
Abstract
Significant effort is spent on monitoring of benthic ecosystems through government funding or indirectly as a cost of business, and metabarcoding of environmental DNA samples has been suggested as a possible complement or alternative to current morphological methods to assess biodiversity. In metabarcoding, a public sequence database is typically used to match barcodes to species identity, but these databases are naturally incomplete. The North Sea oil and gas industry conducts large-scale environmental monitoring programs in one of the most heavily sampled marine areas worldwide and could therefore be considered a “best-case scenario” for macrofaunal metabarcoding. As a test case, we investigated the database coverage of two common metabarcoding markers, mitochondrial COI and the ribosomal rRNA 18S gene, for a complete list of 1802 macrofauna taxa reported from the North Sea monitoring region IV. For COI, species level barcode coverage was 50.4% in GenBank and 42.4% for public sequences in BOLD. For 18S, species level coverage was 36.4% in GenBank and 27.1% in SILVA. To see whether rare species were underrepresented, we investigated the most commonly reported species as a separate dataset but found only minor coverage increases. We conclude that compared to global figures, barcode coverage is high for this area, but that a significant effort remains to fill barcode databases to levels that would make metabarcoding operational as a taxonomic tool, including for the most common macrofaunal taxa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Advances in molecular biology and computer technology have redefined the way most areas of natural sciences are carried out, but assessment of biodiversity has yet to take full advantage of these technology leaps (e.g., Taberlet et al. 2012; Bourlat et al. 2013). In particular, the practice of using estimates of biodiversity change to understand the extent of anthropogenic impact on the environment still routinely uses century-old sampling techniques. In the marine environment, most benthic community impact assessments are based on changes in the macrofauna assemblage sampled by replicate sediment grab samples sieved to retain fauna larger than 0.5 or 1 mm (Bean et al. 2017). This fauna is subsequently determined to lowest possible taxonomical level using morphological characters based on available taxonomic expertise and literature.
Valid and important reasons to continue with this method include the fact that the continuity of data enables tracking changes over time (Obst et al. 2018), but there are major challenges associated with this approach, including increasing lack of taxonomic expertise, lack of taxonomic literature for large areas of the world, subjective and inconsistent taxonomy, and cryptic diversity (Knowlton 1993; Schander and Willassen 2005; Ellingsen et al. 2017). Furthermore, macrofauna is only one part of a community also comprising microeukaryotes and other inconspicuous taxa, such that sampling only macrofauna lowers sensitivity to changes in the community and risks type II errors in impact assessments (Leray and Knowlton 2015; Lanzén et al. 2016). Finally, morphological taxonomy is very time consuming and thereby costly, restricting the possible sampling effort for any given study (Bean et al. 2017).
The implementation of metabarcoding in biodiversity studies—that is inferring community composition through high throughput amplicon sequencing—may be useful to overcome some of the limitations of existing methods (Taberlet et al. 2012; Bourlat et al. 2013). Metabarcoding can be applied to environmental DNA (eDNA) samples from water or sediment (Valentini et al. 2016; Sakata et al. 2020) or DNA extracts from a homogenate of the fauna present in a sample (Taberlet et al. 2012). The resulting barcode sequence reads can subsequently be matched with sequences assigned to taxon names accessed from databases such as NCBI GenBank (Sayers et al. 2020), the Barcode of Life Data System (BOLD) (Ratnasingham and Hebert 2007), or the ribosomal RNA SILVA database (Quast et al. 2012). The most common metazoan species barcode marker is the mitochondrial cytochrome c oxidase subunit I (COI) gene (Hebert et al. 2003). COI is sufficiently variable to distinguish the vast majority of metazoan taxa at the species level and offers the potential to discriminate among cryptic species and enhance diversity measures relative to morphology (Tang et al. 2012; Leray and Knowlton 2016; Sinniger et al. 2016). Due to the lack of truly universal primers, it is often paired with other markers in metabarcoding applications to account for taxonomic groups that do not amplify well such as the 18S rRNA gene (18S). While 18S tends to underestimate macrofaunal diversity relative to morphology due to its lower rate of evolution (Hartmann et al. 2010), conservative primer sites still make it a good candidate for universal eukaryote applications. Though not treated here, other metabarcoding complement or alternative markers have also been suggested, including the ribosomal 28S and mitochondrial 12S RNA genes (Machida and Knowlton 2012; Machida et al. 2012).
