
Abstract
Background
Poly(A)-binding protein interacting protein 1 (PAIP1) is a translational initiation regulatory factor that has been reported as oncogene in multiple malignant diseases. However, its role in hepatocellular carcinoma (HCC) and the potential mechanisms have not been explored.
Methods
PAIP1 expression level in HCC cell lines were detected by real-time quantitative PCR and western blotting. The proliferation and colony formation of HCC cell lines were detected by MTT and colony formation assay. The apoptosis and cell cycle were detected by flow cytometry. The volume and growth rate of the xenograft tumors were observed. The potential mechanism of PAIP1 was analyzed by miRNA Microarray Analysis and TargetScan analysis.
Results
PAIP1 is significantly upregulated in HCC cell lines. PAIP1 knockdown dramatically inhibits cell proliferation and colony formation, induces apoptosis and alters the cell cycle distribution by increasing the G2/M cell percentage. Moreover, PAIP1 knockdown significantly reduces tumorigenesis in a murine transplantation model. Bioinformatics and immunoblotting analysis reveal that PAIP1 knockdown dysregulates cyclin D pathway-related proteins.
Conclusion
PAIP1 plays an oncogenic role in hepatocellular carcinoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Primary liver cancer is the sixth most common malignancy, with hepatocellular carcinoma (HCC) accounting for 80–90% of primary liver cancer cases [1, 2]. Notably, HCC is the second most common cause of cancer mortality following lung cancer [1, 2]. Moreover, researchers estimate an approximately 30% increase in HCC cases over the next decade in most regions worldwide. Unfortunately, 25–70% of HCC patients are diagnosed with incurable advanced-stage disease [1]. These patients have limited therapeutic options, as conventional chemotherapeutic regimens provide minimal clinical benefit [1].
To address this challenge, there is a substantial need for novel, effective molecular-based therapeutics against HCC. To the end, the carcinogenesis of HCC has been connected to various defects in the regulation of translational initiation [3,4,5]. During translational initiation, poly(A)-binding protein (PABP) and eukaryotic translational initiation factor 3 (eIF3) interact cooperatively with the mRNA’s 3’ poly(A) for ribosomal recruitment [6]. One key regulatory factor, PABP interacting protein 1 (PAIP1), directly interacts with PABP and eIF3 to enhance ribosomal recruitment by stabilizing interaction of PABP with eIF4G [6]. Recent work has revealed that oncogenic mTOR-S6K signaling promotes the pro-translational PAIP1-eIF3 interaction [7, 8], while the ubiquitin ligase WWP2 targets PAIP1 for proteasomal degradation, thereby suppressing translation [9]. These findings suggest that PAIP1 expression is necessary to maintain normal translational initiation but PAIP1 hyper-activation may contribute to carcinogenesis.
Of clinical relevance, PAIP1 has been implicated in several human malignancies, including bladder cancer, cervical cancer, and breast cancer [10,11,12,13,14]. Despite the fact that changes in PAIP1 expression have been implicated in several carcinomas, its exact role in human HCC remains unknown. In this study, we investigated the potential role of PAIP1 in human HCC based on in vitro and in vivo experiments. Furthermore, the mechanism of PAIP1 in HCC was explored.
2 Materials and methods
2.1 1Cell culture
The human HCC cell lines HEPG2, HUH-7, and HEP3B were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), and the HCC cell line SMMC-7721 and the normal human hepatocyte cell line L02 were obtained from the Cell Research Institute (Chinese Academy of Sciences, Shanghai, China). Each cell line was independently maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco) and 100 U/ml of penicillin–streptomycin (Gibco). Cultured cells were maintained at 37 °C in a humidified incubator with 5% CO2.
2.2 Lentiviral PAIP1 knockdown
Lentiviral PAIP1 knockdown in HCC cells was performed as previously described with minor modifications [15]. Small interfering RNAs (siRNAs) duplexes for PAIP1 were synthesized by Guangzhou Ribobio Co. (Guangzhou, China) as follows (where dT is deoxyribosylthymine): 5’-CAU GUC GGA CGG UUU CGA UdTdT-3’ and 5’-GCU GCA AAA GGG GAU GAA GdTdT-3’ [16]. The inverted 4E-T siRNA was employed as a negative control siRNA (NC-siRNA) [16]. The siRNA sequences were independently cloned into pLKO.3G containing an eGFP marker, a gift from Christophe Benoist & Diane Mathis (Addgene plasmid #14,748). Using Lipofectamine 2000 (Invitrogen), HEK293T cells were co-transfected with either pLKO.3G-PAIP1-siRNA or pLKO.3G-NC-siRNA along with the packaging plasmid psPAX2 and the envelope plasmid pMD2.G at a 4:3:2 ratio. The viral supernatants were harvested after 48 h, and viral titers were quantified. The lentiviral supernatants were then added to the target HCC cell lines at an MOI of 10 with Enhanced Infection Solution (ENi.S) and polybrene (6 μg/ml, Sigma). Lentiviral infection efficiency was assessed by GFP microscopy 24 h after infection, where we found a lentiviral infection rate exceeding 80% (Additional file 1: Fig. S1).
