
Abstract
Thousand grain weight (TGW) is an important determinant of rice yield, and correlates with grain size, plumpness and grain number per panicle. In rice, there are fewer association mapping studies relating grain weight traits using both SSR and SNP markers. In this study, in order to find robust SSR markers associated with TGW trait and mine elite accessions in rice, we investigated the TGW trait across six environments using a natural population consisted of 462 accessions, and then performed association mapping using both SSR and SNP markers. Using the six datasets from the six environments and their best linear unbiased estimator, we identified eight TGW associated SSR markers, with three environmentally stable and one newly found, on five chromosomes. The associated markers have genetic effect from 3.44% to 20.84%, and two of them carry stable elite allele with positive effect across different environments. Candidate interval association mapping using re-sequencing derived SNP/InDel markers further confirms the TGW-SSR association, and also suggests that 3 TGW-SSR associations were high confident in intervals of size from 176 to 603 kb. These results not only shed more lights on the genetics of TGW trait, but also suggest that the multi-allelic SSR markers should be used as an alternative power tool in gene or QTL mapping.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Current food production is becoming limited because of shortages of cropland, water, and shortages of fertilizers that depend on fossil energy (Pimentel 2012). Today, used as the main food for more than half of the world’s population, rice is expected to give higher yield to meet the needs of the increasing population (Fageria 2007).
Crop yield is a quantitative trait and has complex genetic background. For rice, the panicle number, grain number, and grain weight are three main components of the yield. Beyond the panicle number per unit area and grain number per panicle, the improvement of grain weight is the major way to further increase the yield (Xing and Zhang 2010).
Grain weight is a synthetic reflect of grain length, width, thickness and plumpness. All these traits have been comprehensively studied using bi-parental populations through QTL mapping (Xing et al. 2002; Li et al. 2004, 2020; Weng et al. 2008; Wang et al. 2011; Tang et al. 2013; Xu et al. 2015; Dong et al. 2018; Bazrkar-Khatibani et al. 2019), and hundreds of QTLs related to these traits were reported and stored in the Gramene QTL Database (Gupta et al. 2016).
Based on these or similar studies, many genes relating grain size or weight have been cloned. For example, GS3 (Fan et al. 2006), GL3 (Zhang et al. 2012), GL7 (Wang et al. 2015), GW2 (Song et al. 2007), qSW5 (Shomura et al. 2008), GW5 (Weng et al. 2008; Liu et al. 2017), GS5 (Li et al. 2011), GW8 (Wang et al. 2012), TGW3 (thousand grain weight) (Ying et al. 2018), TGW2 (Ruan et al. 2020) and so on, were reported to regulate grain size and/or weight in rice.
Besides QTL mapping, linkage disequilibrium (LD) based association mapping was used to investigate the genetics of grain weight-related traits in rice, and SNP markers derived from gene chips or genome sequencing were preferred (Huang et al. 2010; Huang et al. 2011; Zhao et al. 2011; Yang et al. 2014; Begum et al. 2015; Liu et al. 2019). However, SSR markers have many advantages comparing with SNP markers, such as highly polymorphic nature, ease of development, and easy-using, and thus is essential for marker assistant selection (MAS) and crop improvement (Collard and MacKill 2008; Miah et al. 2013).
In rice, maybe due to a low automatic level and labor-intensive, there is relatively fewer association mapping studies relating grain weight trait using SSR in large population. In order to find robust SSR markers associated with TGW trait and mine elite accessions in rice, we performed a TGW-SSR association analysis cross six environments, and further confirmed the result using SNP markers. Our study is featured by 1) very high-quality phenotypes of TGW trait, 2) genetic distinct germplasm lines, and 3) super polymorphic SSR markers. The result not only gives invaluable SSR marker which can be used in MAS, but also suggests that combining SSR and SNP markers should be a lower-cost way in finding trait-marker associations.
Results
The Phenotype Evaluation of TGW
All the 462 rice accessions, consisted of 340 lines from China, 1 line from Japan, and 121 lines from Vietnam, were planted in six environments, i.e., three locations in two years (Table S1). In each environment, the lines were planted in four replications and phenotyped separately for each replication. As a result, 24 datasets (3 locations × 2 years × 4 replications) were obtained for TGW (Table S2). To get a good proxy of all the information in the 24 datasets, the BLUE (best linear unbiased estimator) of TGW was calculated (Table S2).
In each environment, the four replications agree well with each other, and the correlation coefficient varies from 0.92 to 0.99 between replications (Table S3). Thus, a total of 7 datasets, including the mean values of the replications in each of the six environments and the BLUE, were used for the following analysis (Table 1; Table S2). The minimum, maximum, mean and median of the 7 datasets are in range of 15.62–17.26, 31.87–35.50, 23.74–24.37 and 23.54–24.37 g, respectively (Table 1).