The use of metabarcoding is limited by the lack of reference sequences in the barcode repositories, reported at an average of 80% to 94% across metazoan groups (Kvist 2013). Many marine shelf, deep water, and polar areas are notoriously undersampled (McClain and Schlacher 2015). As an example, for abyssal plains (54% of Earth’s surface), a staggering 90% of the infauna species found in a typical survey are new to science (Ebbe et al. 2010). Recent work in the central Pacific Ocean—where DNA barcoding has not been implemented to any noteworthy degree—points to the significant effort needed to establish even a preliminary taxonomic baseline for DNA-based species identification (Dahlgren et al. 2016; Glover et al. 2016; Wiklund et al. 2017; Wiklund et al. 2019). In contrast to most deep ocean basins, shelf areas are better understood. In a recent study comparing morphology and COI metabarcoding in the Bay of Biscay, Aylagas et al. found a database coverage of 23% (2016), up from the global 6% as reported by Kvist (2013), but still severely limiting the translation to taxonomy and use of the metabarcode data in understanding biodiversity. It has been suggested that rare or otherwise peripheral taxa are dominating the fraction of species that have not yet been barcoded. The absence of these taxa in the reference databases would thus have a more limited impact on the results from metabarcoding studies than raw percentages would suggest (Hebert et al. 2016). To the extent that fungi are a fit object of comparison, such claims do not seem to hold true, however (Tedersoo et al. 2014, 2017).
Ongoing developments in DNA extraction, sequencing, and analytical methods coupled with an ever-growing body of pertinent scientific publications push for the introduction of metabarcoding to routine environmental monitoring programs (e.g., Bourlat et al. 2013; Aylagas et al. 2014; Bohmann et al. 2014; Pawlowski et al. 2014; Rees et al. 2014; Thomsen and Willerslev 2015; Lanzén et al. 2016; Valentini et al. 2016). While the taxonomic gaps in the global barcode repositories may appear overwhelming, assessments of regional coverage can help understand the progress of database coverage and to identify cases and situations where metabarcoding may be advantageous over other approaches. The North Sea is one of the most heavily sampled marine areas in the world (Hebert et al. 2016). The North and Norwegian Seas are regions where taxonomic work has been carried out for a long time, including 10–15 marine laboratories and another 10 or so in surrounding areas, some of which rank among the oldest in the world (Dean 1893; Lasserre et al. 1994). Dense spatial and temporal sampling of macrofauna due to oil and gas exploitation impact monitoring has also provided unique samples for taxonomic work (e.g., Petersen and George 1991). Though many new species are still found (e.g., Glover et al. 2005; Wiklund et al. 2009a; Strand et al. 2014; Dietrich et al. 2015), the marine macrofauna is well known and documented in published faunas relative to other areas.
The objective of this paper is to investigate the current state of publicly accessible barcode repositories, including GenBank, BOLD, and SILVA, for metabarcode taxon matching—the barcode repository gap—for the COI and 18S genes in marine benthic macrofauna taxa using a large dataset from a densely surveyed area. The North Sea is an area where major effort is applied to monitor environmental impact from the oil and gas extraction industry, and there is substantial stakeholder interest in the performance of metabarcoding technology. We hypothesize that the North Sea is a “best case scenario” in terms of both taxonomic baseline and macrofauna barcode coverage. We further assess this gap for each of the major invertebrate macrofauna phyla Echinodermata, Mollusca, Annelida, and Arthropoda and look at the most common taxa reported in the dataset to investigate the validity of the claim that rare taxa are overrepresented among species lacking barcodes.
Material and methods
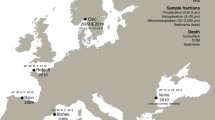
A dataset of macrofauna taxon names was compiled based on survey reports from monitoring Region IV (≈ 20,000 km2) in the northern part of the North Sea (Fig. 1) by downloading taxon lists from the Environmental Monitoring (MOD) database (DNV GL 2020), a central repository of monitoring data for the Norwegian offshore oil and gas industry. The dataset includes around 140 sampling stations (≈ 700 grab samples) at depths ranging between 127 and 385 m from the years 1996, 1999, 2002, 2005, 2008, 2011, and 2014. In accordance with Norwegian regulatory standards (Norwegian Environment Agency 2015), each station was sampled using 4–5 replicate van Veen grabs resulting in 0.5 m2 total sampled area, sieved through a 1 mm sieve. Morphological identifications were originally performed by environmental consultant companies, with most taxa assigned to macrofaunal phyla such as Annelida, Arthropoda, Echinodermata, and Mollusca. Meiofauna < 1 mm such as nematodes are not considered part of the dataset proper for the purposes of assessing biodiversity in these surveys and were reported at a low level or as presence/absence only. To assess the coverage of the most common taxa, a list of the ten most common reported taxa from each sampling station was also compiled as a separate dataset. Taxon names were updated and classified using the World Register of Marine Species (WoRMS) database (Horton et al. 2020). To mitigate differences in taxon names between WoRMS and the sequence databases, both names considered valid by WoRMS and originally reported names from the MOD database (where different) were used as synonyms for searches of the barcode repositories.