2.3 Real-time quantitative polymerase chain reaction (RT-PCR)
Total RNA was extracted from human HCC cell lines using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). To measure mRNA levels, total RNA was reverse transcribed into cDNA using the Sensiscript RT Kit (Qiagen) in accordance with the manufacturer’s instructions. RT-PCR was performed using SYBR Green PCR Master Mix (Roche Applied Science, Indianapolis, IN, USA) on an ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The primers for amplification of PAIP1 were as follows: (forward primer) 5’-TCA GGA TCC GCT AAG CCC CAG GTG GTT-3’ and (reverse primer) 5’-GGC TCG AGT TAC TGT TTT CGC TTA CG-3’ [16]. The expression level of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the endogenous control for sample normalization (forward primer: 5’-CCA CCC ATG GCA AAT TCC ATG GCA-3’; reverse primer, 5’-TCT AGA CGG CAG GTC AGG TCC ACC-3’). Standard thermocycler conditions were employed for data collection, and fold changes were calculated by means of relative quantification (2−ΔΔCT) as previously described by Livak et al. [17].
2.4 Western blotting
Cell lysis was performed using a lysis buffer (Sigma, St. Louis, MO, USA) with a protease inhibitor (Promega, Madison, WI, USA). Cell lysates containing equivalent protein amounts were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE, 10%) and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) via a Mini TransBlot system (Bio-Rad, Hercules, CA, USA). The blots were blocked for 1 h in 5% milk prepared in TBST, and blocked membranes were subsequently incubated at 4 °C overnight with the following antibodies (Cell Signaling Technology, Danvers, MA, USA unless otherwise specified): anti-PAIP1 (diluted 1:5000; cat#ab175211, Abcam, Cambridge, UK), anti-cyclin-dependent kinase 6 (CDK6; diluted 1:200, cat#3136), anti-cyclin D1 (CCND1; diluted 1:500, cat#2978), anti-cyclin D2 (CCND2; diluted 1:500, cat#3741), anti-mouse double minute 2 homolog (MDM2; diluted 1:200, cat#86,934), and anti-GAPDH (diluted 1:1000, cat#2118). The next day, membranes washed and incubated with species-specific secondary antibodies conjugated to horseradish peroxidase for 1 h at room temperature (diluted 1:2000), followed by several washes with TBST to remove unbound antibody. Finally, enhanced chemiluminescence (Pierce, Rockford, IL, USA) was used to visualize the protein expression levels in accordance with the manufacturer's instructions. GAPDH was used as the internal control. Densitometric analysis was performed with Image-J (NIH, Bethesda, MD, USA).
2.5 Cell proliferation assays
For the Cellomics assay, cells were cultured in a 96-well plate for 24 h after lentiviral infection prior to experimentation. Green fluorescence protein (GFP)-expressing cells were quantified once daily for five days using a Cellomics ArrayScan system (Thermo Scientific, Waltham, MA, USA). For the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay, cells were cultured in a 96-well plate for 24 h after lentiviral infection prior to experimentation. At the indicated time points, medium was replaced with fresh medium supplemented with MTT (0.5 mg/ml) (Sigma). Absorbance was measured at a wavelength of 490 nm (OD490).
2.6 Apoptosis assay
Apoptosis was examined using an Apoptosis Detection kit that specifically comprised a FITC-labeled Annexin V/propidium iodide (PI) method (Invitrogen). In brief, cells were infected with the PAIP1-siRNA or the NC-siRNA and cultured for 48 h. Then cells were collected and washed with cold PBS followed by stained with dye buffer containing 10 μl of FITC-A and 10 μl of PI for 15 min at room temperature. Then the stained cells were analyzed by a FACSCalibur (BD Biosciences, San Jose, CA, USA) and analyzed using FLOWJO software (Tree Star, Ashland, OR, USA).