To analyze the stability of TGW under different environments, the correlation coefficient among the six environments was calculated. The result showed the correlation coefficient of TGW has a range of 0.58–0.78 among the six environments, and all the environments, except for NJ14, have a correlation coefficient ≧0.83 with the BLUE (Table 1). Meanwhile, the distribution of TGW trait in the natural population consisted of 462 rice accessions fitted the normal distribution by normal distribution test (P > 0.01) (Table 1, Fig. 1), indicating that the trait is controlled by polygenes. The broad-sense heritability of TGW in our population was 80.44% calculated by using the datasets from the six environments.

The histogram of the TGW trait from the 7 datasets, i.e., a NJ13, b XY13, c YY13, d NJ14, e XY14, f YY14 and g BLUE. The horizontal axis stands for the TGW trait with unit “g”; the vertical axis stands for the count of accessions with the categorized TGW values
The Marker Polymorphism and LD Analysis
To analyze the genetic diversity of the above 462 accessions, a total of 264 evenly distributed SSR markers were genotyped (Tables S4, S5). Of these, 261 markers, with 17 to 29 markers per chromosome, showed polymorphism among the accessions, and a total of 3361 alleles, ranging from 2 to 47 with an average of 12.88 per locus, were found (Table 2).
For the polymorphic markers, the statistical parameters were similar among the 12 chromosomes (Table S4). The major allele frequency has a range of 0.11–0.98, with the median and mean of 0.28 and 0.35 respectively while the minor allele frequency has a range of 0.01–0.41, with the median and mean of 0.14 and 0.16 respectively. These markers were very informative, 219 markers (83.91%) has a PIC (polymorphic information content) value > 0.5, only 6 markers were slightly informative (PIC < 0.2), and three marker (RM129, RM1108 and RM512) do not show polymorphism (PIC = 0) (Table S4). The PIC value was negatively correlated with the major allele frequency (r = -0.98, P < 0.001) and positively correlated with allele number (r = 0.76, P < 0.001).
To evaluate the linkage relationship of the markers, allele-level pairwise linkage disequilibrium for the 261 polymorphic markers in the 462 accessions were analyzed. As a result, 7,521,191 allele pairs were found. Of these, 766,571 (10.19%) pairs, including 64,071 pair within the same chromosome, showed strong LD (P < 0.001) (Table S6). For the allele pairs in strong LD, the minimum, maximum, median and mean of r2 were 0.018, 1.00, 0.06 and 0.13, whereas the allele frequency is 0.001, 0.99, 0.08 and 0.11.
Population Structure Analysis
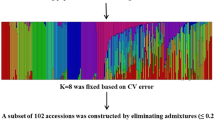
Before conducting the marker and trait association analysis, we estimated the population structure using the polymorphic markers. The most likely number of subpopulations (K) in the 462 accessions was estimated to be 3 based on the delta K value (Fig. 2a). However, there was also a signal on K = 5 (Fig. 2a). To further confirm the population structure of the accessions, a NJ (neighbor joining) tree was constructed using the SSR markers. Four main branches (I, II, III and IV), as well as sub-branches in each main branch, were evident in the tree (Fig. 2b). Since more than 3 branches were observed in the NJ tree, we selected K = 5 for the population structure, in which 172, 48, 98, 96 and 48 accessions were grouped into population 1 to 5 respectively (Fig. 2c). Vietnam accessions were mainly grouped into subpopulation 1, 3 and 4. A striking feature of materials was that their admixture level (the fraction of genome from different subpopulations) was quite low, 446 (92.53%) accessions inherited more than 90% fraction of their genome from their belonging subpopulation (Fig. 2c). Comparing the results of the Structure program and the NJ tree, subpopulation 2, 3 and 5 from the Structure result were mainly in branch I, IV and III, and subpopulation 1 and 4 were scattered in branch I & II, and III & IV respectively (Fig. 2b).

Population genetic architecture analysis of the 462 rice accessions. a rate of change of the likelihood distribution calculated as Delta K b the neighbor joining tree calculated based on the SSR markers in Tassel 3 program; the Roman number I, II, III and IV stands for four main branches c model based population structure defined by Structure program when K = 5
Association Mapping of SSR Marker and TGW
To set the proper cut-off value (significance threshold) for the association mapping, 2000 times permutation tests were performed by reshuffled the original phenotype data and then association mapping. As a result, the 1% and 5% cut-off levels were set for the above 7 phenotype datasets respectively for the real marker-phenotype association (Table 3).