North Sea environmental survey region IV
Three public repositories—one general, one COI specific, and one 18S specific—were used to check taxon barcode coverage at species or higher taxonomic rank: NCBI GenBank, a general, lightly curated sequence database (Sayers et al. 2020); the Barcode of Life Data System (BOLD), a heavily curated COI specific sequence database (Ratnasingham and Hebert 2007); and SILVA SSU v138, a heavily curated, aligned database of small subunit (16S/18S) sequences (Quast et al. 2012). Available COI sequences were confirmed by taxon name batch searches of GenBank and BOLD, while 18S sequences were confirmed by taxon name batch searches of GenBank and the SILVA database. Due to the way searches were returned from BOLD, total taxon records (public and non-public) were recovered at all taxonomic levels, but public sequences were recovered for species level only. Online searches were carried out in January 2020.
Results
The original complete list of survey taxon names at all taxonomic levels comprised 1902 names of which 1568 were identified to species level. Taxa not identified to species level were assigned at various levels from phylum (e.g., “Nematoda” and “Sipuncula”) to genus level by the original taxonomists. The merged list of the ten most common taxa from each survey station comprised 240 taxon names (188 species level names). Validating and updating taxon names and classification in WoRMS reduced the number of valid taxa names to 1802 and valid species names to 1474 (Supplementary Table 1). For the ten most common taxa reported from each station, WoRMS indicated 236 valid taxon names including 184 taxa at the species level (Supplementary Table 2). The independent taxonomic curation of the databases, while lagging behind WoRMS, was found to be relatively updated, even for GenBank, with synonym hits being rare.
Of the 1802 taxa in the total dataset, 56.5% were represented by a publicly available COI barcode in GenBank at the same level as that reported in the morphological dataset. The corresponding figures for 18S were 45.5% for GenBank and 29.2% for SILVA. For the 1474 taxa at the species level, 50.4% were represented by a COI sequence in GenBank and 42.4% by a COI sequence in BOLD. Species coverage exclusive to GenBank was 11.2%, while 3.2% was exclusive to BOLD. Corresponding results for 18S were 36.4% for GenBank and 27.1% for SILVA. Reducing the sensitivity of the search to genus level increased coverage to 69.3% in GenBank for COI and 77.7% in GenBank and 56.2% in SILVA for 18S. Total taxon record coverage in BOLD (rather than publicly available sequences) was 70.1% at the species level and 89.5% at the genus level (Table 1).
For the 236 taxa comprising the ten most common species from each station, the same taxonomic level COI coverage was 61.0% in GenBank, while the 18S coverage was 52.1% in GenBank and 40.7% in SILVA. At species level, COI coverage was 52.2% in GenBank and 47.8% in BOLD. Fourteen COI sequences (7.6%) were only found in GenBank, while six (3.3%) were only found in BOLD. The 18S coverage was 41.3% in GenBank and 33.7% in SILVA. At the genus level, COI coverage in GenBank increased to 83.9% while 18S coverage increased to 77.9% in GenBank and 67.1% in SILVA. Total taxon record coverage in BOLD (rather than publicly available sequences) was 78.8% at the species level and 93.3% at the genus level (Table 2).
Divided by phylum for the most common macroinvertebrate phyla, coverage was roughly similar (45.0–51.1% at species level for the whole dataset in GenBank), except for Echinodermata, where coverage was substantially higher, with a COI coverage of 77% at species level for the whole dataset in GenBank. For the 18S gene, coverage between phyla was more uneven, with 10–20% lower coverage for Arthropoda compared to other phyla in the complete dataset (Tables 1 and 2).
Discussion
For taxonomy-based metabarcoding to be effective as a supplement to classic monitoring methods, open access barcode repositories that cover a significant fraction of the biota are a necessity. A global analysis of the paucity of metazoan COI barcodes in data repositories was recently estimated to 85% including both public and private entries (Kvist 2013), suggesting that a major effort is needed to fill this gap. In a study from the Philippines, only 715 species were identified as barcoded out of an estimated 50,000 species native to the islands (Fontanilla et al. 2014). Even a study on a more restricted dataset (n = 138) of marine benthic macrofauna at similar depths from the Bay of Biscay found a paucity of 77% (Aylagas et al. 2016). In contrast, our study, based on a dataset of 1474 benthic marine macrofauna species at shelf depth in the North Sea, shows that COI DNA barcodes were available for 50.4% of macrofauna species through GenBank and 42.4% through the publicly accessible part of the BOLD database. In other words, if a metabarcoding project managed to sample and successfully sequence the COI barcode gene from macrofauna species so far recorded in this region, around half could be determined to species level. This increased coverage highlights the critical importance of the cumulative effort of taxonomy and systematics research and well-funded barcode initiatives, including active Barcode of Life programs with marine components in surrounding countries.