2.7 Colony formation assay
Cells were infected with the PAIP1-siRNA or the NC-siRNA and cultured for 24 h. The cells were then collected and plated in six-well plates at a density of 5 × 102 cells per well. After 10 days, cells were washed with phosphate-buffered saline, followed by fixation with methanol/acetic acid (3:1; v:v), and finally stained with Giemsa [18]. The number of colonies were counted under a microscope [19].
2.8 Cell cycle analysis
For cell cycle analysis, cells were first resuspended in ice-cold phosphate buffered saline. To measure DNA content, cells were stained with PI solution (50 µg/ml PI, 100 µg/ml RNase A, 0.05% Triton X-100 in NaCl/Pi) and then incubated in the dark at 37 °C for 30 min. DNA content was observed by flow cytometry using a FACSCalibur (BD Biosciences, San Jose, CA, USA) and analyzed using FLOWJO software (Tree Star, Ashland, OR, USA).
2.9 Subcutaneous xenograft tumor model
Female BALB/c nude mice (4–6 weeks of age) were purchased from Shanghai SLAC Laboratory Animal Co. (Shanghai, China). PAIP-siRNA-infected SMMC-7721 cells (5 × 106; KD group) or NC-siRNA-infected SMMC-7721 cells (5 × 106; NC group) were injected subcutaneously (s.c.) into nude mice via their dorsal flanks. Each group contained 10 mice. Tumor volumes (V) were measured daily by calipers and were calculated by the following formula: V = 1/2 × S2 × L, where S and L are the shortest and longest diameter of the tumor, respectively. Mice were sacrificed four weeks after injection, and final tumor weights were measured.
2.10 miRNA microarray analysis
Total RNA was extracted from the foregoing SMMC-7721 xenograft tumors using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). RNA (2 µg) was labelled and hybridized to a Genechip miRNA v.2.0 microarray (Affymetrix, CA, USA). Then, the raw data were quartile-normalized with the miRNAQC software (Affymetrix) prior to exporting the intensity data into Genespring 7.2 software (Agilent, CA, USA). In order to perform the unsupervised cluster analysis according to the Pearson correlation algorithm, the non-hybridized probes were removed by filtering probes with the flag = “TRUE” function. Thereafter, an analysis of variance (ANOVA) was applied to identify differentially-expressed mature human miRNAs between the NC and KD samples. P-values were then corrected with the Benjamini-Hogberg method. Processing of each sample was repeated in triplicate.
2.11 KEGG pathway analysis
TargetScan software (http://www.targetscan.org) was then applied to predict putative target genes for the differentially-expressed mature human miRNAs between the NC and KD samples. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database was then used to identify the significantly-enriched biological pathways for these putative target genes. P-values (via two-sided Fisher’s exact testing) and q-values were calculated to select significantly-enriched pathways and control for the false discovery rate (FDR), respectively [20].
2.12 Statistical analysis
Data are expressed as mean ± standard deviation (SD). Differences between groups were analyzed using either the two-tailed paired or unpaired Student’s t-test. A P-value of less than 0.05 was considered statistically significant.
3 Results
3.1 PAIP1 expression was upregulated in human HCC cell lines
First, PAIP1 mRNA expression levels were analyzed in four human HCC cell lines (SMMC-7721, HEPG2, HUH-7, HEP3B) relative to the normal human hepatocyte cell line L02. PAIP1 transcript levels were upregulated in all four HCC cell lines, and the relative PAIP1 transcript levels were similar in all four HCC cell lines (P < 0.01, Fig. 1A). Then, PAIP1 protein expression levels were analyzed in the human HCC cell lines relative to the normal human hepatocyte cell line L02. PAIP1 protein levels were upregulated in all four HCC cell lines, and the relative PAIP1 protein levels were similar in all four HCC cell lines (P < 0.01, Fig. 1B).