In total, 19 marker-TGW association pairs, with marker R2 ranged from 3.44% -20.84%, were found, and 8 markers distributed in 5 chromosomes were involved (Table 4, Fig. 3). Of these association pairs, 5 were significant at the 1% level, with the rest at the 5% level. Of the 7 datasets, no marker-TGW association pairs was found in dataset the NJ14, while 1 to 5 pairs in the other datasets. This result was consistent with the phenotype analysis result that the dataset NJ14 has a lower quality suggested by the correlation analysis. Of the 8 associated markers, both RM566 and RM3600 were identified in 4 datasets, and thus should be stable; and RM259 had the largest effect with R2 ranged from 12.38% in dataset YY14 to 20.84% in XY13 (Table 4).

The manhattan plot of the TGW-SSR marker association in dataset a NJ13, b XY13, c YY13, d NJ14, e XY14, f YY14 and g BLUE. The blue and red line indicates the cut-off values at 1% and 5% level respectively
To mine elite alleles for TGW trait, the allele effect of the above associated markers was predicted (Table S7). As a result, 108 negative values were detected, comparing 42 positive values, suggesting that most alleles give a negative effect. Interestingly, there were 2 alleles, i.e. 237/237 bp (base pair) in marker M566, and 150/150 bp in marker RM3600, give stable positive effect in different environments, and there are 102 and 29 accessions carrying the 2 alleles respectively. The average TGW of the 102 and 29 accessions were 24.87 (calculated from the datasets XY13, XY14 and YY14) and 24.61 (calculated from the datasets XY13, YY13 and YY14) respectively, and both were significantly larger than the global average value 24.01 (student t-test, P < 1E-03).
Candidate Interval Based Association Mapping of TGW Trait using SNP Markers
To further confirm the association mapping result above and give a higher resolution of the associated loci, the association mapping between the SNP/InDel variants flanking the target markers and TGW trait was performed using 57 accessions (Table 5). Of the 8 associated markers, 7 (except RM244) were properly aligned to the reference genome, and the sequences with length ± 500 kb (kilo bases) flanking the markers were retrieved. Thus, a total of 7 MB genome segment were analyzed, and total of 98,599 variants, were identified (Table 5, Table S8). Based on cut-off values from permutations (Table 3), a total of 1,313 loci were found to significantly associated with TGW in at least one of the 7 datasets at the significance 1% or 5% (Table 5, Table S9, Fig S1). The association mapping result showed that significant association signals exist in each of the intervals flanking the 7 markers, but most loci were located in the interval flanking the marker RM486, RM566 and RM255, while less were found in the interval flanking the rest of the markers (Table 5, Table S9). For the marker RM486, RM225 and RM566, most significant loci were located in the interval 34,840–35,443 (603 kb in length), 3,009–3,292 (283 kb), and 14,602–14,778 kb (176 kb), respectively (Table S9).
Discussion
Despite of the controversy over the origins of rice (Fuller et al. 2007), the Asian-cultivated rice accessions occupy an important position, and is grown worldwide and comprises the staple food for half of the global population. Of the 462 accessions used in this study, part was from Vietnam (latitudes below 17°N), and part was from northeastern China and Japan, (latitudes above 45°N) (Table S1). We observed little genetic admixture among the accessions (Fig. 1), and this situation agrees well with previous studies (Liu et al. 2015; Dang et al. 2016; Edzesi et al. 2016; Liu et al. 2017). This result may partly because that a fewer marker used in inferring the population structure, and some admixture may be missed out. However, the distinct genetic background is a true reflection of the feature of our materials. The 2 parts of our materials belongs to the subspecies indica (accessions from Vietnam) and japonica (accessions from northeastern China and Japan) respectively. The two subspecies were rarely hybridized due to different flowering time (Liu et al. 1996). Therefore, these germplasm lines are invaluable for genetic studies and rice breeding. The high polymorphic feature of our materials was also reflected by our SSR result. We found 2 to 47 alleles with an average of 12.88 per locus, and the average PIC for all markers was 0.72. The polymorphic level of our population is higher than that from Garris et al. (2005), Agrama et al. (2007) Vanniaraja et al. (2012) and Dang et al. (2015).
TGW is an important trait for many crops, such as maize, rice, soybean and wheat, and is quantitative trait which can be easily affected by environmental factors. In order to find robust SSR markers associated with TGW trait and mine elite lines in rice, we used 462 rice accessions and phenotyped them in 6 environments. A total of 24 datasets (including replications) were obtained, and an incredible consistency among these datasets indicating by high correlate coefficient was observed (Table 1, Tables S2, S3). There were some studies that studied TGW gene mapping, but usually used datasets from 3 to 4 environments (Dang et al. 2015; Qiu et al. 2015; Feng et al. 2016). Given the development of the high-throughput and low-cost genotyping technology, phenotyping will become more and more important. In our association mapping, 7 datasets were used, and all the datasets fit a normal distribution well (Table 1, Fig. 1). Comparing with other studies, our high quality phenotype datasets would lay a good foundation for the following QTL mapping.
