
Abstract
Background
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a rare neurological degenerative disorder caused by the mutations of MLC1 or GLIALCAM with autosomal recessive or autosomal dominant inheritance and a different prognosis, characterized by macrocephaly, delayed motor and cognitive development, and bilateral abnormal signals in cerebral white matter (WM) with or without cysts on magnetic resonance imaging (MRI). This study aimed to reveal the clinical and genetic features of MLC patients with GLIALCAM mutations and to explore the brain pathological characteristics and prognosis of mouse models with different modes of inheritance.
Methods
Clinical information and peripheral venous blood were collected from six families. Genetic analysis was performed by Sanger sequencing of GLIALCAM. GlialcamArg92Trp/+ and GlialcamLys68Met/Thr132Asn mouse models were generated based on mutations from patients (c.274C>T(p.Arg92Trp) (c.203A>T(p.Lys68Met), and c.395C>A (p.Thr132Asn))). Brain pathologies of the mouse models at different time points were analyzed.
Results
Six patients were clinically diagnosed with MLC. Of the six patients, five (Pt1–Pt5) presented with a heterozygous mutation in GLIALCAM (c.274C>T(p.Arg92Trp) or c.275G>C(p.Arg92Pro)) and were diagnosed with MLC2B; the remaining patient (Pt6) with two compound heterozygous mutations in GLIALCAM (c.203A>T (p.Lys68Met) and c.395C>A (p.Thr132Asn)) was diagnosed with MLC2A. The mutation c.275C>G (p.Arg92Pro) has not been reported before. Clinical manifestations of the patient with MLC2A (Pt6) progressed with regression, whereas the course of the five MLC2B patients remained stable or improved. The GlialcamArg92Trp/+ and GlialcamLys68Met/ Thr132Asn mouse models showed vacuolization in the anterior commissural WM at 1 month of age and vacuolization in the cerebellar WM at 3 and 6 months, respectively. At 9 months, the vacuolization of the GlialcamLys68Met/ Thr132Asn mouse model was heavier than that of the GlialcamArg92Trp/+ mouse model. Decreased expression of Glialcam in GlialcamArg92Trp/+ and GlialcamLys68Met/ Thr132Asn mice may contribute to the vacuolization.
Conclusions
Clinical and genetic characterization of patients with MLC and GLIALCAM mutations revealed a novel mutation, expanding the spectrum of GLIALCAM mutations. The first Glialcam mouse model with autosomal recessive inheritance and a new Glialcam mouse model with autosomal dominant inheritance were generated. The two mouse models with different modes of inheritance showed different degrees of brain pathological features, which were consistent with the patients’ phenotype and further confirmed the pathogenicity of the corresponding mutations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Megalencephalic leukoencephalopathy with subcortical cysts (MLC, OMIM 604004) is an infantile-onset autosomal recessive or dominant neurodegenerative disease characterized by cerebral white matter (WM) edema and cyst formation [1, 2]. The classic phenotype of MLC is macrocephaly within the first year of life, moderate to severe motor development delay, normal or mild mental development delay, motor deterioration with ataxia, seizures, spasticity in subsequent years, and extrapyramidal outcomes [1, 2]. Magnetic resonance imaging (MRI) displays bilateral symmetric diffuse abnormal signals in WM with edema, with or without subcortical cysts in the anterior-temporal and fronto-parietal lobes; the internal capsule, cerebellum, and brainstem are occasionally involved [1, 3]. The lifespan ranges from toddler age to seventies [4,5,6,7,8]. MLC1 (OMIM 605908) was the first gene found to be mutated in MLC in 2001 [9]. Mutations in MLC1 lead to classic MLC, which is diagnosed as MLC1 (OMIM 604004) and account for 75% of patients with MLC. The clinical and genetic characteristics and natural history of MLC1 have been completely studied [10,11,12,13,14,15]. The second gene GLIALCAM was found mutated in 2011 [4]. The phenotype associated with GLIALCAM is named as MLC2. MLC2 can be further divided into two types: MLC2A with autosomal recessive and MLC2B with autosomal dominant inheritance, and MLC2 patients accounted for a total of 20% of MLC patients. MLC2A and MLC2B present with different clinical symptoms and prognosis [4]. So far, only 89 patients with MLC and GLIALCAM mutations have been reported in five papers [4, 8, 11, 12, 16]. The brain tissues of patients with MLC are rare for MLC1 mutations and unobtainable for GLIALCAM mutations. Therefore, MLC mouse models are important for studying the brain pathophysiology of this disorder. In the present study, we aimed to reveal the clinical characteristics, including those in long-term follow-up studies, genetic features, and phenotype–genotype relationship in MLC patients with GLIALCAM mutations and generate Glialcam mouse models with autosomal recessive and autosomal dominant inheritance to clarify the disease course and understand the brain pathological features of mouse models with different modes of inheritance.