The phylum with the highest COI coverage was Echinodermata, at 77.0% species level coverage. This is likely both a result of the taxon being represented by relatively few (n = 74) and large species that are easy to recognize and identify, but also due to an ongoing effort to barcode all Norwegian echinoderms as part of a broad initiative to map and barcode the entire Norwegian fauna (Bakken 2009). In contrast, the global number for echinoderm barcode database representation is only 5% (Kvist 2013). Sponges and benthic cnidarians have been identified as taxa where COI sequence data frequently fails to produce species-level resolution (Mcfadden et al. 2011; Vargas et al. 2012; Kvist 2013), but the low presence of these taxa in the North Sea dataset rules out any claim that this is a significant cause of the identified barcode gap.
While GenBank generally has higher COI coverage than BOLD, sequences are uncurated and lack morphological metadata, raising concerns regarding the accurate taxonomic assignment of sequences in this database and inflated diversity estimates for metabarcoding datasets. While substantial error rates have been shown for certain groups, such as a 20% species level misidentification of fungal sequences (Nilsson et al. 2006), metazoan sequences in particular seem to be surprisingly accurate, with a reported likely error rate less than 1% at genus level (Leray et al. 2019).
Total BOLD record coverage is significantly higher than BOLD records containing public barcodes: 15–40% higher for the different phyla in the complete list of species. While not all records signify the existence of actual sequence data for the recorded taxon, records usually indicate at least a barcoding attempt on the record in question. BOLD is a curated database with more stringent metadata and supplementary information requirements than, e.g., GenBank, meaning that records could represent all of taxa where barcoding was attempted but failed, taxa that are still being processed, and taxa that are withheld until after publication in a journal or project completion. In any case, the large discrepancy between reported taxon records and actually publicly available barcode sequences suggests that coverage of public sequences in BOLD will increase as more sequences are released into the public part of the database by data contributors.
The 18S gene evolves more slowly than mitochondrial COI and is therefore suitable to resolve higher rank relationships (Hillis and Dixon 1991) while COI saturates at family rank in most taxa (Wiklund et al. 2009b), and the 18S gene has been used to infer higher-rank diversity in metabarcoding projects using phylogenetic approaches (Lanzén et al. 2016; Fonseca et al. 2017). The 18S gene has also been extensively used to resolve higher metazoan phylogenies and is one of the most widely sequenced genetic markers among metazoans. This facilitates assignment of specimens belonging to taxonomic groups with poor COI coverage such as meiofauna (Kvist 2013).
At species level in our dataset, the 18S gene (36.4%) had lower GenBank coverage than COI (50.4%), with even lower coverage (27.1%) in SILVA. In contrast, GenBank 18S genus level coverage (77.7%) was actually higher than COI (69.3%) (SILVA: 56.2%). However, most available sequences only cover part of the approximately 1800 base pair length of the 18S gene, meaning that actual sequence searches will only return a portion of the sequences identified as 18S in the databases. Still, the ability to resolve higher level taxa makes the 18S marker even more valuable in cases with lower database coverage at low taxonomic level. Compared to GenBank, SILVA 18S coverage was around 10–20% lower, probably due to the more stringent requirements and additional curation of the SILVA database than of the more lightly curated GenBank.
It has been suggested that lack of barcode coverage could be partly alleviated if rare and peripheral species are overrepresented among species lacking barcodes (Hebert et al. 2016). The publicly available COI barcode coverage for the list of the most common species in our dataset was 52.2% in GenBank and 47.8% in BOLD. For 18S, the corresponding GenBank coverage was 41.3%. We also note that of the 27 arthropod species recorded in the list of the most common taxa, only 4 (14.7%) where represented with an 18S sequence in GenBank. Our results indicate that the coverage of the most common species is only slightly higher than for all reported species, and thus, we could not see that rare species were underrepresented in the barcode repositories for our data.