PAIP1 Upregulation in HCC Cell Lines and PAIP1 Knockdown by siRNA. A Real-time polymerase chain reaction (RT-PCR) and B Western blotting were used to determine PAIP1 expression in the four HCC cell lines relative to that of the normal hepatocyte cell line L02. C RT-PCR and D Western blotting validated siRNA-mediated PAIP1 knockdown in the SMMC-7721 cell line. The HEPG2 cell line displayed similar siRNA-mediated PAIP1 knockdown (data not shown). GAPDH served as the loading control in all experiments. Results are displayed as means and standard deviations (SDs) from three independent experiments. **P < 0.01
3.2 Efficient paip1 knockdown by lentivirally-delivered siRNA
The SMMC-7721 and HEPG2 cell lines were infected with lentivirus carrying a PAIP1-siRNA or a negative control-siRNA (NC-siRNA). Lentiviral infection efficiency was assessed by GFP expression 24 h after infection, where we found a lentiviral infection rate exceeding 80% (Additional file 1: Fig. S1). PAIP1 mRNA expression was significantly decreased after siRNA-mediated PAIP1 knockdown (P < 0.01, Fig. 1C). Accordingly, Western blotting confirmed that PAIP1 protein expression levels were notably decreased following siRNA-mediated PAIP1 knockdown (Fig. 1D).
3.3 PAIP1 knockdown inhibited HCC cell proliferation
In Cellomics assay, PAIP1 knockdown significantly suppressed cell proliferation in SMMC-7721 cells, as demonstrated by decreased GFP + cell counts over a five-day time course (P < 0.01, Fig. 2A). Specifically, cell counts decreased by 43% and 46% in the PAIP1-siRNA cells on days four and five, respectively (Fig. 2A). Similarly, PAIP1 knockdown significantly suppressed cell proliferation in HEPG2 cells (P < 0.01, Fig. 2B). Specifically, cell counts decreased by 44% and 45% in the PAIP1-siRNA cells on days four and five, respectively (Fig. 2B).

Effect of PAIP1 Knockdown on HCC Cell Proliferation. A, B Cellomics experiments for A SMMC-7721 cell proliferation and B HEPG2 cell proliferation. Representative images of green fluorescence protein (GFP)-expressing cells over time (left panels, image magnification: 10 ×). Proliferation curves (right panels) were constructed based on GFP expression. The cell count/fold represents the fold-change of the cell count for each respective day normalized to the original cell count on day one. C, D MTT experiments for C SMMC-7721 cell proliferation and D HEPG2 cell proliferation. OD490/fold represents the fold-change of the OD490 value for each respective day normalized to the original OD490 value on day one. Results are displayed as means and standard deviations (SDs) from three independent experiments. *P < 0.05, **P < 0.01
In the MTT assay, PAIP1-siRNA SMMC-7721 cells showed significantly less proliferation over the five-day time course relative to their control counterparts (P < 0.01, Fig. 2C). Specifically, cell absorbance decreased by 24% and 26% in the PAIP1-siRNA cells on days four and five, respectively (Fig. 2C). Similarly, PAIP1-siRNA HEPG2 cells showed significantly less proliferation over the five-day time course relative to their control counterparts (P < 0.05, Fig. 2D). Specifically, cell absorbance decreased by 11% and 11% in the PAIP1-siRNA cells on days four and five, respectively (Fig. 2D).
3.4 PAIP1 knockdown increased HCC cell apoptosis and reduced colony formation
PAIP1 knockdown in SMMC-7221 cells induced a 95% increase in apoptosis rate (P < 0.01, Fig. 3A). In addition, PAIP1 knockdown in HEPG2 cells induced a 54% increase in apoptosis rate (P < 0.01, Fig. 3B).

Effect of PAIP1 Knockdown on HCC Cell Apoptosis and Colony Formation. A, B Annexin V/PI-FACS analysis of apoptosis levels in A SMMC-7721 and B HEPG2 cells. C, D Assessment of colony formation ability in C SMMC-7721 and D HEPG2 cells. Cells were plated in a six-well plate (500 cells/well) for a period of 10 days. Clones were counted after staining with Giemsa. Results are displayed as means and standard deviations (SDs) from three independent experiments. *P < 0.05, **P < 0.01
Then, PAIP1 knockdown resulted in a 52% decrease in clone formation rates in SMMC-7221 cells (P < 0.01, Fig. 3C). In addition, PAIP1 knockdown resulted in a 68% decrease in clone formation rates in HEPG2 cells (P < 0.01, Fig. 3D).
3.5 PAIP1 knockdown induced G2/M phase shift in HCC cells
To investigate whether PAIP1 silencing leads to cell cycle defects, SMMC-7721 and HEPG2 cells lentivirally-infected with either PAIP1-siRNA or NC-siRNA were subjected to cell cycle analysis by staining with PI and subsequent FACS analysis. PAIP1 knockdown resulted in a 45% increase in the G2/M phase cell percentage in SMMC-7221 cells (P < 0.01, Fig. 4A). In addition, PAIP1 knockdown resulted in a 90% increase in the G2/M phase cell percentage in HEPG2 cells (P < 0.01, Fig. 4B).