Methods
Clinical analysis
Patients were enrolled from the Pediatric Neurology Department, Peking University First Hospital from 2007 to 2017 in accordance with the following criteria: (1) diagnosed with MLC clinically; (2) no pathogenetic mutation in MLC1 was detected by sanger sequencing. Clinical information, such as visiting age, sex, onset age, presenting symptoms, and MRI results, was collected. The study was approved by the Ethics Committee of Peking University First Hospital, and written informed consents was obtained from the patients’ legal guardians.
Follow-up study data, including head circumference (HC), motor and mental development or retrogress, cranial MRI, head trauma histories, and other related symptoms, were collected by telephone follow-up survey. Motor function was assessed using the Chinese version of Gross Motor Function Classification System (GMFCS) for children with cerebral palsy [12]. The classification from GI to GV was represented as slightly to severe limited motor ability based on the patients’ age and motor function. Cognitive function was evaluated by word expression. The classification from C0 to C3 represented different levels of cognitive capacity from high to low according to their scores.
Genetic analysis
Peripheral venous blood samples were collected from six patients and their family members. DNA extraction, amplification, and sequencing of GLIALCAM were performed as previously reported [11, 12]. The sequence variants were screened and analyzed using a flexible function converged online tool for detailed annotation Variant Effect Predictor (VEP, https://asia.ensembl.org/Homo_sapiens/Tools/VEP) [17]. Finally, a comprehensive prediction was analyzed in accordance with the 2013 ACMG standards and guidelines [18].
Animals
Mutations were derived from Chinese MLC patients with GLIALCAM mutations c.274C>T, p.Arg92Trp, c.203A>T(p.Lys68Met), and c.395C>A (p.Thr132Asn). Adult GlialcamArg92Trp/+, GlialcamLys68Met/+, and GlialcamThr132Asn/+ point mutation mice were obtained from the Beijing Vitalstar Biotechnology Co., Ltd. using a CRISPR/Cas9-based knock-in strategy in C57Bl/6N mice (Fig. S2A). Then, we crossbred heterozygous mutant mice between GlialcamLys68Met/+ and GlialcamThr132Asn/+ mice to obtain the compound heterozygous mouse GlialcamLys68Met/Thr132Asn. The study was approved by the Animals Ethical Committee of Peking University First Hospital, and animal housekeeping and experiments were in compliance with the guidelines of the committee. All mice were housed in an animal care facility (SPF) with a 12-h light–dark schedule and with freely available food and water. The DNA of the tail was extracted using the KAPA Express Extract kit, and genotype identification of these mice was performed by PCR and Sanger sequencing using the following primers: forward primer 5ʹ-AGGGCTTGAAG TTGGAAATGGGCTA-3ʹ; reverse primer 5′-AATTACCACTCAGCCTTTGCGTTGC-3ʹ. Table S1 records the sex and generations of mice used for the experiment.
HE staining and vacuole quantification
Mice were anesthetized by 0.5% pentobarbital sodium at the dosage of 50 mg/kg, perfused by 0.9% saline and 4% paraformaldehyde. Brains were then perfusion-fixed with 4% paraformaldehyde and embedded in paraffin. Tissue slices were stained with hematoxylin and eosin (HE). Slice series from brain sagittal sections stained with HE were obtained through scanning by NanoZoomer RS 2.0.0 (HAMAMATSU, Japan) at 40× objective. Three slices of each animal brain were obtained, and three different animals of each genotype at four different time points were analyzed. Blind quantification of the vacuoles in the cerebellar and anterior commissural WM was performed to the genotype using ImageJ.