Conclusions
The general state of marine fauna barcode coverage is poor, and in this regard, the North Sea stands out in a positive way: roughly half of the 1474 marine macrofauna shelf species from the North Sea dataset analyzed in this study had a COI barcode in a public repository (42.4–50.4%). Missing barcodes were not limited to lesser known or rare taxa, however: barcode coverage for the most common species was only slightly higher than for all recorded species (47.8–52.2%). 18S coverage (27.1–36.4%), while lower than COI, was still substantial. 18S is able to resolve higher level taxon groups, and also targets the meio- and microeukaryote community, which makes it a good general complement to COI for metabarcoding studies.
While a substantial repository gap remains even in the North Sea, this gap is shrinking, and the release of currently non-public sequences, together with targeting common marine macrofaunal species for barcoding, could make taxonomic assignment of most North Sea macrofauna viable within the foreseeable future. Worldwide, this is an atypical situation, however: In most areas, being able to resolve marine macrofauna identity in metabarcoding applications is a more remote prospect.
References
Aylagas E, Borja A, Rodríguez-Ezpeleta N (2014) Environmental status assessment using DNA metabarcoding: towards a genetics based marine biotic index (gAMBI). PLoS One 9:e90529–e90511. https://doi.org/10.1371/journal.pone.0090529
Aylagas E, Borja A, Irigoien X, Rodríguez-Ezpeleta N (2016) Benchmarking DNA metabarcoding for biodiversity-based monitoring and assessment. Front Mar Sci 3:1809–1812. https://doi.org/10.3389/fmars.2016.00096
Bakken T (2009) Biodiversity-increasing knowledge. Fauna Norvegica 29:1–2
Bean TP, Greenwood N, Beckett R, Biermann L, Bignell JP, Brant JL, Copp GH, Devlin MJ, Dye S, Feist SW, Fernand L, Foden D, Hyder K, Jenkins CM, van der Kooij J, Kröger S, Kupschus S, Leech C, Leonard KS, Lynam CP, Lyons BP, Maes T, Nicolaus EEM, Malcolm SJ, McIlwaine P, Merchant ND, Paltriguera L, Pearce DJ, Pitois SG, Stebbing PD, Townhill B, Ware S, Williams O, Righton D (2017) A review of the tools used for marine monitoring in the UK: combining historic and contemporary methods with modeling and socioeconomics to fulfill legislative needs and scientific ambitions. Front Mar Sci 4:263. https://doi.org/10.3389/fmars.2017.00263
Bohmann K, Evans A, Gilbert MTP, Carvalho GR, Creer S, Knapp M, Yu DW, de Bruyn M (2014) Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol Evol 29:358–367. https://doi.org/10.1016/j.tree.2014.04.003
Bourlat SJ, Borja A, Gilbert J, Taylor MI, Davies N, Weisberg SB, Griffith JF, Lettieri T, Field D, Benzie J, Glöckner FO, Rodríguez-Ezpeleta N, Faith DP, Bean TP, Obst M (2013) Genomics in marine monitoring: new opportunities for assessing marine health status. Mar Pollut Bull 74:19–31. https://doi.org/10.1016/j.marpolbul.2013.05.042
Dahlgren TG, Wiklund H, Rabone M, Amon D, Ikebe C, Watling L, Smith C, Glover A (2016) Abyssal fauna of the UK-1 polymetallic nodule exploration area, Clarion-Clipperton Zone, central Pacific Ocean: Cnidaria. Biodivers Data J 4:e9277–e9231. https://doi.org/10.3897/BDJ.4.e9277
Dean B (1893) Notes on the marine biological laboratories of Europe. Am Nat 27:697–707. https://doi.org/10.1086/275775
Dietrich A, Hager T, Bönsch R, Winkelmann C, Schmidt A, Nygren A (2015) A new species of Myrianida (Autolytinae, Syllidae, Annelida) from the North Sea, with short notes on the distribution and habitat of Northeast Atlantic autolytines. Mar Biol Res 11:804–813. https://doi.org/10.1080/17451000.2015.1016971
DNV GL (2020) The Environmental Monitoring Database MOD. https://projects.dnv.com/MOD/MODKart/. Accessed 15 Feb 2020. Access for research granted after contacting DNV GL, OSLO.
