GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23:1846–1847. https://doi.org/10.1093/bioinformatics/btm254
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W et al (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. https://doi.org/10.1093/nar/gkv007
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28:882–883. https://doi.org/10.1093/bioinformatics/bts034
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287. https://doi.org/10.1089/omi.2011.0118
Horvath S, Dong J (2008) Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol 4:e1000117. https://doi.org/10.1371/journal.pcbi.1000117
Sticht C, De La Torre C, Parveen A, Gretz N (2018) miRWalk: an online resource for prediction of microRNA binding sites. PLoS ONE 13:e0206239. https://doi.org/10.1371/journal.pone.0206239
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. https://doi.org/10.1101/gr.1239303
Friedman J, Hastie T, Tibshirani R (2010) Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33:1–22
Wang RY, Yang YR, Chang HC (2022) The SDF1-CXCR4 axis is involved in the hyperbaric oxygen therapy-mediated neuronal cells migration in transient brain ischemic rats. Int J Mol Sci 23. https://doi.org/10.3390/ijms23031780
Song L, Mu L, Wang H (2022) MicroRNA-489-3p aggravates neuronal apoptosis and oxidative stress after cerebral ischemia-reperfusion injury. Bioengineered 13:14047–14056. https://doi.org/10.1080/21655979.2022.2062534
Hong Y, Liu Q, Peng M, Bai M, Li J, Sun R et al (2020) High-frequency repetitive transcranial magnetic stimulation improves functional recovery by inhibiting neurotoxic polarization of astrocytes in ischemic rats. J Neuroinflammation 17:150. https://doi.org/10.1186/s12974-020-01747-y
Acknowledgements
The authors would like to express their appreciation to Tiande Huang, Lanying Huang, and Kewen Wang (The First Affiliated Hospital of Guangxi Medical University) for their support on the experiment.
Funding
This work was supported by the Joint Funds for the Innovation of Science and Technology, Fujian Province (Grant numbers: 2023Y9133, 2020Y9110) and National Natural Science Foundation of China (Grant number: 82172531).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. JMC and XLL were responsible for experimental design. Data collection and analysis were performed by YY and QFC. XLL and QFC were responsible for the creation of new software used in the work. XLL and YY performed the experimental validation. JMC and XLL have been involved in drafting the manuscript. JMC, QFC and JN provided financial support. The first draft of the manuscript was written by JMC. JN reviewed and revised the manuscript. All authors read and approved final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
The part involving human subjects in this study was approved by ethical committee of the First Affiliated Hospital of Guangxi Medical University (No. 2022-K138-01) and also identified in the Chinese Clinical Trial Registry (registration no. ChiCTR2200067102) on December 27, 2022. All experiments were performed in accordance with relevant guidelines and regulations such as the Declaration of Helsinki and the patients signed the informed consent form and agreed to be published. All animal care and experimental procedures in this study were approved by the Guangxi Medical University Animal Research Ethics Committee (No. 202210004).
Consent for Publication
All authors agree to the publication of this manuscript.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Q., Li, X., Yang, Y. et al. Combined Analysis of Human and Experimental Rat Samples Identified Biomarkers for Ischemic Stroke. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04512-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04512-x