Effect of PAIP1 Knockdown on HCC Cell Cycle Progression. PI-FACS analysis of cell cycle progression in A SMMC-7721 and B HEPG2 cells. Results are displayed as means and standard deviations (SDs) from three independent experiments. *P < 0.05, **P < 0.01
3.6 PAIP1 knockdown suppressed SMMC-7721 xenograft tumor growth in nude mice
SMMC-7721 cells with/without PAIP1 knockdown were s.c. injected into the dorsal flank of nude mice (Fig. 5A). Remarkably, the average xenograft tumor volume was strikingly reduced by 97% in the KD group on day 17 post-inoculation (P < 0.01, Fig. 5B). At 28 days post-inoculation, KD mice carried tumors that were 97% less massive than their NC counterparts (P < 0.01, Fig. 5C).

Effect of PAIP1 Knockdown on In Vivo SMMC-7721 Xenograft Tumorigenicity. A SMMC-7721 cells were subcutaneously (s.c.) injected into the dorsal flanks of nude mice. B Graph of xenograft tumor volumes over the initial 17-day period. C Final tumor weights were measured 28 days after s.c. injection. NC negative control tumor group, KD PAIP1-knockdown tumor group. *P < 0.05, **P < 0.01 means significant difference between NC and KD group from the beginning day
3.7 PAIP1 knockdown induced cyclin D pathway dysregulation
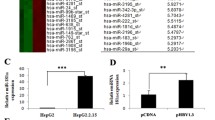
miRNAs regulate their target genes by decreasing their mRNA levels [21]. Thus, to better understand the molecular mechanism(s) underlying PAIP1’s effects in HCC cells, we examined the expression of differentially-expressed mature human miRNAs between the NC and KD samples using a miRNA microarray analysis. Cluster analysis was able to clearly distinguish the miRNA microarray profiles of NC and KD tumors (Fig. 6A).

Cyclin D Pathway Dysregulation Induced by PAIP1 Knockdown. A Cluster analysis of miRNA expression in PAIP1-knockdown (KD) and negative control (NC) SMMC-7721 xenograft tumor cells. Each row represents a unique miRNA, and each column represents an individual tumor sample (n = 3 tumors in each group). Red or green coloring represents elevated or decreased expression, respectively. B Results of the KEGG pathway analysis showing the top ten highest-ranking KEGG pathways. The ‘focal adhesion’ and ‘pathways in cancer’ pathways have been circled in red. C Representative Western blots of four key cyclin D pathway genes. GAPDH served as loading control. D The relative expression of each protein is graphically displayed. Results are displayed as means and standard deviations (SDs) from three independent experiments. *P < 0.05, **P < 0.01
TargetScan analysis was then applied to predict putative target genes for the differentially-expressed mature human miRNAs between the NC and KD tumors. We then analyzed the enrichment of these putative target genes in the KEGG pathway database. The resulting KEGG pathway analysis produced several noteworthy biological pathways that have been previously associated with oncogenesis, including ‘focal adhesion’ and ‘pathways in cancer’ (Fig. 6B) [22,23,24].
Based upon the TargetScan analysis and the significant dysregulation in the KEGG ‘focal adhesion’ and ‘pathways in cancer’ pathways, we identified the focal adhesion/cancer-associated cyclin D pathway as a putative target pathway of PAIP1 dysregulation in HCC cells [25,26,27]. On this basis, we sought to validate whether PAIP1 knockdown significantly affects the expression of key cyclin D pathway proteins (i.e., CDK6, CCND1, CCND2, and MDM2). Applying Western blotting analysis on xenograft tumors (Fig. 6C), we demonstrated that the expression of CDK6 – a direct target of one of the differentially-expressed miRNAs – is significantly upregulated in KD tumors (P < 0.01, Fig. 6D). On the other hand, we observed significant downregulation in CCND1, CCND2, and MDM2 in KD tumors (P < 0.01, Fig. 6D).