Ebbe B, Billet DSM, Brandt A, Ellingsen K, Glover A, Keller S, Malyutina M, Martinez Arbizu P, Molodtsova T, Rex MA, Smith C, Tselepides A (2010) Chapter 8. Diversity of Abyssal Marine Life. Life in the Worlds Oceans. Humana Press, Totowa, pp 1–22
Ellingsen KE, Yoccoz NG, Tveraa T, Hewitt JE, Thrush SF (2017) Long-term environmental monitoring for assessment of change: measurement inconsistencies over time and potential solutions. Environ Monit Assess 189:595. https://doi.org/10.1007/s10661-017-6317-4
Fonseca VG, Sinniger F, Gaspar JM, Quince C, CREER S, Power DM, Peck LS, Clark MS (2017) Revealing higher than expected meiofaunal diversity in Antarctic sediments: a metabarcoding approach. Sci Rep 7:6094. https://doi.org/10.1038/s41598-017-06687-x
Fontanilla IKC, Torres AF, Canasa JAD, Yap SL, Ong PS (2014) State of animal DNA barcoding in the Philippines: a review of COI sequencing of Philippine native fauna. Phillippine Sci Lett 7:104–137
Glover AG, Källström B, Smith CR, Dahlgren TG (2005) World-wide whale worms? A new species of Osedax from the shallow north Atlantic. P R Soc B 272:2587–2592. https://doi.org/10.1098/rspb.2005.3275
Glover AG, Wiklund H, Rabone M, Amon D, Smith C, O'Hara T, Mah C, Dahlgren TG (2016) Abyssal fauna of the UK-1 polymetallic nodule exploration claim, Clarion-Clipperton Zone, central Pacific Ocean: Echinodermata. Biodivers Data J 4:e7251–e7248. https://doi.org/10.3897/BDJ.4.e7251
Hartmann M, Howes CG, Abarenkov K, Mohn WW, Nilsson RH (2010) V-Xtractor: an open-source, high-throughput software tool to identify and extract hypervariable regions of small subunit (16S/18S) ribosomal RNA gene sequences. J Microbiol Methods 83:250–253. https://doi.org/10.1016/j.mimet.2010.08.008
Hebert PDN, Ratnasingham S, deWaard JR (2003) Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc Lond B (Suppl) 270:S96–S99. https://doi.org/10.1098/rsbl.2003.0025
Hebert PDN, Hollingsworth PM, Hajibabaei M (2016) From writing to reading the encyclopedia of life. Philos Trans R Soc Lond B 371:20150321–20150329. https://doi.org/10.1098/rstb.2015.0321
Hillis DM, Dixon M (1991) Ribosomal DNA: molecular evolution and phylogenetic inference. Q Rev Biol 66:411–453. https://doi.org/10.1086/417338
Horton T et al. (2020) World register of marine species. Available from https://www.marinespecies.org at VLIZ. Accessed 2020-01-15. https://doi.org/10.14284/170
Knowlton N (1993) Sibling species in the sea. Annu Rev Ecol Syst 24:189–216
Kvist S (2013) Barcoding in the dark?: a critical view of the sufficiency of zoological DNA barcoding databases and a plea for broader integration of taxonomic knowledge. Mol Phylogenet Evol 69:39–45. https://doi.org/10.1016/j.ympev.2013.05.012
Lanzén A, Lekang K, Jonassen I, Thompson EM, Troedsson C (2016) High-throughput metabarcoding of eukaryotic diversity for environmental monitoring of offshore oil-drilling activities. Mol Ecol 25:4392–4406. https://doi.org/10.1111/mec.13761
Lasserre P, Mcintyre AD, Ogden JC, Ray GC, Grassle JF (1994) Marine laboratory networks for the study of the biodiversity, function and management of marine ecosystems. Biol Int 31:1–34
Leray M, Knowlton N (2015) DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. P Natl Acad Sci USA 112:2076–2081. https://doi.org/10.1073/pnas.1424997112
Leray M, Knowlton N (2016) Censusing marine eukaryotic diversity in the twenty-first century. Philos Trans R Soc Lond Ser B Biol Sci 371:20150331. https://doi.org/10.1098/rstb.2015.0331
Leray M, Knowlton N, Ho S-L, Nguyen BN, Machida RJ (2019) GenBank is a reliable resource for 21st century biodiversity research. Proc Natl Acad Sci 116(45):22651–22656. https://doi.org/10.1073/pnas.1911714116
Machida RJ, Knowlton N (2012) PCR primers for metazoan nuclear 18S and 28S ribosomal DNA sequences. PLoS One 7(9):e46180. https://doi.org/10.1371/journal.pone.0046180