Robust marker-trait association is essential in MAS. In this study, we found 8 SSR markers associating with TGW trait (Table 4). Of the 8 markers, RM3600 was a newly found; while the other 7 markers, i.e., RM259, RM486, RM218, RM225, RM314, RM566 and RM244, were reported to associate with TGW by previous studies (Moncada et al. 2001; Cui et al. 2002; Hua et al. 2002; Zhuang et al. 2002; Gao et al. 2004; Jiang et al. 2004. However, none of these previous studies covered all the markers in one studies. Interestingly, these markers were also reported to associated with grain number (marker RM259) (Hua et al. 2003), grain yield (marker RM486 and RM314) (Moncada et al. 2001), and biomass yield (marker RM225) (Lian et al. 2005). Therefore, this result genetically explained why these trait correlates with each other.
Of the 462 accessions, there are 2 accessions, i.e. Z7238 and Z5164, have higher TGW values, and are slightly out-lied of others (Fig. 1, Fig S2). The TGW phenotype of the 2 outliers is stable cross different environments (Fig S2), and thus should be controlled by genetical factors. In order to test whether our association result differ with or without the 2 outliers, we performed the SSR-TGW association after removing the 2 outliers. As a result, the result was very similar: 6 of the 8 markers, except RM314 and RM244, were detected (Table S10). This may partly because that an inappropriate cut-off (i.e., the cut-off listed in Table 3, which were derived from the permutation of all the samples) was used. To sum up, our result is robust to a few outliers, because there are at least 10 accessions for each allele.
Of the 8 TGW associated markers, 3 markers, i.e. RM259, RM566 and RM3600, were identified in 3 out of the 6 environments, and thus could be regarded as environmentally stable. Luckily, based on the stable markers, we identified 2 alleles, i.e. 237/237 bp in marker M566 and 150/150 bp in marker RM3600, give stable positive effect in different environments, and 102 and 29 elite accessions carrying the 2 alleles were found respectively (Table S7).
The re-sequencing data of 57 accessions was used for validating the SSR association mapping result and getting a higher resolution of the associated loci. The size of ± 500 kb was used because it is a safe size for linkage disequilibrium is rice (Mather et al. 2007). We performed a permutation in testing SNP/InDel-TGW association using the whole genome-wide SNPs (11.54 Million) in our re-sequenced population (57 samples), few significant signals were detected (data not shown). However, when using the SNP/InDel variants in our candidate intervals, significant SNP-TGW associations were detected (Table 5). Due to LD of variants in association studies, the variants close to the true associated loci usually give strong association signals (Mather et al. 2007). Therefore, for our SNP/InDel-TGW association result, we proposed that the number of significant TGW-associated loci in the target interval was a good proxy to validate the SSR-TGW association result. We found 974, 123 and 189 unique loci in the intervals flanking the marker RM486, RM225 and RM566 respectively (Table 5). This result suggests that true TGW-associated loci should exist in the intervals. Furthermore, this result narrowed down the candidate intervals to Chr1:34,840–35,443, Chr1:3,009–3,292, and Chr9:14,602–14,778 kb for marker RM486, RM225 and RM566 respectively (Table S9). Whereas the marker RM259, RM218 and RM3600, few associated loci were found in their intervals (Table 5), and the probable explanations could be 1) there is no true TGW-associated loci in the intervals, 2) the re-sequencing population is not large enough to get a significant result, and 3) the true TGW-associated loci is outside of the intervals.
In this study, we also planned to test that multi-allelic SSR markers have a higher discrimination power than bi-allelic SNP markers in association mapping. However, we tested the intervals flanking the 7 markers, and got significant association signals in every interval when using SNP markers (Table 5). It seems that the low level polymorphism limitation of SNP markers was compensated by their abundance in chromosomes. On the other hand, if in strong LD block, SNP markers indeed have lower discrimination power, which is a common and annoying problem in gene or QTL mapping (Tsykun et al. 2017). Of the 8 TGW associated markers we found, RM566 was in strong LD with 7 markers (i.e., RM3600, RM3533, RM6570, RM201, OSR28, RM1013, and RM3912-2), and RM244 was in strong LD with 9 markers (i.e., RM216, RM311, RM1125, RM258, RM269, RM6160, RM590, RM333 and RM6646) (Table S6). We successfully ruled out the LD markers from the truly associated ones in our result (Table 4). This result may suggest that the multi-allelic SSR markers were an alternative tool to conquer LD in gene or QTL mapping. However, further comprehensive simulations should be performed to prove this.
Methods
Plant Materials and Phenotyping of TGW
A total of 462 rice accessions, stored and supplied by State Key Laboratory of Crop Genetics and Germplasm Enhancement (Nanjing Agricultural University), were used as the plant materials. Of these, 121 were from Vietnam, and 340 were from China, 1 was from Japan (Table S1). These rice lines were representative germplasm resources of Asia and have been widely using in breeding research in China.