4 Discussion
Here, we identified PAIP1 as an oncogene in HCC cells and provided a basis for the potential application of PAIP1 in targeted–therapy of cancer. Our initial experiments revealed significantly elevated expression of PAIP1 in four HCC cell lines. Furthermore, our results displayed that PAIP1 knockdown inhibits proliferation, induces apoptosis, reduces colony formation, and promotes G2/M phase shift in these HCC cell lines. But PAIP1 knockdown caused S phase arrest in SMMC7721 cells but not in HepG2 cells. This might be due to the difference between these two cell lines. Cell cycle distribution was not the major target of PAIP1 in HepG2 cells. Furthermore, depletion of PAIP1 expression suppressed SMMC-7721 xenograft tumorigenesis in a nude mouse model. Bioinformatics analysis and immunoblotting on SMMC-7721 xenograft tumor cells revealed that PAIP1 knockdown dysregulates several cyclin D pathway proteins. These findings suggest that PAIP1 may be a critical oncogene in HCC.
MiRNA microarray was a common method to explore the mechanism in cancer. mRNAs are generally the target of miRNAs, so miRNA array could reflect the expression pattern of mRNAs. Even, miRNA assay might give us more information than mRNA array. Although more research is required to reveal the underlying mechanism of PAIP1 in tumorigenesis, there is a substantial body of evidence linking dysregulations in PAIP1-associated translational initiation factors with oncogenesis [28]. Kratze et al. has reported that many malignancies display hyper-activation of eIF4 proteins (i.e., eIF4A, eIF4E, eIF4G, and 4E-BP proteins), resulting in enhanced binding to the 5’ cap and preferential translation of cancer-associated genes, including cyclin D1, c-myc, and ornithine decarboxylase [29]. As PAIP1 stabilizes PABP’s interaction with eIF4G [6], the PAIP1 upregulation in HCC cells observed here may promote the translation of oncogenic mRNAs by promoting eIF4G hyper-activation. In miRNA microarray assay, we identified the cyclin D pathway as a putative target pathway of PAIP1 dysregulation in HCC cells [25,26,27]. We then demonstrated that PAIP1 significantly affects the expression of four cyclin D pathway genes (namely, CDK6, CCND1, CCND2, and MDM2).
Most notably, we found that CCND1 and CCND2 were significantly downregulated upon PAIP1 knockdown in SMMC-7721 xenograft tumor cells, suggesting that PAIP1 promotes CCND1, CCND2, and MDM2 expression in HCC cells. The cyclin D proteins CCND1 and CCND2 have been positively linked to HCC oncogenesis [30,31,32]. Mechanistically, CCND1 and CCND2 interact with CDK6 and CDK4, forming the cyclin D/CDK6 and cyclin D/CDK4 complexes. These complexes hyper-phosphorylate retinoblastoma (Rb), thereby facilitating the G1-S phase transition [33]. Thus, many cancer cells depend on elevated cyclin D/CDK activity to transition through this regulatory checkpoint [33]. Consistently,l PAIP1 knockdown downregulates CCND1 and CCND2 expression in HCC cells, thereby restricting cyclin D/CDK activity and producing the observed anti-malignant effects. Based on Kratze et al.’s proposed model [29], we speculate that PAIP1 expression in HCC cells promotes eIF4G hyper-activation, thereby promoting preferential translation of CCND1 and CCND2 transcripts.
Interestingly, we also found that CDK6 was significantly upregulated upon PAIP1 knockdown in SMMC-7721 xenograft tumor cells, suggesting that PAIP1 negatively regulates CDK6 expression in HCC cells. In this regard, PAIP1 surprisingly behaves as a tumor suppressor. PAIP1’s target PABP has been shown to promote the formation and/or stabilization of nuclear TAR-DNA binding protein of 43 kDa (TDP-43), a negative regulator of CDK6 and its downstream targets Rb and Rb2/p130 [34, 35]. Therefore, PAIP1 may promote PABP’s interaction with TDP-43, thereby negatively regulating the oncogenic CDK6-Rb pathway.
Then, we found that MDM2 was significantly downregulated upon PAIP1 knockdown in SMMC-7721 xenograft tumor cells, suggesting that PAIP1 promotes MDM2 expression in HCC cells. MDM2 functions as a negative regulator of the tumor suppressor p53 and may be responsible for inactivation of p53 in HCC tumors [36]. Mechanistically, MDM2 catalyzes p53 ubiquitination, targeting the p53 protein for degradation [37]. Normal p53 activity reroutes cells that have undergone oncogenic damage to G1 phase arrest via inhibiting cyclin D/CDK4 [37]. Disruption of p53 activity by MDM2 eliminates p53-mediated inhibition of cyclin D/CDK4, enabling oncogenes to drive uncontrolled cell proliferation [37]. Consistently, PAIP1 knockdown downregulates MDM2 expression in HCC cells, thereby maintaining p53 activity and producing the observed anti-malignant effects. However, much more experiments are necessary to further support the foundation in this study. For example, in miRNA microarray assay, cytokine-cytokine receptor interaction was another signaling pathway dominantly affected by PAIP1. The interaction of cytokines with their receptor are important in processes such as s cell proliferation, migration, EMT, and so on. In addition, the data suggested that PAIP1 was an oncogene in liver cancer. Then, if PAIP1 could be considered as a potential drug target? What’s role of PAIP1 in chemotherapy of patients with liver cancer? These questions will be a critical issue in our following study.