Fluorescence immunohistochemistry
Fluorescence immunohistochemistry staining of Glialcam and Gfap were used to check the expression and localization of Glialcam among different group. For each genotype, three brain sections each of three different mice were analyzed by the Olympus confocal microscope and FV3000 system (Tokyo, Japan).
Statistical analyses
WM vacuolization was analyzed using one-way ANOVA using SPSS version 19 (Armonk, USA) and GraphPad Prism 5 (San Diego, Canada). A P value < 0.05 was considered as statistically significant.
Results
Clinical and genetic findings of patients with MLC and GLIALCAM mutations
Clinical and follow-up findings
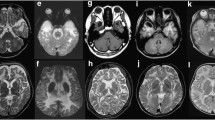
Six patients (Pt1–Pt6) enrolled in this study were clinically diagnosed with MLC (Table 1). One patient (Pt6) has been reported twice and three patients (Pt1–Pt3) have been reported once in two follow-up studies [11, 12]. The two patients (Pt5 and Pt6) were new-coming, and another three follow-up studies were performed for all six patients. The clinical characteristics of four patients were macrocephaly, earlier normal motor development, and classic MRI. Three patients were diagnosed as MLC2B and one as MLC2A. The patient with MLC2A showed motor retrogress, while one patient with MLC2B had an improvement in the MRI. For all six patients, the onset age ranged from birth to 7 months, with a median of 4 months. Macrocephaly was noted in five patients (Pt1–Pt4 and Pt6) with HC above the 97th percentile. Pt5 presented with HC > 1 SD history. In all six patients, the development of earlier motor and a rapid HC growth milestones, such as head control, rolling over, and sitting alone, were all normal (Table 2). However, the patients spent more time from walking unstably to walking stably. Motor developmental delay was observed in Pt4 and Pt6. Cognitive development was normal in all patients, except in Pt4, at the first visit (Table 3). Autism and language development delay were found in Pt4. Epilepsy occurred in Pt5. Transient macrocephaly was found in the mothers of Pt1 and Pt4 before 1 year old. Hypotonia was found in Pt2 and Pt3. Motor retrogress after head trauma was found in Pt6. Bilateral diffuse cerebral abnormal signals in swollen WM with or without cysts were found in the MRIs of all patients, expect Pt5, at the first visit. In Pt5, only subcortical WM was involved in the MRI. Posterior limb of the internal capsule was involved in Pt1 and Pt3.
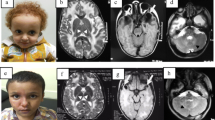
In the follow-up study, one patient (Pt2) was missing and the remaining five patients attended for 5 (Pt1), 4 (Pt3), 2 (Pt4), 2 (Pt5), and 5 (Pt6) times of follow-up study. HC above the 97th percentile of normal children was found in Pt1, Pt3, Pt4, and Pt6, and a normal HC was found in Pt5 at the first visit (Fig. S1). The HC showed a rapid growth and then slowed down after 2 years in Pt5. GMFCS classification improved in Pt1, Pt3, and Pt5 from GI to normal, stabilized in Pt5 at GI, and declined in Pt6 from GII to GIII (Table 2). The cognitive classification declined in four patients, except Pt1 (Table 2); Pt3 and Pt5–Pt6 declined from normal to C1, whereas Pt4 declined from C1 to C3. School performance was above average for those who attended school, and Pt6 dropped out of school at 11 years because of motor disability. Additionally, language absence and autism only appeared in Pt4. Focal epilepsy occurred four times in 2 months and was controlled by sodium valproate in Pt5. Pt6 had two unconsciousness episodes after head trauma with motor deterioration. The MRI was typical in four patients (Pt1–Pt3 and Pt6), with bilateral diffuse abnormal signals in swollen cerebral WM with or without subcortical cysts, and atypical with only subcortical WM involved in Pt5; the involvement of the posterior limbs of the internal capsule (PLIC) was discovered in the MRI in Pt1 and Pt3, no anterior limbs of internal capsule (ALIC) were found. Thorough MRI improvement was detected in Pt1 (Fig. 1).