Machida RJ, Kweskin M, Knowlton N (2012) PCR primers for metazoan mitochondrial 12S ribosomal DNA sequences. PLoS One 7(4):e35887. https://doi.org/10.1371/journal.pone.0035887
McClain CR, Schlacher TA (2015) On some hypotheses of diversity of animal life at great depths on the sea floor. Mar Ecol 36:849–872. https://doi.org/10.1111/maec.12288
Mcfadden CS, Benayahu Y, Pante E, Thoma JN, Nevarez PA, France SC (2011) Limitations of mitochondrial gene barcoding in Octocorallia. Mol Ecol Resour 11:19–31. https://doi.org/10.1111/j.1755-0998.2010.02875.x
Nilsson RH, Ryberg M, Kristiansson E, Abarenkov K, Larsson KH, Kõljalg U (2006) Taxonomic reliability of DNA sequences in public sequence databases: a fungal perspective. PLoS One 1(1):e59. https://doi.org/10.1371/journal.pone.0000059
Norwegian Environment Agency (2015) Environmental monitoring of petroleum activities on the Norwegian continental shelf. M-408
Obst M, Vicario S, Lundin K, Berggren M, Karlsson A, Haines R, Williams A, Goble C, Mathew C, Güntsch A (2018) Marine long-term biodiversity assessment suggests loss of rare species in the Skagerrak and Kattegat region. Mar Biodivers 48:2165–2176. https://doi.org/10.1007/s12526-017-0749-5
Pawlowski J, Esling P, Lejzerowicz F, Cedhagen T, Wilding TA (2014) Environmental monitoring through protist next-generation sequencing metabarcoding: assessing the impact of fish farming on benthic foraminifera communities. Mol Ecol Resour 14(6):1129–1140. https://doi.org/10.1111/1755-0998.12261
Petersen ME, George JD (1991) A new species of Raricirrus from northern Europe, with notes on its biology and a discussion of the affinities of the genus (Polychaeta: Ctenodrilidae). In Systematics, Biology and Morphology of World Polychaeta. Proceedings of the 2nd International Polychaete Conference, Copenhagen 1986 (pp. 185-208). Ophelia Publications
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. https://doi.org/10.1093/nar/gks1219
Ratnasingham S, Hebert PDN (2007) BOLD: the barcode of life data system (http://www.barcodinglife.org). Mol Ecol 7:355–364 doi:https://doi.org/10.1111/j.1471-8286.2006.01678.x
Rees HC, Maddison BC, Middleditch DJ, Patmore JRM, Gough KC (2014) The detection of aquatic animal species using environmental DNA - a review of eDNA as a survey tool in ecology. J Appl Ecol 51:1450–1459. https://doi.org/10.1111/1365-2664.12306
Sakata MK, Yamamoto S, Gotoh RO, Miya M, Yamanaka H, Minamoto T (2020) Sedimentary eDNA provides different information on timescale and fish species composition compared with aqueous eDNA. Environ DNA 66:081257–081214. https://doi.org/10.1002/edn3.75
Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch-Mizrachi I (2020) GenBank. Nucleic Acids Res 47:D94–D93. https://doi.org/10.1080/17451000510018962
Schander C, Willassen E (2005) What can biological barcoding do for marine biology? Mar Biol Res 1:79–83. https://doi.org/10.1080/17451000510018962
Sinniger F, Pawlowski J, Harii S, Gooday AJ, Yamamoto H, Chevaldonné P, Cedhagen T, Carvalho G, Creer S (2016) Worldwide analysis of sedimentary DNA reveals major gaps in taxonomic knowledge of deep-sea benthos. Front Mar Sci 3:92. https://doi.org/10.3389/fmars.2016.00092
Strand M, Herrera-Bachiller A, Nygren A, Kånneby T (2014) A new nemertean species: what are the useful characters for ribbon worm descriptions? JMBE 94(2):317–330. https://doi.org/10.1017/S002531541300146x
Taberlet P, Coissac E, Pompanon F, Brochmann C, Willerslev E (2012) Towards next-generation biodiversity assessment using DNA metabarcoding. Mol Ecol 21:2045–2050. https://doi.org/10.1111/j.1365-294X.2012.05470.x
Tang CQ, Leasi F, Obertegger U, Kieneke A, Barradough TG, Fontaneto D (2012) The widely used small subunit 18S rDNA molecule greatly underestimates true diversity in biodiversity surveys of the meiofauna. PNAS 109:16208–16212.