To evaluate the TGW phenotype, all the accessions were planted in 3 location for 2 years, i.e., Nanjing (the Experiment Farm, Nanjing Agricultural University, 32.01°N, 118.64°S) (short for NJ13 and NJ14), Xinyang (32.12°N, 114.067°S) (short for XY13 and XY14) and Yuanyang (35.06°N, 113.94°S) (short for YY13 and YY14) in 2013 and 2014. In each of the field experiments, 4 replications in randomized block design were used, and every line was planted in 5 rows, with 8 plants each row, 20 cm between rows and 17 cm between each plant. The plants were grown in normal rice growing season condition (from May to October each year) until totally mature. All the plants of each accession were harvested together, and the seed were dried for 4 days under sun. For the TGW phenotyping, 1000 plump grains were random selected and weighted. This phenotyping process was repeated for 3 times, and the average value was used for further analysis.
SSR Marker Genotyping and Variants Detection
The genomic DNA of each accession was extracted from young leaf tissue using DNeasy Plant Mini Kits (QIAGEN, Hilden, Germany) according to the kit manual instruction. A total of 264 SSR markers were selected from the Gramene database (https://archive.gramene.org/) according to their distribution on the rice 12 chromosomes (Tello-Ruiz et al. 2018) (Table S4). Primers were synthesized in Generay Biotech (Shanghai) Co., Ltd. (Shanghai, China). A 10 μl PCR reaction system was used: 10 mM tris–HCl (pH 9.0), 50 mM KCl, 1.5 mM MgCl2, 0.1% triton X-100, 0.5 nM dNTPs, 0.14 pM primers, 0.5U Taq polymerase, and 20 ng template DNA. The amplification was performed using a PTC-100™ Peltier thermal cycler (MJ research™ Incorporated, The USA) based on a standard PCR condition. The PCR products were run on 8% polyacrylamide gel at 150 V for 1–1.5 h, and visualized using silver staining. The size of PCR product was calculated by the software Quantity One.
The genome re-sequencing was performed on an Illumina Hiseq2000 platform (Illumina, San Diego, USA) according to its standard sequencing protocol, and pair-end library with insert length of 300 bp was used. The rice genome assembly IRGSP-1.0 (c.v. Nipponbare) was downloaded from NCBI (https://www.ncbi.nlm.nih.gov/assembly/GCF_001433935.1/) and used as the reference genome. Sequencing reads of the individuals were first filtered using Trimmomatic 0.33 (Bolger et al. 2014) and then mapped to the reference using BWA (Burrows Wheeler Aligner) 0.7.17 (Li and Durbin 2009). In both programs, default settings were used. After mapping, duplicates marking, base quality re-calibrating, SNP/InDel joint calling were performed according to GATK (Genome Analysis Toolkit) best practice in GATK 3.8 (Van der Auwera et al. 2013). The resulting variants were filtered in Tassel5 (Bradbury et al. 2007) with the following parameter: minimum sample counts cut-off 20%, minor allele frequency cut-off 0.05. The location of the SSR markers was determined based on alignment of their primer sequences and the reference genome, in which the tool short-blastn in BLAST 2.7 (Camacho et al. 2008) was used.
Phenotype Statistical Analysis
All the description statistical analyses were performed in R language. The broad sense heritability was computed using the lme4 package (Douglas et al. 2014) using the formula H = Vg/(Vg + Vge/L + Ve/RL), where Vg is the genetic variance, Vge is variance of genetic by environment, Ve is the error variance, and L is number of environment and R is the number of repeats in each environment. The BLUE value of each accession was calculated in the lme4 package based on the year × location × replication matrix, with the accession as fix effect and year, location as random effect (Douglas et al. 2014).
Marker Diversity and Population Structure Analysis
The diversity of the SSR markers was analyzed in PowerMarker 3.25. PIC was calculated for each marker using the following formula: PIC = 1—Σ(i from 1 to n)Pi2—Σ(i from 1 to n-1)Σ(j from i+1 to n) 2 × Pi2Pj2, where Pi and Pj are the allele frequencies at alleles i and j, respectively, and n is the number of alleles (Botstein et al. 1980). A higher PIC indicates that a marker is more useful for distinguishing individuals and understanding relationships among them. The major and minor allele frequencies refer to the frequency at which the first and second most common allele occurs in a given population, respectively (Florez 2013). Due to lacking of polymorphism, three markers, i.e., RM129, RM1108 and RM512, were discarded in further analysis. For measuring SNP/InDel variants diversity, nucleotide diversity, defined by Nei and Li (1979), was calculated in VCFTOOLS 0.1.17.