MRI of Pt1 and Pt5. T2WI, T2 weighted-image. a–i MRI of Pt1. a–c MRI of Pt1 at 7 m; a Bilateral diffuse abnormal signals in subcortical and central white matter with abnormal signals (arrows) in the partial posterior limb of the internal capsule in axial T2WI; b temporal cysts were detected in axial T2WI (arrows); c temporal cysts in sagittal T2WI (arrows); d–f MRI of Pt1 at 7 years 9 months. Normal white matter signals without temporal cysts; g–i MRI of Pt1’s mother at 35 years 1 month, normal signals in white matter without temporal subcortical cysts; j–o MRI of Pt5. j–l MRI of Pt5 at 2 years 4 months; j bilateral abnormal signals in subcortical white matter (arrows) with normal central white matter and without cysts in axial T2WI; k, l abnormal signals in subcortical white matter in axial and sagittal T1WI; m–o MRI of Pt5 at 2 years 6 months. No improvement was seen in cerebral white matter
Genetic findings
Four promising variants in GLIALCAM were detected in six patients were all genetically diagnosed (Table 4). One patient (Pt6) presented with compound heterozygous variants c.203A>T (p.Lys68Met) and c.395C>A (p.Thr132Asn) in GLIALCAM, while the five remaining patients (Pt1–Pt5) harbored only one variant in GLIALCAM. Of the five patients with mono-allelic variant in GLIALCAM, four patients (Pt1–Pt3, and Pt5) shared the same variant c.274C>T(p.Arg92Trp), which has been reported in patients with MLC before [4], while Pt4 had a novel variant c.275G>C(p.Arg92Pro) (Fig. S3), which has not been reported before. Except the variant in Pt3 arising de novo, variants in Pt1, Pt2, Pt4, and Pt5 were all inherited from their mothers. The mothers of Pt1 and Pt4 had a history of transient macrocephaly.
All identified variants in GLIALCAM were four missense variants. All changed sites were analyzed to be highly conservative by GERP. All of the four variants were unrecorded in gnomAD database and predicted to be deleterious by Polyphen2, SIFT, Mutation Taster, and Model.
According to the ACMG guideline [18], the variants c.274C>T (p.Arg92Trp), c.275G>C(p.Arg92Pro), c.203A>T (p.Lys68Met), and c.395C>A (p.Thr132Asn) were classified as “pathogenic,” “likely pathogenic,” “likely pathogenic,” and “uncertain significance,” respectively.
Brain histopathological findings
Validation of the GLIALCAM point mutation in mouse models
Genotype verification was performed by PCR and Sanger sequencing at postnatal 7 days for every mouse born from the crossbred of GlialcamArg92Trp/+ with C57Bl/6N wild-type mice or GlialcamLys68Met/+ and GlialcamThr132Asn /+ mice (Fig. S2B).
Pathological results
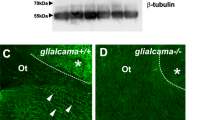
Vacuoles were detected in the WM of GlialcamArg92Trp/+ and GlialcamLys68Met/Thr132Asn mice through brain histopathology. The vacuolization of anterior commissural WM started within 1 month of age in GlialcamArg92Trp/+ and GlialcamLys68Met/Thr132Asn models and showed a decreased tendency in subsequent months (Fig. 2a). GlialcamLys68Met/Thr132Asn mice presented a heavier degree of vacuolization than GlialcamArg92Trp/+ mice at 9 months in anterior commissure. No vacuoles were detected in the anterior commissural WM of wild-type mice. The vacuoles in GlialcamArg92Trp/+ murine cerebellum were detected at 3 months, prominent at 6 months, and increased at 9 months, while the vacuoles in GlialcamLys68Met/Thr132Asn were first detected at 6 months, increased at 9 months (Fig. 2b), and became more prominent than GlialcamArg92Trp/+ mice at 9 months. Quantification analysis showed these results (Fig. 2c, d). Glialcam expression and localization of astrocytes along blood vessel in the cerebellum at 9 months were shown (Fig. 3), indicating decreased Glialcam fluorescent in the brains of GlialcamArg92Trp/+ and GlialcamLys68Met/Thr132Asn mice comparing with wild-type mice; and no obvious mislocalization of Glialcam and Gfap were found.