In summary, the present study showed that PAIP1 plays an oncogene role in the tumorigenesis of HCC, and its oncogenic effects may interact with cyclin D signaling pathway. Our results provide informational support for using PAIP1 as potential therapeutic target for HCC therapy.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Park JW, Chen M, Colombo M, Roberts LR, Schwartz M, Chen PJ, et al. Global patterns of hepatocellular carcinoma management from diagnosis to death: the BRIDGE Study. Liver Int. 2015;35:2155–66.
McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis. 2015;19:223–38.
Zhu Q, Qiao G, Zeng X, Li Y, Yan J, Duan R, et al. Elevated expression of eukaryotic translation initiation factor 3H is associated with proliferation, invasion and tumorigenicity in human hepatocellular carcinoma. Oncotarget. 2016;7:49888–901.
Liu R, Lv Y, Tang Y, Wang Y, Chen X, Zheng X, et al. Eukaryotic translation initiation factor 5A2 regulates the migration and invasion of hepatocellular carcinoma cells via pathways involving reactive oxygen species. Oncotarget. 2016;7:24348–60.
Wang Z, Zheng H, Gao W, Han J, Cao J, Yang Y, et al. eIF5B increases ASAP1 expression to promote HCC proliferation and invasion. Oncotarget. 2016;7:62327–39.
Thakor N, Smith MD, Roberts L, Faye MD, Patel H, Wieden H-J, et al. Cellular mRNA recruits the ribosome via eIF3-PABP bridge to initiate internal translation. RNA Biol. 2017;14:553–67.
Tavares MR, Pavan IC, Amaral CL, Meneguello L, Luchessi AD, Simabuco FM. The S6K protein family in health and disease. Life Sci. 2015;131:1–10.
Martineau Y, Wang X, Alain T, Petroulakis E, Shahbazian D, Fabre B, et al. Control of Paip1-eukayrotic translation initiation factor 3 interaction by amino acids through S6 kinase. Mol Cell Biol. 2014;34:1046–53.
Lv Y, Zhang K, Gao H. Paip1, an effective stimulator of translation initiation, Is targeted by WWP2 for ubiquitination and degradation. Mol Cell Biol. 2014;34:4513–22.
Scotto L, Narayan G, Nandula SV, Subramaniyam S, Kaufmann AM, Wright JD, et al. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol Cancer. 2008;7:58.
Narayan G, Bourdon V, Chaganti S, Arias-Pulido H, Nandula SV, Rao PH, et al. Gene dosage alterations revealed by cDNA microarray analysis in cervical cancer: identification of candidate amplified and overexpressed genes. Genes Chromosomes Cancer. 2007;46:373–84.
Network CGAR. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22.
Payton S. Bladder cancer: Genetic studies mark change in fortune for bladder cancer. Nat Rev Urol. 2014;11:123.
Piao J, Chen L, Jin T, Xu M, Quan C, Lin Z. Paip1 affects breast cancer cell growth and represents a novel prognostic biomarker. Hum Pathol. 2017;73:33–40.
Shen J, Lin H, Li G, Jin R-A, Shi L, Chen M, et al. TR4 nuclear receptor enhances the cisplatin chemo-sensitivity via altering the ATF3 expression to better suppress HCC cell growth. Oncotarget. 2016;7:32088–99.
Martineau Y, Derry MC, Wang X, Yanagiya A, Berlanga JJ, Shyu A-B, et al. Poly (A)-binding protein-interacting protein 1 binds to eukaryotic translation initiation factor 3 to stimulate translation. Mol Cell Biol. 2008;28:6658–67.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001;25:402–8.
Musumeci G. Past, present and future: overview on histology and histopathology. J Histol Histopathol. 2014;1:5.
Lu J, He M-L, Wang L, Chen Y, Liu X, Dong Q, et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 2011;71:225–33.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300.