The linkage disequilibrium between each pair of alleles from each pair of markers and their significance levels were analyzed in Arlequin 3.5.2 (Excoffier et al. 2005). The r2 (r2 = DA × DB/pA(1-pA)pB(1-pB), whereas D = pAB-pA × pB) value was used to measure the level of LD between markers and the significance level (P-value) was calculated using an extension of Fisher exact probability test on contingency tables (Excoffier et al. 2005). The allele pairs with P-value < 0.0001 were recorded as strong LD.
In the following analysis that using SSR markers, all the alleles that exist in with less than 10 accessions were set to missing. The number of subpopulations (K) of the materials was determined in Structure 2.3.4 (Pritchard et al. 2000). The admixture model was used, and the length of burn-in period and the number of MCMC (Markov Chain Monte Carlo) replications after burn-in were set to 100,000 and 500,000 respectively. K equals 2 to 10 was tested and 5 independent runs were made for each value of K. The log-likelihood mean value of the 5 runs at each K was used. The structure result was submitted to Structure Harvester (http://taylor0.biology.ucla.edu/structureHarvester/), and the optimal K value was determined based on the ΔK (ΔK = mean(|L’’K|)/sd[L(K)]) due to the mean log-likelihood value increased over increased K (Evanno et al. 2005). The population structure matrix (Q) was generated based on the optimal K. The SSR marker based phylogenetic neighbor joining tree was calculated in Tassel 3 (Bradbury et al. 2007) and FigTree 1.4.3 was used to output the figure.
Association Mapping of TGW Trait
The associations between trait and SSR markers, and the effect of marker on the phenotype were calculated in Tassel 3 following the method described by Emon et al (2015) with slightly modified, and the MLM (mixed linear model) was used. The heterozygous loci were removed in the calculation. The population structure Q matrix was from the calculation above, and the Kinship (K) matrix was calculated in Tassel 3 Two thousands of permutation tests were used to help define the cut-off value (significance threshold) (Churchill and Doerge 1994). In the permutation, the original phenotype data was reshuffled and then performed association analysis in Tassel 3. Since no real associations between the SNPs and the ‘simulated’ phenotypes were expected, a threshold could be set based on these fake association mapping results.
In order to validate the SSR association mapping result and give a higher resolution of the associated loci, 57 of the 462 accessions were randomly selected and re-sequenced (Table S8), and their SNP/InDel variants were called. The associations between the trait and SNP/InDel variants were calculated in Tassel 5 (Bradbury et al. 2007), and the GLM (general linear model) was used. In the SNP/InDel association mapping, we only focused on the region near the significant SSR markers identified in the SSR association analysis. An interval size of ± 500 kb flanking the target SSR markers was used. Similarly to the SSR-TGW association mapping above, the cut-off values of the SNP/InDel variants and TGW trait association were done using 2000 times permutation in Tassel 5.
Data Availability
The datasets generated during and/or analysed during the current study are available from the supplementary materials.
References
Agrama HA, Eizenga GC, Yan W (2007) Association mapping of yield and its components in rice cultivars. Mol Breeding 19:341–356
Bazrkar-Khatibani L, Fakheri BA, Hosseini-Chaleshtori M, Mahender A, Mahdinejad N et al (2019) Genetic mapping and validation of quantitative trait loci (QTL) for the grain appearance and quality traits in rice (Oryza sativa L.) by using recombinant inbred line (RIL) population. Int J Genomics 3160275. https://doi.org/10.1155/2019/3160275
Begum H, Spindel JE, Lalusin A, Borromeo T, Gregorio G, Hernandez J et al (2015) Genome-wide association mapping for yield and other agronomic traits in an elite breeding population of tropical rice (Oryza sativa). PLoS ONE 10:e0119873
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y et al (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 2:2633–2635
Botstein M, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulo SJ et al (2008) BLAST+: architecture and applications. BMC Bioinformatics 10:4215
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Collard BCY, MacKill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci 363:557–572. https://doi.org/10.1098/rstb.2007.2170
Cui H, Peng B, Xing Z, Xu G, Yu B et al (2002) Molecular dissection of seedling-vigor and associated physiological traits in rice. Theor Appl Genet 105:745–753
Dang XJ, Liu EB, Liang YF, Liu QM, Breria CM et al (2016) QTL detection and elite alleles mining for stigma traits in Oryza sativa by association mapping. Frontiers in Plant Science. 7:1188. https://doi.org/10.3389/fpls.2016.01188