White matter vacuolization in GlialcamArg92Trp/+ and GlialcamLys68Met/Thr132Asn mice in the anterior commissural and cerebellar WM. a, b HE staining of mice aged 1 month, 3 months, 6 months, and 9 months shows vacuolization in white matter. In the anterior commissure, vacuolization starts from 1 month in GlialcamArg92Trp/+ and GlialcamLys68Met/Thr132Asn mice, and GlialcamLys68Met/Thr132Asn has more vacuoles than GlialcamArg92Trp/+ mice at 9 months. In cerebellum white matter, vacuolization in GlialcamArg92Trp/+ mice started from 3 months; in GlialcamLys68Met/Thr132Asn mice, vacuolization started from 6 months, and the degree of vacuolization was more prominent than that in GlialcamArg92Trp/+ mice at 9 months. Scale bars 50 µm. c, d Quantification confirms anterior commissural vacuolization in GlialcamArg92Trp/+, GlialcamLys68Met/Thr132Asn has more prominent vacuoles than the wild type with a decreased tendency, and GlialcamLys68Met/Thr132Asn has more vacuolization than GlialcamArg92Trp/+ mice at 9 months. c, d show significantly more prominent white matter cerebellar vacuolization of GlialcamLys68Met/Thr132Asn mice than GlialcamArg92Trp/+ mice at 9 months with an increased tendency. P < 0.05, †P < 0.01 and ‡P < 0.001. Graph bars represent the standard error of the mean

Glialcam expression decreased in astrocyte along blood vessels in cerebellum at 9 months. Fluorescence immunohistochemistry staining of Glialcam (green) and the astrocytic cytoskeletal protein Gfap (red) in the white matter of the cerebellum. Arrows (white) point to nucleus (blue) of endothelial cell, and arrows (yellow) point to the Glialcam protein in astrocyte along blood vessels. It seemed that Glialcam showed a decreased red fluorescent in GlialcamArg92Trp/+ and GlialcamLys68Met/Thr132Asn mice brain. Scale bars 20 μm
Discussion
In this study, macrocephaly, normal or slightly delayed in motor and cognitive development, and classic MRI features were found in five of the six patients evaluated. Therefore, the five patients were clinically diagnosed. The other patient had HC > 1 SD and a rapidly increased macrocephaly history during the infant period, with only subcortical WM involved; this patient was clinically diagnosed as atypical MLC. Four missense variants c.274C>T (p.Arg92Trp), c.275G>C(p.Arg92Pro), c.203A>T (p.Lys68Met) and c.395C>A (p.Thr132Asn) were detected in the six patients. One patient (Pt6) had two compound heterozygous mutations, and the five other patients had one mono-allelic mutation (Pt1–Pt5). Therefore, the six patients were genetically diagnosed. Five patients (Pt1–Pt5) had MLC2B, and one patient (Pt6) had MLC2A.
Previous studies reported that macrocephaly is the most common presenting symptom in the patients with MLC [1, 4, 8]; in the present study, it appeared in five of the six patients. In the long term, HC growth varied among the patients. Rapid growth after 2 years was found in Pt3 and Pt5 and was first discovered in patients with GLIALCAM mutations. The HC of four patients with MLC2B (Pt1–Pt4) were always above 97th of the control at the fifth follow-up study, but macrocephaly was normalized in one of the patient with MLC2B in the follow-up studies. Pt5 presented with normal HC all the time and with a rapid HC growth in the infant period, which was reported previously [4, 8, 12, 17]. The motor development of five patients with MLC2B developed stably or faster, indicating an improvement; meanwhile, that of patient with MLC2A declined because of motor retrogress. Early motor development is normal before walking [4]. The changes in the GMFCS of the patients indicate that motor development was kept stable or improved in patients with MLC2B and deteriorated in the patient with MLC2A. Early cognitive milestones were almost normal, but autism and the absence of language appeared in one patient with MLC2B (Pt4), which was reported previously [8]. In the follow-up study, the cognitive development was almost slightly delayed by word expression, and the school performance was almost normal. Almost all patients with MLC2A and MLC2B were detected with classic MRI, but atypical MRI involving subcortical WM only was found in one patient with MLC2B (Pt5). One patient with MLC2B showed improved MRI; this phenomenon was also reported in literature [8]. A double line throughout involvement was the most common abnormal signal of the internal capsule in patients with MLC1 and MLC2A [1, 8]. The present study is the first to report the involvement of the posterior limb of the internal capsule in the patients (Pt1 and Pt3). Regarding other neurological dysfunction, autistic behavior was frequently found in patients with MLC2B and rare in patients with MLC1; thus far, autistic behavior has not been reported in patients with MLC2A [4, 8, 16]. This finding could be used to distinguish MLC2B in patients with MLC1 and MLC2A clinically. Therefore, combining the clinical and genetic results revealed that the compound heterozygous mutations of GLIALCAM caused deteriorated progress, while dominant mutations lead to improved phenotype with macrocephaly and delayed development with or without macrocephaly. Atypical presentations including the involvement of subcortical white matter on MRI, a normalized head circumference on follow-up, or the absence of language and autism appear in patients with MLC2B.