Tedersoo L, Bahram M, Põlme S, Kõljalg U, Yorou NS, Wijesundera R, Villarreal Ruiz L, Vasco-Palacios AM, Thu PQ, Suija A, Smith ME, Sharp C, Saluveer E, Saitta A, Rosas M, Riit T, Ratkowsky D, Pritsch K, Poldmaa K, Piepenbring M, Phosri C, Peterson M, Parts K, Paertel K, Otsing E, Nouhra E, Njouonkou AL, Nilsson RH, Morgado LN, Mayor J, May TW, Majuakim L, Lodge DJ, Lee SS, Larsson K-H, Kohout P, Hosaka K, Hiiesalu I, Henkel TW, Harend H, Guo L-D, Greslebin A, Grelet G, Geml J, Gates G, Dunstan W, Dunk C, Drenkhan R, Dearnaley J, De Kesel A, Dang T, Chen X, Buegger F, Brearley FQ, Bonito G, Anslan S, Abell S, Abarenkov K (2014) Global diversity and geography of soil fungi. Science 346:1078
Tedersoo L, Bahram M, Puusepp R, Nilsson RH, James TY (2017) Novel soil-inhabiting clades fill gaps in the fungal tree of life. Microbiome. https://doi.org/10.1186/s40168-017-0259-5
Thomsen PF, Willerslev E (2015) Environmental DNA–an emerging tool in conservation for monitoring past and present biodiversity. Biol Conserv 183:4–18. https://doi.org/10.1016/j.biocon.2014.11.019
Valentini A, Taberlet P, Miaud C, Civade R, Herder J, Thomsen PF, Bellemain E, Besnard A, Coissac E, Boyer F, Gaboriaud C, Jean P, Poulet N, Roset N, Copp GH, Geniez P, Pont D, Argillier C, Baudoin J-M, Peroux T, Crivelli AJ, Olivier A, Acqueberge M, Le Brun M, Møller PR, Willerslev E, Dejean T (2016) Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol Ecol 25:929–942. https://doi.org/10.1111/mec.13428
Vargas S, Schuster A, Sacher K, Büttner G, Schätzle S, Läuchli B, Hall K, Hooper JNA, Erpenbeck D, Wörheide G (2012) Barcoding sponges: an overview based on comprehensive sampling. PLoS One 7:e39345–e39347. https://doi.org/10.1371/journal.pone.0039345
Wiklund H, Glover AG, Dahlgren TG (2009a) Three new species of Ophryotrocha (Annelida: Dorvilleidae) from a whale-fall in the North-East Atlantic. Zootaxa 2228:43–56. https://doi.org/10.11646/zootaxa.2228.1.3
Wiklund H, Glover AG, Johannessen PJ, Dahlgren TG (2009b) Cryptic speciation at organic-rich marine habitats: a new bacteriovore annelid from whale-fall and fish farms in the North-East Atlantic. Zool J Linn Soc-Lond 155:774–785. https://doi.org/10.1111/j.1096-3642.2008.00469.x
Wiklund H, Taylor JD, Dahlgren TG, Todt C, Ikebe C, Rabone M, Glover AG (2017) Abyssal fauna of the UK-1 polymetallic nodule exploration area, Clarion-Clipperton Zone, central Pacific Ocean: Mollusca. ZooKeys 707:1–46. https://doi.org/10.3897/zookeys.707.13042
Wiklund H, Neal L, Glover AG, Drennan R, Rabone M, Drennan R, Dahlgren TG (2019) Abyssal fauna of polymetallic nodule exploration areas, eastern Clarion-Clipperton Zone, central Pacific Ocean: Annelida: Capitellidae, Opheliidae, Scalibregmatidae and Travisiidae. ZooKeys 883:1–82. https://doi.org/10.3897/zookeys.883.36193
Acknowledgments
The METAMON team, including Aud Larsen, Christofer Troedsson, Anders Lanzén, Eric Thompson, Katrine Sandnes Skaar, Christian Collin-Hansen, Juliette Diouma-Leyris, and Jessica Ray, is acknowledged for discussions regarding metabarcoding in marine benthic environments. The manuscript was improved by comments from the editor and three anonymous reviewers.
Funding
Open Access funding provided by NORCE Norwegian Research Centre AS. This work was funded by Statoil ASA (now Equinor ASA) as a delivery of the METAMON pre-project. Additional funding was received from the Norwegian Biodiversity Information Centre (Artsdatabanken).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The funders had no role in data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Sampling and field studies
All necessary permits for sampling and observational field studies have been obtained by the authors from the competent authorities and are mentioned in the acknowledgements, if applicable.
Data availability
All data used in this research is available in the associated supplementary files.
Author contribution
TGD and AGG conceived and designed research. JTH, EE, TGD and RHN conducted data analyses. POJ contributed morphology based taxon lists from the North Sea oil and gas fields. TGD, JHT and RHN wrote the manuscript. All authors read and approved the manuscript.
Additional information
Communicated by K. Kocot
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 2448 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hestetun, J.T., Bye-Ingebrigtsen, E., Nilsson, R.H. et al. Significant taxon sampling gaps in DNA databases limit the operational use of marine macrofauna metabarcoding. Mar. Biodivers. 50, 70 (2020). https://doi.org/10.1007/s12526-020-01093-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12526-020-01093-5