Dang XJ, Tran Thi TG, Edzesi WM, Liang LJ, Liu QM et al (2015) Population genetic structure of Oryza sativa in East and Southeast Asia and the discovery of elite alleles for grain traits. Scientific Reports 5: 11254. https://doi.org/10.1038/srep11254
Dong Q, Zhang ZH, Wang LL, Zhu YJ, Fan YY et al (2018) Dissection and fine-mapping of two QTL for grain size linked in a 460-kb region on chromosome 1 of rice. Rice (N Y) 11:44
Douglas B, Martin M, Ben B, Steven W (2014) lme4: Linear mixed-effects models using Eigen and S4. R package version 1.0–6
Edzesi WM, Dang XJ, Liang LJ, Liu EB, Zaid IU et al (2016) Genetic diversity and elite allele mining for grain traits in rice (Oryza sativa L.) by association mapping. Frontiers in Plant Science. 7:787. https://doi.org/10.3389/fpls.2016.00787
Emon RM, Islam MM, Halder J, Fan Y (2015) Genetic diversity and association mapping for salinity tolerance in Bangladeshi rice landraces. The Crop Journal 3:440–444
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinforma 1:47–50
Fageria N (2007) Yield physiology of rice. J Plant Nutr 30:843–879
Fan C, Xing Y, Mao H, Lu T, Han B et al (2006) GS3, a major QTL for grain ength and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112:1164–1171
Feng Y, Lu Q, Zhai R, Zhang M, Xu Q (2016) Genome wide association mapping for grain shape traits in indica rice. Planta 244:819–830
Florez JC (2013) Genomic and personalized medicine, 2nd edn. Academic Press, The USA
Fuller DQ, Harvey E, Qin L (2007) Presumed domestication? Evidence for wild rice cultivation and domestication in the fifth millennium BC of the Lower Yangtze region. Antiquity 81:316–331. https://doi.org/10.1017/S0003598X0009520X
Gao YM, Zhu J, Song YS, He CX, Shi CH et al (2004) Analysis of digenic epistatic effects and QE interaction effects QTL controlling grain weight in rice. J Zhejiang Univ Sci 5:371–377
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch S (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169:1631–1638
Gupta P, Naithani S, Tello-Ruiz MK, Chougule K, D’Eustachio P et al (2016) Gramene database: navigating plant comparative genomics resources. Curr Plant Biol 7–8:10–15
Hua J, Xing Y, Wu W, Xu C, Sun X et al (2003) Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 100:2574–2579
Hua JP, Xing YZ, Xu CG, Sun XL, Yu SB et al (2002) Genetic dissection of an elite rice hybrid revealed that heterozygotes are not always advantageous for performance. Genetics 162:1885–1895
Huang X, Wei X, Sang T, Zhao Q, Feng Q et al (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–969
Huang X, Zhao Y, Wei X, Li C, Wang A et al (2011) Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet 44:32–41
Jiang GH, Xu CG, Li XH, He YQ (2004) Characterization of the genetic basis for yield and its component traits of rice revealed by doubled haploid population. Acta Genet. Sin. 31:63–72
Li X, Wei Y, Li J, Yang F, Chen Y et al (2020) Identification of QTL TGW12 responsible for grain weight in rice based on recombinant inbred line population crossed by wild rice (Oryza minuta) introgression line K1561 and indica rice G1025. BMC Genet 21:10. https://doi.org/10.1186/s12863-020-0817-x
Li Y, Fan C, Xing Y, Jiang Y, Luo L et al (2011) Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet 43:1266-U134
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li J, Xiao J, Grandillo S, Jiang L, Wan Y et al (2004) QTL detection for rice grain quality traits using an interspecific backcross population derived from cultivated Asian (O. sativa L.) and African (O. glaberrima S.) rice. Genome 47:697–704
Lian X, Xing Y, Yan H, Xu C, Li X et al (2005) QTLs for low nitrogen tolerance at seedling stage identified using a recombinant inbred line population derived from an elite rice hybrid. Theor Appl Genet 112:85–96
Liu C, Chen K, Zhao X, Wang X, Shen C et al (2019) Identification of genes for salt tolerance and yield-related traits in rice plants grown hydroponically and under saline field conditions by genome-wide association study. Rice (N Y) 12:88. https://doi.org/10.1186/s12284-019-0349-z
Liu EB, Liu XL, Zeng SY, Zhao KM, Zhu CF et al (2015) Time-course association mapping of the grain-filling rate in rice (Oryza sativa L.). Plos One 10 (3):e0119959. https://doi.org/10.1371/journal.pone.0119959
Liu J, Chen J, Zheng X, Wu F, Lin Q et al (2017) GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat Plants 3:17043. https://doi.org/10.1038/nplants.2017.43
Liu K, Zhou Z, Xu C, Zhang Q, Maroof M (1996) An analysis of hybrid sterility in rice using a diallel cross of 21 parents involving indica, japonica and wide compatibility varieties. Euphytica 90:275–280
Mather KA, Caicedo AL, Polato NR, Olsen KM, McCouch S et al (2007) The extent of linkage disequilibrium in Rice (Oryza sativa L.). Genetics 177:2223–2232