Thus far, 19 mutations have been reported in GLIALCAM, including 6 mutations associated with MLC2B and 13 mutations associated with MLC2A. One novel mutation c.275G>C (p.Arg92Pro) was found in the present study, expanding the spectrum of GLIALCAM mutations. Interestingly, another substitution at the same nucleotide site [c.275G>A (p.Arg92Gln)] was detected in a patient with MLC2A [4]. The different inheritance patterns and clinical presentation of p.Arg92Pro and p.Arg92Gln, possibly resulting from the replacement by different amino acid residues from basic hydrophilic (Arg) to neutral hydrophobic (Pro) and neutral hydrophilic (Gln), might alter the secondary structure and affect the function of the protein. However, the mechanism needs to be explored further. The mutation c.274C>T (p.Arg92Trp) accounted for four of the five patients with MLC2B. A previous study also detected this mutation, which accounted for 28.5% of the reported patients with MLC2B [4]. Thus, c.274C>T (p.Arg92Trp) was supposed to be a hot-spot mutation.
Glialcam-null mice and GlialcamGly89Ser/+ mouse models were generated previously [19,20,21]. GLIALCAM mutations with autosomal dominant inheritance result in an improved phenotype, while GLIALCAM mutations with autosomal recessive inheritance lead to motor deterioration and mental decline in patients with MLC. Glialcam mutation mouse models can be used to investigate brain histopathological differences and prognosis of different inheritances of MLC. Here, we generated the first GlialcamLys68Met/Thr132Asn mouse model with an autosomal recessive inherited manner and a new GlialcamArg92Trp/+mouse model with an autosomal dominant inherited manner. No MRI abnormality was observed during the entire period in GlialcamArg92Trp/+ and wild-type mice (data not shown). The HE staining on the brain tissue slices was conducted to observe the pathological changes. Vacuoles were detected in the anterior commissural WM in GlialcamArg92Trp/+ and GlialcamLys68Met/Thr132Asn mice at 1 month, indicating that the pathological change started from the infant period, consistent with findings in human patients. Thereafter, the vacuoles showed a decreased tendency at subsequent time points, a similar phenomenon observed in the corpus callosum of Glialcam-null mice [21]. No vacuoles were found in wild-type mice. In the cerebellar WM, GlialcamArg92Trp/+mice showed pathological changes earlier than GlialcamLys68Met/Thr132Asn, both with increased tendency but more severe pathological changes appeared in GlialcamLys68Met/Thr132Asn than GlialcamArg92Trp/+ at middle age. Thus, the WM vacuoles appeared in both mouse models within the infant period and increased in the cerebellum during the disease course. GlialcamLys68Met/Thr132Asn mice showed more severe vacuolization in the anterior commissure and cerebellum than GlialcamArg92Trp/+and wild-type mice at middle ages, indicating the more severe involvement of brain tissues in the former. No improvement occurred in GlialcamArg92Trp/+mice at the end of the observation, which are different from the results in some patients with GLIALCAMArg92Trp/+ mutation. The difference in the vacuolization timeline in white matter between patients and mice may be due to their different development times or anatomic structures. The maximum age observed is 9 months, and whether the remitting of cerebellar WM vacuolization appears or not remains to be seen. Therefore, the pathology in mice with bi-allelic mutations in Glialcam was more severe than that with mono-allelic mutation over time, consistent with the prognosis of human patients. GlialcamArg92Trp/+ mice did not show remitting in contrast to some patients with MLC2B and the same mutation. The expression of Glialcam in astrocytes along blood vessels of cerebellar white matter at 9 months were decreased, indicating that the decreased expression of Glialcam may contribute to the vacuolization, but Western Blot are needed to verify this in the future. There was no obvious difference of localization between Glialcam and Gfap in three group.