Yan X-L, Fu C-J, Chen L, Qin J-H, Zeng Q, Yuan H-F, et al. Mesenchymal stem cells from primary breast cancer tissue promote cancer proliferation and enhance mammosphere formation partially via EGF/EGFR/Akt pathway. Breast Cancer Res Treat. 2012;132:153–64.
Liu X, He M, Hou Y, Liang B, Zhao L, Ma S, et al. Expression profiles of microRNAs and their target genes in papillary thyroid carcinoma. Oncol Rep. 2013;29:1415–20.
Van der Auwera I, Yu W, Suo L, Van Neste L, Van Dam P, Van Marck EA, et al. Array-based DNA methylation profiling for breast cancer subtype discrimination. PLoS ONE. 2010;5: e12616.
Li T, Wan B, Huang J, Zhang X. Comparison of gene expression in hepatocellular carcinoma, liver development, and liver regeneration. Mol Genet Genomics. 2010;283:485–92.
Yang J, Wang N. Analysis of the molecular mechanism of osteosarcoma using a bioinformatics approach. Oncol Lett. 2016;12:3075–80.
Wang D, Zhang Q, Jin Y. miRNAs associated with the malignant transformation of non-small cell lung cancer. Acta biochim Biophys Sin. 2016;48:395–8.
Kim HS, Kim SC, Kim SJ, Park CH, Jeung H-C, Kim YB, et al. Identification of a radiosensitivity signature using integrative metaanalysis of published microarray data for NCI-60 cancer cells. BMC Genomics. 2012;13:1.
Loreni F, Mancino M, Biffo S. Translation factors and ribosomal proteins control tumor onset and progression: how? Oncogene. 2014;33:2145–56.
Kratzke RA. Targeting eukaryotic protein translation in mesothelioma. Transl Lung Cancer Res. 2017;6:343–9.
Zeng Z, Tu J, Cheng J, Yao M, Wu Y, Huang X, et al. Influence of CCND1 G870A polymorphism on the risk of HBV-related HCC and cyclin D1 splicing variant expression in Chinese population. Tumor Biol. 2015;36:6891–900.
Andrisani OM, Studach L, Merle P. Gene signatures in hepatocellular carcinoma (HCC). Semin Cancer Biol. 2011;21:4–9.
Tomoda T, Nouso K, Sakai A, Ouchida M, Kobayashi S, Miyahara K, et al. Genetic risk of hepatocellular carcinoma in patients with hepatitis C virus: a case control study. J Gastroenterol Hepatol. 2012;27:797–804.
Geleta B, Makonnen E, Abay S. Cyclic dependent kinase (CDK): role in cancer pathogenesis and as drug target in cancer therapeutics. J Cancer Sci Ther. 2016;8:160–7.
McGurk L, Virginia ML, Trojanowksi JQ, Van Deerlin VM, Lee EB, Bonini NM. Poly-A binding protein-1 localization to a subset of TAR DNA-binding protein of 43 kDa inclusions in amyotrophic lateral sclerosis occurs more frequently in patients harboring an expansion in C9orf72. J Neuropath Exp Neur. 2014;73:837–45.
Ayala YM, Misteli T, Baralle FE. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci U S A. 2008;105:3785–9.
Qiu S-J, Ye S-L, Wu Z-Q, Tang Z-Y, Liu Y-K. The expression of the mdm2 gene may be related to the aberration of the p53 gene in human hepatocellular carcinoma. J Cancer Res Clin Oncol. 1998;124:253–8.
Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12.
Acknowledgements
We thank the colleagues from Meijiayouke Biomed Inc for English editing and grammar correction.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81760427).
Author information
Authors and Affiliations
Contributions
NZ and XC conceived and designed this study. NZ performed the experiments, analyzed the data and drafted the manuscript. NZ revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The experimental protocols were approved by the Ethics Committee of the Second Affiliated Hospital of Nanchang University (Reference 20210201) and all experiments were performed in accordance with the 1964 Helsinki Declaration and its later amendments. This paper has not been published elsewhere in whole or in part. All authors have read and approved the content, and agree to submit it for consideration for publication in your journal. There are no ethical/legal conflicts involved in the article.
Competing interests
The authors have declared no conflicts of interest in this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Lentiviral Infection Efficiency in SMMC-7721 Cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, N., Chen, X. PAIP1 is a novel oncogene in human hepatocellular carcinoma. Discov Onc 13, 132 (2022). https://doi.org/10.1007/s12672-022-00530-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-022-00530-0