Miah G, Rafii MY, Ismail MR, Puteh AB, Rahim HA et al (2013) A review of microsatellite markers and their applications in rice breeding programs to improve blast disease resistance. Int J Mol Sci 14:22499–22528
Moncada P, Martinez CP, Borrero J, Chatel M, Gauch H et al (2001) Quantitative trait loci for yield and yield components in an Oryza sativa X Oryza rufipogon BC2F2 population evaluated in an upland environment. Theor Appl Genet 102:41–42
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Pimentel D (2012) World overpopulation Environ Dev Sustain 14:151–152
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qiu X, Pang Y, Yuan Z, Xing D, Xu J et al (2015) Genome-wide association study of grain appearance and milling quality in a worldwide collection of Indica rice germplasm. PLoS ONE 10:e0145577. https://doi.org/10.1371/journal.pone.0145577.eCollection2015
Ruan B, Shang L, Zhang B, Hu J, Wang Y et al (2020) Natural variation in the promoter of TGW2 determines grain width and weight in rice. New Phytol [Epub ahead of print]
Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H et al (2008) Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet 40:1023–1028
Song X, Huang W, Shi M, Zhu M, Lin HA (2007) QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39:623–630
Tang SQ, Shao GN, Wei XJ, Chen ML, Sheng ZH et al (2013) QTL mapping of grain weight in rice and the validation of the QTL qTGW3.2. Gene 527:201–206. https://doi.org/10.1016/j.gene.2013.05.063
Tello-Ruiz MK, Naithani S, Stein JC, Gupta P, Campbell M et al (2018) Gramene 2018: unifying comparative genomics and pathway resources for plant research. Nucleic Acids Res 46:D1
Tsykun T, Rellstab C, Dutech C, Sipos G, Prospero S (2017) Comparative assessment of SSR and SNP markers for inferring the population genetic structure of the common fungus Armillaria cepistipes. Heredity 119:371–380. https://doi.org/10.1038/hdy.2017.48
Van der Auwera GA, Carneiro MO, Hart C, Poplin R, Del AG et al (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11. https://doi.org/10.1002/0471250953.bi1110s43
Vanniarajan C, Vinod KK, Pereira A (2012) Molecular evaluation of genetic diversity and association studies in rice (Oryza sativa L.). J Genet 91:1–11
Wang S, Wu K, Yuan Q, Liu X et al (2012) Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet 44:950–954
Wang Y, Xiong G, Hu J, Jiang L, Yu H et al (2015) Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat Genet 47:944–948
Wang L, Wang A, Huang X, Zhao Q, Dong G et al (2011) Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor Appl Genet 122:327–340
Weng J, Gu S, Wan X, Gao H, Guo T et al (2008) Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res 18:1199–1209
Xing Z, Tan F, Hua P, Sun L, Xu G et al (2002) Characterization of the main effects, epistatic effects and their environmental interactions of QTLs on the genetic basis of yield traits in rice. Theor Appl Genet 105:248–257
Xing Y, Zhang Q (2010) Genetic and molecular bases of rice yield. Annu Rev Plant Biol 61:421–442
Xu F, Sun X, Chen Y, Huang Y, Tong C et al (2015) Rapid identification of major QTLs associated with rice grain weight and their utilization. PLoS ONE 10:e0122206. https://doi.org/10.1371/journal.pone.0122206.eCollection2015
Yang W, Guo Z, Huang C, Duan L, Chen G et al (2014) Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nature Commun 5:5087. https://doi.org/10.1038/ncomms6087
Ying JZ, Ma M, Bai C, Huang XH, Liu JL et al (2018) TGW3, a major QTL that negatively modulates grain length and weight in rice. Mol Plant 11:750–753. https://doi.org/10.1016/j.molp.2018.03.007
Zhang X, Wang J, Huang J, Lan H, Wang C et al (2012) Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proc Natl Acad Sci USA 109:21534–21539
Zhao K, Tung C, Eizenga G, Wright M, Ali M et al (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun 2:467
Zhuang JY, Fan YY, Rao ZM, Wu JL, Xia YW et al (2002) Analysis on additive effects and additive-by-additive epistatic effects of QTLs for yield traits in a recombinant inbred line population of rice. Theor Appl Genet 105:1137–1145
Acknowledgements
We thank Dr. Qiangming Liu for software assistance. We thank Miss Siqi Chen and Mr. Yulong Li for phenotyping assistance.
Funding
This study was funded by grants from the National Natural Science Foundation of China (31571743 and 31671658).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by [Xiangong Chen], [Xiaojing Dang] and [Ya Wang]. The first draft of the manuscript was written by [Xiangong Chen] and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by: Yann-Rong Lin
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, X., Dang, X., Wang, Y. et al. Association Mapping of Thousand Grain Weight using SSR and SNP Markers in Rice (Oryza sativa L.) Across Six Environments. Tropical Plant Biol. 14, 143–155 (2021). https://doi.org/10.1007/s12042-021-09282-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-021-09282-7