The clinical and genetic characteristics of patients with MLC and GLIALCAM mutations were revealed. One novel mutation was identified, expanding the spectrum of GLIALCAM mutations. The first Glialcam mouse models with autosomal recessive inheritance and a new Glialcam mouse model with autosomal dominant inheritance were generated. The two mouse models with different inheritance patterns showed different degrees of brain pathological features, which were consistent with patients’ phenotype and further confirmed the pathogenicity of the corresponding mutations.
References
van der Knaap MS, Barth PG, Stroink H, van Nieuwenhuizen O, Arts WFM, Hoogenraad F, et al. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol. 1995;37:324–34.
Boor PKI, De Groot K, Mejaski-Bosnjak V, Brenner C, van Der Knaap MS, Scheper GC, et al. Megalencephalic leukoencephalopathy with subcortical cysts: an update and extended mutation analysis of MLC1. Hum Mutat. 2006;27:505–12.
Roy U, Joshi B, Ganguly G. Van der Knaap disease: a rare disease with atypical features. BMJ Case Rep. 2015;2015:bcr2015209831. https://doi.org/10.1136/bcr-2015-209831.
López-Hernández T, Ridder MC, Montolio M, Capdevila-Nortes X, Polder E, Sirisi S, et al. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet. 2011;88:422–32.
Singhal BS, Gursahani RD, Udani VP, Biniwale AA. Megalencephalic leukodystrophy in an Asian Indian ethnic group. Pediatr Neurol. 1996;14:291–6.
Pascual-Castroviejo I, van der Knaap MS, Pronk JC, García-Segura JM, Gutiérrez-Molina M, Pascual-Pascual SI. Vacuolating megalencephalic leukoencephalopathy: 24 year follow-up of two siblings. Neurologia. 2005;20:33–40.
Saijo H, Nakayama H, Ezoe T, Araki K, Sone S, Hamaguchi H, et al. A case of megalencephalic leukoencephalopathy with subcortical cysts (van der Knaap disease): molecular genetic study. Brain Dev. 2003;25:362–6.
Hamilton EMC, Tekturk P, Cialdella F, van Rappard DF, Wolf NI, Yalcinkaya C, et al. Megalencephalic leukoencephalopathy with subcortical cysts: characterization of disease variants. Neurology. 2017;90:e1395–e14031403.
Leegwater PAJ, Yuan BQ, van der Steen J, Mulders J, Könst AAM, Boor PKI, et al. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet. 2001;68:831–8.
Kariminejad A, Rajaee A, Ashrafi MR, Alizadeh H, Tonekaboni SH, Malamiri RA, et al. Eight novel mutations in MLC1 from 18 Iranian patients with megalencephalic leukoencephalopathy with subcortical cysts. Eur J Med Genet. 2015;58:71–4.
Guo M, Jiang Y, Xie H, Wu Y, Shang J, Gu Q, et al. Analysis of HEPACAM mutations in a Chinese family with megalencephalic leukoencephalopathy with subcortical cysts. Zhonghua Er Ke Za Zhi. 2012;50:895–8 (in Chinese).
Cao B, Yan H, Guo M, Xie H, Gu Q, Xiao J, et al. Ten novel mutations in Chinese patients with megalencephalic leukoencephalopathy with subcortical cysts and a long-term follow-up research. PLoS ONE. 2016;11:e0157258.
Choi SA, Kim SY, Yoon J, Choi J, Park SS, Seong MW, et al. A unique mutational spectrum of MLC1 in Korean patients with megalencephalic leukoencephalopathy with subcortical cysts: P.Ala275Asp founder mutation and maternal uniparental disomy of chromosome 22. Ann Lab Med. 2017;37:516–21.
Vellarikkal SK, Jayarajan R, Verma A, Ravi R, Senthilvel V, Kumar A, et al. A founder mutation MLC1 c.736delA associated with megalencephalic leukoencephalopathy with subcortical cysts-1 in north Indian kindred. Clin Genet. 2018;94:271–3.
Montagna G, Teijido O, Eymard-Pierre E, Muraki K, Cohen B, Loizzo A, et al. Vacuolating megalencephalic leukoencephalopathy with subcortical cysts: functional studies of novel variants in MLC1. Hum Mutat. 2006;27:292.
Arnedo T, Aiello C, Jeworutzki E, Dentici ML, Uziel G, Simonati A, et al. Expanding the spectrum of megalencephalic leukoencephalopathy with subcortical cysts in two patients with GLIALCAM mutations. Neurogenetics. 2014;15:41–8.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17:122.
Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–47.
Hoegg-Beiler MB, Sirisi S, Orozco IJ, Ferrer I, Hohensee S, Auberson M, et al. Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat Commun. 2014;5:3475.
van der Knaap MS, Lai V, Köhler W, Salih MA, Fonseca MJ, Benke TA, et al. Megalencephalic leukoencephalopathy with cysts without MLC1 defect. Ann Neurol. 2010;67:834–7.
Bugiani M, Dubey M, Breur M, Postma NL, Dekker MP, Ter Braak T, et al. Megalencephalic leukoencephalopathy with cysts: the Glialcam-null mouse model. Ann Neurol. 2017;4:450–65.
Acknowledgements
We thank all the patients and their families for their cooperation and contribution. This work was supported by the National Natural Science Foundation of China (Grant Number: 81741053, 81501123); by the Beijing Natural Science Foundation (Grant Number: 7151010, 7172217); by the Beijing Municipal Science & Technology Commission (Grant Number: Z161100000216133, Z161100004916169); by the Beijing Institute for Brain Disorders Foundation (Grant Number: BIBDPXM2014_014226_000016) and the Beijing Municipal Natural Science Key Project (Grant Number 15G10050); by Beijing key laboratory of molecular diagnosis and study on pediatric genetic diseases (Grant Number BZ0317); by the National Key Research and Development Program of China (Grant Number: 2016YFC1306201, 2016YFC0901505); by the Fundamental Research Funds for the Central Universities (Grant Number: BMU2017JI002).
Funding
This work was funded by the National Natural Science Foundation of China (Grant Number: 81741053, 81501123); by the Beijing Natural Science Foundation (Grant Number: 7151010, 7172217); by the Beijing Municipal Science & Technology Commission (Grant Number: Z161100000216133, Z161100004916169); by the Beijing Institute for Brain Disorders Foundation (Grant Number: BIBDPXM2014_014226_000016) and the Beijing Municipal Natural Science Key Project (Grant Number 15G10050); by Beijing key laboratory of molecular diagnosis and study on pediatric genetic diseases(Grant Number BZ0317); by the National Key Research and Development Program of China (Grant Number: 2016YFC1306201, 2016YFC0901505); by the Fundamental Research Funds for the Central Universities (Grant Number: BMU2017JI002).
Author information
Authors and Affiliations
Contributions
JMW and YWJ study design and revision of the manuscript; ZS animal experiments, data analysis and draft of the manuscript; HFY animal experiments and data analysis; B-BC, MMG, KG:genetic analysis; HX, YLY, HX, QG, ML, YW follow-up patient’s information and revision of the manuscript; JXX analysis for MRI. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
The study had been approved by Ethics Committee of Peking University First Hospital for patients, the animal study had been approved by the Animals Ethical Committee of Peking University First Hospital.
Conflict of interest
No financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shi, Z., Yan, HF., Cao, BB. et al. Identification in Chinese patients with GLIALCAM mutations of megalencephalic leukoencephalopathy with subcortical cysts and brain pathological study on Glialcam knock-in mouse models. World J Pediatr 15, 454–464 (2019). https://doi.org/10.1007/s12519-019-00284-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-019-00284-w